

Abstract
HIF prolyl-4-hydroxylase 2 (PHD2) is a non-heme Fe, 2-oxoglutarate (2OG) dependent dioxygenase that regulates the hypoxia inducible transcription factor (HIF) by hydroxylating two conserved prolyl residues in N-terminal oxygen degradation domain (NODD) and C-terminal oxygen degradation domain (CODD) of HIF-1α. Prior studies have suggested that the substrate preference of PHD2 arises from binding contacts with the β2β3 loop of PHD2. In this study we tested the substrate selectivity of PHD2 by kinetic competition assays, varied ionic strength, and global protein flexibility using amide H/D exchange (HDX). Our results revealed that PHD2 preferred CODD by 20-fold over NODD and that electrostatics influenced this effect. Global HDX monitored by mass spectrometry indicated that binding of Fe(II) and 2OG stabilized the overall protein structure but the saturating concentrations of either NODD or CODD caused an identical change in protein flexibility. These observations imply that both substrates stabilize the β2β3 loop to the same extent. Under unsaturated substrate conditions NODD led to a higher HDX rate than CODD due to its lower binding affinity to PHD2. Our results suggest that loop closure is the dominant contributor to substrate selectivity in PHD2.
Keywords: HIF, hypoxia, non-heme iron, α-ketoglutarate, HIF-prolyl hydroxylase
1. Introduction
Oxygen homeostasis in humans and other metazoans is transcriptionally regulated by the hypoxia inducible factor-1α (HIF-1α)[1, 2], which governs cellular responses to hypoxia, such as the balance of aerobic/anaerobic metabolism, angiogenesis, and erythropoiesis. HIF-1α stability is controlled by the HIF-prolyl hydroxylases (PHD1, PHD2 and PHD3 in humans)[3, 4], which hydroxylate specific Pro residues found in the N- and C-terminal O2-dependent degradation domains of HIF-1α: NODD (Pro402) and the CODD (Pro564), leading to proteasomal degradation of HIF-1α[5, 6]. Hydroxylation of either Pro402 or Pro564 is sufficient for HIF-1α to bind tightly to pVHL[5, 6], leading to proteasomal degradation [7]. A key question in this field is substrate selectivity of PHD isoforms toward the ODD domains, as this determines the stability of HIF-1α. As PHD2 is thought to be the primary O2-sensor under normoxic conditions [8], detailed enzymological studies of PHD2 are important both to understand the chemical mechanisms of prolyl hydroxylation, as well as to identify the basis for substrate selectivity in PHD2 and related enzymes.
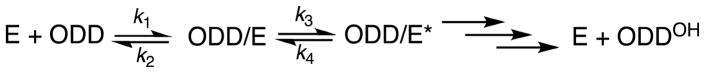
PHD2 is a non-heme Fe(II), 2-oxoglutarate (2OG)-dependent oxygenase that is thought to follow the consensus mechanism for related oxygenases (Scheme 1) in which substrates bind in the order of 2OG, primary substrate (ODD), then O2 [9–11]. Binding of the primary substrate triggers O2 binding by inducing the formation of a 5-coordinate Fe(II) cofactor [10, 12], and associated changes to local contacts within the active site. Oxidative decarboxylation leads to formation of a putative Fe(IV)O intermediate [13–15] (Scheme 1), which effects hydroxylation of the target Pro residue (CODD, Pro564; NODD, Pro402) to complete the cycle. As the half-reactions are sequential, oxidative decarboxylation that is uncoupled from hydroxylation can occur [16], leading to excessive consumption of 2OG in some cases.
Scheme 1.

Consensus mechanism and ODD sequences.
Cell-based assays have shown that the PHD isoforms exhibit differential selectivity toward the ODD domains, with PHD2 reacting more rapidly with CODD than NODD in HIF-1α constructs [17]. Immunoblotting showed CODD hydroxylation prior to NODD hydroxylation in cell-based assays suggesting some level of synergy between the two sites [18], however binding assays of very long ODD constructs revealed that CODD bound to purified PHD2 with roughly ten-fold increased affinity relative to NODD [19], suggesting that intrinsic binding affinity may explain the apparent hydroxylation order. Due to the complicated relationship between local and extended ODD sequences on PHD2 activity [18, 20], we used short peptides (~20 residues) containing the local ODD sequences to test for local effects on the substrate selectivity of PHD2.
There are conflicting reports as to the substrate selectivity of PHD2 toward the two ODD domains, as the reactions of PHD2 with NODD and CODD have been reported with widely varying rates (kcat/KM) and Michaelis constants (KM). Early reports based on 2OG consumption assays were widely divergent in terms of reported KM values. Assays using PHD2 within crude cell lysates indicated a strong preference for CODD (KM(NODD) = 130 μM, KM(CODD) = 7 μM) [21]; whereas assays using purified PHD2 indicated essentially equivalent substrate preferences: (KM(NODD) = 44 μM, KM(CODD) = 37 μM) [19]. In contrast, O2-consumption assays using purified PHD2 indicated a marked preference for CODD (KM(NODD) = 24 μM, KM(CODD) = 2 μM), differing from the results of the 2OG assay [22]. Although the biochemical data indicate that CODD is preferred, the wide variation in KM presents a challenge to understanding how the rates relate to substrate binding for PHD2.
