

Abstract
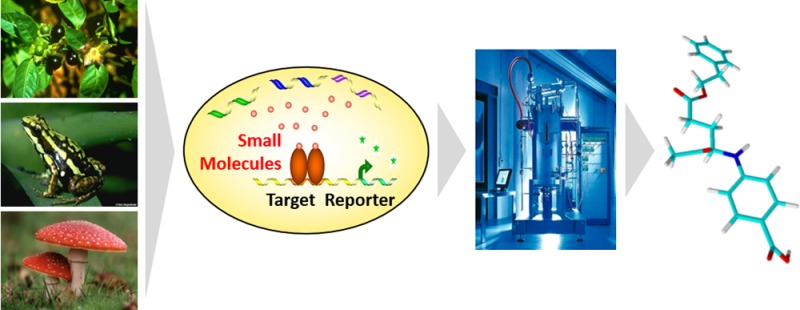
Synthetic biology has been heralded as a new bioengineering platform for the production of bulk and specialty chemicals, drugs, and fuels. Here, we report for the first time a series of 74 novel compounds produced using a combinatorial genetics approach in baker’s yeast. Based on the concept of “coevolution” with target proteins in an intracellular primary survival assay, the identified, mostly scaffold-sized (200–350 MW) compounds, which displayed excellent biological activity, can be considered as prevalidated hits. Of the molecules found, >75% have not been described previously; 20% of the compounds exhibit novel scaffolds. Their structural and physicochemical properties comply with established rules of drug- and fragment-likeness and exhibit increased structural complexities compared to synthetically produced fragments. In summary, the synthetic biology approach described here represents a completely new, complementary strategy for hit and early lead identification that can be easily integrated into the existing drug discovery process.
Keywords: synthetic biology, yeast, drug discovery, novel structures, diversity, scaffold-sized
The deciphering of the human genome and the new knowledge on genes and their functions has created plenty of new target opportunities for the pharmaceutical industry.1 However, traditional approaches toward hit and lead finding, such as high-throughput screening (HTS) and fragment-based screening (FBS), still only cover a fraction of the potential “chemical space”. Small molecule compound collections are often a reflection of former drug targets,2−4 thus covering previously explored chemical space. Also, the creation of chemical libraries is limited by feasible chemical reactions.2 For this reason and although HTS has been a valuable hit/lead source for historically tractable target classes, its success rate lags behind for many novel or traditionally undruggable targets, for instance inhibitors of protein–protein interactions, Class B GPCRs or phosphatases.2
HTS and FBS are two common approaches used in the pharmaceutical industry for the identification of hits which deliver almost antithetic chemical starting points. In HTS, millions of compounds are initially screened to assess biological activity against a target. There is an increasing awareness, however, regarding the quality of HTS libraries, and compound collections nowadays follow more closely the ideal of screening lead-like compounds rather than drug-like or drug-unlike compounds. Nevertheless, many compounds in these corporate collections still lack structural diversity and possess a relatively high lipophilicity and poor thermodynamic solubility.1,2,5−7
Compared to HTS, FBS samples the smaller chemical space of fragment-sized molecules more efficiently. This efficiency is borne out by a higher probability of fragments matching a receptor due to their reduced complexity. However, binding events are harder to detect owing to the inferior affinities of low molecular weight fragments for the target. Consequently, fragment hits require more medicinal chemistry efforts to transform them into lead compounds with acceptable affinity.8,9 In addition, FBS in general requires targets to be isolatable, and the availability of structural information is often critical to the success of fragment-based drug discovery.8,9
In this paper, we describe a synthetic biology (SB)-based hit-finding approach (Figure 1) that is complementary to existing strategies and extendable to novel or difficult therapeutic targets. The approach combines the use of biosynthetic reactions for compound generation with advantages from FBS (small, scaffold-sized molecules), natural products (NPs) (diversity and structural complexity, “prevalidated” by nature), and HTS (higher potency hits, initial SAR, generation of chemical probes). The compounds resulting from this process possess favorable structural and physicochemical properties, are amenable to synthetic modifications, and exhibit a high degree of novelty. Being produced by enzymes and selected on the basis of an intracellular functional assay, SB hits explore biologically relevant chemical space with “privileged”, NP-like fragments.10−13
Figure 1.

