

Abstract

A frontier area in the development of activatable (turn-on) fluorescence-based probes is that concerned with rapid and selective stimulus triggering of probe activation so as to allow for biomarker identification and cellular imaging. The work here is concerned with a cloaked fluorophore composed of a reporter whose fluorescence is efficiently quenched by it being bound to an activatable trigger group through a novel self-immolative linker. Highly selective and rapid activation of the trigger group is achieved by chemical and enzymatic means that result in activated trigger group detachment from the self-immolative linker, with the latter subsequently cleaved from the reporter autonomously, thereby unmasking intense, red-shifted fluorescence emission. To achieve this success, we used a trimethyl-locked quinone propionic acid trigger group and an N-methyl-p-aminobenzyl alcohol self-immolative linker attached to the reporter. Delineated here are the synthesis and characterization of this cloaked fluorophore and the evaluation of its triggered turning on in the presence of an up-regulated enzyme in human cancer cells, NAD(P)H:quinone oxidoreductase-1 (NQO1, DT-diaphorase, EC 1.6.99.2).
The success of the ever-growing field of in vivo and ex vivo fluorescence imaging of diseased cells for diagnosis, pathology, and treatment applications rests upon the existence of disease-specific molecules whose spectroscopic signal is readily differentiated from that of the background of surrounding normal cells.1 Molecular probes whose cloaked fluorescence reporter signal is turned on by endogenous enzymes offer distinct advantages over always-on reporters, in particular, higher signal-to-background (positive-to-negative) values.2 Critical for cancer imaging is the development of libraries of turn-on probes that will allow for the collection of real-time information on the diseased tissue cell microenvironment and the high-fidelity definition of diseased and healthy tissue borders during fluorescence-assisted surgical resections.3−5
Unfortunately, the number of pro-fluorogenic probes that can have their fluorescence output selectively and quickly revealed by the presence of cancer-associated enzyme is extremely small, as is the type of disease-associated activating enzyme. Also, routes to improving the positive-to-negative value are limited. There are three extant probes that passively target cancer cells and rapidly (<30 min) identify the diseased cells by reporter fluorescence that is initiated by intracellular enzyme action; only three distinct cancer-linked enzymes have been targeted to date.5−7 Enzyme-initiated dequenching of probe fluorescence has been used to improve the signal-to-background value of the resulting reporter in diseased (positive) cells in comparison to that of unactivated probe or unaffected (negative) cells. Probe fluorescence is quenched via fluorescence resonance energy transfer (FRET) or photoinduced electron-transfer (PeT) mechanisms, and reporter fluorescence comes about by enzyme-initiated disassociation of the FRET quencher/donor pair upon enzyme binding7 or cleavage of the quencher (trigger group) from the probe.5,6 However, modest signal-to-background (SBR)/positive-to-negative (PNR) or tumor-to-background (TBR) ratios have been reported.5,6
Ideal probe characteristics include rapid cell uptake and highly selective activation to yield the reporter, low fluorescence efficiency (Φprobe) of the unactivated probe but high fluorescence efficiency of the reporter (Φreport), and high-quality retention of the reporter inside the diseased cells. Furthermore, the probe–reporter pair will exhibit large differences in energies of absorption maxima (λabsprobe vs λabs) and emission maxima (λemisprobe vs λemis), so significantly enhanced PNR/TBR values arise from the low absorptivity of the probe at the reporter’s maximum excitation wavelength.
A highly promising approach that incorporates many of the above desired probe/reporter qualities is one wherein the probe is tripartite, being composed of a self-immolative linker8 between the enzyme substrate/trigger group and the reporter moiety in the fluorescently silent probe. As a result of enzyme activation, the trigger group is first removed, and then the self-immolative linker is autonomously eliminated to yield the now highly fluorescent reporter. It is anticipated that by the careful selection of linker characteristics, the optical properties of the probe can be made distinct from those of the reporter, and the linker can be quickly cleaved from the reporter subsequent to trigger group activation.
Scheme 1. Turn-On Self-Immolative Profluorophore 1.