The large variation in reported values for the Michaelis constants, with KM(NODD) values ranging from 20 – 50 μM and KM(CODD) values ranging from 1 – 30 μM [17, 19, 23], is largely a reflection of three factors that introduce variation into the kinetic data, two of which are common to the broad class of 2OG oxygenases. First is the inherent difficulty of measuring turnover for the 2OG oxygenases, due to the relatively slow rate of turnover and the absence of chromophoric substrates. This has led to a reliance in many reports on the decarboxylation assay that measures the release of 14CO2 from [1-14C]2OG. Second is the tendency for 2OG oxygenases to uncouple oxidative and reductive half-reactions, leading to decarboxylation without substrate hydroxylation [24–26]. This in turn greatly impacts assays that rely on the rate of CO2 liberation or O2 consumption, and certainly leads to over-estimation of turnover rates. Third is the pKA = 7.2 of the Fe2+-OH2 group in (Fe+2OG)PHD2 [23], leading to a significant decrease in activity as pH is increased above the pKa [27]. Based on these divergent Michaelis constants, and the potential for uncoupled oxidative decarboxylation to inflate the observed rates, we felt that the substrate preference and rates could be better defined by directly measuring the formation of hydroxylated product (ODDOH) using MALDI-TOF [28, 29].
It appears that PHD2 may discriminate NODD from CODD due to a combination of electrostatics and a conformational change of the β2β3 loop (Fig. 1). Closure of the β2β3 loop is essential for tight ODD binding, as shown by the crystal structure of CODD/(Mn+NOG)PHD2 [30], and activity assays with chimeric PHD enzymes [7]. Sequence comparison shows that CODD contains six acidic residues, compared to three for NODD (Scheme 1), suggesting that electrostatics may be the origin of differential binding – this is supported by the sequence preference of PHD2 (–AP(Y/F)(I/L)X4(D/E)–) obtained from yeast two-hybrid binding assays [31]. Further, protease susceptibility assays showed that CODD/PHD2 was greatly stabilized relative to NODD/PHD2, both in terms of the length of time required for proteolysis as well as in the proteolytic site on the β2β3 loop stabilized by ODD binding [32].
Fig. 1.
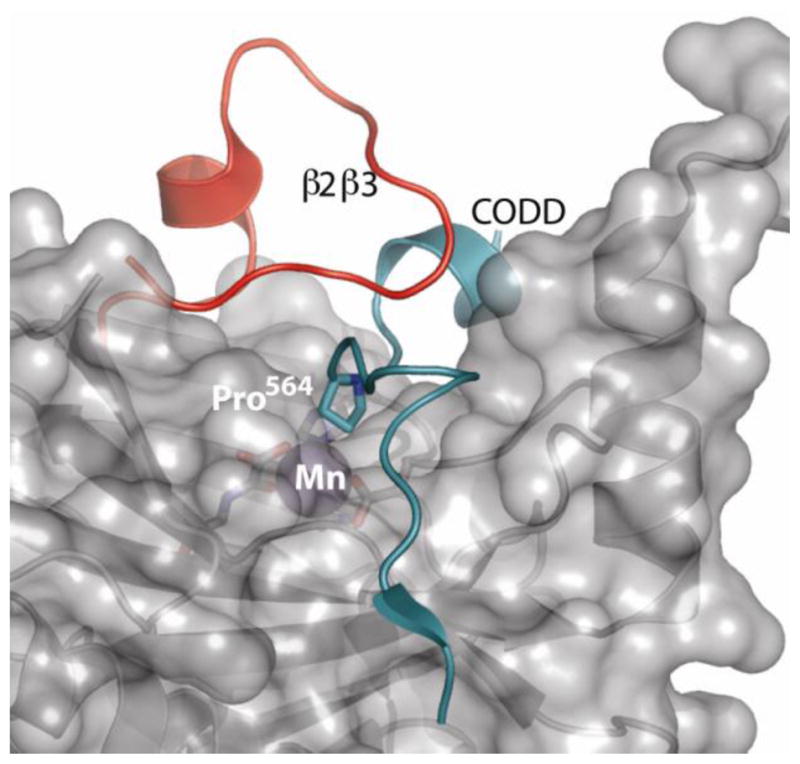
Structure of (Mn+NOG)PHD2 bound to CODD [30] showing the β2β3 loop (red) and CODD (cyan).
The contribution of ODD/PHD2 binding to the catalytic efficiency of PHD2 (kcat/KM) is not fully defined, despite its importance to O2 sensing. Catalytic efficiency dictates the rate constant under conditions of low substrate concentration, and provides insight into the basis for discriminating NODD from CODD. Microscopic steps contributing to kcat/KM include those steps between the diffusional collision with ODD through the first irreversible step – as a consequence, weaker binding of NODD relative to CODD could constitute the entire basis for substrate selectivity of PHD2. Furthermore, binding equilibria can contribute to overall rate-limitation, making understanding the binding ODD/PHD2 process a key factor in understanding the chemistry of O2-sensing.
