

Abstract
The role of protein dynamics in the reaction catalyzed by dihydrofolate reductase from the hyperthermophile Thermotoga maritima (TmDHFR) has been examined by enzyme isotope substitution (15N, 13C, 2H). In contrast to all other enzyme reactions investigated previously, including DHFR from Escherichia coli (EcDHFR), for which isotopic substitution led to decreased reactivity, the rate constant for the hydride transfer step is not affected by isotopic substitution of TmDHFR. TmDHFR therefore appears to lack the coupling of protein motions to the reaction coordinate that have been identified for EcDHFR catalysis. Clearly, dynamical coupling is not a universal phenomenon that affects the efficiency of enzyme catalysis.
Kinetic isotope effect (KIE) studies with isotopically labeled substrates are a well-established method to probe the mechanisms of enzymatic reactions.1−7 More recently, kinetic studies have been performed where entire enzymes have been isotopically substituted with all 14N, 12C, and nonexchangeable 1H atoms replaced by heavier stable isotopes.8−13 Such complete enzyme isotopic substitution slows protein motions ranging from femtosecond bond vibrations to millisecond structural changes, while the electrostatic properties are unaffected.14,15 Comparing the kinetic behavior of “heavy” enzymes (isotopically labeled with 15N, 13C, and 2H) with that of “light” enzymes (with natural isotope abundance) can therefore reveal information about the relationship between enzyme catalysis and protein dynamics.8,9
Isotope substitution in dihydrofolate reductase from Escherichia coli (EcDHFR) and one of its mutants,10,11 purine nucleoside phosphorylase,8 HIV protease,9 alanine racemase,13 and pentaerythritol tetranitrate reductase12 causes noticeable changes in the rates of the chemical steps, demonstrating that protein motions have a small but measurable effect on the catalyzed reactions. DHFR catalyzes the formation of tetrahydrofolate (H4F) by transferring hydride from C4 of NAPDH to C6 of dihydrofolate (H2F) and adding a proton to N5 of H2F. It has long been used as a model to examine the effects of protein dynamics on enzyme catalysis.10,11,16−27 In the case of EcDHFR, “promoting motions” had been hypothesized to enhance hydride transfer.28−32 In contrast, combined experimental and computational analyses of complete enzyme isotopic substitution indicated that while protein motions do couple to the reaction coordinate, they do not drive tunneling or modulate the barrier of the chemical transformation.10,11 Rather, the reactivity difference between the “light” and “heavy” enzymes is due to a change in the frequency of dynamical recrossing, nonproductive trajectories that do not remain on the product side of the transition-state dividing surface.33 Interestingly, these studies indicated that the dynamical coupling to the chemical step is enhanced in a catalytically compromised mutant.10 Recrossing coefficients for enzyme-catalyzed reactions tend to be closer to unity than for their counterparts in solution,34−37 and it has been shown that compression of the reaction coordinate can in fact be anticatalytic in enzymes.38 These observations suggest that efficient enzymes may be characterized by reduced dynamical coupling to the reaction coordinate relative to the uncatalyzed reactions.
EcDHFR is a relatively flexible monomeric enzyme that contains several mobile segments, namely, the M20, FG, and GH loops (Figure 1).40 These loops control the physical steps of substrate binding and product release by switching the enzyme between the “closed” and “occluded” conformations.40 In contrast, DHFR from the hyperthermophile Thermotoga maritima (TmDHFR) forms a very stable dimer, and the FG loop is locked in the dimer interface (Figure 1).39,41 These structural features contribute to the exceptional thermostability of TmDHFR (Tm = 83 °C). On the other hand, TmDHFR appears to be fixed in an open conformation and shows catalytic activity considerably lower than that of EcDHFR.39,41,42 The KIE on the TmDHFR-catalyzed hydride transfer was found to be highly temperature-dependent below 25 °C but largely temperature-independent at elevated temperatures.42 The reaction proceeds with a contribution from quantum-mechanical tunneling, particularly at low temperatures, but this is not promoted by long-range protein motions.20−23,43 To investigate whether the environmental coupling to hydride transfer observed in EcDHFR10,11 also applies to an enzyme with less conformational flexibility, a kinetic comparison of “heavy” TmDHFR with its “light” counterpart was performed.
Figure 1.
