

Abstract
Background
In fleshy fruit, abscission of fully ripe fruit is a process intimately linked to the ripening process. In many fruit-tree species, such as olive (Olea europaea L. cv. Picual), there is a coupling of the full ripening and the activation of the abscission-zone (AZ). Although fully ripe fruit have marked physiological differences with respect to their AZs, dissimilarities in gene expression have not been thoroughly investigated. The present study examines the transcriptome of olive fruit and their AZ tissues at the last stage of ripening, monitored using mRNA-Seq.
Results
Roche-454 massive parallel pyrosequencing enabled us to generate 397,457 high-quality EST sequences, among which 199,075 were from ripe-fruit pericarp and 198,382 from AZ tissues. We assembled these sequences into 19,062 contigs, grouped as 17,048 isotigs. Using the read amounts for each annotated isotig (from a total of 15,671), we identified 7,756 transcripts. A comparative analysis of the transcription profiles conducted in ripe-fruit pericarp and AZ evidenced that 4,391 genes were differentially expressed genes (DEGs) in fruit and AZ. Functional categorization of the DEGs revealed that AZ tissue has an apparently higher response to external stimuli than does that of ripe fruit, revealing a higher expression of auxin-signaling genes, as well as lignin catabolic and biosynthetic pathway, aromatic amino acid biosynthetic pathway, isoprenoid biosynthetic pathway, protein amino acid dephosphorylation, amino acid transport, and photosynthesis. By contrast, fruit-enriched transcripts are involved in ATP synthesis coupled proton transport, glycolysis, and cell-wall organization. Furthermore, over 150 transcripts encoding putative transcription-factors (TFs) were identified (37 fruit TFs and 113 AZ TFs), of which we randomly selected eight genes and we confirmed their expression patterns using quantitative RT-PCR.
Conclusion
We generated a set of EST sequences from olive fruit at full ripening, and DEGs between two different olive tissues, ripe fruit and their AZ, were also identified. Regarding the cross-talk between fruit and AZ, using qRT-PCR, we confirmed a set of TF genes that were differentially expressed, revealing profiles of expression that have not previously been reported, this offering a promising beginning for studies on the different transcription regulation in such tissues.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-14-866) contains supplementary material, which is available to authorized users.
Keywords: Abscission, Gene expression, Olea europaea, 454 Pyrosequencing, Ripening, Transcriptomic comparative
Background
Olive (Olea europaea L.), of worldwide economic importance, has high intra-specific genetic variation with a genome size of about 1,800 Mb [1]. This feature serves to analyze biological processes of biotechnological interest such as phenolic and lipid metabolism during fruit development [2–4] as well as terpenoids and sterols [5]. Directly or indirectly, these processes all affect the quality of olive oil as well as its nutritional profile. The genomic data on olive is augmenting through advances in mapping the olive genome [6, 7], and the DNA of the whole plastome of ‘Frantoio’, an Italian cultivar, has been sequenced [8]. Also, sequencing of the olive genome has been undertaken in Italy through the project OLEA (http://www.oleagenome.org/). Concomitantly, a number of large datasets of expressed sequence tag (EST) datasets have recently been reported for olive, generating 261,485 ESTs [2] and 443,811 ESTs [9] employing the 454 pyrosequencing technologies, an additional 1,132 ESTs with the use of suppression subtractive hybridization [3], as well as 2 million ESTs using Sanger and 454 pyrosequencing technologies [10], this being important for extending the catalog of olive transcripts in order to facilitate gene discovery, functional analysis, and molecular breeding.
Fruit ripening, abscission and senescence are key physiological events that occur during the growth and development of higher plants. These bear commercial implications both for the plant and the harvest. In agricultural research, the manipulation of genes governing these phenomena is key in order to develop varieties that can produce fruits with longer shelf lives as well as crops that tolerate greater environmental stress. Given that several genes are involved in these processes, the manipulation of complex traits such as ripening, abscission, and senescence is not feasible using single genes, and therefore efforts are being focused on specific transcription factors (TFs) that control entire pathways [11].The development of olive fruit involves complex processes following a double sigmoidal growth curve which lasts for 4-5 months and is influenced by numerous factors, including genotype [12, 13]. Olive-fruit properties at the time of harvest, including the final mix of primary and secondary metabolites that accumulate during ripening, largely determine the quality of the resulting oil and fruit. Recent transcriptomic and metabolic studies have demonstrated changes taking place during the development of the olive-fruit and the beginning of ripening [3, 4]. Progress in determining the transcriptome of the olive in terms of functional annotation and the assignment of gene ontology have made it possible to accurately describe of differences in gene expression between olive tissues [2, 3]. However, transcriptome information of the olive fruit at full ripening has not yet been determined.
After fruit ripening, many fruit-tree species undergo massive natural fruit abscission. In olive, abscission of mature fruit depends on the activation of the abscission zone (AZ) located between the pedicel and fruit, and the patterns of mature fruit abscission differ between cultivars [14, 15]. In some olive cultivars (cv. Picual), fruit ripening associated events lead finally to the abscission of the ripe fruit from the pedicel, this taking place at 217 days post-anthesis (DPA) [14, 15]. In a previous study, we reported the comparison of the Picual fruit AZ transcriptomes at two different stages (pre-abscission vs. abscission) using the RNA-Seq technique; 148 Mb of sequences (443,811 good-quality sequence reads) resulted and 4,728 differentially expressed genes were identified from these two samples [9]. Among the 70 TF genes induced during mature-fruit abscission in the olive AZ, the classes that are well represented included bZIP proteins, MYB proteins, and homeobox domain proteins [9]. The comparison between AZ and fruit allow us to restrict the set of genes putatively related to the abscission, and in this direction the results may hold worthwhile perspectives for the study of this process. Cross-talk between the two tissues may involve different components of the signaling network, such as TFs and other signaling molecules, playing either direct or indirect roles. However, molecular-genetic information on the relationship between ripe fruit and AZ is still very limited. In this study, using 454 pyrosequencing technology, we analyzed the overall transcriptional profile of olive (cv. Picual) fruit pericarp at full ripening to significantly expand the olive transcript catalog. We focused on comparing the transcriptomes generated from pericarp and AZ tissues of ripe fruit to establish the divergences as well as similarities in transcriptional networks, and especially to characterize the biological processes and transcriptional regulators enriched in gene clusters that are differentially regulated. Here, we found a total of 397,457 ESTs assembled into 17,048 isotigs, for which we made extensive annotations. In total, we identified 4,391 differentially expressed genes (DEGs) in ripe fruit and AZ, and characterized their biological functions using gene ontology (GO) annotation and KEGG pathway analysis. The results from this study show that distinct patterns of transcriptional regulation occurs among ripe fruit and their AZ in olive, identifying common and distinct TFs that have not been previously related to fruit ripening or abscission.
Results and discussion
454 sequencing of olive transcriptomes
To characterize olive transcriptomes and generate expression profiles between fruit ripening and abscission, Roche/454 GS-FLX (Titanium) pyrosequencing technology was used to sequence two cDNA samples from fruit pericarp and the AZ, which were collected from olive (cv. Picual) fruits at the ripe stage (217 DPA), when abscission occurs (Figure 1). After the cDNA libraries were prepared, their pyrosequencing was finished, and initial quality filtering was performed with the default parameters. The runs gave a total of 199,075 high-quality sequence reads for fruit pericarp, and 198,382 high-quality sequence reads for AZ (Additional file 1). Thus, a total of 397,457 high-quality ESTs were found for the two study samples. Additional file 2 offers a general view of the sequencing and assembly processes which provides the length distribution for these high-quality reads. Although many reads were very short (<100), over 80% were 300 to 500 bp in length. We assembled these sequences into 19,062 contigs (Additional file 2) grouped into 17,048 isotigs (7,003 for fruit, and 10,045 for AZ, respectively) (Additional file 1; Additional file 2). The average length of the contigs was around 500 bases and most of the contigs had fewer than 10 reads (Additional file 2). We assembled most of the high-quality reads (55%) into longer contigs, implying high coverage for these sequencing data. We then found over 10,000 UniProt identities using BLAST analysis on the sequences assembled (Additional file 1). Some 40% of the isotigs failed to map to UniProt identities, thus constituting a source to discover new genes.
Figure 1.

Tissues of olive ( Olea europaea L. Picual) at 217 DPA used in sequencing: pericarp (epicarp and mesocarp) of ripe fruit and AZ. DPA: days post-anthesis.
Comparison of olive transcriptomes between fruit and AZ tissues
To investigate ripening-abscission distinctions, we compared the transcriptomes of olive fruit and AZ at full ripening (fruit-pericarp vs. fruit-AZ at 217 DPA). Read amounts for each of the 15,671 annotated isotigs (6,533 for fruit, and 9,138 for AZ) lead to the identification of 7,756 transcripts in our experiment (Additional file 3), which 4,391 were differentially expressed genes (DEGs); hereafter, these are called group I (P < 0.01), whereas the other genes (43%) having either low read abundance or non differential representation are called group II (Figure 2A). Thus, the comparative analysis of the transcription profiles conducted in pericarp and AZ of ripe fruit evidenced that a huge number of genes are differentially expressed in fruit and AZ. Of these 4,391 DEGs (Additional file 4), 1,482 showed a higher expression in the fruit pericarp, while 2,909 were overexpressed in the AZ at 217 DPA (Additional file 5; Additional file 6). A comparison of the DEGs indicated that 1,265 genes of these were common in both tissues, whereas 936 DEGs were expressed only in fruit (fruit genes), and 2,190 DEGs were expressed exclusively in AZ at 217 DPA (AZ genes) (Figure 2B). Thus, we identified a large number of fruit and AZ genes, implying that they participate in physiological processes exclusive to certain tissues.
Figure 2.
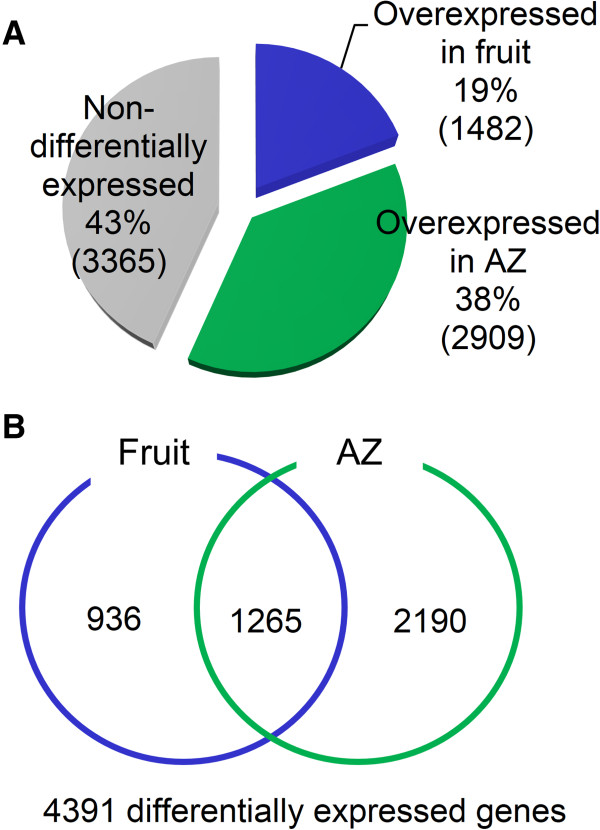
Distribution of genes differentially expressed between olive ripe fruit and its AZ at 217 DPA. (A) The number and percentages of overexpressed genes by tissue: olive fruit and AZ at late stage of ripening. (B) Overlap of overexpressed fruit genes and overexpressed AZ genes. This figure shows the number the transcripts that were specific for each tissue.
To determine which cell processes might be critical in the last stage of fruit ripening in both tissues, we grouped transcripts by their expression signatures in both samples. For group I genes, hierarchical cluster analysis enabled us to identify 2 major clusters, called A and B. Cluster A had the 1,482 most abundant transcripts in fruit-pericarp at 217 DPA, while cluster B bore the 2,909 most abundant transcripts in fruit-AZ at 217 DPA. Subsequently, we split these two clusters into two subclusters, (A1, A2) and (B1, B2), respectively (Additional file 7). We present volcano plots for each hierarchal cluster group and identify gene with both high fold change and significance (Figure 3, Additional file 7). Subcluster A1 had 555 transcripts, which were more abundant in the fruit-pericarp sample with lower expression levels in the fruit-AZ sample at 217 DPA (“fruit-enriched genes”). Meanwhile, cluster A2 contained the 936 expressed transcripts exclusively in the fruit-pericarp sample at 217 DPA (“fruit genes”). In the fruit-AZ sample, cluster B1 had the 710 most abundant transcripts and lower expression levels in the fruit-pericarp sample at 217 DPA (“AZ-enriched genes”), whereas cluster B2 included the 2,190 exclusively expressed transcripts in the fruit-AZ sample at 217 DPA (“AZ genes”).