Here we tested the origin of substrate selectivity of PHD2 by direct competition assays, increased ionic strength, and global protein flexibility for PHD2 upon binding ODD. Our results indicate that PHD2 prefers CODD by 20-fold over NODD, and that electrostatics accounts for a portion of this effect. We also performed global hydrogen deuterium exchange experiments (HDX) with the aim of monitoring possible structural changes upon binding of each substrate to the enzyme. Our global HDX data reveals that binding of Fe(II) and 2OG to the enzyme active site stabilizes the overall enzyme structure as reported previously by proteolysis studies [32], however we noted a crucial difference from that earlier report. At saturating concentrations of substrate, CODD and NODD gave indistinguishable HDX profiles, which strongly suggests that both substrates stabilize the β2β3 loop to the same extent. This implicates loop closure as the dominant contributor to selectivity in PHD2.
2. Experimental Procedures
2.1 Materials
All chemicals were purchased from commercial vendors, and used without purification with the exception of NODD. The sequences of the peptide substrates were derived from the native HIF-1α395–414 (NODD) and HIF-1α556–574 (CODD) sequences. The sequence of the NODD peptide used in this work was DALTLLAP402AAGDTIISLDYG and the CODD peptide was DLDLEALAP564YIPADDDFQL. The underlined residues were changed from the native sequences as follows: NODD (F413Y), CODD (M561A, M568A). The NODD peptide (desalted) was purchased from EzBiolab (Carmel, IN, USA) and purified by C18 column reversed phase-HPLC using an H2O/CH3CN gradient (5 – 95% CH3CN, 0.1% TFA), whereas the CODD peptide (99 % purity) was purchased from GL Biochem LTD (Shanghai).
2.2 Protein expression and purification
Recombinant human PHD2 (corresponding to residues 178–426 of the full-length PHD2 sequence) was expressed and purified as previously reported [23]. PHD2 was expressed with an N-terminal GST tag (GST-PHD2) in E. coli BL21(DE3) cells using a pGEX-4T-1 vector (Stratagene). The GST-PHD2 fusion was purified using an affinity column (GSTrap, GE Bioscience), then the GST tag was removed by incubating GST-PHD2 with restriction grade thrombin for 16 hours at 4 °C. Thrombin was removed by a Hitrap Benzamidine column (GE Bioscience), and un-cleaved GST-PHD2 and GST was removed with a GSTrap column. PHD2 was treated with 50 mM EDTA then buffer exchanged into 50 mM HEPES pH 7.00 and stored at − 20.0 °C. PHD2 purity was checked by SDS-PAGE, and the molecular weight determined using a QStar-XL hybrid quadrupole-TOF mass spectrometer (Applied Biosystems).
2.3 Steady-state kinetic assays
All activity assays were performed with saturating concentrations of 2OG (100 μM), (NH4)2Fe(SO4)2 (20 μM), ascorbic acid (1 mM), and 1 μM PHD2 in 50 mM HEPES pH 7.00 at 37.0 °C. Reactions were quenched at different time points with 2:1 ratio of acetonitrile and 0.2 % trifluoroacetic acid saturated with 4-α-cyano hydroxycinnamic acid. Samples were then spotted onto a target plate and analyzed by a Bruker Daltonics Omniflex MALDI-TOF mass spectrometer. Initial rates (v0) were obtained from the temporal change in mol fraction of hydroxylated ODDOH (χODDOH), as v0 = [ODD]0 χODDOH /t.
2.4 Global hydrogen deuterium exchange (HDX) kinetics of PHD2
Global HDX experiments were measured with an ABI QStar-XL mass spectrometer. PHD2 (50 μM) was pre-incubated in 10 mM NH4OAc at pH 7.00 on ice with the cofactor and substrates as indicated: MnSO4 (200 μM), 2OG (1.0 mM), and CODD or NODD (0 – 500 μM). Mn2+ was used as a structural mimic of Fe2+ in order to avoid spurious oxidation. The HDX reactions were initiated by diluting 3 μL of PHD2 samples with 27 μL 10 mM NH4OAc (in D2O) pD 7.00 at 25 °C, with aliquots quenched by injecting onto an HPLC loop at various time points from 15 second to 60 minutes. Solution pD was determined by adding 0.4 to the pH meter reading (pD = pHmeter + 0.4). HDX aliquots (20 μL) were injected and buffer exchanged using a C18 HPLC column with a 0.1 % formic acid (pH 2.5) mobile phase. The HPLC column, injection loop, and mobile phases were kept in an ice bath to minimize amide back exchange during sample workup. PHD2 was eluted with a 10 minute gradient, (H2O/CH3CN, 0.1% formic acid), and was directly sprayed into an ABI QStar-XL for determination of global deuterium incorporation level. Data was analyzed using the Bioanalyst software.
During analysis of HDX samples some amide hydrogens can back exchange with the HPLC solvent, leading to an apparent loss of deuteration. In order to correct for back exchange occurring during the LC steps, the deuterium content of PHD2 at each quench point was corrected through comparison with the mass of a non-deuterated sample (m0) and the mass of a fully deuterated PHD2 sample (m100). The fully deuterated protein was prepared by incubation in D2O for a day at 37.0 °C followed by 10 minutes at 70 °C. The number of exchangeable backbone amides (D) was then calculated by subtracting the number of Pro residues (12) and the N-terminus from of the total number of exchangeable amides on PHD2 (N = 236).