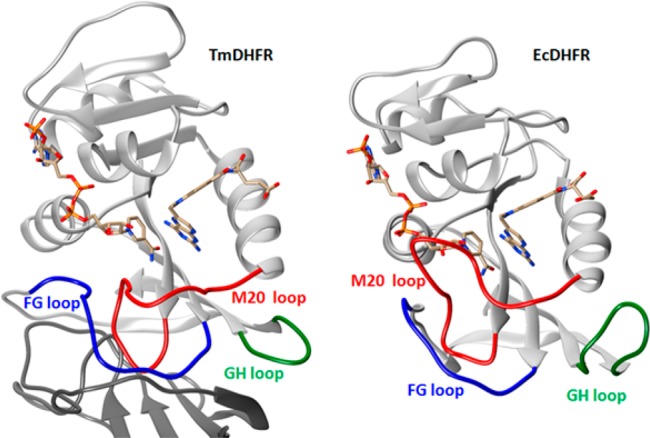
Cartoon representation of (left) TmDHFR (PDB entry 1D1G)39 and (right) EcDHFR (PDB entry 1DRE).40 Only one subunit and the dimer interface of TmDHFR are shown. The ligands NADPH and methotrexate are shown as sticks. The M20 loop (red) is shown in its closed conformation in EcDHFR and in the open conformation in TmDHFR. The FG and GH loops are highlighted in blue and green, respectively.
“Heavy” TmDHFR was produced in minimal medium containing only 15N-, 13C-, and 2H-labeled ingredients [see the Supporting Information (SI)]. Purified “heavy” TmDHFR showed a molecular weight increase of 10.6%, indicating that over 98% of the nonexchangeable atoms had been replaced by the corresponding heavy isotopes (Figure S1 in the SI). Circular dichroism spectra of the “light” and “heavy” enzymes were essentially superimposable (Figure S2), suggesting that isotope substitution does not significantly affect the secondary structure of TmDHFR.
The reactivities of “light” and “heavy” TmDHFR were first characterized at pH 7 under steady-state conditions, where hydride transfer is only partially rate-limiting.42 The steady-state rate constants for “light” TmDHFR, kcatLE, are higher than those for its “heavy” counterpart, kcat (Figure 2 and Table S1 in the SI). Between 15 and 65 °C, the magnitude of the enzyme KIEcat (kcatLE/kcat ≈ 1.35) is largely unchanged, but it increases to 1.73 ± 0.01 at 7 °C (Figure 2 and Table S2). Interestingly, the temperature dependence of the enzyme KIEcat in TmDHFR greatly differs from that of the EcDHFR KIEcat, which increases steadily from 1.04 ± 0.03 at 10 °C to 1.16 ± 0.01 at 35 °C.11
Figure 2.

Experimental TmDHFR data for steady-state and hydride transfer (pre-steady-state) rate constants at pH 7. (A) Steady-state kinetic data; (B) pre-steady-state kinetic data. Data points and Arrhenius fits are shown for “light” (red circles) and “heavy” (blue triangles) TmDHFR. (C, D) Enzyme KIEs (ratio of rate constants for “light” and “heavy” TmDHFR, kLE/kHE) under steady-state and pre-steady-state conditions, respectively.