Figure 3.
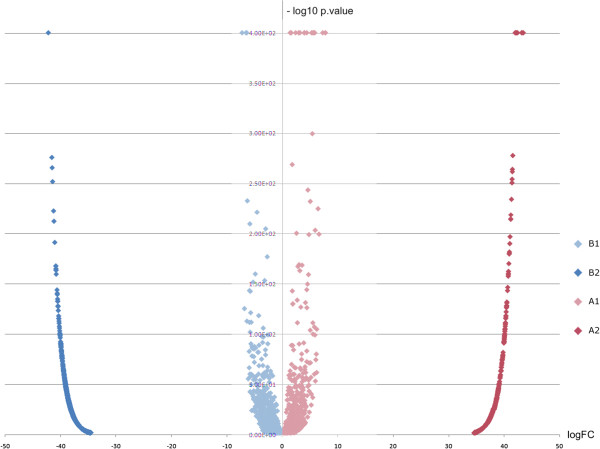
Volcano blots show significant changes in gene expression between fruit and AZ tissues at 217 DPA. Dispersion graph of the–log10 p value (y axis) against the logFC (x axis) corresponding to the genes clustered by their differential expression: A1 (fruit-enriched genes), A2 (fruit genes), B1 (AZ-enriched genes) and B2 (AZ genes). Fold changes and their associated P values for all probe sets can be found in Additional file 7.
For each cluster, the most abundant transcripts appear in Table 1. For the fruit-enriched transcripts, the greatest differential expression was found for a transcript participating in abscisic acid (ABA) stress ripening (coding for an abscission stress ripening-like protein), and a transcript coding for β-glucosidase involved in carbohydrate metabolic process, suggesting that such ripening processes as cell-wall alterations occur in fruit-pericarp at the last stages of olive ripening. Also, a significantly higher expression in ripe fruit vs. AZ tissues was found for an ACO1 (1-aminocyclopropane-1-carboxylic acid oxidase 1) and ETR1 (ethylene receptor 1) involved in ethylene biosynthesis and perception, respectively, suggesting that ACO1 as well as ETR1 may be instrumental in balancing ethylene biosynthesis needs with ethylene signaling requirements to full ripening in olive-pericarp. Another transcript coding for thaumatin-like protein, which is developmentally regulated particularly in fruits during ripening, but is also induced in response to biotic or abiotic stress [16] revealed a fruit-enriched expression pattern. Also, tubulins beta chain revealed a fruit-enriched expression pattern, signifying that activation of vesicle trafficking involving these tubulins may take part in fruit-pericarp during fruit ripening. On the other hand, the genes that encode anthocyanidin synthase, 6,7-dimethyl-8-ribityllumazine synthase, and alpha-expansin 8 (EXP8) were the genes most highly expressed among those expressed exclusively in olive fruit compared to AZ (Table 1). A key component in the riboflavin pathway, 6,7-dimethyl-8-ribityllumazine synthase or CORONATINE INSENSITIVE1 SUPPRESSOR (COS1) is involved in jasmonic acid mediated signaling pathway [17]. This suggests that COS1 may participate in jasmonate signaling to regulate olive ripening, but not to regulate abscission of mature fruit. Previous works have shown that in many crops (e.g. grape [18], apple [19], litchi [20], and Chinese bayberry [21]) the anthocyanin content in fully ripe fruit correlates well with the cumulative expression of anthocyanin biosynthetic genes. In the present study, it was found that expression of anthocyanidin synthase was up-regulated in fruit-pericarp at full ripe stage, suggesting the regulation of anthocyanin biosynthesis by anthocyanidin synthase in the late olive-ripening stage. In addition, the strong up-regulation of EXP8 indicates that this expansin plays a major role in cell-wall alterations involved in olive ripening.
Table 1.
The most abundant transcripts in fruit (Cluster A) and AZ (Cluster B) at the last stage of olive ripening
| Unigene ID | UniProt ID | Fruit | AZ | p-value | Description |
|---|---|---|---|---|---|
| Cluster A | |||||
| Cluster A1 | Enriched in fruit | ||||
| OL006944 | Q2TUW1 | 20742.30 | 1196.08 | 0.00E + 00 | Abscisic stress ripening-like protein = Glycine max |
| OL007219 | Q8GVD0 | 5033.88 | 214.76 | 0.00E + 00 | Beta-glucosidase, Bglc = Olea europaea subsp. europaea |
| OL001156 | B9H1F2 | 5022.65 | 95.79 | 0.00E + 00 | Uncharacterized protein = Populus trichocarpa |
| OL006467 | E0CU96 | 2920.45 | 169.91 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL001418 | B9R8J3 | 8902.90 | 1040.58 | 0.00E + 00 | Phosphoprotein ECPP44 = Ricinus communis |
| OL007236 | Q8H159 | 4247.26 | 1430.34 | 0.00E + 00 | Polyubiquitin 10 [Cleaved into: Ubiquitin], UBQ10, At4g05320 |
| OL003644 | C6KMJ4 | 4794.95 | 1423.76 | 0.00E + 00 | ACC oxidase, ACO1 = Boea hygrometrica |
| OL006886 | Q0WLP3 | 5085.83 | 870.26 | 0.00E + 00 | Uncharacterized protein = Arabidopsis thaliana |
| OL006727 | P29512 | 1812.59 | 180.74 | 0.00E + 00 | Tubulin beta-2/beta-3 chain, TUBB2 TUB2, At5g62690; TUBB3 TUB3, At5g62700 |
| OL006856 | Q06R56 | 1174.95 | 18.56 | 0.00E + 00 | Acetyl-CoA carboxylase beta subunit accD, FEC0159 = Forsythia europaea |
| OL006553 | O04111 | 1437.34 | 5.97 | 0.00E + 00 | Chalcone synthase, CHS, Perilla frutescens |
| OL002387 | B9S382 | 949.24 | 22.02 | 0.00E + 00 | Tubulin beta chain = Ricinus communis |
| OL000027 | A2IBF9 | 1329.71 | 22.64 | 0.00E + 00 | Flavanone-3-hydroxylase = Gossypium hirsutum |
| OL002907 | B9SLE5 | 3731.27 | 15.50 | 0.00E + 00 | Peptidase = Ricinus communis |
| OL000014 | A1E4D3 | 610.81 | 13.51 | 0.00E + 00 | Ethylene receptor, ETR1 = Coffea canephora |
| OL003708 | D5LY28 | 609.68 | 3.53 | 0.00E + 00 | Soluble acid invertase 1, SAI1 = Orobanche ramosa |
| OL001944 | B9RP00 | 1995.12 | 42.28 | 9.70E-301 | Uncharacterized protein = Ricinus communis |
| OL007516 | Q9LLB7 | 4587.02 | 1203.54 | 5.39E-270 | Thaumatin-like protein = Vitis vinifera |
| OL001075 | B7U8J4 | 1418.63 | 52.49 | 1.22E-244 | Expansin, CDK3 = Diospyros kaki |
| OL005738 | D7U0E8 | 538.58 | 14.77 | 2.73E-233 | Uncharacterized protein = Vitis vinifera |
| OL007398 | Q9AXU0 | 1854.70 | 19.23 | 5.46E-226 | Major latex-like protein = Prunus persica |
| OL000584 | A5BN70 | 585.33 | 8.71 | 2.72E-204 | Uncharacterized protein = Vitis vinifera |
| OL006621 | O49877 | 1057.22 | 162.37 | 2.53E-201 | CYP1 (Cysteine protease TDI-65) |
| OL004008 | D7SNI5 | 615.01 | 5.64 | 2.08E-200 | Uncharacterized protein = Vitis vinifera |
| OL007235 | Q8H145 | 432.58 | 14.77 | 5.11E-200 | Putative elongation factor (Fragment), At1g56075 |
| Cluster A2 | Fruit genes | ||||
| OL003887 | D7SKG3 | 9684.90 | 0.00 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL004525 | D7T2N4 | 5878.45 | 0.00 | 0.00E + 00 | 6,7-dimethyl-8-ribityllumazine synthase = Vitis vinifera |
| OL000028 | A2ICC9 | 2823.47 | 0.00 | 0.00E + 00 | Anthocyanidin synthase, ANS = Vitis vinifera |
| OL006333 | E0CQN9 | 1291.93 | 0.00 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL002413 | B9S4E4 | 2111.11 | 0.00 | 0.00E + 00 | Alpha-expansin 8 = Ricinus communis |
| OL004078 | D7SQ46 | 928.57 | 0.00 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL002282 | B9S053 | 1262.49 | 0.00 | 0.00E + 00 | ATP synthase alpha subunit mitochondrial = Ricinus communis |
| OL003892 | D7SKJ8 | 1091.87 | 0.00 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL004267 | D7SVD2 | 1810.56 | 0.00 | 5.65E-279 | Uncharacterized protein = Vitis vinifera |
| OL006945 | Q2UYU6 | 619.52 | 0.00 | 3.12E-265 | Flavonoid-3′-hydroxylase = Vitis vinifera |
| OL003801 | D7SI22 | 693.27 | 0.00 | 7.63E-263 | Uncharacterized protein = Vitis vinifera |
| OL005180 | D7TJ49 | 903.07 | 0.00 | 2.23E-255 | Uncharacterized protein = Vitis vinifera |
| OL007481 | Q9FXL4 | 642.40 | 0.00 | 8.53E-252 | Elicitor inducible beta-1,3-glucanase, NtEIG-E76 = Nicotiana tabacum |
| OL004529 | D7T2X5 | 1244.84 | 0.00 | 2.22E-235 | Uncharacterized protein = Vitis vinifera |
| OL004452 | D7T0N0 | 444.07 | 0.00 | 1.16E-219 | Uncharacterized protein = Vitis vinifera |
| OL001743 | B9RI89 | 699.45 | 0.00 | 1.76E-215 | Serine-threonine protein kinase = Ricinus communis |
| OL007506 | Q9LIC2 | 398.86 | 0.00 | 3.50E-215 | Multispanning membrane protein-like, At3g13772 |
| OL005327 | D7TN33 | 2085.55 | 0.00 | 7.29E-198 | Uncharacterized protein = Vitis vinifera |
| OL004599 | D7T4I1 | 790.51 | 0.00 | 5.34E-191 | Uncharacterized protein = Vitis vinifera |
| OL007004 | Q40168 | 880.38 | 0.00 | 1.21E-182 | Floral homeotic protein AGAMOUS, TAG1 = Solanum lycopersicum |
| OL001261 | B9I6M7 | 505.91 | 0.00 | 2.94E-181 | Uncharacterized protein = Populus trichocarpa |
| OL007205 | Q84V57 | 368.45 | 0.00 | 1.16E-180 | Pectinesterase = Nicotiana benthamiana |
| OL006690 | P14721 | 454.41 | 0.00 | 4.27E-171 | Dihydroflavonol-4-reductase, DFRA = Antirrhinum majus |
| OL006603 | O24329 | 532.05 | 0.00 | 2.50E-163 | Putative uncharacterized protein = Ricinus communis |
| OL007050 | Q45QI7 | 831.88 | 0.00 | 3.09E-161 | Chalcone-flavonone isomerase, CHI = Camellia sinensis |
| Cluster B | |||||
| Cluster B1 | Enriched in AZ | ||||
| OL007063 | Q53U35 | 466.67 | 39974.19 | 0.00E + 00 | Similar to pathogenesis-related protein, STH-2 = Solanum lycopersicum |
| OL004910 | D7TBW7 | 92.31 | 12814.10 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL000784 | A5C4X8 | 19.48 | 1499.53 | 0.00E + 00 | Uncharacterized protein = Vitis vinifera |
| OL005534 | D7TTS3 | 9.40 | 677.78 | 7.69E-234 | Uncharacterized protein = Vitis vinifera |
| OL001130 | B9GQM0 | 24.67 | 528.28 | 1.89E-222 | Glycosyltransferase, CAZy family GT8 = Populus trichocarpa |
| OL001048 | B3Y023 | 6.44 | 337.01 | 1.24E-210 | Arginine decarboxylase, PpADC, Prunus persica |