The number of deuterons retained (D) was analyzed as a function of exchange time by fitting D to three pools of amides (A) that exchange at fast (kfast = 20 min−1), medium (kmed = 1 min−1), and slow (kslow = 0.05 min−1) rates. Amides that were not exchangeable on the 60 min timescale were taken as the frozen pool of amides (Afrozen = N – Atot).
3. Results
3.1 PHD2 shows a preference for CODD over NODD in the presence of both substrates
PHD2 hydroxylates Pro402(NODD) and Pro564(CODD) of HIF-1α, however, reports conflict as to the relative substrate preference. Some reports indicated that both Pro residues were hydroxylated at a similar rate, whereas other reports suggested that CODD was hydroxylated at a faster rate than NODD. In order to determine the substrate selectivity of PHD2 activity assays were performed under competitive conditions in the presence of 20 μM total substrate, but with a varying ratio of NODD:CODD. As we directly monitored hydroxylation via MALDI, both CODD and NODD could be measured simultaneously. The initial rates for hydroxylation of both ODDs were plotted as a function of varied mole fraction of NODD (Figure 2). As χNODD was increased, the initial rate for CODD hydroxylation decreased from 1.3 min−1 to 0.8 min−1; conversely, the initial rate for NODD hydroxylation increased from 0.04 min−1 to 0.6 min−1. The initial rates at χNODD = 0.5 for CODD (v0/E = 0.76 ± 0.02 min−1) and NODD (v0/E = 0.03 ± 0.02 min−1) exhibit a high level of precision. This data shows that PHD2 preferentially hydroxylated CODD in the presence of NODD even at elevated mole fraction of NODD (Figure 2).
Fig. 2. Substrate selectivity of PHD2 under competitive condition.

PHD2 (1 μM), 2OG (100 μM), ODD (2–18 μM; 20 μM total), (NH4)2Fe(SO4)2 (20 μM) and ascorbic acid (1 mM) in 50 mM HEPES pH 7.00 at 37 °C. (CODD (○), NODD (●)).
3.2 Columbic forces are essential in substrate preference of PHD2
Crystallography showed the ODD-binding surface of PHD2 contained several basic residues whereas the ODD substrates contain acidic residues flanking the LXXLAP motif suggesting that electrostatics may be important in binding substrates to PHD2. In order to test the role of electrostatics in PHD2 substrate recognition, activity assays were performed in the presence of varied NaCl concentration. The initial rates of PHD2 were measured as a function of varying ODD concentration in the presence and in the absence of 100 mM NaCl, then the rate constants kcat and kcat/KM were determined by fitting the data to the Michaelis-Menten equation (Table 1).
Table 1.
Steady state kinetic constants for PHD2 using CODD or NODD
| Substrate peptide | kcat (min−1) | kcat/KM (μM−1min−1) | KM (μM) |
|---|---|---|---|
| CODD (0 mM NaCl) | 1.13 ± 0.05 | 1.0 ± 0.1 | 1.0 ± 0.3 |
| CODD (+ 100 mM NaCl) | 1.2 ± 0.1 | 0.30 ± 0.06 | 4.2 ± 1.2 |
| NODD (0 NaCl) | 0.61 ± 0.06 | 0.04 ± 0.01 | 14 ± 3 |
| NODD (+ 100 mM NaCl) | 0.62 ± 0.04 | 0.05 ± 0.01 | 11 ± 3 |
Fixed concentrations of 2OG (100 μM), (NH4)2Fe(SO4)2 (20 μM), ascorbic acid (1 mM), and ambient O2 (217 μM), but varied ODD (0 – 50 μM) in 50 mM HEPES pH 7.00 at 37.0 °C
In the absence of added NaCl, the kcat/KM value for CODD (1.0 μM−1min−1) was 25 fold higher than kcat/KM for NODD (0.04 μM−1min−1) showing that PHD2 was more reactive toward CODD than NODD (Fig. 3 and Table 1). The addition of 100 mM NaCl did not alter the kcat/KM value of NODD (0.05 μM−1min−1) but decreased the kcat/KM for CODD (0.3 μM−1min−1), indicating that attractive columbics were operative in CODD/PHD2 binding, but not in NODD/PHD2 binding. A fuller investigation of the ionic strength dependence was not pursued.
Fig. 3. The effect of 100 mM NaCl on the kinetics of ODD hydroxylation by PHD2.

PHD2 was incubated with saturating 2OG (100 μM), (NH4)2Fe(SO4)2 (20 μM), and ascorbic acid (1 mM), but varied ODD (0 – 50 μM) in 50 mM HEPES pH 7.00 at 37.0 °C. CODD, (○) no NaCl added and (●) 100 mM NaCl added. NODD, (△) no NaCl added and (▲) 100 mM NaCl added.
3.3 NODD and CODD bind equivalently to PHD2
Global amide HDX was used to test solvent protection of PHD2 upon binding NODD and CODD. Apo PHD2 (no added substrate or cofactor) and (Mn+2OG)PHD2 were analyzed as reference states, in which Mn(II) was used as a isosteric replacement for Fe(II) as was done previously for PHD2 crystallography [30]. Solvent protection of NODD/(Mn+2OG)PHD2 and CODD/(Mn+2OG)PHD2 was measured under conditions of both low (20 μM) and high (50 μM) ODD concentrations to observe the effects of the ODD/PHD2 binding equilibrium. Amide HDX timecourses (Fig. 4) for PHD2 in different cofactor/substrate bound states were fitted by binning the amides into three pools (An) based on average exchange rates: fast (kfast = 20 min−1), intermediate (kmed = 1 min−1), slow (kslow = 0.05 min−1); the remaining amides were inaccessible to D2O solvent.