Scheme of the synthetic biology combinatorial genetics approach in baker’s yeast.
Results and Discussion
Synthetic Biology Platform, Screening, and Compound Isolation
The detailed methodology of the SB platform used here has been described previously.14 In short, the technology combines horizontal gene transfer and expression with stringent screening technologies in a system where the same yeast cell is used to first generate the compounds and second to screen for activity of these compounds against an internal or external assay. The basis for producing novel compounds is genetic material sourced from (i) known biochemical pathways producing drug-relevant chemical scaffolds, (ii) organisms with unknown genetic diversity that can provide new enzymatic activities, and (iii) organisms reported to have a medicinal effect or that can tolerate different types of infection (e.g., bacterial or viral). In the present study, a functional Brome Mosaic Virus (BMV) replication assay was selected as the primary screen (see Methods section). The assay setup and compound isolation procedures are described in the online Methods section. We investigated yeast strains containing YACs (yeast artificial chromosomes) expressing combinations of pathway and cDNA genes deriving from 8 pathways and 14 libraries containing cDNA (Supporting Information, Table S1 and S4).
A total of 74 YAC-dependent compounds were isolated from 35 yeast crude extracts (clones). Twenty-eight compounds (38%) exhibited activity in a secondary BMV assay. All compounds were isolated by preparative HPLC, and their structures were elucidated by means of mass spectrometry and one- and two-dimensional homo- and heteronuclear NMR spectroscopy. Figure 2 shows selected examples of compounds (1–34) produced by this SB approach.
Figure 2.

Examples of compounds produced by synthetic biology. Compounds 1–34 were isolated from yeast. Ribavirin was used as reference in secondary activity assays. The entire list of 74 compounds is shown in Supporting Information Figure S2.
Synthetic Biology Compound Properties
The 74 isolated compounds were assessed for their physicochemical properties and their quality as starting points for lead optimization. These data are summarized in Table 1. The SB approach described here produces mainly fragment- (110 < MW[Da]<250) to scaffold-sized (250 < MW[Da]<350) molecules.9 Average MW, clogP, and HBD values are in line with Congreve’s rule-of-three (Ro3) criteria for fragments.15 HBA, ROT, and PSA values are above recommended values.15,16 Depending on the calculation algorithm used, only 3–8% of the isolated compounds have a high calculated logP (clogP > 5). Less than 10% of the compounds produced by SB give rise to one or more violations in the rule-of-five (Ro5).12,17,18 Overall, the SB compounds possess fragment- to scaffold-like molecular weights.
Table 1. Physicochemical, Structural, and Shape-Based Properties of Synthetic-Biology-Derived Compounds Compared to Fragment- and Drug-Like Compoundsa.
| type of compound | fragment-like | synthetic biology | drug-like |
|---|---|---|---|
| rule | rule-of-three15 threshold | average value | rule-of-five17 threshold |
| MW | <300 | 300 | ≤500 |
| clogP | ≤3 | 2.5 | ≤5 |
| HBD | ≤3 | 2.4 | ≤5 |
| HBA | ≤3 | 5.1 | ≤10 |
| ROT | ≤3c | 5.6 | ≤10d |
| PSA | ≤60c | 83 | ≤140d |
| HAC | ≈15b | 22 | ≈38b |
| pIC50 | 4.4b | 5.2 | 8b |
| shape complexity Csp3/(Csp3+Csp2) | 0.28 | 0.38 | 0.41 |
| shape complexity core skeleton Csp3/(Csp3+Csp2) | 0.15 | 0.53 | 0.23 |
| stereochemical complexity (Cstereogenic/Ctotal) | 0.02 | 0.07 (0.05)e | 0.08 |
Synthetic biology values are average values. Shape and stereochemical complexity are average values determined using a diverse 13 190 fragment library (Key Organics Ltd.) and a set of 870 marketed drugs of MW < 500 Da (extracted from DRUGBANK) for fragment- and drug-like molecules, respectively. Otherwise, values refer to thresholds of Ro3 and Ro5 or recommended in literature for fragment- and drug-like molecules. Abbreviations: MW, molecular weight; clogP, calculated logarithm of the octanol–water partition coefficient; HBD, number of hydrogen bond donors; HBA, number of hydrogen bond acceptors; ROT, number of rotatable bonds; PSA, polar surface area; HAC, number of non-hydrogen atoms.