| OL001934 | B9RNU7 | 144.97 | 1060.02 | 1.01E-205 | Protein phosphatase 2c = Ricinus communis |
| OL007508 | Q9LJU7 | 141.11 | 842.87 | 6.39E-178 | Purple acid phosphatase 18, PAP18 PAP30, At3g20500 |
| OL000621 | A5BSF5 | 14.55 | 385.83 | 8.80E-161 | Uncharacterized protein = Vitis vinifera |
| OL004617 | D7T4X3 | 86.61 | 712.16 | 3.01E-154 | Uncharacterized protein = Vitis vinifera |
| OL000020 | A1X877 | 6.01 | 209.08 | 1.26E-152 | NRC1 = Solanum lycopersicum |
| OL002350 | B9S255 | 11.95 | 656.04 | 2.81E-144 | Uncharacterized protein = Ricinus communis |
| OL002844 | B9SJN1 | 4.75 | 235.99 | 1.57E-143 | Transcription factor hy5 = Ricinus communis |
| OL000814 | A5C762 | 3.65 | 376.25 | 2.47E-126 | Uncharacterized protein = Vitis vinifera |
| OL003935 | D7SLN3 | 4.44 | 211.35 | 3.03E-122 | Uncharacterized protein = Vitis vinifera |
| OL003232 | B9SWQ3 | 15.25 | 265.80 | 2.37E-119 | Serine/threonine protein kinase = Ricinus communis |
| OL000971 | A9PCV7 | 6.58 | 498.02 | 7,68E-114 | Uncharacterized protein = Populus trichocarpa |
| OL004147 | D7SS09 | 8.28 | 365.56 | 8,33E-113 | Uncharacterized protein = Vitis vinifera |
| OL003339 | B9T0K9 | 6.41 | 353.47 | 1.06E-112 | Plasminogen activator inhibitor 1 RNA-binding protein, putative = Ricinus communis |
| OL007507 | Q9LJI5 | 50.33 | 494.78 | 6.20E-107 | V-type proton ATPase subunit d1, VHA-D1, At3g28710 |
| OL000585 | A5BN72 | 2.24 | 113.57 | 6.46E-103 | Uncharacterized protein = Vitis vinifera |
| OL001014 | B1PK08 | 114.23 | 616.10 | 3.23E-100 | Putative polygalacturonase = Olea europaea |
| OL007154 | Q6RYA0 | 51.28 | 584.61 | 1,32E-98 | Salicylic acid-binding protein 2 = Nicotiana tabacum |
| OL003709 | D5M8I6 | 22.52 | 216.21 | 3.46E-98 | Uncharacterized protein = Vitis vinifera |
| OL000614 | A5BR22 | 108.46 | 1156.08 | 1.18E-92 | Uncharacterized protein = Vitis vinifera |
| Cluster B2 | AZ genes | ||||
| OL007111 | Q68V46 | 0.00 | 1349.85 | 0.00E + 00 | Beta-1,3-glucanase, glu-4 = Olea europaea |
| OL001027 | B2M153 | 0.00 | 517.74 | 4.51E-277 | Putative laccase = Rosa hybrid cultivar |
| OL002714 | B9SF95 | 0.00 | 614.34 | 7.75E-267 | Nitrate transporter = Ricinus communis |
| OL006675 | O98664 | 0.00 | 576.60 | 5.46E-253 | Ribulose bisphosphate carboxylase large chain, rbcL = Kigelia africana |
| OL007711 | Q9XEL8 | 0.00 | 396.80 | 8.65E-224 | 3-hydroxy-3-methylglutaryl-coenzyme A reductase 2, HMGR2 = Capsicum annuum |
| OL000602 | A5BPW9 | 0.00 | 602.46 | 2.97E-213 | Uncharacterized protein = Vitis vinifera |
| OL001338 | B9NAX4 | 0.00 | 3264.55 | 3.51E-192 | Uncharacterized protein = Populus trichocarpa |
| OL000148 | A5AN11 | 0.00 | 396.34 | 1.06E-168 | Uncharacterized protein = Vitis vinifera |
| OL004086 | D7SQA7 | 0.00 | 273.23 | 1.06E-168 | Uncharacterized protein = Vitis vinifera |
| OL003142 | B9STR3 | 0.00 | 277.60 | 5.43E-166 | Endosomal P24A protein = Ricinus communis |
| OL002860 | B9SK95 | 0.00 | 489.81 | 8.68E-165 | 12-oxophytodienoate reductase opr = Ricinus communis |
| OL005126 | D7THY5 | 0.00 | 222.22 | 1.74E-164 | Uncharacterized protein = Vitis vinifera |
| OL007151 | Q6RH27 | 0.00 | 570.32 | 1.42E-160 | NAC domain protein, SlNAC1 = Solanum lycopersicum |
| OL004686 | D7T6Y2 | 0.00 | 305.72 | 6.41E-145 | Uncharacterized protein = Vitis vinifera |
| OL007397 | Q9AXR6 | 0.00 | 248.90 | 1.31E-141 | ATP:citrate lyase = Capsicum annuum |
| OL000367 | A5B7F7 | 0.00 | 319.83 | 2.10E-140 | Uncharacterized protein = Vitis vinifera |
| OL002951 | B9SML0 | 0.00 | 214.99 | 6.88E-136 | Lyase = Ricinus communis |
| OL007180 | Q7XE16 | 0.00 | 176.98 | 8.81E-134 | Cell division cycle protein 48 = Oryza sativa subsp. japonica |
| OL005047 | D7TFE6 | 0.00 | 357.56 | 1.76E-133 | Uncharacterized protein = Vitis vinifera |
| OL000444 | A5BDC8 | 0.00 | 382.41 | 5.77E-129 | Uncharacterized protein = Vitis vinifera |
| OL002899 | B9SL31 | 0.00 | 245.52 | 2.31E-128 | Transcription factor = Ricinus communis |
| OL003084 | B9SRT5 | 0.00 | 96.17 | 1.89E-124 | Phospholipid-transporting atpase = Ricinus communis |
| OL007255 | Q8LAH7 | 0.00 | 341.40 | 4.96E-119 | 12-oxophytodienoate reductase 1, AtOPR1, At1g76680 |
| OL001800 | B9RJM7 | 0.00 | 394.51 | 1.27E-116 | Uncharacterized protein = Ricinus communis |
| OL002929 | B9SM03 | 0.00 | 296.74 | 2.54E-116 | Uncharacterized protein = Ricinus communis |
The sequences were selected at p < 0.01 and were sorted by p-value. The table shows the total read count in RPKMx1000 for each gene after normalization across the 2 samples: (a) Fruit at 217 DPA, (b) AZ at 217 DPA.
Among the most abundant AZ-enriched transcripts, we identified a homolog of STH-2 (Similar to pathogenesis-related protein 2) (Table 1), encoding a pathogenesis-related protein (PR), which are observed in the olive AZ during the induction of mature-fruit abscission [9]. However, further work is necessary to ascertain the biological significance of pathogenesis-related gene expression in the olive AZ during abscission. In pea, there is an accumulation of STH2 homologs during late embryogenesis [22], and in Craterostigma plantagineum during rehydration of desiccated plants [23]. In addition, a homolog of PAP18 (At3g20500), encoding a purple acid phosphatase (PAP) induced to phosphate limitation [24], and a homolog of glutamine synthetase, were very significantly expressed in fruit-AZ compared to fruit-pericarp tissue, indicating a role for these proteins in intercellular transport during mature-fruit abscission. PAPs, metallophosphoesterases that contain a bimetal nucleus in their active center [25], were involved in plant tolerance to phosphate limitation [24]. Previous experiments showed that, in phloem companion cells, glutamine synthetase activity affects proline levels [26]. The predominant expression of glutamine synthetase suggests redistribution of proline within the AZ during abscission. Among the most abundant AZ genes (Table 1, Cluster B2), cell wall-related genes were detected. This was expected because the main changes in texture related to cell separation result from enzyme-mediated structural and compositional changes in the cell wall. This includes, for example, a beta-1,3-glucanase, which catalyze the hydrolysis of β-1,3-glucan linkages of callose, as well as participating in many processes including cell-wall remodeling, secondary-wall formation, and phytohormone activation [27]. Reportedly, abscission induction is accompanied by the marked up-regulation of a gene that encodes β-1,3-glucanase, as well as the down-regulation of a gene that encodes a callose synthase in the fruit-AZ [9]. This activation of beta-1,3-glucanase was stronger in olive AZ, showing that this phenomenon is related to fruit abscission in olive. Also, one gene associated with nitrate transport is among AZ genes, suggesting the function of nitrate as an important ion for fruit abscission.
Gene ontology functional enrichment analysis of differentially expressed genes
To provide a general view on the functions and processes that change in fruit and AZ at the last stage of ripening, we classified the differentially expressed genes using the Gene Ontology (GO) database. In addition, based on their sequence similarities, we assigned GO accessions to the differentially expressed genes to identify the proteins in the UniProt database annotated with GO accessions in addition to the InterPro and Pfam domains they contained. Among the 15,671 annotated isotigs, 7,433 were designated at least one GO term (Additional file 1, Additional file 8). The GO terms “Oxidation reduction”, “Oxidoreductase activity”, and “Membrane” were the most represented ones among the biological process (Figure 4), molecular function (Figure 5), and cellular component categories (Figure 6), respectively.
Figure 4.

Comparison of GO “biological process” term frequencies in overexpressed unigenes. Comparison of the occurrence frequencies of the GO “biological process” terms in the GO annotations of the unigenes of the 1,491 overexpressed unigenes in olive fruit and the 2,900 overexpressed transcripts in olive AZ at 217 DPA. The number of occurrences is given for the most frequent terms.
Figure 5.

Comparison of GO “molecular function” term frequencies in overexpressed unigenes. Comparison of the occurrence frequencies of the GO “molecular function” terms in the GO annotations of the unigenes of the 1,491 overexpressed unigenes in olive fruit and the 2,900 overexpressed transcripts in olive AZ at 217 DPA. The number of occurrences is given for the most frequent terms.
Figure 6.
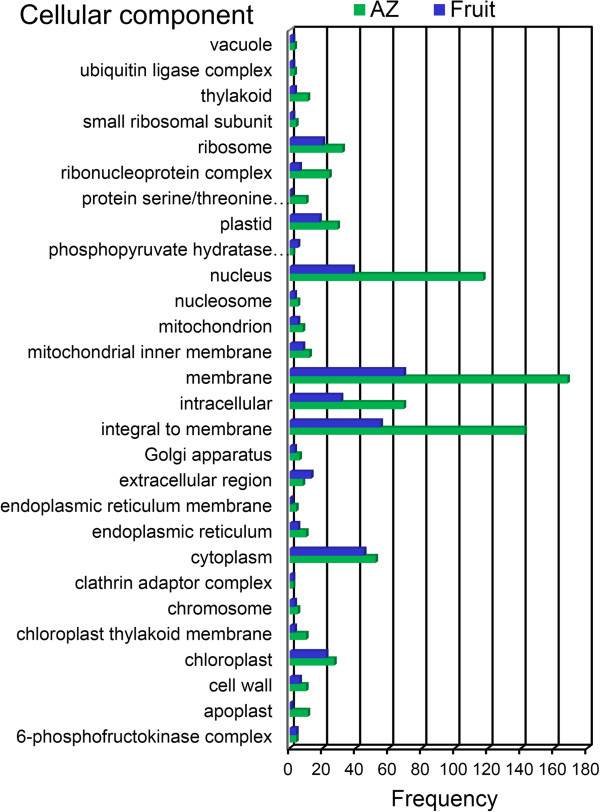
Comparison of GO “cellular component” term frequencies in overexpressed unigenes. Comparison of the occurrence frequencies of the GO “cellular component” terms in the GO annotations of the unigenes of the 1,491 overexpressed unigenes in olive fruit and the 2,900 overexpressed transcripts in olive AZ at 217 DPA. The number of occurrences is given for the most frequent terms.