The Michaelis constants (KM) of TmDHFR were found to be mildly temperature-dependent. The KM values for both NADPH and DHF are ∼1 μM at 45 °C and identical within the expermental error for “light” and “heavy” TmDHFR (Table S3); they decrease to <0.5 μM at 10 and 20 °C. It is unclear whether there is a difference between the KM values for “light” and “heavy” TmDHFR at low temperature, since these low values were difficult to measure accurately. Nevertheless, the nature of tight ligand binding in TmDHFR remains unchanged upon enzymatic isotope substitution.42
The effect of heavy isotope substitution on the TmDHFR-catalyzed hydride transfer was measured in single-turnover experiments. At pH 7.0, the rate constants of the chemical step for “light” and “heavy” TmDHFR are essentially identical, giving an enzyme KIEH (kHLE/kH) of ∼1 at all temperatures (Figure 2 and Tables S1 and S2). In contrast, for EcDHFR the enzyme KIEH increases from 0.93 ± 0.02 at 10 °C to 1.18 ± 0.09 at 40 °C, leading to an activation energy difference (ΔEa) of 5.78 ± 1.61 kJ mol–1.11 The apparent pKa values for hydride transfer for “light” and “heavy” TmDHFR are also similar (Table S4). As the enzyme KIEH does not change significantly with pH for either TmDHFR (Figure S3 and Table S4) or EcDHFR,11 the difference in the pKa values of the two enzymes is unlikely to be significant to our discussion, and comparison of the enzyme KIEH values at a single pH value is appropriate. To the best of our knowledge, TmDHFR is to date the only enzyme for which no noticeable “heavy” enzyme KIE has been observed for the chemical step.8−13
Under steady-state conditions, heavy isotope substitution causes a strong reactivity difference for TmDHFR. The absence of an effect on hydride transfer on the other hand suggests that protein motions play a role only in the physical steps during TmDHFR catalysis. This finding is in agreement with previous investigations of the solvent effects, which showed no sign of long-range coupled motions.17,19,22 Many TmDHFR variants with disrupted dimer interfaces show a larger decrease in steady-state turnover than in hydride transfer rate constants.20,43 Hence, the reduced kcatHE is likely due to an isotope effect on the intra- and/or intersubunit motions that are important in the physical steps of catalysis.20,43,44 In addition, the enzyme KIEcat for EcDHFR increases with temperature, but for the double mutant EcDHFR-N23PP/S148A, KIEcat remains constant (∼1),10,11 consistent with the observation that the release of the product tetrahydrofolate is rate-limiting in the wild-type enzyme and involves a large conformational change11,40 while the release of NADP+ is rate-limiting in the mutant and most likely involves only a small conformational change.10,28 Conformational changes in TmDHFR appear to be minimal.39,43 Hence, the magnitude of the enzyme KIEcat (∼1.35) in TmDHFR is relatively constant at most temperatures. In turn, the abrupt increase in the enzyme KIEcat at low temperatures could be caused by a switch in the conformational equilibrium favorable for reaction. This needs to be verified by additional studies, such as binding studies on isotopically labeled transition-state analogues and/or protein segments.
There has been continuing controversy over the potential role of protein motions in “promoting” enzymatic hydrogen tunneling at physiologically relevant temperatures.10,11,16,28,45−54 On the basis of our previous calculations performed on EcDHFR,10,11 the enzyme kinetic isotope effects on hydride transfer (KIEH) reported here imply the absence of dynamical coupling to the reaction coordinate in TmDHFR at all temperatures examined. We have shown previously that the dynamical coupling to the chemical step is enhanced in a catalytically compromised mutant of EcDHFR.10,11 TmDHFR may derive a slight benefit from the lack of dynamical coupling. Conformational and structural constraints imposed by dimerization are instead the major cause of TmDHFR’s low activity. In EcDHFR, formation of the closed DHFR–substrate complex excludes solvent molecules from the active site and allows the formation of a geometric and electrostatic environment conducive to hydride transfer, thus lowering the reorganization energy (the energy required to reorient the reactants during the reaction).40 In TmDHFR, such conformational sampling is prevented by the enzyme’s dimeric structure, generating a DHFR–substrate complex in which the active site is exposed to solvent interactions. This compromises the electrostatic preorganization (the enzyme’s ability to arrange the substrates with “product-like” geometry and electrostatics), leading to an increase in the reorganization energy and a relatively low rate constant for hydride transfer.
In summary, the enzyme KIEH of ∼1 observed for TmDHFR reveals much information about the structural and dynamical properties of the enzyme. If the enzyme KIE reports on recrossing events in TmDHFR in the same way as it does in EcDHFR,10,11 then it appears that recrossing events in TmDHFR are unaffected by protein dynamics. Previous studies of EcDHFR and its variant indicated that dynamical effects contribute only a small change to the activation free energy.10,11 Therefore, the low activity of TmDHFR is most likely due to poor electrostatic preorganization and is unrelated to dynamical coupling. The inability of TmDHFR to form a closed conformation favorable for reaction outweighs any potential small benefit from the reduction in recrossing events. These characteristics of TmDHFR may reflect an evolutionary trade-off between catalytic activity and thermal stability. The relationship between dynamics and barrier crossing/recrossing must be examined further by experimentation and calculation.
Acknowledgments
This work was supported by Grant BB/J005266/1 (R.K.A.) from the UK Biotechnology and Biological Sciences Research Council (BBSRC). The authors express their gratitude to Iñaki Tuñón and Vicent Moliner for their insightful comments on the manuscript.