Typical value, taken from ref (16),
ref (16),
ref (23).
In brackets: stereochemical complexity of deglycosylated synthetic biology compounds.
Shape complexity describes the fraction of sp3 hybridized carbon atoms compared to the sum of sp3 and sp2 hybridized carbons. Stereochemical complexity relates the number of chiral carbons to the total number of carbons in any given compound.19−21 Both values are useful descriptors to describe the structural complexity of a molecule. Complexity, namely, a higher degree of saturation in molecules, increases the likelihood of higher solubility and lower melting points, physical properties that are associated with successful drugs.19 In addition, this property has been shown to be beneficial by increasing the probability of appropriate complementary interactions between ligand and target, resulting in greater selectivity and less off-target effects.19 Molecules rich in sp3-configured centers also have the capacity of covering larger portions of chemical space.8,9
Shape and stereochemical complexity of SB compounds were compared to “drug-” and “fragment-like” compound sets. Data sets were sourced as follows. A “drug-like” data set of 870 marketed drugs was extracted from DRUGBANK using a MW ≤ 500 Da criteria. Also, two sets of fragment-like compounds were sourced: a diverse 13 190 fragment set from Key Organics Ltd. (Full BIONET Fragments Library July 2013, http://www.keyorganics.net/services/bionet-products/fragment-libraries/) and a diverse fragment library containing 7849 compounds from ChemBridge, Inc. (Fragment Library Database—SDF Format Last Modified 8/1/2013, http://www.chembridge.com/internal/index.php). The SB compounds shape and stereochemical complexity values rank above the diversity oriented fragment set and are close to the drug-like molecule set (Table 1). Interestingly, the average shape complexity of the core scaffold is higher than that of the whole compounds, indicating a high degree of structural complexity connected to the core scaffolds of the SB compounds (Table 1). Stereochemical complexity, however, remains comparatively low.
Optimizability is a key attribute of successful hits in a discovery program and is often used to rank prospective hits resulting from a screen.22 Interestingly, many of the compounds have at least two points of chemical variability to fit with existing chemistry approaches. In cases where synthesis is difficult, the compounds may be produced in larger quantities in yeast, using metabolic engineering approaches, because this is how they originated.
The described compounds can be assessed for suitability as hits for drug discovery. A phenolic group is present in several of the compounds. Although phenols can represent valuable starting points for lead identification or lead optimization (LI/LO), their presence in a final drug is usually undesired as it is often associated with a clearance liability via glucoronidation. In these cases, common strategies can be used to either replace this feature (for example with acidic N–H groups) or to produce prodrug formulations. In addition, some of the compounds contain anilinic groups which are often flagged as a potential toxicophore. However, the most common motif observed is p-anthranilic acid (PABA), which is known to have little toxicity and is of minimal concern. After all, it should be considered that these are hit structures. During LI/LO, possible problems could be addressed—in the case of a genotoxic aniline, a solution could be to replace it with the corresponding amino-pyridine, which has a lower tendency to be genotoxic.
There is no doubt that compounds produced by this method have an inherent tendency toward a more challenging synthesis compared to compounds which have originated in a chemical library. However, for this set of compounds, synthetic routes could quickly be envisaged for many. Therefore, it is expected that during an LI/LO process, the molecular properties and target affinity could be optimized rather rapidly. Examples for clearly tractable hit structures are 32, 33, and 34, which have been resynthesized. Conceptually, a hybrid approach may also be considered to optimize SB compounds in which some of the enzymes which give rise to the hits are used in vitro (biocatalysis) and combined with conventional synthetic chemistry. This would enable scalability and rapid optimization demanded by the LO process.