Also GO terms were identified in the category of biological processes that proved to be over-represented in the lists of genes that showed higher expression in ripe fruit and AZ tissues, respectively (Figure 4). These GO terms constitute indicators of different biological processes that two different tissues underwent in the last stage of ripening. A number of GO classifications proved to be over-represented in genes which had augmented transcript accumulation in fruit at the last stage of ripening. The over-represented group in fruit at 217 DPA having the greatest number among the differentially expressed genes was “Oxidation reduction”, “Metabolic process”, “Transport”, “Transmembrane transport”, “Protein amino acid phosphorylation”, “Glycolysis” and “Carbohydrate metabolic process” (Figure 4). Remarkably, the AZ at 217 DPA also bore a significant representation of transcripts associated with “Metabolic process”, “Oxidation reduction”, “Regulation of transcription”, “Transmembrane transport”, “Transport”, and “Protein amino acid phosphorylation” (Figure 4). Thus, GO terms including “Oxidation reduction”, “Transport”, “Transmembrane transport”, “Protein amino acid phosphorylation”, and “Carbohydrate metabolic process”, were enriched in both lists of genes (Figure 4), indicating that the same biological processes might necessitate different gene sets in two different tissues during full ripening and abscission to support their activities. Sharp differences nevertheless appeared between the two lists of enriched GO terms. Notably, GO terms associated with aromatic amino acid family biosynthetic process, lignin catabolic and biosynthetic process, isoprenoid biosynthetic process, protein amino acid dephosphorylation, amino acid transport, photosynthesis, auxin signaling pathway, apoptosis, defense responses, and responses to stresses were highly enriched in genes more highly expressed in the olive AZ, while differences with respect to other enriched GO terms included ATP synthesis coupled proton transport, glycolysis, and plant-type cell-wall organization which underwent enrichment in genes of higher expression in ripe fruits, suggesting that such biological processes may be associated with ripening-abscission distinctions.
The profile of abundant transcripts in olive ripe fruit (217 DPA) indicates a predominant expression of proteins related to “Oxidoreductase activity”, “Catalytic activity”, “Transferase activity”, “Hydrolase activity”, as well as, “Nucleotide binding”, “Metal-ion binding”, and “ATP binding”, while the “Catalytic activity”, “Transferase activity”, and “Metal-ion binding” GO term was the most over-represented term for the genes in the olive AZ at 217 DPA (Figure 5). Differences of other enriched GO terms included 2-alkenal reductase activity, acyltransferase activity, amino acid transmembrane transporter activity, antiporter activity, drug transmembrane transporter activity, phosphoprotein phosphatase activity, ATP binding, calcium-ion binding, DNA binding, heme binding, and zinc-ion binding which proved to be enriched in genes that showed higher expression in AZ, while acetyl-CoA carboxylase activity, cysteine-type endopeptidase activity, and hydrogen ion transmembrane transporter activity, which were found to be enriched in genes more abundantly expressed in ripe fruit.
Finally, within the “Cellular compartment” category, the “Membrane”, “Integral to membrane” and “Cytoplasm” GO terms constituted the most overrepresented category for the genes with increased transcript accumulation in ripe fruit at 217 DPA (Figure 6). The distribution of gene functions (according to GO assignment) in the fruit and the AZ transcriptomes were largely similar, especially in the categories of molecular function and metabolism, but also different gene functions. These annotations constitute a useful resource for research on gene function, cellular structures, and processes in the two tissues studied.
Metabolic pathways in the last stage of fruit ripening
The olive transcriptomes at the last stage of fruit ripening from our experiment provide the means to examine metabolic and other pathways which differ between the two tissues during this process. GO enrichment identified metabolic pathways that may be key to the last stage of fruit ripening and abscission. To delineate these metabolic pathways further, we mapped the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg) [28] database to the annotations in our transcript data. Of the 10,139 detected proteins in our experiment, 1,442 were annotated with 1,034 Enzyme Commission (EC) codes and mapped to 137 different KEGG pathways (Additional file 9).
GO term representation of all differentially expressed genes between fruit and AZ tissues at 217 DPA is shown in Figure 7. This revealed significantly enriched pathways: biosynthesis of secondary metabolites (101 enzymes represented), microbial metabolism in diverse environments (59), starch and sucrose metabolism (20, Additional file 10, Table 2), amino sugar and nucleotide sugar metabolism (18, Additional file 11, Table 2), cysteine and methionine metabolism (17, Additional file 12, Table 2), methane metabolism (15, Additional file 13), glycolysis/gluconeogenesis (15, Additional file 14), glycine, serine, and threonine metabolism (13, Additional file 15), and arginine and proline metabolism (13, Additional file 16).
Figure 7.
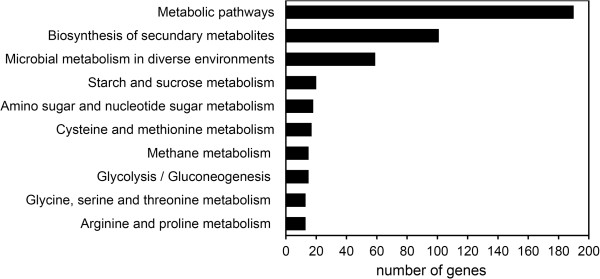
Histogram illustrating pathway enrichment analyses. Distribution of the number of differentially expressed genes between ripe fruit and AZ tissues in different metabolic pathways.
Table 2.
List of olive transcripts from KEGG pathway maps with EC numbers, Unigene ID numbers, UniProt ID numbers, normalized expression values and annotated gene description
| KEGG PATHWAY | EC | Unigene ID | UniProt ID | Fruit | AZ | p-value | Description |
|---|---|---|---|---|---|---|---|
| STARCH AND SUCROSE METABOLISM | |||||||
| Cluster A | |||||||
| Cluster A1 | Enriched in fruit | ||||||
| 3.2.1.26 | OL003708 | D5LY28 | 610 | 4 | 0.00E + 00 | Beta-fructofuranosidase | |
| 2.4.1.13 | OL000884 | A7IZK5 | 244 | 85 | 1.61E-36 | Sucrose synthase | |
| 3.2.1.39 | OL002642 | B9SCU1 | 111 | 94 | 5.10E-22 | Glucan endo-1,3-beta-D-glucosidase | |
| Cluster A2 | Fruit genes | ||||||
| 2.4.1.1 | OL002542 | B9S939 | 250 | 0 | 3.20E-96 | 1,4-alpha-glucan phosphorylase | |
| 2.4.1.12 | OL002104 | B9RUD8 | 5 | 0 | 3.91E-03 | Cellulose synthase | |
| 2.7.7.27 | OL000035 | A3KCF8 | 10 | 0 | 6.10E-05 | Glucose-1-phosphate adenylyltransferase | |
| 2.7.7.9 | OL002654 | B9SD97 | 5 | 0 | 1.95E-03 | UDP glucose pyrophosphorylase | |
| 3.1.1.11 | OL001166 | B9H3W4 | 10 | 0 | 3.91E-03 | Pectin methylesterase | |
| 5.1.3.6 | OL007529 | Q9LPC1 | 8 | 0 | 9.77E-04 | UDP-glucuronate 4-epimerase | |
| 5.3.1.9 | OL003650 | C6TGC6 | 6 | 0 | 1.95E-03 | Glucose-6-phosphate isomerase | |
| Cluster B | |||||||
| Cluster B1 | Enriched in AZ | ||||||
| 3.2.1.2 | OL006254 | E0AE02 | 18 | 32 | 1.82E-03 | Beta-amylase | |
| 4.1.1.35 | OL001047 | B3VDY9 | 102 | 168 | 2.19E-06 | UDP-glucuronate decarboxylase | |
| 2.4.1.21 | OL001761 | B9RIR1 | 5 | 28 | 3.24E-08 | ADP-glucose synthase | |
| Cluster B2 | AZ genes | ||||||
| 3.2.1.4 | OL007034 | Q43149 | 0 | 20 | 9.31E-10 | Cellulase | |
| 2.7.1.4 | OL006635 | O65583 | 0 | 9 | 2.44E-04 | Fructokinase | |
| 3.1.3.12 | OL002988 | B9SNT9 | 0 | 4 | 1.95E-03 | Trehalose 6-phosphate phosphatase | |
| 3.2.1.1 | OL007022 | Q42678 | 0 | 17 | 2.38E-07 | Alpha-amylase | |
| 3.2.1.15 | OL000895 | A7PZL3 | 0 | 10 | 3.05E-05 | Polygalacturonase | |
| 3.2.1.20 | OL007491 | Q9LEC9 | 0 | 12 | 2.91E-11 | Alpha-glucosidase | |
| CYSTEINE AND METHIONINE METABOLISM | |||||||
| Cluster A | |||||||
| Cluster A1 | Enriched in fruit | ||||||
| 2.6.1.1 | OL001190 | B9HAW0 | 35 | 8 | 3.18E-06 | Aspartate transaminase | |
| 2.7.1.100 | OL002217 | B9RY82 | 64 | 24 | 1.36E-05 | S-methyl-5-thioribose kinase | |
| Cluster A2 | Fruit genes | ||||||
| 2.1.1.14 | OL002466 | B9S6C1 | 81 | 0 | 1.29E-26 | 5methyltetrahydropteroyltriglutamate-homocysteine S-methyltransferase | |
| 2.1.1.37 | OL001007 | B0FPD7 | 6 | 0 | 1.95E-03 | DNA (cytosine-5-)-methyltransferase | |
| 2.5.1.47 | OL002734 | B9SFU8 | 52 | 0 | 4.34E-19 | Cysteine synthase | |
| 2.5.1.6 | OL007215 | Q8GTL5 | 10 | 0 | 2.44E-04 | Methionine adenosyltransferase | |
| Cluster B | |||||||
| Cluster B1 | Enriched in AZ | ||||||
| 2.5.1.48 | OL002235 | B9RYU1 | 3 | 23 | 1.94E-06 | Cystathionine gamma-synthase | |
| 2.8.1.2 | OL001730 | B9RHZ9 | 7 | 57 | 2.12E-13 | 3-mercaptopyruvate sulfurtransferase | |
| 3.1.3.77 | OL006405 | E0CSI1 | 5 | 17 | 5.08E-04 | Acireductone synthase | |
| 3.3.1.1 | OL006738 | P35007 | 239 | 308 | 2.02E-06 | Adenosylhomocysteinase | |
| 5.3.1.23 | OL002002 | B9RR88 | 12 | 74 | 7.52E-15 | S-methyl-5-thioribose-1-phosphate isomerase | |
| Cluster B2 | AZ genes | ||||||
| 1.1.1.27 | OL007383 | Q96569 | 0 | 191 | 9.54E-07 | L-lactate dehydrogenase | |
| 1.14.17.4 | OL006733 | P31237 | 0 | 67 | 1.36E-20 | ACC oxidase | |
| 2.3.1.30 | OL006733 | B9S9Q4 | 0 | 27 | 1.16E-10 | Serine O-acetyltransferase | |
| 2.5.1.16 | OL003685 | D2K8S6 | 0 | 52 | 5.55E-17 | Spermidine synthase | |
| 4.1.1.50 | OL000082 | A5AFT0 | 0 | 133 | 2.80E-45 | Adenosylmethionine decarboxylase | |
| 2.7.2.4 | OL005688 | D7TYU1 | 0 | 5 | 7.81E-03 | Aspartate kinase | |
| AMINO SUGAR AND NUCLEOTIDE SUGAR METABOLISM | |||||||
| Cluster A | |||||||
| Cluster A1 | Enriched in fruit | ||||||
| 5.3.1.8 | OL001163 | B9H303 | 77 | 5 | 1.21E-21 | Mannose-6-phosphate isomerase | |
| 5.3.1.9 | OL001141 | B9GV29 | 16 | 4 | 7.53E-04 | Glucose-6-phosphate isomerase | |
| Cluster A2 | Fruit genes | ||||||
| 1.1.1.271 | OL002645 | B9SCY0 | 17 | 0 | 3.05E-05 | GDP-L-fucose synthase | |
| 2.7.1.4 | OL004560 | D7T3P0 | 12 | 0 | 6.10E-05 | Fructokinase | |
| 2.7.7.27 | OL001390 | B9R7X6 | 7 | 0 | 9.77E-04 | Glucose-1-phosphate adenylyltransferase | |
| 2.7.7.64 | OL007403 | Q9C5I1 | 11 | 0 | 1.91E-06 | UTP-monosaccharide-1-phosphate uridylyltransferase | |
| 2.7.7.9 | OL002654 | B9SD97 | 5 | 0 | 1.95E-03 | UTP-glucose-1-phosphate uridylyltransferase | |
| 5.1.3.6 | OL007529 | Q9LPC1 | 8 | 0 | 9.77E-04 | UDP-glucuronate 4-epimerase | |
| Cluster B | |||||||
| Cluster B1 | Enriched in AZ | ||||||
| 3.2.1.14 | OL007471 | Q9FS45 | 10 | 134 | 5.46E-30 | Chitinase | |
| 4.1.1.35 | OL001047 | B3VDY9 | 102 | 168 | 2.19E-06 | UDP-glucuronate decarboxylase | |
| Cluster B2 | AZ genes | ||||||
| 3.2.1.55 | OL002630 | B9SCF3 | 0 | 6 | 2.44E-04 | Alpha-N-arabinofuranosidase | |
| 5.1.3.12 | OL002629 | B9SQF3 | 0 | 6 | 3.91E-03 | UDP-glucuronate 5”-epimerase | |
| 5.4.2.8 | OL003424 | B9T3D2 | 0 | 5 | 9.77E-04 | Phosphomannomutase | |
The table shows the total read count in RPKMx1000 for each gene after normalization across the 2 samples: (a) Fruit at 217 DPA, (b) AZ at 217 DPA. We selected the sequences at p < 0.01.