Fig. 4. Global amide HDX kinetics of PHD2.
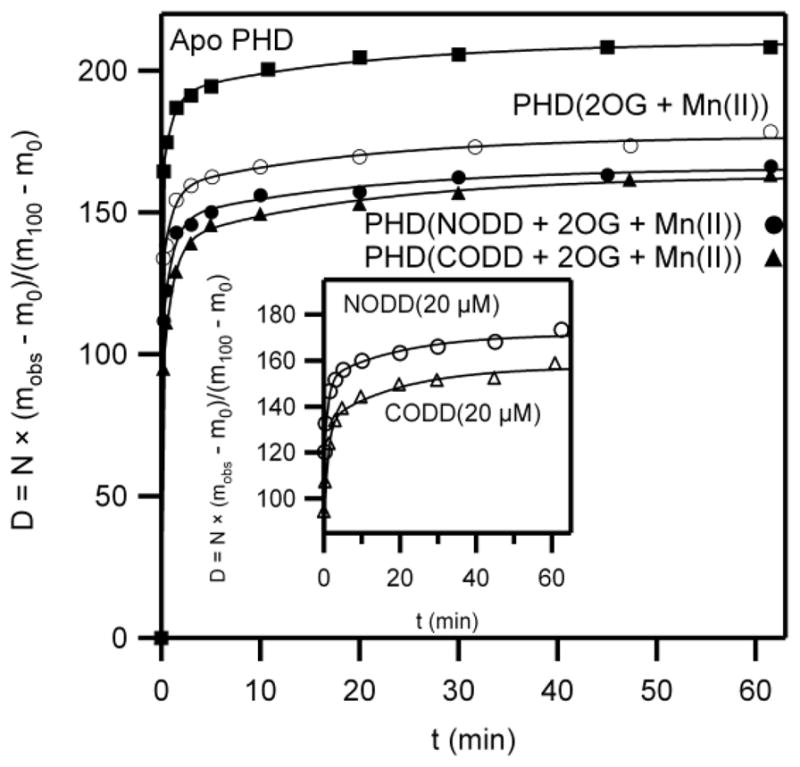
(■) PHD2 (5 μM) in 10 mM NH4OAc pH 7.0, (○) PHD2 (5 μM), MnSO4 (20 μM), and 2OG (100 μM) (●) PHD2 (5 μM), MnSO4 (20 μM) 2OG (100 μM), and NODD (50 μM), (▲) PHD2 (5 μM), Mn(SO4) (20 μM), 2OG (100 μM), and CODD (50 μM). Inset: low concentration conditions, [NODD] or [CODD] = 20 μM.
Amide exchange occurs from solvent-exposed amides, which are those located in transiently unfolded or unstructured regions of the protein. Solvent-protected amides are those that are either buried within a tightly structured core of the protein, or else are located at a macromolecular binding surface. The key pool for interpreting the PHD2 conformational change are the inaccessible amides (Afrozen), as these are either buried in the folded core of the PHD2, or protected from solvent by ODD binding.
Solvent protection for PHD2 increased upon cofactor binding, as seen comparing (Mn+2OG)PHD2 to apo PHD2. The frozen amide population increased from Afrozen = 26 in Apo PHD2 to Afrozen = 59 in (Mn+2OG)PHD2, indicating that the core of the PHD2 had become more tightly structured upon binding cofactor (Table 2). This is likely due to the stabilizing effect of metal and 2OG binding, which connects several strands of the β-barrel [30, 33].
Table 2.
| Apo | (Mn+2OG) | 20 μM NODD (Mn+2OG) | 20 μM CODD (Mn+2OG) | 50 μM NODD (Mn+2OG) | 50 μM CODD (Mn+2OG) | |
|---|---|---|---|---|---|---|
| Atot | 210 ± 1 | 177 ± 1 | 171 ± 1 | 157 ± 1 | 166 ± 1 | 163 ± 1 |
| Afast | 160 ± 2 | 128 ± 2 | 116 ± 3 | 85 ± 3 | 105 ± 3 | 87 ± 3 |
| Amed | 31 ± 2 | 31 ± 2 | 36 ± 3 | 48 ± 3 | 42 ± 3 | 53 ± 3 |
| Aslow | 19 ± 1 | 18 ± 2 | 20 ± 2 | 24 ± 2 | 18 ± 2 | 24 ± 2 |
| Afrozen | 26 | 59 | 65 | 79 | 70 | 73 |
Accessible amides: total, Atot; slow, Aslow, (kslow = 0.05 min−1); intermediate, Amed (kmed = 1 min−1); fast, Afast (kfast = 20 min−1).
PHD2 (5 μM) in 10 mM NH4OAc pH 7.00, MnSO4 (20 μM) and 2OG (100 μM); NODD and CODD as indicated.