Synthetic Biology Compound Novelty Analysis
SciFinder, PubChem, ChemSpider, and ChEMBL were searched for the presence of the SB originated structures. Figure 3a shows the percentage of compounds new to each database. Overall, no match was found for more than 75% of the compounds in either SciFinder, PubChem, or ChemSpider. Similarity and substructure searches were carried out in SciFinder. Figure 3b shows a summary of the SciFinder structural similarity scores of the SB compounds isolated in this work, with 100% similarity designating previously reported structures and ≥99% structural similarity relating to different or previously not assigned relative stereochemistry or close analogues differing only in the length of aliphatic side chains (e.g., methyl/ethyl). These similarity and substructure searches also helped in defining core scaffolds of the isolated compounds, for which side chains were removed. Scaffolds were defined as novel if both the parent compound and the core skeleton exhibited SciFinder similarity scores of less than 90% compared to any other entry. Interestingly, scaffold novelty often derived from new combinations of known natural product-derived substructures. Figure 4 shows examples of novel scaffolds derived from SB compounds. Overall ≈20% of the compounds represented novel scaffolds, although some redundancies within the set were observed. Forty percent of these compounds exhibited activity in the secondary BMV assay, therefore relating novelty to biologically relevant chemical space.
Figure 3.

Novelty and similarity of synthetic biology compounds. (a) Percentage of synthetic biology compounds new to different databases. (b) Similarity scores of synthetic biology compounds in SciFinder. (c) PubChem nearest neighbor molecular quantum numbers city-block distance (CBDMQN) difference of synthetic biology compounds. (d) GDB-17 CBDMQN difference of synthetic biology compounds.
Figure 4.

Novel synthetic biology scaffolds.
Synthetic Biology Compound Diversity Analysis
Studies were conducted to determine diversity within the SB compound set. One common way to ascertain diversity is to fingerprint compounds by a composition method (e.g., Daylight or MACCS keys) and then measure the distance between different library members according to a Tanimoto or other similarity metric.24 Compounds above a similarity threshold can be assigned to the same “cluster”, whereas those more dissimilar are not. To this end, compounds were fingerprinted with MACCS keys, and a Tanimoto similarity value of 0.85 was used to define clusters. The average cluster size across the limited 74 SB compound set is 1.27, and thus, the majority of compounds exist in their own cluster and can be described as singletons. The largest cluster size is 3, of which there are four such clusters (Supporting Information, Table S2).
These data were compared with distributions calculated for two “diverse” libraries of commercially available fragments. In these compound collections (ChemBridge and Key Organics), the average cluster sizes across the compound sets are 1.36 and 1.30, respectively. This shows that the SB set has fewer average compounds per cluster (i.e., is more diverse) than the fragment collections examined. Of course, there is a significant difference in size of the compound sets analyzed. However, the results obtained for the ChemBridge and Key Organics sets nevertheless exemplify typical distributions observed across libraries that were constructed with diversity as a fundamental objective.
As the size of the SB libraries increases, the issue of compound redundancy may have to be addressed. However, diversity of the SB compounds is controlled by the nature of the functional screen used for isolating the yeast transformants. Thus, use of a different screen would be expected to give rise to new compounds. Furthermore, retention times and high-resolution MS data can be used to dereplicate active fractions. In addition, one of the important aspects of this approach is that if a limitation on scaffold diversity was observed, further sets of genes could be introduced into the yeast, expanding the chemical repertoire, and thus, to some extent, diversity could also be controlled in this manner.