Transcription factors in olive fruit at the late stage of ripening
Of 4,391 differentially expressed genes, 150 genes putatively encoding TF of diverse families were differentially expressed in olive AZ compared to fruit at 217 DPA (P < 0.01). The majority of these were induced in AZ (Figure 8, Additional file 17). Overall, 37 genes had peak read amounts within cluster A (the set of fruit-induced genes), and 113 genes within cluster B (the set of AZ-induced genes). Within cluster A, the most abundant TFs proved to be a MADS-box domain protein (AG1) detected within subcluster A2 (Additional file 17). Indeed, MADS-box proteins were the most abundant TFs in ripe fruit, two in subcluster A1 (TAGL2 and AGL9) and one in subcluster A2 (AG1), implying coordinated regulation of this class of TFs in ripe fruit (217 DPA). However, in cluster A the well-represented classes included homeobox domain proteins, zinc finger (ZF) proteins, basic helix-loop-helix (bHLH) proteins, and Basic Leucine Zipper (bZIP) proteins. Cluster A1 is enriched in the MADS-box and ZF TF families (Figure 9A, Additional file 17), whereas cluster A2 was rich in the bHLH, homeobox, ZF and bZIP families (Figure 9B, Additional file 17). The control of fleshy-fruit ripening involves many different TFs. In climacteric as well as non-climacteric fruits, a number of MADS-box genes reportedly regulate fruit development and ripening [29]. Master regulators in tomato are HB-box (LeHB-1), MADS-box (SEP4-like, RIN, TDR4, TAG1, TAGL1), SBP-box (CNR), and NAC genes [30]. A series of TFs, homologous to several of these master regulators, appear in ripe olive fruit (Additional file 17).
Figure 8.
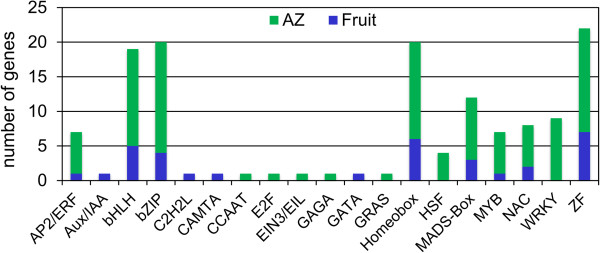
Differentially expressed TF genes and classification of TF families. Comparison of significantly overexpressed TF transcripts between fruit (blue) and AZ (green) at 217 DPA. Number of transcripts related to TFs in each TF family. The 150 TF genes were classified into 20 TF families.
Similarly, the well represented classes in AZ tissue at the late stage of ripening (Cluster B) included ZF proteins, homeobox domain proteins, bHLH proteins, and bZIP proteins (Figure 8). Cluster B1 is enriched in ZF proteins and homeobox domain proteins (Figure 9C), whereas cluster B2 was found to be rich in the bHLH and bZIP families (Figure 9D). Thus, although two clusters containing members from several TF families, in each cluster, clearly significant difference was found in the proportion of families. Moreover, there are distinct TF families in each cluster: the Aux/IAA, C2H2L, CAMTA families in cluster A, and the HSF, GRAS, GAGA-binding protein, EIN3/EIL, E2F/DP, CCAAT-binding protein and WRKY families in cluster B (Figure 9). The enrichment of sequence elements in different gene groups from each cluster in combination with data on transcript abundance offer a tenable set of TFs which could bind these elements and that could be examined in future research.
Among the AZ-overexpressed TF types, HSF proteins, GRAS proteins, GAGA-binding protein, E2F/DP protein, and WRKY proteins were abundantly represented in the olive AZ during mature-fruit abscission [9]. The diversification and functional interaction of HSFs is known, as is their integration into the complex stress signaling and response networks of plants [31], and, a HSF-like TF, TBF1, have been identified as a key molecular mechanism for plant growth-to-defense transition [32]. In our analysis, 4 HSF TFs were exclusively overexpressed in olive-AZ (Additional file 17), supporting the idea that an increase of these HSF genes might be associated with mature-fruit abscission in olive AZ. Transcriptional regulators belonging to the GRAS family have been related to plant growth and development, as well as to biotic and abiotic stress [33]. Also, we report that several GRAS TFs, including homologs of GRA1, GRAS4, GRAS6, and GRAS10 (Solanum lycopersicum), are exclusively overexpressed in the olive AZ (Additional file 17), suggesting that these GRAS TFs probably mediate abscission-responsive transcription. Ever since GAGA-binding proteins were identified and characterized in plants, few advances have been made in explaining their function. Another up-regulated gene in olive-AZ was a homolog of BBR/BPC1 (Vitis vinifera), a GAGA-binding transcriptional activator (Additional file 17), indicating that this family control transcriptional activation of homeotic genes, probably started by ethylene, which potentially leads to the activation of abscission-related proteins in the olive AZ. E2F/DP family of TFs having critical and antagonistic functions in pathways involved in DNA repair, cell division, and differentiation. In olive, E2F3, encoding a key component of the cyclin D/retinoblastoma/E2F pathway that is a potent activator of E2F-responsive genes in Arabidopsis [34], was highly expressed during mature-fruit abscission in the AZ [9]. Here, we also identified one member of E2F family exclusively overexpressed in the AZ (Additional file 17). WRKY proteins are known to have a key part in plant defense against several types of biotic stress, developmental processes, and certain signal-transduction processes that are plant-hormone mediated (e.g. GA, ABA, or SA) [35]. Notably, our analyses have revealed that 9 WRKY genes (Additional file 17) are exclusively over-regulated in the olive AZ, which it is consistent with previous studies where the expression of some WRKY genes are induced during floral abscission [36] and mature-fruit abscission [9]. Thus, our data corroborate that, in the olive AZ, TFs belonging to these families may potentially help trigger the transcriptional cascade. Further study would be needed to reveal the molecular basis of gene expressional regulation.
Among the 37 TF genes induced in ripe fruit (Cluster A), 25 were exclusively expressed in fruit (Cluster A2, Additional file 17). We found it useful to consider these “fruit TFs” (Figure 9B) separately from 12 “fruit-enriched” TFs (Figure 9A), which were upregulated in ripe fruit compared to AZ at 217 DPA. The 25 genes encode 6 ZF proteins, 5 homeobox proteins, 5 bHLD domain class TFs, 3 bZIP, one MADS-box TF (AG1), one MYB TF (MYBA22), one NAC TF, one Aux/IAA (IAA1) protein, one CAMTA TF, and one C2H2L TF (Figure 9B, Additional file 17). This finding suggests that TFs from these families have potentially important roles in mediating late events during olive ripening. Similarly, among the 113 TF genes induced in the AZ at 217 DPA (Cluster B, Additional file 17), most of them (94) were exclusively expressed in the AZ compared to the ripe fruit (AZ TFs, cluster B2). These genes encoding 14 bZIP family TFs, 12 bHLH family TFs, 12 ZF proteins, 9 MADS-box family TFs, 9 homeobox family TFs, 9 WRKY family TFs, 5 NAC family TFs, 5 AP2/ERF family TFs, 5 MYB family TFs, 4 Heat shock factor (HSF) proteins, 3 GRAS proteins, one EIN3/EIL protein, one E2F protein and one CCAAT protein, among others (Figure 9D). The 10 most differentially overexpressed genes in the olive AZ encoding TFs were MYBPA1 (Vitis vinifera), one WRKY (Ricinus communis), MYB108-like protein 1 (Vitis vinifera), one ZF (Ricinus communis), one MYB (Arabidopsis thaliana At3g06490), one bZIP (Vitis vinifera), NAC1 TF (Solanum lycopersicum), one HSF (Vitis vinifera), WRKY30 protein (Vitis aestivalis) and SHORT VEGETATIVE PHASE MADS-box protein (Arabidopsis thaliana At2g22540, SVP) (Additional file 17). Abundant genes encoding putative TFs in the AZ support the contention that a key role is played by transcription regulation during abscission in olive [9]. Thus, among all TF genes expressed differentially between the two tissues; only 25 genes were found to be expressed preferentially in ripe fruit and 94 genes in AZ (Additional file 17).
Figure 9.
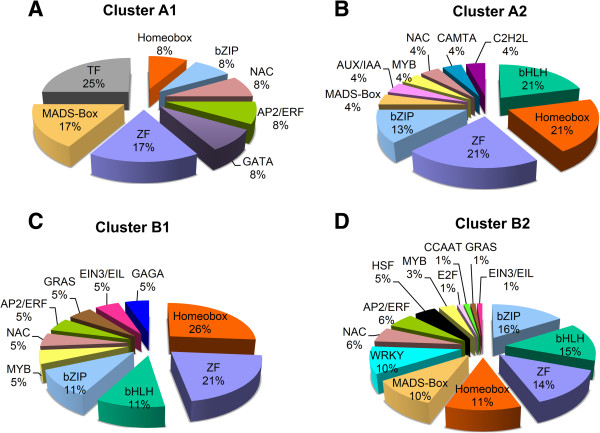
Distribution of olive TF genes expressed at 217 DPA in fruit or AZ tissues, based on their family membership. Fruit-(Cluster A1) or AZ-enriched (Cluster B1) and fruit (Cluster A2) or AZ (Cluster B2) TFs at the last stage of olive fruit ripening. (A) Fruit-enriched (Cluster A1) or (B) fruit (Cluster A2) TFs and (C) AZ-enriched (Cluster B1) or (D) AZ (Cluster B2) TFs at the last stage of olive fruit ripening.
A total of 24 ZF proteins within our analysis show this class of TF to be among the most represented both in ripe fruit and in AZ tissues (Figure 8). Indeed, a ZP gene, AtZFP2[37], reportedly has delayed flower senescence as well as abscission, but AtZFP2 has been shown to participate with DNA BINDING WITH ONE FINGER (AtDOF4.7) in suppressing PGAZAT expression [20]. According to our data, 16 of out 24 ZF genes (Additional file 17) are among the over-regulated TFs in the olive AZ, supporting the coordinated action of ZF proteins in the AZ during fruit abscission. The majority of bHLH proteins identified to date have been functionally characterized in arabidopsis, but, in other plant species, a low number of bHLH genes have been functionally characterized [38]. These genes serve to regulate carpel, anther, and epidermal-cell development, as well as flavonoid biosynthesis, phytochrome signaling, hormone signaling, stress responses, and fruit dehiscence [38]. Gene transcription is known to be regulated by MYB transcription factors in combination with bHLH proteins, which include certain MYC transcription factors. In this sense, MYB and MYC (bHLH) proteins interact to form multi-protein complexes [39]. Reportedly, MYB and bHLH proteins in arabidopsis, cooperate in TTG1-dependent transcriptional regulation [40]. Also, our results demonstrate over-regulation in the olive AZ of 4 out of 5 MYB genes identified (Additional file 17), and 15 out of 20 bHLH genes identified (Additional file 17). We cannot rule out the possibility that these bHLH proteins, including MYC2 (Vitis vinifera), constitute an interaction partner for these MYB TFs for the regulation of genes needed for processes downstream in the AZ during fruit abscission. Further research is necessary to ascertain whether these bHLH TFs act together with MYB proteins in the olive AZ. In this context, homo-and heterodimers formed by bZIP transcription factors are key in the regulation of development and defense responses [41]. Also, bZIP TFs are members of TFs families abundantly represented in the olive AZ (Figure 8). Among those are HY5 and RF2a genes, which were induced in the olive AZ compared with ripe fruit (Additional file 17), and were induced also in melon AZ during early induction of mature-fruit abscission [42]. HY5 is known to mediate the light response [43], whereas RF2a and RF2b functions may be involved in biotic or abiotic stress response or signaling [44]. Three TGA-type bZIP genes have been proposed as governing abscission and regulating abscission-related gene expression [45] as well as up-regulation of the genes bZIP16, bZIP17, bZIP44, bZIP45, bZIP53, and VIP1 in the olive AZ during mature-fruit abscission [9]. In this light, bZIP proteins appear to be positive regulators in abscission signaling. In addition, most NAC proteins were also overexpressed in the olive AZ in comparison with ripe fruit (Additional file 17). Previously, we have found that 5 genes homologous to NAC TFs (ANAC029, ANAC002, ANAC022, ANAC091, and ANAC042) showed enhanced expression during mature-fruit abscission [9], as also reported during the immature-fruit abscission in apple [46]. This finding is noteworthy because transcriptome analyses have recently demonstrated regulation by a NAC transcription factor family. This is not restricted to biotic and abiotic stress responses, but also affects numerous other processes, including senescence, ABA signaling and fruit ripening [28, 47].