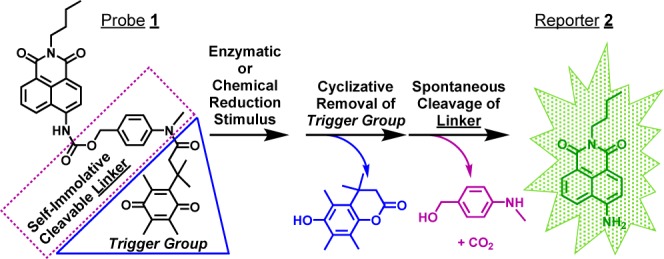
To that end, we developed probe 1,9 consisting of a trimethyl-locked quinone propionic acid (Q3PA) trigger group10 linked to a fluorescently masked naphthalimide (reporter 2) by N-methyl-p-aminobenzyl alcohol, NMPABA. The Q3PA trigger group was selected due to its known rapid and highly selective reduction10 by NAD(P)H:quinone oxidoreductase-1 (NQO1),11,12 an enzyme intimately associated with cancer13 and overexpressed 2- to 50-fold in the cytosol of numerous human tumor cells (e.g., colon, breast, pancreas, and nonsmall cell lung).11 The previously unreported NQO1-selective, turn-on fluorescence probe 1 relies on tuning the push–pull internal charge transfer (ICT)14 of the naphthalimide scaffold via its attachment to the self-immolative NMPABA linker using an electron-withdrawing carbamate connection. We show here that upon initiation of trigger group removal from the tripartite probe, subsequent rapid cleavage of linker occurs so as to afford reporter 2, which has distinct spectral properties such that NQO1-positive cancer cells can be imaged and identified with a positive-to-negative ratio of ∼500:1.
As seen in Figure 1, the spectroscopic properties of probe 1 and reporter 2 in buffered aqueous media are strikingly different, and these differences are attributed to the presence of the quinone propionic acid trigger group (Q3PA), as well as the carbamate connection between the N-methyl-p-aminobenzyl alcohol, NMPABA, linker, and the naphthalimide fluorophore. The λabsreport is red shifted roughly 50 nm in comparison to λabs (432 nm vs 380 nm), as is λemisreport vs λemis (532 nm vs 480 nm), with both observations being in accord with the presence of the electron-deficient carbamate14 in probe 1. Interestingly, the absorptivity of probe 1 at the maximum absorption wavelength of reporter 2 is ∼5 times lower than that for reporter 2. The Φprobe value of 0.002, which is 95 times less than Φreport = 0.19,9 is due to photoinduced electron transfer (PeT)15 quenching of the naphthalimide reporter by the Q3PA group. This conclusion is supported by outcomes based on voltammetric and spectroscopic data for probe 1, reporter 2, and Q3PA;9 the free energy of the PeT process is −0.98 eV, indicating ready electron transfer from the excited naphthalimide to the electron-poor Q3PA group of probe 1. In comparison to the previously reported probe Q3NI,5 there is a 3-fold-larger change in fluorescence efficiency for probe 1 upon NQO1 action to yield the corresponding reporter (Φreport/Φprobe), and the fluorescence energy range of reporter 2 is more attractive for possible imaging applications.
Figure 1.

Absorbance and emission spectra of 2 μM probe 1 and reporter 2 in pH 7.40, 0.1 M phosphate buffer. T = 25 °C.
We next wanted to demonstrate rapid turning on of reporter 2 by the self-immolative cleavage of the NMPABA linker subsequent to the reduction of probe 1. Dithionite reduction of the Q3PA moiety is complete within 1 s, and the half-life of the lactonization reaction is less than 2 min.16 Thus, sodium dithionite was added to aqueous solutions of probe 1, and the resulting fluorescence from reporter 2 was used to determine the extent of probe 1 conversion to reporter 2 (Figure 2). Reporter 2 is quickly turned on (t1/2 ∼6 min) under physiologically relevant conditions, pointing to the rapid self-immolative cleavage of the N-methyl-p-aminobenzyl alcohol and CO2 loss8 to yield reporter 2; this occurs because of the electron-rich character of the N-methyl-p-aminobenzyl alcohol vs that of the more typically encountered p-aminobenzyl alcohol linker.8,17 Conversion was confirmed by mass spectrometry analysis of probe 1 solutions treated with dithionite.9 On the basis of the relatively short time for the turn-on process, the use of probe 1 should allow for detecting the presence of NQO1 enzyme activity.
Figure 2.

Reduction-stimulated turning on of reporter 2. Time-course fluorescence (λex = 432 nm, λemis = 540 nm) from 10 μM probe 1 (pH 7.40, 0.1 M phosphate buffer) after reduction by 16 μM sodium dithionite. The inset contains time-dependent spectra of 2 μM probe 1 upon reduction. T = 25 °C.