Binding of NODD or CODD under saturating conditions (50 μM) led to equivalent increases in the froze amide pool when compared with (Mn+2OG)PHD2 (Table 2). The 50 μM NODD/(Mn+2OG)PHD2 sample exhibited an appreciable increase in the frozen amide pool (Afrozen = 70), which is most likely due to solvent protection at the binding interface. A nearly identical increase in the frozen amide pool was observed for the 50 μM CODD/(Mn+2OG)PHD2 sample (Afrozen = 73). The small difference in the frozen amide pool between the NODD and CODD samples (ΔAfrozen = 3) is within experimental uncertainty, and strongly suggests that both NODD and CODD bind to the same surface of PHD2, stabilizing the β2β3 loop to equivalent extents.
In contrast, the binding of NODD or CODD at low concentrations (20 μM) led to different frozen amide pools relative to (Mn+2OG)PHD2 (inset, Fig. 4). The large difference in frozen amide population for 20 μM NODD binding relative to 20 μM CODD binding (ΔAfrozen = 14) is well outside of experimental uncertainty, and indicates a difference in binding equilibrium. This is most likely due to the stronger binding of CODD (KM = 1 μM) relative to NODD (KM = 14 μM), and the position of the ODD + PHD2 binding equilibrium.
One observation worth noting is the change in the number of amides undergoing exchange on particular timescales (Afast and Amed) upon ODD binding. It was found that saturating [ODD] had the general effect of redistributing amides from faster pools into slower pools – in particular, incubating (Mn+2OG)PHD2 with 50 μM ODD decreased the number of fast amides (Afast), but increased the number of amides undergoing intermediate exchange (Amed).
4. Discussion
The substrate selectivity of PHD2 has been the subject of confusing literature reports, which have been clarified with the present work using purified PHD2 and short ODD peptides. Most prior reports indicated that CODD was preferred over NODD, however the Michaelis constants (KM) and catalytic efficiency of PHD2 (kcat/KM) have been subject to variations of ~10-fold. These variations likely arose from complications with the various assays used to measure turnover. We used kinetic assays that directly measure the formation of hydroxylation product (ODDOH) to avoid potential complications from uncoupled oxidations or side-reactions that may impede the 2OG assay. Our amide HDX approach directly measured solvent protection upon binding NODD or CODD, in order to compare the binding footprints of these two substrates.
Our kinetics data indicate that PHD2 prefers the CODD substrate by ~20-fold over the NODD substrate (Table 1, kcat/KM). Further, this substrate preference is sufficient to explain the relative order of hydroxylation observed in longer ODD constructs [17, 18, 20], without the need to invoke any sort of sequential cross-communication between the ODD segments in HIF-1α. These observations were in good agreement with the KM values obtained by the O2-consumption assay [22], but differed significantly from those obtained by the 2OG assay [19, 21]. It appears that the 2OG assays suffer from non-ideal behavior at low substrate concentrations leading to inflated KM. This may be due to uncoupling of oxidative decarboxylation or from non-enzymatic decarboxylation, contributing to the observed rates of 2OG turnover. By measuring ODDOH formation directly, the MALDI assay used herein avoided these complications, and is recommended for steady-state kinetics assays.
Direct competitive experiments under varying CODD:NODD ratios (Fig. 2) indicated that PHD2 prefers to hydroxylate CODD, even at low CODD:NODD ratio – consequently, CODD should be hydroxylated first under physiological conditions. This is as expected based solely on the catalytic efficiency of PHD2 toward these two substrates (Table 1). These results are in accord with a prior competition assay [19], however our results were obtained at low concentrations of substrate ([NODD] + [CODD] = 20 μM), corresponding to conditions appropriate for discussing catalytic efficiency.
The substrate preference of PHD2 is a direct product of the catalytic efficiency (kcat/KM) of this enzyme, which is largely a result of differential substrate binding. A minimal kinetic model for catalysis at sub-saturating ODD includes diffusional encounter and release (k1 and k2), a kinetically coupled β2β3 loop closure (k3 and k4), and then subsequent steps including the oxidative decarboxylation. Our results, combined with existing structural and protease susceptibility data [30, 32], indicate that there are two coupled steps necessary for substrate binding: diffusional encounter (k1) and β2β3 loop closure (k3).
As CODD has 6 negatively charged residues and NODD has only 3 negatively charged residues, we thought it likely that electrostatic attraction to PHD2 would be more pronounced for CODD than for NODD. We observed that adding 100 mM NaCl decreased the catalytic efficiency of PHD2 toward CODD (kcat/KM) by ~ 3-fold, but had no effect on the reaction with NODD (Table 1). This is most likely due to an increase in the rate of substrate release (k2) for CODD at elevated [NaCl]; nevertheless, this did not equalize the substrate affinity. We note that CODD remained ~ 7 fold better as a substrate than NODD, even at 100 mM NaCl, which suggests that a subsequent step may further discriminate these two substrates. This subsequent step is most likely to be closure of the β2β3 loop to adopt a catalytically competent conformation, as seen crystallographically [30]. Notably, PHD2 binding kinetics using substrates very similar to those used herein showed that NODD and CODD bound to PHD2 with identical rates, but NODD release was ~ 8 times faster than CODD in the presence of elevated [NaCl] [19]. This difference in binding affinity is in excellent agreement with the difference in catalytic efficiency toward the two substrates, which indicates that binding is the principle origin of substrate selectivity in PHD2.