To validate our RNA-seq results, we performed quantitative real time PCR (qRTPCR) to determine the levels of expression in eight olive genes taken from the list of TF genes differentially expressed across ripe fruit and AZ. Three genes, bHLH (UniProt ID: D7T931), AG1 (UniProt ID: Q40168) and ZF (UniProt ID: B9H0X4), were identified as being overexpressed in ripe fruit in RNA-seq data analysis and thus were designated for further confirmation (Figure 10A). Similarly, 5 genes, ERF3 (UniProt ID: Q9LW49), MYBPA1 (UniProt ID: A4F4L3), MYB108 (UniProt ID: C3W4Q3), NAC (UniProt ID: Q6RH27) and MYB/At3g06490 (UniProt ID: Q6R095), were identified as being overexpressed in AZ in RNA-seq data analysis and were assigned to further confirmation (Figure 10B).
Figure 10.
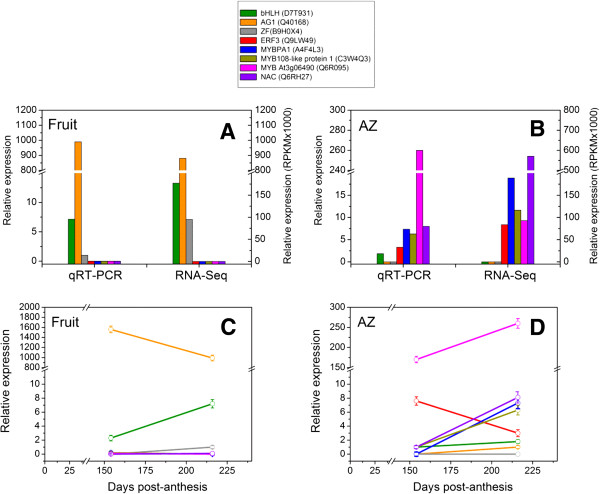
Validation of pyro-sequencing data. A total of eight TF genes showing differential expression in our pyro-sequencing experiment were selected and their relative expression determined using qRT-PCR in two olive tissues: (A) fruit pericarp at 217 DPA and (B) fruit-AZ at 217 DPA. (C) qRT-PCR analysis of eight TF genes in olive fruit during fruit ripening. (D) qRT-PCR analysis of eight TF genes in olive AZ during abscission of mature-fruit. In the Materials and Methods section, genes and their primers are indicated. Relative expression values were normalized to the lowest expression value taken as 1. The data represent the mean values (±SEs) of duplicate experiments from three independent biological samples.
The qRT-PCR analysis confirmed the enrichment bHLH, AG1 and ZF genes in ripe fruit and the enrichment of ERF3, MYBPA1, MYB108, NAC and MYB/At3g06490 genes in the olive AZ. Notably, the expression of ERF3, MYBPA1, MYB108, NAC and MYB/At3g06490 were not detected in fruit (Figure 10A), and the expression of bHLH, AG1 and ZF were not detected in AZ (Figure 10B). Thus, the qRT-PCR expression results correlated with the RNA-seq expression data for the genes tested. In addition, we used qRT-PCR analysis for the expression profiles of eight TFs in olive fruit and AZ during fruit ripening and abscission (between 154 and 217 DPA). The expression of bHLH and ZF increased 3-fold and 1-fold in olive fruit, respectively, during ripening, while AG1 expression decreased 1.6-fold during ripening (Figure 10C), implying that these genes are involved in ripening events. On the other hand, transcripts of MYBPA1, MYB108, NAC and MYB/At3g06490 accumulated during abscission in olive AZ, whereas the expression of ERF3 was decreased in olive AZ during abscission (Figure 10D). Hence, the expression pattern of some genes in olive fruit or AZ, performed by qRT-PCR, are shown to represent the transcriptome related to fruit ripening or the transcriptome related to the activation of abscission.
Conclusion
We performed 454 transcriptome sequencing and de novo assembly for two tissues, ripe fruit and AZ, of Olea europaea. As a result, we describe transcriptomic differences between the ripe fruit and this AZ occurring at last stage of ripening in olive as well as potential new genes generated. Changes in gene transcripts were accompanied by changes in expression of TFs, especially those in the TFs MADS-box, ZF, homeobox domain proteins, bHLH, and bZIP families, that putatively may trigger the cross-talk between fruit and AZ. Our results indicate that genes encoding members of Aux/IAA, C2H2L, and CAMTA families were preferentially transcribed in ripe fruit. By contrast, TF genes of the HSF, GRAS, GAGA-binding protein, EIN3/EIL, E2F/DP, CCAAT-binding protein, and WRKY families were preferentially transcribed in AZ. Furthermore, by quantitative real-time PCR analysis, we confirmed the mRNA-Seq results for eight TF genes. This result implies that the study of those TFs associated with the expression pattern observed in ripe fruit could open major biological pathways governing gene-expression regulation in ripe fruit. These data supply the first comprehensive and comparative molecular information for understanding the expression differences in these tissues.
Methods
Plant material and RNA isolation
20-year-old olive trees (Olea europaea L. cv. Picual) in an orchard near Badajoz (Spain) grown under drip irrigation and fertirrigation (irrigation with suitable fertilizers in the solution) were studied. Picual olive flowers were tagged on the day of pollination and the fruit-pericarp (fruit mesocarp and epicarp) and fruit-AZ samples were collected from olive fruits subsequently harvested at last stage of ripening (217 days post-anthesis, DPA), at which time they abscise (Figure 1). The fruit AZs, located between the pedicel and fruit, were manually dissected from longitudinal sections of the samples with a razor blade into pieces to a maximum width of 1 mm on each side of the abscission fracture plane [15]. Fruit-AZ wings containing pericarp or pedicel/calyx-like tissues were discarded. Fresh samples (fruit-pericarp and fruit-AZ at 217 DPA), using 300 fruits, were immediately frozen in liquid nitrogen and stored at-80°C for RNA isolation.
Total RNA was extracted from fruit-pericarp and-AZ tissues at 217 DPA using the Spectrum Plant Total RNA Kit (Sigma-Aldrich) according to the manufacturer’s instructions and eluted with nuclease-free water. After DNaseI (Ambion) treatment, RNA quality was gel verified and quantified spectrophotometrically (NanoDrop, ThermoScientific, http://http://www.thermofisher.com/). Messenger RNA was isolated twice with Dynabeads Oligo (dT)25 (Dynal Biotech ASA, Dynal Invitrogen, http://www.invitrogen.com) to minimize rRNA contamination. One microgram of mRNA per sample was used as template for first-strand cDNA synthesis using SMART technology (Clontech Laboratories Inc, http://www.clontech.com/) to favor full-length synthesis. Double-stranded cDNA was made by 13 cycles of longdistance PCR. Complementary DNA was purified with QIAquick columns (Qiagen, http://www.qiagen.com/) to eliminate oligo-dT and enzymes. The cDNA quality was verified with an Agilent 2100 Bioanalyzer (Nimblegen, http://www.nimblegen.com/).
Library preparation for pyro-sequencing
Three micrograms of each cDNA sample were nebulized to produce fragments of a mean size between 400 and 800 bp. Preparation of cDNA fragment libraries and emulsion PCR conditions were performed as described in the Roche GS FLX manual. Pyro-sequencing was performed on a Roche Genome Sequencer FLX instrument (454LifeScience-Roche Diagnostics, http://www.454.com/) at Lifesequencing S.L. (Valencia, Spain).
Trimming and assembly of pyro-sequenced reads
The quality of the reads was assessed with PERL scripts developed at Lifesequencing for trimming and validation of high-quality sequences. Adaptor sequences used for library preparation were entered in an adaptor-trimming database to the PERL Program. New SFF output files were generated with the sfftools (454 Life Science/Roche), keeping the largest starting trimpoint and the smallest ending trimpoint. Trimmed reads were assembled with NEWBLER version 2.3 (454 Life Science/Roche) with default parameters. Following quality control, when performing the assembly, some reads were removed due to short quality for the reads to be used.
Annotation
We selected a wide set of reference proteins from taxonomically related organisms. We included all proteins form eudicotyledons with annotations for the terms: carbohydrate metabolic process, secondary metabolic process, cell-wall, cell-wall organization, and phytohormones, in order to have a complete reference protein representation for these specific aspects probably related with ripening and abscission process. The total number of reference proteins was 125,428. The inclusion of proteins from taxonomically distant organisms with rich functional annotations such as Vitis vinifera or Ricinus communis, allowed us to annotate new proteins that could be lost if we include proteins only from close organisms. To obtain a high quality annotation we chose a very restrictive level of similarity between the isotig and the annotator reference protein. The similarity required must be high to sufficiently support the inference of function from the reference protein. In this work, BLAST E value lower than 10-20 was required for function inference. It is important to note that the smaller the E value is, the higher similarity between sequences is, and thus, the greater the confidence of the function assignment is. The massive BLASTX of all isotigs against the 125,428 reference proteins was performed using a cloud computing environment (Amazon web services).
Quantification of the expression levels
The reference proteins were proteins representative of UniRef90 clusters. This strategy fixed a minimum similarity distance between reference proteins and was the basis of our clustering of isotigs for obtaining unigenes and quantifying their expression levels. The name of each unigene was inferred from the name of the UniRef90 representative proteins that annotated each unigene. We quantified the expression for these unigenes, here defined as clusters of isotigs annotated by the same reference protein. The number of reads assigned to each isotig was calculated taking into account that the reads of each contig were counted only one time. Given that isotigs represent transcribed isoforms, it could be possible that different isotigs sharing some contigs were clustered within the same unigene. In those cases, the reads of each contig was counted only one time. The normalization of the absolute values of the number of reads was done based on [48]. We obtained the RPKM (Reads Per Kilobase of exon model per Million mapped reads). In this case, we used the length of the reference protein in nucleotides since we were working without a reference genome and then without exon models. This normalization allows the comparison of the expression values between unigenes from the same or from different samples [48].
Differential expression analysis
The method used for the analysis of differential expression in this work was edger [49], a Bioconductor package for differential expression analysis of digital gene-expression data able to account for biological variability.
EdgeR models count data using on overdispersed Poisson model, and use an empirical Bayes procedure to moderate the degree of over-dispersion across genes. For the analysis of the differential expression with Edge R the input was a table of counts, with rows corresponding to genes/proteins and columns to samples. EdgeR models the data as negative binomial (NB) distributed, Ygi ~ NB(Mipgj, Фg) for gene g and sample i. Here Mi is the library size (total number of reads), Фg is the dispersion, and pgj is the relative abundance of gene g in experimental group j to which sample i belongs. The NB distribution reduces to Poisson when Фg = 0. This is an especially appropriate method to be used in RNA-Seq projects [50, 51]. In this work, an isotig was considered differentially expressed when it exhibited highly significant difference in read abundance at P < 0.01.
GO annotations
GO annotations [52] were obtained from Uniprot and inferred from the GO annotations of the proteins representative of each unigene. GO Terms coming from the 3 different GO ontologies (Biological process, Molecular function and Cellular component) were analyzed separately. We found the number of proteins annotated with each term. In the GOSlim analysis, every GO term was translated into a GO Term taken from a set of selected general GO Terms in order to provide a more general and homogeneous perspective of the GO Terms found in a sample. To perform the GOSlim analysis, we selected the GOSlim terms proposed by the European Institute of Bioinformatics (EBI) as GO Terms selected for studies in Plants. The GO-slim studies were developed using Bio4j (http://www.bio4j.com/), a graph database that integrates all Uniprot, GO, taxonomy, RefSeq and Enzyme database elements in nodes connected by edges that represent their relationships. We selected a subset of terms to gain a broad functional overview and, using bio4j at the back-end, we obtained the GO-slim results. At this selected granularity level we obtained the functional profile of GO-slim terms that allowed us to highlight general features.