Supporting Information Available
Full experimental procedures; mass spectra of purified proteins; circular dichroism spectra; tabulated experimental data for kH, kcat, and enzyme KIEs; and pH dependence of kH. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Schramm V. L. Acc. Chem. Res. 2003, 36, 588. [DOI] [PubMed] [Google Scholar]
- Garcia-Viloca M.; Truhlar D. G.; Gao J. Biochemistry 2003, 42, 13558. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. J. Biol. Chem. 2003, 278, 51975. [DOI] [PubMed] [Google Scholar]
- Park H.; Girdaukas G. G.; Northrop D. B. J. Am. Chem. Soc. 2006, 128, 1868. [DOI] [PubMed] [Google Scholar]
- O’Leary M. H. Acc. Chem. Res. 1988, 21, 450. [Google Scholar]
- Seravalli J.; Huskey W. P.; Schowen K. B.; Schowen R. L. Pure Appl. Chem. 1994, 66, 695. [Google Scholar]
- Tai C.-H.; Cook P. F. Acc. Chem. Res. 2000, 34, 49. [DOI] [PubMed] [Google Scholar]
- Silva R. G.; Murkin A. S.; Schramm V. L. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipp D. R.; Silva R. G.; Schramm V. L. J. Am. Chem. Soc. 2011, 133, 19358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Pernia J. J.; Luk L. Y. P.; García-Meseguer R.; Martí S.; Loveridge E. J.; Tuñón I.; Moliner V.; Allemann R. K. J. Am. Chem. Soc. 2013, 135, 18689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk L. Y. P.; Ruiz-Pernia J. J.; Dawson W. M.; Roca M.; Loveridge E. J.; Glowacki D. R.; Harvey J. N.; Mulholland A. J.; Tuñón I.; Moliner V.; Allemann R. K. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 16344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pudney C. R.; Guerriero A.; Baxter N. J.; Johannissen L. O.; Waltho J. P.; Hay S.; Scrutton N. S. J. Am. Chem. Soc. 2013, 135, 2512. [DOI] [PubMed] [Google Scholar]
- Toney M. D.; Castro J. N.; Addington T. A. J. Am. Chem. Soc. 2013, 135, 2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born M.; Oppenheimer R. Ann. Phys. 1927, 389, 457. [Google Scholar]
- Carpenter B. K. In Quantum Tunnelling in Enzyme-Catalysed Reactions; Allemann R. K., Scrutton N. S., Eds.; Royal Society of Chemistry: Cambridge, U.K., 2009. [Google Scholar]
- Loveridge E. J.; Behiry E. M.; Guo J.; Allemann R. K. Nat. Chem. 2012, 4, 292. [DOI] [PubMed] [Google Scholar]
- Loveridge E. J.; Tey L.-H.; Behiry E. M.; Dawson W. M.; Evans R. M.; Whittaker S. B.-M.; Gunther U. L.; Williams C.; Crump M. P.; Allemann R. K. J. Am. Chem. Soc. 2011, 133, 20561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveridge E. J.; Allemann R. K. ChemBioChem 2011, 12, 1258. [DOI] [PubMed] [Google Scholar]
- Loveridge E. J.; Tey L.-H.; Allemann R. K. J. Am. Chem. Soc. 2010, 132, 1137. [DOI] [PubMed] [Google Scholar]
- Loveridge E. J.; Allemann R. K. Biochemistry 2010, 49, 5390. [DOI] [PubMed] [Google Scholar]
- Loveridge E. J.; Maglia G.; Allemann R. K. ChemBioChem 2009, 10, 2624. [DOI] [PubMed] [Google Scholar]
- Loveridge E. J.; Evans R. M.; Allemann R. K. Chem.—Eur. J. 2008, 14, 10782. [DOI] [PubMed] [Google Scholar]
- Pang J. Y.; Pu J. Z.; Gao J. L.; Truhlar D. G.; Allemann R. K. J. Am. Chem. Soc. 2006, 128, 8015. [DOI] [PubMed] [Google Scholar]
- Sikorski R. S.; Wang L.; Markham K. A.; Rajagopalan P. T. R.; Benkovic S. J.; Kohen A. J. Am. Chem. Soc. 2004, 126, 4778. [DOI] [PubMed] [Google Scholar]
- Watney L. B.; Agarwal P. K.; Hammes-Schiffer S. J. Am. Chem. Soc. 2003, 125, 3745. [DOI] [PubMed] [Google Scholar]