To evaluate the ability of human NAD(P)H:quinone oxidoreductase-1 (hNQO1) to initiate the decloaking of probe 1 and reveal free reporter 2 upon Q3PA trigger group activation, we utilized the fluorescent product formation technique.18 Under in vitro conditions, solutions of probe 1 (2–60 μM) incubated with hNQO1 (40 μg) and its cofactor NADH (100 μM) exhibited steady increases in fluorescence, suggesting a relatively fast rate of reporter 2 production; control experiments with NADH alone did not yield significant changes in fluorescence, a result in accord with that previously shown for the Q3PA trigger group.10 In addition, the Q3PA trigger group is stable to reduction/addition reactions in the presence of glutathione and ascorbate as well as dithiothreitol.5 Furthermore, hNQO2 plus its cofactor13 is ineffective at releasing reporter 2. From the hNQO1 enzyme experiments, the initial rate of reporter 2 formation, V (μmol min–1 mg hNQO1–1), was calculated and then plotted as a function of probe 1 concentration (Figure 3). Apparent kinetic parameters were obtained by fitting the data in Figure 3 to Michaelis–Menten kinetics,19 namely, the Michaelis constant (Km) = 10.4 ± 1.0 μM, maximum velocity (Vmax) = 0.00225 ± 0.00008 μmol min–1 mg hNQO1–1, catalytic constant (kcat) = 0.068 ± 0.002 min–1, and catalytic efficiency (kcat/Km) = 6.52 ± 0.67 × 103 M–1 min–1. Also, there is no apparent inhibition of hNQO1 activity with the [probe 1] values studied. Thus, substantial reporter 2 release occurs upon probe 1 interaction with hNQO1 under physiologically relevant conditions.
Figure 3.

Kinetics of human NQO1 (40 μg) with probe 1 (2–60 μM) in pH 7.4, 0.1 M phosphate buffer. Values (n = 3) are the average ±1 sample standard deviation. The curve is the best fit to the average data. T = 25 °C.
To investigate the possibility of differentiating types of cancer cells based on hNQO1 content (positive vs negative), we exposed live hNQO1-positive and hNQO1-negative cells to probe 1 under typical culture conditions (37 °C/CO2 atmosphere). Confocal fluorescence microscopy images of hNQO1-positive colorectal cancer cells (HT29) incubated with probe 1 indicated significant probe 1 uptake and activation, resulting in intense intracellular reporter 2 production. However, hNQO1-negative nonsmall cell lung cancer cells (H596) treated with probe 1 revealed minimal signal (Figure 4), pointing to the intracellular stability of probe 1 in NQO1-negative cells, similar to what we have observed for another amide-linked napthalimide reporter, Q3NI.5 Wide-field fluorescence micrographs yielded similar results. Upon statistical evaluation of the fluorescence intensities in confocal images of hNQO1-positive (35 samples) and hNQO1-negative (29 samples) cells like those in Figure 4, a value of 11 was found for the positive-to-negative ratio (PNR).9 Similarly, for wide-field fluorescence images (33 positive samples/20 negative samples), a PNR of 510 was observed.9 As a direct point for comparison, a PNR of 23 was obtained for the Q3NI probe using the same HT29(NQO1-positive)/H596(NQO1-negative) cells during wide-field microscopy imaging experiments.5 Thus, the PNR value of ∼500 obtained with probe 1/reporter 2 is unprecedented in the context of our previous work with identical cell lines, and this value can be improved in wide-field measurements by the use of a filter set (BCECF) that more completely encompasses the emission profile of reporter 2. It is clear that the outcomes discussed here come about from the exceedingly large difference in fluorescence efficiency of reporter 2 vs probe 1 (95-fold increase) that results from the hNQO1-initiated removal of the trigger group quencher and self-immolative linker, the very high fluorescence efficiency of reporter 2, the significantly energy-shifted absorption maxima, and the correspondingly lower molar absorptivity of probe 1 vs reporter 2 at the excitation maximum of reporter 2. Also, the energies over which probe 1/reporter 2 is excited/emits light are shifted to the red by roughly 100 nm when compared to those of the previously documented Q3NI probe,5 leading to a significantly decreased background (negative) signal.
Figure 4.
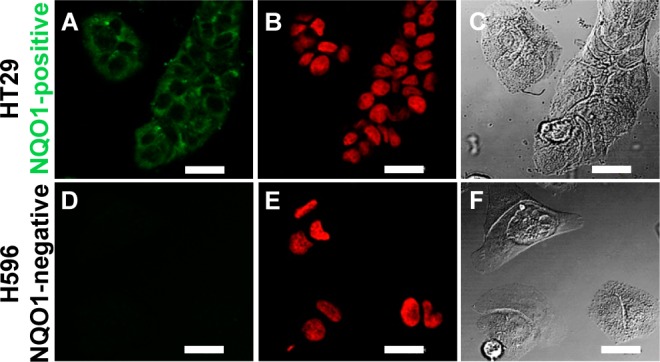
Microscopy images of hNQO1-positive HT29 colon (A, B, C) and hNQO1-negative H596 lung (D, E, F) cancer cells after incubation at 37 °C with 20 μM probe 1. Confocal images in A, B, D, and E; differential interference contrast in C and F. DRAQ5 nuclear stain was used for B and E. Scale bar = 20 μm.