Previously, protease susceptibility revealed greater protection of the β2β3 loop from protease digestion for CODD/PHD2 than for NODD/PHD2 [32], which was interpreted to indicate that CODD bound to a larger surface of PHD2 than NODD. In contrast, our global amide HDX data shows that both NODD and CODD protect PHD2 to identical extents, suggesting that these substrates bind to identical surfaces. This discrepancy arises, in our opinion, from the weaker binding equilibrium for NODD/PHD2 than for CODD/PHD2, as shown by the amide protection data shown in Fig. 4. It is likely that the NODD/PHD2 binding equilibrium was not saturated in the earlier report [32], leading to a detectable fraction of substrate-free PHD2 during the protease digestion assay.
The closed conformation of the β2β3 loop in the CODD/PHD2 structure [30] suggested that loop closure plays a role in catalysis. We tested the relative ability of CODD and NODD to induce the closed conformation of this loop at low and high [ODD] concentrations with (Mn2++2OG) constituted PHD2 using global amide HDX/MS. The reasons for using Mn2+ in the place of Fe2+ were several-fold: Mn2+ was used as an isosteric replacement for Fe2+ in an earlier crystallographic study [30]; both metal ions possess a similar charge/size ratio; and Mn2+ is air-stable, thereby avoiding any potential oxidation of the metal center during the amide HDX experiment. Amide HDX is uniquely capable of testing ODD/PHD2 binding, reporting on changes in solvent access upon ODD binding. NODD or CODD led to identical solvent protection when both were present at saturating concentrations (50 μM) (Fig. 4). The simplest interpretation of this data is that closing of the β2β3 loop of PHD2 is kinetically coupled to ODD binding.
In contrast, the use of moderate ODD concentrations (20 μM) revealed differences in solvent protection (Fig. 4). These differences are simply the result of the tighter binding of CODD (KM = 1.2 μM), than of NODD (KM = 14 μM); as CODD/PHD2 is saturated under this condition, whereas NODD/PHD2 is not saturated – CODD/PHD2 is fully protected from solvent whereas NODD/PHD2 is only partially protected. As both NODD and CODD induce identical amide protection when fully saturating, it is very likely that the substrate selectivity of PHD2 does not arise from altered ODD/PHD2 contacts, as previously suggested [32], but rather from CODD being able to push the equilibrium for β2β3 loop closure forward.
The number of amides exchanging on an intermediate timescale (Amed) increased from ~ 30 to ~ 48 when ODD was bound to (Mn+2OG)PHD2 (Table 2). Notably, this appears to be more than a simple slowing of a large population of amides, as the pool undergoing slow exchange (Aslow) increases to an insignificant extent upon ODD binding. We propose that a conformational change in the PHD2/ODD adduct that occurrs on this intermediate timescale (kconf ~ kmed = 1 min−1), leading to the increased pool of intermediate amides. As this timescale is remarkably similar to the rate of turnover under saturation (kcat = 1.1 min−1), such a conformational change could be partially rate-limiting. When considered in light of the conformational change by the β2β3 loop induced by substrate binding [30], we think it very likely that a decrease in the rate of β2β3 loop closure leads to this change in amide exchange.
Although the rate-limiting step for PHD2 turnover remains to be identified, this step precedes decarboxylation when ODD is saturating, as no oxidized intermediates were observed in the pre-steady state [34], and a slightly inverse solvent isotope effect on kcat was observed [23]. The results reported herein suggest that the kinetically coupled closing of the β2β3 loop may limit the rate of turnover under conditions of low ODD concentration. This has important ramifications for the physiological function of PHD2, for two key reasons. First, it indicates that CODD is hydroxylated before NODD simply due to the catalytic efficiency of PHD2 toward these substrates. Second, it suggests the potential to regulate PHD2 function by post-translational modifications that alter the charge of either the ODDs or the β2β3 loop of PHD2 (eg: phosphorylation), as this would directly impact the ability PHD2 to bind the ODD.
Supplementary Material
Scheme 2.
Highlights.
PHD2 prefers the CODD substrate by 20-fold over the NODD substrate
Substrate selectivity can be explained by the relative rate constants for each substrate
Electrostatics accounts for a small part of the substrate selectivity
Both NODD and CODD induce the same conformational change in PHD2
Acknowledgments
We thank the NIH for funding (R01-GM077413). SP was partially supported by the Turkish ministry of national education.