- Rod T. H.; Radkiewicz J. L.; Brooks C. L. III Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 6980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehr D. D.; McElheny D.; Dyson H. J.; Wright P. E. Science 2006, 313, 1638. [DOI] [PubMed] [Google Scholar]
- Bhabha G.; Lee J.; Ekiert D. C.; Gam J.; Wilson I. A.; Dyson H. J.; Benkovic S. J.; Wright P. E. Science 2011, 332, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Tharp S.; Selzer T.; Benkovic S. J.; Kohen A. Biochemistry 2006, 45, 1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P. K.; Billeter S. R.; Rajagopalan P. T. R.; Benkovic S. J.; Hammes-Schiffer S. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron C. E.; Benkovic S. J. Biochemistry 1997, 36, 15792. [DOI] [PubMed] [Google Scholar]
- Francis K.; Stojković V.; Kohen A. J. Biol. Chem. 2013, 288, 35961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pu J.; Gao J.; Truhlar D. G. Chem. Rev. 2006, 106, 3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca M.; Moliner V.; Tuñón I.; Hynes J. T. J. Am. Chem. Soc. 2006, 128, 6186. [DOI] [PubMed] [Google Scholar]
- Ruiz-Pernía J. J.; Tuñón I.; Moliner V.; Hynes J. T.; Roca M. J. Am. Chem. Soc. 2008, 130, 7477. [DOI] [PubMed] [Google Scholar]
- Kanaan N.; Ferrer S.; Martí S.; Garcia-Viloca M.; Kohen A.; Moliner V. J. Am. Chem. Soc. 2011, 133, 6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca M.; Oliva M.; Castillo R.; Moliner V.; Tuñón I. Chem.—Eur. J. 2010, 16, 11399. [DOI] [PubMed] [Google Scholar]
- Liu H.; Warshel A. J. Phys. Chem. B 2007, 111, 7852. [DOI] [PubMed] [Google Scholar]
- Dams T.; Auerbach G.; Bader G.; Jacob U.; Ploom T.; Huber R.; Jaenicke R. J. Mol. Biol. 2000, 297, 659. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R.; Kraut J. Biochemistry 1997, 36, 586. [DOI] [PubMed] [Google Scholar]
- Dams T.; Bohm G.; Auerbach G.; Bader G.; Schuring H.; Jaenicke R. Biol. Chem. 1998, 379, 367. [PubMed] [Google Scholar]
- Maglia G.; Javed M. H.; Allemann R. K. Biochem. J. 2003, 374, 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loveridge E. J.; Rodriguez R. J.; Swanwick R. S.; Allemann R. K. Biochemistry 2009, 48, 5922. [DOI] [PubMed] [Google Scholar]
- Guo J.; Loveridge E. J.; Luk L. Y. P.; Allemann R. K. Biochemistry 2013, 52, 3881. [DOI] [PubMed] [Google Scholar]
- Pisliakov A. V.; Cao J.; Kamerlin S. C. L.; Warshel A. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 17359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou D.; Caratzoulas S.; Kalyanaraman C.; Mincer J. S.; Schwartz S. D. Eur. J. Biochem. 2002, 269, 3103. [DOI] [PubMed] [Google Scholar]
- Knapp M. J.; Klinman J. P. Eur. J. Biochem. 2002, 269, 3113. [DOI] [PubMed] [Google Scholar]
- Hay S.; Scrutton N. S. Nat. Chem. 2012, 4, 161. [DOI] [PubMed] [Google Scholar]
- Scrutton N. S.; Basran J.; Sutcliffe M. J. Eur. J. Biochem. 1999, 264, 666. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. Chem. Rev. 2006, 106, 3095. [DOI] [PubMed] [Google Scholar]
- Warshel A. Proc. Natl. Acad. Sci. U.S.A. 1984, 81, 444.6582500 [Google Scholar]
- Glowacki D. R.; Harvey J. N.; Mulholland A. J. Nat. Chem. 2012, 4, 169. [DOI] [PubMed] [Google Scholar]
- Kohen A.; Cannio R.; Bartolucci S.; Klinman J. P. Nature 1999, 399, 496. [DOI] [PubMed] [Google Scholar]
- Oyeyemi O. A.; Sours K. M.; Lee T.; Resing K. A.; Ahn N. G.; Klinman J. P. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 10074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.