In summary, we have presented the synthesis, properties, and biological evaluation of a novel turn-on fluorescent probe for the cancer-linked enzyme NQO1. The discrimination of human cells possessing NQO1, from those that do not, is achieved in a highly selective fashion, which results from the dramatically increased fluorescence efficiency of the red-shifted reporter 2 that is generated only upon hNQO1 action on the trigger group of probe 1. We are now using the ICT-tunable probe 1 and its variants to develop methods, including multiphoton imaging routes,5 that allow for the identification of hard-to-detect NQO1-expressing cancerous tissues, as well as the diversification of disease targets20,21 for the activation of new trigger groups. With regard to longer-term impacts, enzyme-activatable probes will play important roles as “intelligent” imaging agents applied to the fluorescence-assisted resection of cancerous tissues22 and efficacy studies of personalized chemotherapy.23
Acknowledgments
This material is based upon work supported by the U.S. National Institutes of Health (5R21CA135585, P42ES013648) and the U.S. National Science Foundation under grant CHE-0910845.
Supporting Information Available
Synthesis conditions and spectral properties of probe 1 and reporter 2, voltammetric data, and PNR data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Frangioni J. V. J. Clin. Oncology 2008, 26, 4012–4021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H.; Choyke P. L. Acc. Chem. Res. 2011, 44, 83–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen Q. T.; Olson E. S.; Aguilera T. A.; Jiang T.; Scadeng M.; Ellies L. G.; Tsien R. Y. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 4317–4322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S.; Zhang E.; Su Y.; Cheng T.; Shi C. Biomaterials 2011, 32, 7127–7138. [DOI] [PubMed] [Google Scholar]
- Silvers W. C.; Prasai B.; Burk D. H.; Brown M. L.; McCarley R. L. J. Am. Chem. Soc. 2013, 135, 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdoes M.; Oresic Bender K.; Segal E.; van der Linden W. A.; Syed S.; Withana N. P.; Sanman L. E.; Bogyo M. J. Am. Chem. Soc. 2013, 135, 14726–14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Fan J.; Wang J.; Dou B.; Zhou F.; Cao J.; Qu J.; Cao Z.; Zhao W.; Peng X. J. Am. Chem. Soc. 2013, 135, 17469–17475. [DOI] [PubMed] [Google Scholar]
- Erez R.; Shabat D. Org. Biomol. Chem. 2008, 6, 2669–2672. [DOI] [PubMed] [Google Scholar]
- See Supporting Information.
- Mendoza M. F.; Hollabaugh N. M.; Hettiarachchi S. U.; McCarley R. L. Biochemistry 2012, 51, 8014–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danson S.; Ward T. H.; Butler J.; Ranson M. Can. Treat. Rev. 2004, 30, 437–449. [DOI] [PubMed] [Google Scholar]
- Ernster L. Chem. Scr. 1987, 27A, 1–13. [Google Scholar]
- Siegel D.; Yan C.; Ross D. Biochem. Pharmacol. 2012, 83, 1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srikun D.; Miller E. W.; Domaille D. W.; Chang C. J. J. Am. Chem. Soc. 2008, 130, 4596–4597. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R.Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, 2006. [Google Scholar]
- Perera I. Master’s Thesis. Louisiana State University, 2012. [Google Scholar]
- Robbins J. S.; Schmid K. M.; Phillips S. T. J. Org. Chem. 2013, 78, 3159–3169. [DOI] [PubMed] [Google Scholar]
- Beija M.; Afonso C. A.; Martinho J. M. Chem. Soc. Rev. 2009, 38, 2410–2433. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. Methods Enzymol. 1979, 63, 103–138. [DOI] [PubMed] [Google Scholar]
- Li L.; Zhang C.-W.; Chen G. Y. J.; Zhu B.; Chai C.; Xu Q.-H.; Tan E.-K.; Zhu Q.; Lim K.-L.; Yao S. Q. Nat. Commun. 2014, 5, 3276. [DOI] [PubMed] [Google Scholar]
- Wang R.; Chen L.; Liu P.; Zhang Q.; Wang Y. Chem.—Eur. J. 2012, 18, 11343–11349. [DOI] [PubMed] [Google Scholar]
- Choi H. S.; Gibbs S. L.; Lee J. H.; Kim S. H.; Ashitate Y.; Liu F.; Hyun H.; Park G.; Xie Y.; Bae S.; Henary M.; Frangioni J. V. Nat. Biotechnol. 2013, 31, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiyama H.; Rauenzahn S.; Shim J. S.; Karikari C. A.; Feldmann G.; Hua L.; Kamiyama M.; Schuler F. W.; Lin M.-T.; Beaty R. M.; Karanam B.; Liang H.; Mullendore M. E.; Mo G.; Hidalgo M.; Jaffee E.; Hruban R. H.; Jinnah H. A.; Roden R. B. S.; Jimeno A.; Liu J. O.; Maitra A.; Eshleman J. R. Clin. Cancer Res. 2013, 19, 1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.