Abbreviations
- 2OG
2-oxoglutarate
- CODD
C-terminal transactivation domain
- ESI-MS
electrospray ionization mass spectrometry
- FIH, FIH-1
the factor inhibiting HIF
- GST
glutathione S-transferase
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HIF
Hypoxia Inducible Factor
- MALDI-TOF
matrix-assisted laser desorption time of flight
- NODD
N-terminal transactivation domain
- NOG
N-oxalyl glycine
- PHD2
HIF prolyl hydroxylase domain 2
- pVHL
von-Hippel Lindau protein
- SIE
solvent isotopic effect
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang GL, Jiang BH, Rue E, Semenza GL. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Semenza GL. Physiology. 2004;19:176–182. doi: 10.1152/physiol.00001.2004. [DOI] [PubMed] [Google Scholar]
- 3.Bruick RK. Genes Dev. 2003;17:2614–2623. doi: 10.1101/gad.1145503. [DOI] [PubMed] [Google Scholar]
- 4.Bruick RK, McKnight SL. Science. 2001;294:1337–1340. doi: 10.1126/science.1066373. [DOI] [PubMed] [Google Scholar]
- 5.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 6.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 7.Villar D, Vara-Vega A, Landazuri MO, Del Peso L. Biochem J. 2007;408:231–240. doi: 10.1042/BJ20071052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hausinger RP. Crit Rev Biochem Mol Biol. 2004;39:21–68. doi: 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]
- 10.Pavel EG, Zhou J, Busby RW, Gunsior M, Townsend CA, Solomon EI. J Am Chem Soc. 1998;120:743–753. [Google Scholar]
- 11.Hanauske-Abel HM, Gunzler V. J Theor Biol. 1982;94:421–455. doi: 10.1016/0022-5193(82)90320-4. [DOI] [PubMed] [Google Scholar]
- 12.Hoffart LM, Barr EW, Guyer RB, Bollinger JM, Jr, Krebs C. Proc Natl Acad Sci USA. 2006;103:14738–14743. doi: 10.1073/pnas.0604005103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krebs C, Fujimori DG, Walsh CT, Bollinger JM. Acc Chem Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Price JC, Barr EW, Tirupati B, Bollinger JM, Jr, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 15.Grzyska PK, Ryle MJ, Monterosso GR, Liu J, Ballou DP, Hausinger RP. Biochemistry. 2005;44:3845–3855. doi: 10.1021/bi048746n. [DOI] [PubMed] [Google Scholar]
- 16.Myllyla R, Majamaa K, Gunzler V, Hanauske-Abel HM, Kivirikko KI. J Biol Chem. 1984;259:5403–5405. [PubMed] [Google Scholar]
- 17.Pappalardi MB, McNulty DE, Martin JD, Fisher KE, Jiang Y, Burns MC, Zhao HZ, Ho T, Sweitzer S, Schwartz B, Annan RS, Copeland RA, Tummino PJ, Luo LS. Biochem J. 2011;436:363–369. doi: 10.1042/BJ20101201. [DOI] [PubMed] [Google Scholar]
- 18.Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Mol Cell Biol. 2005;25:6415–6426. doi: 10.1128/MCB.25.15.6415-6426.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flashman E, Bagg EAL, Chowdhury R, Mecinovic J, Loenarz C, McDonough MA, Hewitson KS, Schofield CJ. J Biol Chem. 2008;283:3808–3815. doi: 10.1074/jbc.M707411200. [DOI] [PubMed] [Google Scholar]
- 20.Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J. J Biol Chem. 2006;281:28712–28720. doi: 10.1074/jbc.M604628200. [DOI] [PubMed] [Google Scholar]
- 21.Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J. J Biol Chem. 2003;278:30772–30780. doi: 10.1074/jbc.M304982200. [DOI] [PubMed] [Google Scholar]
- 22.Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, Schofield CJ. Biochem J. 2007;401:227–234. doi: 10.1042/BJ20061151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Flagg SC, Giri N, Pektas S, Maroney MJ, Knapp MJ. Biochemistry. 2012;51:6654–6666. doi: 10.1021/bi300229y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen YH, Comeaux LM, Eyles SJ, Knapp MJ. Chem Commun. 2008:4768–4770. doi: 10.1039/b809099h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Henshaw TF, Feig M, Hausinger RP. J Inorg Biochem. 2004;98:856–861. doi: 10.1016/j.jinorgbio.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 26.Liu A, Ho RY, Que L, Jr, Ryle MJ, Phinney BS, Hausinger RP. J Am Chem Soc. 2001;123:5126–5127. doi: 10.1021/ja005879x. [DOI] [PubMed] [Google Scholar]
- 27.Dao JH, Kurzeja RJM, Morachis JM, Veith H, Lewis J, Yu V, Tegley CM, Tagari P. Anal Biochem. 2009;384:213–223. doi: 10.1016/j.ab.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 28.Flagg SC, Martin CB, Taabazuing CY, Holmes BE, Knapp MJ. J Inorg Biochem. 2012;113:25–30. doi: 10.1016/j.jinorgbio.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saban E, Flagg SC, Knapp MJ. J Inorg Biochem. 2011;105:630–636. doi: 10.1016/j.jinorgbio.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chowdhury R, McDonough MA, Mecinovic J, Loenarz C, Flashman E, Hewitson KS, Domene C, Schofield CJ. Structure. 2009;17:981–989. doi: 10.1016/j.str.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Landazuri MO, Vara-Vega A, Viton M, Cuevas Y, del Peso L. Biochem Biophys Res Commun. 2006;351:313–320. doi: 10.1016/j.bbrc.2006.09.170. [DOI] [PubMed] [Google Scholar]
- 32.Stubbs CJ, Loenarz C, Mecinovic J, Yeoh KK, Hindley N, Lienard BM, Sobott F, Schofield CJ, Flashman E. J Med Chem. 2009;52:2799–2805. doi: 10.1021/jm900285r. [DOI] [PubMed] [Google Scholar]
- 33.Hangasky JA, Taabazuing CY, Valliere MA, Knapp MJ. Metallomics. 2012 doi: 10.1039/c3mt20153h. Accepted for “Microbial Metallomics” special issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Krebs C, Schofield CJ. FEBS J. 2010;277:4089–4099. doi: 10.1111/j.1742-4658.2010.07804.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.