Quantitative RT-PCR
Total RNA (2 μg) was reverse-transcribed with random hexamers and Superscript III (Invitrogen), according to the manufacturer’s instructions. Purified cDNA (2 ng) was used as a template for qRTPCR. qRT-PCR assays were performed with gene-specific primers. Primer sequences were 5′- CATGTCAGAGCAAAGAGAGGGCAA-3′ (forward) and 5′-ACTCGCTGCTGATAGTTTCAT-3′ (reverse) for bHLH (UniProt ID: D7T931); 5′-ATGGCATTGCAGAGTGATCAATCA-3′ (forward) and 5′-TTGAAGAGGTGGTTGATCTTG-3′ (reverse) for AG1 (UniProt ID: Q40168); 5′-AATGAGGGAATCTGCCATACT-3′ (forward) and 5′-CTCTCTAGCCACGTGGCCAGA-3′ (reverse) for ZF (UniProt ID: B9H0X4); 5′-AATGGCGTTAAGGAGGTCCACTAC-3′ (forward) and 5′-AGGTAAAGGGAAGTTAGTTTTAGC-3′ (reverse) for ERF3 (UniProt ID: Q9LW49); 5′-ATGGGAAGGTCTCCTTGTTGTTCA-3′ (forward) and 5′-CTTGATCTCATTGTCGGTTCGACC-3′ (reverse) for MYBPA1 (UniProt ID: A4F4L3); 5′-TATTTACGCCCAGACGTTCGTCGA-3′ (forward) and 5′-TCTCTCAACCAATCGTGGCATCCA-3′ (reverse) for MYB108 (UniProt ID: C3W4Q3); 5′-CTTGATGATTGGGTGTTGTGCCGA-3′ (forward) and 5′-TTGATCATTGTACTGCATTTGAGA-3′ (reverse) for NAC (UniProt ID: Q6RH27); 5′-G TATTTACGCCCAGACGTTCGTCGA-3′ (forward) and 5′-TCTCTCAACCAATCGTGGCATCCA-3′ (reverse) for MYB transcription factor At3g06490 (UniProt ID: Q6R095). The cDNA was amplified using SYBRGreen-PCR Master kit (Applied Biosystems, Foster City, CA, USA) containing an AmpliTaq Gold polymerase on an iCycler (BioRad Munich, Germany), following the protocol provided by the supplier. Samples were subjected to thermal cycling conditions of DNA polymerase activation at 94°C, 45 s at 55°C, 45 s at 72°C, and 45 s at 80°C; a final elongation step of 7 min at 72°C was performed. The melting curve was designed to increase 0.5°C every 10 s from 62°C. The amplicon was analyzed by electrophoresis and sequenced once for identity confirmation. qRT-PCR efficiency was estimated via a calibration dilution curve and slope calculation. Expression levels were determined as the number of cycles needed for the amplification to reach a threshold fixed in the exponential phase of the PCR (CT). The data were normalized for the quantity of O. europaea ubiquitin (OeUB) gene [53]. Duplicates from three biological replicates were used in two independent experiments.
Electronic supplementary material
Additional file 1: Results for the 454 sequencing runs. (DOCX 11 KB)
Additional file 2: Summary of parameters used for the sequencing and assembly in the study of the olive transcriptomes: fruit (blue bars) and AZ (green bars) at 217 DPA. (A) Read-length distribution. A total of 443,811 good-quality sequence reads were obtained from the 2 samples. (B) Contig-length distribution. A total of 19,062 contigs were assembled from 199,075 redundant reads obtained after clustering and assemblage. The average contig length was around 500 bases. (C) Contig-read total distribution from fruit and AZ 454 sequencing data. (D) Isotig-length distribution. (TIFF 1 MB)
Additional file 3: List of 7,756 transcripts with Unigene ID in our experiment. (XLSX 1 MB)
Additional file 4: List of 4,391 differentially expressed genes in our experiment (P < 0.01, group I). (XLSX 784 KB)
Additional file 5: Genes overexpressed in the fruit pericarp in our experiment (P < 0.01, group I: Cluster A). (XLSX 297 KB)
Additional file 6: Genes overexpressed in the fruit AZ in our experiment (P < 0.01, group I: Cluster B). (XLSX 525 KB)
Additional file 7: Subcluster A1, A2, B1 and B2. (XLSX 782 KB)
Additional file 8: Proportion of annotated isotigs in each of the samples, and the proportion of annotated isotigs that present functional annotations of Gene Ontology (GO) or that are found annotated with the enzyme commission (EC) number. (TIFF 327 KB)
Additional file 9: Pathways identified through KEGG mapping. (XLSX 13 KB)
{kind=link}
Additional file 10: Graphic representation of the starch and sucrose metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the starch and sucrose metabolism pathway. (PNG 40 KB)
{kind=link}
Additional file 11: Graphic representation of the amino sugar and nucleotide sugar metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the amino sugar and nucleotide sugar metabolism pathway. (PNG 66 KB)
{kind=link}
Additional file 12: Graphic representation of the cysteine and methionine metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the cysteine and methionine metabolism pathway. (PNG 45 KB)
{kind=link}
Additional file 13: Graphic representation of the methane metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the methane metabolism pathway. (PNG 72 KB)
{kind=link}
Additional file 14: Graphic representation of the glycolysis/gluconeogenesis pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the glycolysis/gluconeogenesis pathway. (PNG 26 KB)
{kind=link}
Additional file 15: Graphic representation of the glycine, serine and threonine metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the glycine, serine and threonine metabolism pathway. (PNG 42 KB)
{kind=link}
Additional file 16: Graphic representation of the arginine and proline metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the arginine and proline metabolism pathway. (PNG 64 KB)
Additional file 17: Fruit-or AZ-enriched transcription factors at the last stage of olive fruit ripening. Sequences were selected after establishing a P < 0.01. The table shows the total read count in RPKMx1000 for each gene after normalization across the 2 samples: (a) fruit at 217 DPA, (b) AZ at 217 DPA. (DOCX 43 KB)
Acknowledgements
This work was supported by the ‘Ministerio de Ciencia e Innovación’, Spain (BFU2010-18116). We thank J.L. Grosson for free access to the plant material and R. Tobes (Era7) for technical assistance.
Abbreviations
- AZ
Abscission zone
- cDNA
Complementary deoxyribonucleic acid
- DPA
Days post-anthesis
- qRT-PCR
Quantitative real time polymerase chain reaction
- FC
Fold change
- GO
Gene ontology
- EST
Expressed sequence tag
- RPKM
Reads per kilobase of exon per million mapped reads.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RP collected plant samples, isolated the RNA samples, contributed to transcriptome sequencing, and real time PCR. MAP help in sample collection and other experiments. IMSC contributed to manuscript revision. MCGJ conceived and organized the experiments, performed transcriptome sequencing experiments, bioinformatics analyses, and preparation and manuscript critical edition. All authors read and approved the final manuscript.
Contributor Information
Ruben Parra, Email: rparraes@alumnos.unex.es.
Miguel A Paredes, Email: mparedes@unex.es.
Isabel M Sanchez-Calle, Email: imscalle@ugr.es.
Maria C Gomez-Jimenez, Email: mcgomez@unex.es.
References
- 1.Loureiro J, Rodriguez E, Costa A, Santos C. Nuclear DNA content estimations in wild olive (Olea europaea L. ssp. europaea var. sylvestris Brot.) and Portuguese cultivars of O. europaea using flow cytometry. Genet Resour Crop Evol. 2007;54:21–25. doi: 10.1007/s10722-006-9115-3. [DOI] [Google Scholar]
- 2.Alagna F, D’Agostino N, Torchia L, Servili M, Rao R, Pietrella M, Giuliano G, Chiusano ML, Baldoni L, Perrotta G. Comparative 454 pyrosequencing of transcripts from two olive genotypes during fruit development. BMC Genomics. 2009;10:399. doi: 10.1186/1471-2164-10-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galla G, Barcaccia G, Ramina A, Collani S, Alagna F, Baldoni L, Cultrera NG, Martinelli F, Sebastiani L, Tonutti P. Computational annotation of genes differentially expressed along olive fruit development. BMC Plant Biol. 2009;9:128. doi: 10.1186/1471-2229-9-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bianco L, Alagna F, Baldoni L, Finnie C, Svensson B, Perrotta G. PLoS ONE. 2013. Proteome regulation during Olea europaea fruit development. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stiti N, Triki S, Hartmann MA. Formation of triterpenoids throughout Olea europaea fruit ontogeny. Lipids. 2007;42:55–67. doi: 10.1007/s11745-006-3002-8. [DOI] [PubMed] [Google Scholar]
- 6.De la Rosa R, Angiolillo A, Guerrero M, Pellegrini M, Rallo L, Besnard G, Bervillé A, Martin A, Baldoni L. A first linkage map of olive (Olea europaea L.) cultivars using RAPD, AFLP, RFLP and SSR markers. Theor Appl Genet. 2003;106:1273–1282. doi: 10.1007/s00122-002-1189-5. [DOI] [PubMed] [Google Scholar]
- 7.Wu S, Collins G, Sedgley M. A molecular linkage map of olive (Olea europaea L.) based on RAPD, microsatellite, and SCAR markers. Genome. 2004;47:26–35. doi: 10.1139/g03-091. [DOI] [PubMed] [Google Scholar]
- 8.Mariotti R, Cultrera NGM, Munoz-Diez C, Baldoni L, Rubini A. Identification of new polymorphic regions and differentiation of cultivated olives (Olea europaea L.) through platome sequence comparison. BMC Plant Biol. 2010;10:211. doi: 10.1186/1471-2229-10-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gil-Amado JA, Gomez-Jimenez MC. Transcriptome analysis of mature fruit abscission control in olive. Plant Cell Physiol. 2013;54(2):244–269. doi: 10.1093/pcp/pcs179. [DOI] [PubMed] [Google Scholar]
- 10.Muñoz-Mérida A, González-Plaza JJ, Cañada A, Blanco AM, Del García-López MC, Rodríguez JM, Pedrola L, Sicardo MD, Hernández ML, De la Rosa R, Belaj A, Gil-Borja M, Luque F, Martínez-Rivas JM, Pisano DG, Trelles O, Valpuesta V, Beuzón CR. De novo assembly and functional annotation of the olive (olea europaea) transcriptome. DNA Res. 2013;20(1):93–108. doi: 10.1093/dnares/dss036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nath P, Sane AP, Trivedi PK, Sane VA, Asif MH. Role of transcription factors in regulating ripening, senescence and organ abscission in plants. Stewart Postharvest Rev. 2007;3:1–14. doi: 10.2212/spr.2007.2.6. [DOI] [Google Scholar]
- 12.Conde C, Delrot S, Gerós H. Physiological, biochemical and molecular changes occurring during olive development and ripening. J Plant Physiol. 2008;165(15):1545–1562. doi: 10.1016/j.jplph.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 13.Gomez-Jimenez MC, Paredes MA, Gallardo M, Fernandez-Garcia N, Olmos E, Sanchez-Calle IM. Tissue-specific expression of olive S-adenosyl methionine decarboxylase and spermidine synthase genes and polyamine metabolism during flower opening and early fruit development. Planta. 2010;232(3):629–647. doi: 10.1007/s00425-010-1198-6. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Jimenez MC, Paredes MA, Gallardo M, Sanchez-Calle IM. Mature fruit abscission is associated with up-regulation of polyamine metabolism in the olive abscission zone. J Plant Physiol. 2010;167(17):1432–1441. doi: 10.1016/j.jplph.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 15.Parra-Lobato MC, Gomez-Jimenez MC. Polyamine-induced modulation of genes involved in ethylene biosynthesis and signalling pathways and nitric oxide production during olive mature fruit abscission. J Exp Bot. 2011;62(13):4447–4465. doi: 10.1093/jxb/err124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu K, Xu Q, Da X, Guo F, Ding Y, Deng X. Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis) BMC Genomics. 2012;13:10. doi: 10.1186/1471-2164-13-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xiao S, Dai L, Liu F, Wang Z, Peng W, Xie D. COS1: an Arabidopsis coronatine insensitive1 suppressor essential for regulation of jasmonate-mediated plant defense and senescence. Plant Cell. 2004;16:1132–1142. doi: 10.1105/tpc.020370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castellarin S, Di Gaspero G. Transcriptional control of anthocyanin biosynthetic genes in extreme phenotypes for berry pigmentation of naturally occurring grapevines. BMC Plant Biol. 2007;7(1):46. doi: 10.1186/1471-2229-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Espley RV, Hellens RP, Putterill J, Stevenson DE, Kutty-Amma S, Allan AC. Red colouration in apple fruit is due to the activity of the MYB transcription factor, MdMYB10. Plant J. 2007;49(3):414–427. doi: 10.1111/j.1365-313X.2006.02964.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wei YZ, Hu FC, Hu GB, Li XJ, Huang XM, Wang HC. Differential expression of anthocyanin biosynthetic genes in relation to anthocyanin accumulation in the pericarp of Litchi Chinensis Sonn. PLoS One. 2011;6(4):e19455. doi: 10.1371/journal.pone.0019455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Niu SS, Xu CJ, Zhang WS, Zhang B, Li X, Lin-Wang K, Ferguson IB, Allan AC, Chen KS. Coordinated regulation of anthocyanin biosynthesis in Chinese bayberry (Myrica rubra) fruit by a R2R3 MYB transcription factor. Planta. 2010;231(4):887–899. doi: 10.1007/s00425-009-1095-z. [DOI] [PubMed] [Google Scholar]
- 22.Barrat D, Clark J. Proteins arising during the late stages of embryogenesis in Pisum sativum L. Planta. 1991;184:14–23. doi: 10.1007/BF00208230. [DOI] [PubMed] [Google Scholar]
- 23.Suarez-Rodriguez MC, Edsga D, Hussain SS, Alquezar D, Rasmussen M, Gilbert T, Nielsen BH, Bartels D, Mundy J. Transcriptomes of the desiccation-tolerant resurrection plant Craterostigma plantagineum. Plant J. 2010;63:212–228. doi: 10.1111/j.1365-313X.2010.04243.x. [DOI] [PubMed] [Google Scholar]
- 24.Wang L, Li Z, Qian W, Guo W, Gao X, Huang L, Wang H, Zhu H, Wu JW, Wang D, Liu D. The Arabidopsis purple acid phosphatase AtPAP10 is predominantly associated with the root surface and plays an important role in plant tolerance to phosphate limitation. Plant Physiol. 2011;157(3):1283–1299. doi: 10.1104/pp.111.183723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barford D, Das AK, Egloff MP. The structure and mechanism of protein phosphatases: insights into catalysis and regulation. Annu Rev Biophys Biomol Struct. 1998;27:133–164. doi: 10.1146/annurev.biophys.27.1.133. [DOI] [PubMed] [Google Scholar]
- 26.Brugiere N, Dubois F, Limami AM, Lelandais M, Roux Y, Sangwan RS, Hirel B. Glutamine synthetase in the phloem plays a major role in controlling proline production. Plant Cell. 1999;11(10):1995–2012. doi: 10.1105/tpc.11.10.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu CT, Bradford KJ. Class I chitinase and beta-1,3-glucanase are differentially regulated by wounding, methyl jasmonate, ethylene, and gibberellin in tomato seeds and leaves. Plant Physiol. 2003;133(1):263–273. doi: 10.1104/pp.103.024687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 1999;27:29–34. doi: 10.1093/nar/27.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fabi JP, Seymour GB, Graham NS, Broadley MR, May ST, Lajolo FM, Cordenunsi BR, Oliveira do Nascimento JR. Analysis of ripening-related gene expression in papaya using an Arabidopsis-based microarray. BMC Plant Biol. 2012;12:242. doi: 10.1186/1471-2229-12-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Klee HJ, Giovannoni JJ. Genetics and control of tomato fruit ripening and quality attributes. Annu Rev Genet. 2011;45:41–59. doi: 10.1146/annurev-genet-110410-132507. [DOI] [PubMed] [Google Scholar]
- 31.Scharf KD, Berberich T, Ebersberger I, Nover L. The plant heat stress transcription factor (Hsf) family: structure, function and evolution. Biochim Biophys Acta. 1819;2012:104–119. doi: 10.1016/j.bbagrm.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 32.Pajerowska-Mukhtar KM, Wang W, Tada Y, Oka N, Tucker CL, Fonseca JP, Dong X. The HSF-like transcription factor TBF1 is a major molecular switch for plant growth-to-defense transition. Curr Biol. 2012;22:103–112. doi: 10.1016/j.cub.2011.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mayrose M, Ekengren SK, Melech-Bonfil S, Martin GB, Sessa G. A novel link between tomato GRAS genes, plant disease resistance and mechanical stress response. Mol Plant Pathol. 2006;7(6):593–604. doi: 10.1111/j.1364-3703.2006.00364.x. [DOI] [PubMed] [Google Scholar]
- 34.Xu G, Livingston DM, Krek W. Multiple members of the E2F transcription factor family are the products of oncogenes. Proc Natl Acad Sci U S A. 1995;92:1357–1361. doi: 10.1073/pnas.92.5.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rushton PJ, Somssich IE, Ringler P, Shen QJ. WRKY transcription factors. Trends Plant Sci. 2010;15(5):247–258. doi: 10.1016/j.tplants.2010.02.006. [DOI] [PubMed] [Google Scholar]
- 36.Meir S, Philosoph-Hadas S, Sundaresan S, Selvaraj KSV, Burd S, Ophir R, Kochanek B, Reid MS, Jiang CZ, Lers A. Microarray analysis of the abscission-related transcriptome in the tomato flower abscission zone in response to auxin depletion. Plant Physiol. 2010;154:1929–1956. doi: 10.1104/pp.110.160697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai S, Lashbrook CC. Stamen abscission zone transcriptome profiling reveals new candidates for abscission control: enhanced retention of floral organs in transgenic plants overexpressing Arabidopsis ZINC FINGER PROTEIN2. Plant Physiol. 2008;146:1305–1321. doi: 10.1104/pp.107.110908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feller A, Machemer K, Braun EL, Grotewold E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011;66(1):94–116. doi: 10.1111/j.1365-313X.2010.04459.x. [DOI] [PubMed] [Google Scholar]
- 39.Wolberger C. Multiprotein-DNA complexes in transcriptional regulation. Annu Rev Biophys Biomol Struct. 1999;28:29–56. doi: 10.1146/annurev.biophys.28.1.29. [DOI] [PubMed] [Google Scholar]
- 40.Zimmermann IM, Heim MA, Weisshaar B, Uhrig JF. Comprehensive identification of Arabidopsis thaliana MYB transcription factors interacting with R/B-like BHLH proteins. Plant J. 2004;40:22–34. doi: 10.1111/j.1365-313X.2004.02183.x. [DOI] [PubMed] [Google Scholar]
- 41.Nijhawan A, Jain M, Tyagi AK, Khurana JP. Genomic survey and gene expression analysis of the basic leucine zipper transcription factor family in rice. Plant Physiol. 2008;146:333–350. doi: 10.1104/pp.107.112821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corbacho J, Romojaro F, Pech JC, Latché A, Gomez-Jimenez MC. Transcriptomic events involved in melon mature-fruit abscission comprise the sequential induction of cell-wall degrading genes coupled to a stimulation of endo and exocytosis. PLoS ONE. 2013;8(3):e58363. doi: 10.1371/journal.pone.0058363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Osterlund MT, Hardtke CS, Wei N, Deng XW. Targeted destabilization of HY5 during light-regulated development of Arabidopsis. Nature. 2000;405(6785):462–466. doi: 10.1038/35013076. [DOI] [PubMed] [Google Scholar]
- 44.Liu Y, Dai S, Beachy RN. Role of the C-terminal domains of rice (Oryza sativa L.) bZIP proteins RF2a and RF2b in regulating transcription. Biochem J. 2007;405(2):243–249. doi: 10.1042/BJ20061375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tucker ML, Whitelaw CA, Lyssenko NN, Nath P. Functional analysis of regulatory elements in the gene promoter for an abscission-specific cellulase from bean and isolation, expression, and binding affinity of three TGA-type basic leucine zipper transcription factors. Plant Physiol. 2002;130:1487–1496. doi: 10.1104/pp.007971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Botton A, Eccher G, Forcato C, Ferrarini A, Begheldo M, Zermiani M, Moscatello S, Battistelli A, Velasco R, Ruperti B, Ramina A. Signaling pathways mediating the induction of apple fruitlet abscission. Plant Physiol. 2011;155:185–208. doi: 10.1104/pp.110.165779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olsen AN, Ernst HA, Leggio LL, Skriver K. NAC transcription factors: structurally distinct, functionally diverse. Trends Plant Sci. 2005;10:79–87. doi: 10.1016/j.tplants.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 48.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 49.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8(6):469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 51.Vijay N, Poelstra JW, Künstner A, Wolf JB. Challenges and strategies in transcriptome assembly and differential gene expression quantification: a comprehensive in silico assessment of RNA-seq experiments. Mol Ecol. 2013;22(3):620–634. doi: 10.1111/mec.12014. [DOI] [PubMed] [Google Scholar]
- 52.Gene ontology consortium: The Gene Ontology in 2010: extensions and refinementsNucleic Acids Res 2010,38(Database issue):D331-D335. [DOI] [PMC free article] [PubMed]
- 53.Gil-Amado JA, Gomez-Jimenez MC. Regulation of polyamine metabolism and biosynthetic gene expression during olive mature-fruit abscission. Planta. 2012;235(6):1221–1237. doi: 10.1007/s00425-011-1570-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Results for the 454 sequencing runs. (DOCX 11 KB)
Additional file 2: Summary of parameters used for the sequencing and assembly in the study of the olive transcriptomes: fruit (blue bars) and AZ (green bars) at 217 DPA. (A) Read-length distribution. A total of 443,811 good-quality sequence reads were obtained from the 2 samples. (B) Contig-length distribution. A total of 19,062 contigs were assembled from 199,075 redundant reads obtained after clustering and assemblage. The average contig length was around 500 bases. (C) Contig-read total distribution from fruit and AZ 454 sequencing data. (D) Isotig-length distribution. (TIFF 1 MB)
Additional file 3: List of 7,756 transcripts with Unigene ID in our experiment. (XLSX 1 MB)
Additional file 4: List of 4,391 differentially expressed genes in our experiment (P < 0.01, group I). (XLSX 784 KB)
Additional file 5: Genes overexpressed in the fruit pericarp in our experiment (P < 0.01, group I: Cluster A). (XLSX 297 KB)
Additional file 6: Genes overexpressed in the fruit AZ in our experiment (P < 0.01, group I: Cluster B). (XLSX 525 KB)
Additional file 7: Subcluster A1, A2, B1 and B2. (XLSX 782 KB)
Additional file 8: Proportion of annotated isotigs in each of the samples, and the proportion of annotated isotigs that present functional annotations of Gene Ontology (GO) or that are found annotated with the enzyme commission (EC) number. (TIFF 327 KB)
Additional file 9: Pathways identified through KEGG mapping. (XLSX 13 KB)
Additional file 10: Graphic representation of the starch and sucrose metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the starch and sucrose metabolism pathway. (PNG 40 KB)
Additional file 11: Graphic representation of the amino sugar and nucleotide sugar metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the amino sugar and nucleotide sugar metabolism pathway. (PNG 66 KB)
Additional file 12: Graphic representation of the cysteine and methionine metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the cysteine and methionine metabolism pathway. (PNG 45 KB)
Additional file 13: Graphic representation of the methane metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the methane metabolism pathway. (PNG 72 KB)
Additional file 14: Graphic representation of the glycolysis/gluconeogenesis pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the glycolysis/gluconeogenesis pathway. (PNG 26 KB)
Additional file 15: Graphic representation of the glycine, serine and threonine metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the glycine, serine and threonine metabolism pathway. (PNG 42 KB)
Additional file 16: Graphic representation of the arginine and proline metabolism pathway by KEGG. Boxes colored in red represent the EC number of the enzymes encoded by differentially expressed genes generated by this study (fruit at 217 DPA vs. AZ at 217 DPA) that are homologous to genes involved in the arginine and proline metabolism pathway. (PNG 64 KB)
Additional file 17: Fruit-or AZ-enriched transcription factors at the last stage of olive fruit ripening. Sequences were selected after establishing a P < 0.01. The table shows the total read count in RPKMx1000 for each gene after normalization across the 2 samples: (a) fruit at 217 DPA, (b) AZ at 217 DPA. (DOCX 43 KB)