Chemical Space Classification by Molecular Quantum Numbers
Reymond and co-workers have described a classification system that places organic molecules in chemical space on the basis of 42 molecular quantum numbers (MQN).25,26 The MQNs are defined as value descriptors of structural elements by simply counting atoms of certain types, bonds, polar groups, and topological features. Using the MQN webserver (http://reymond.dcb.unibe.ch/), we calculated the similarity of the isolated SB compounds to nearest neighbors in the PubChem (Figure 3c) and GDB-17 databases (Figure 3d). GDB-17 stands for a chemical universe database of organic molecules of up to 17 heavy atoms (C, N, O, S, and Cl) virtually created following simple chemical stability and feasibility rules. It contains 166 billion small molecules.27 Similarity in MQN-space is measured in city-block distance (CBDMQN) by summing up the absolute values of the differences between the MQN-values of the SB compounds and the nearest neighbor in the reference database. CBDMQN for SB molecules with less than 17 heavy atoms (HAC ≤ 17) and their nearest neighbor in the GDB-17 database were calculated, giving rise to distance (d) values between d = 2 and d = 11 (Figure 3d). A similar result was obtained calculating CBDMQN between all 74 SB structures and their closest neighbor in the PubChem database, leading to distances between d = 0 and d = 13 (Figure 3c). Although the magnitude of the d values does not indicate a high diversity to known or theoretically predicted structures,28 SB compounds are clearly novel within the vast chemical space explored in these databases.
Chemical Space Analysis Using ChemGPS-NP
Having established that the SB compounds were generally diverse and novel, it was of interest to know whether these compounds are more similar to natural products (NP) or commercial screening compounds/synthetic small molecules.
One convenient system to describe the NP-likeness of a set of compounds is provided by the ChemGPS-NP approach.29,30 This method converts relevant molecule descriptors (associated with concepts related to NP-likeness, such as number of chiral centers, ratio of O vs N atoms, etc.) into principal component space and provides a standardized system to “map” sets of new compounds onto these coordinates. It is useful to use such a system, because it has already been shown to be relevant in separating natural from non-natural compounds.29,30
A web server generating the raw principal component analysis (PCA) scores for compounds was utilized to calculate values for three data sets, a compound set comprising 20 000 randomly selected compounds extracted from the Asinex Platinum screening library,31 a natural product set comprising 5150 compounds from Analyticon (MEGx library)32 and the 74 synthetic biology compounds identified in yeast.
Figure 5 shows the resulting map of chemical space between the libraries. According to this PCA mapping, there is a clear chemical space separation between the screening library and the natural product library. Furthermore, the SB compounds span both chemical spaces. The majority (65 compounds) fall into the space occupied by the screening compounds, but a significant number (9 compounds, 12%) sit outside this, in the coordinate space occupied by natural products.
Figure 5.
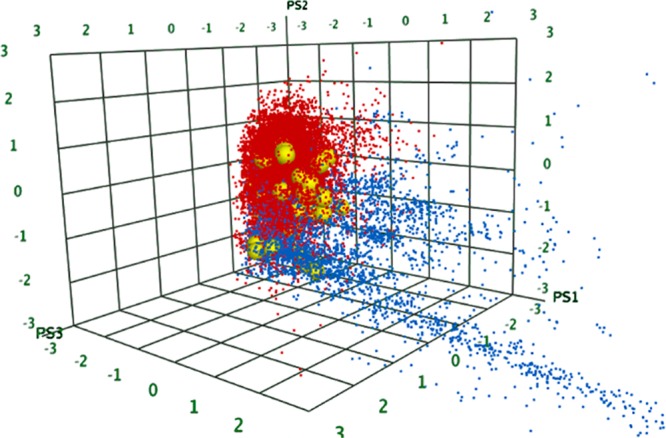
PCA mapping of different libraries. Subset of Asinex screening library (red), Analyticon NP library (blue), and the SB compounds (yellow). The three axes represent the three first principal components as generated by the ChemGPS-NP webserver.
Given the relatively small number of SB compounds being analyzed, we wanted to evaluate the statistical likelihood of achieving this result. An experiment was thus conducted by selecting 100 randomly picked sets of compounds with the same molecular weight distribution (each to the nearest whole MW value) to the synthetic biology set from an internal database containing over 20 million commercially available compounds. The internal database only contained low numbers of natural product compounds (<0.4%). ChemGPS-NP scores were generated for each of these sets, and the numbers of compounds falling within the natural product volume were recorded. We assumed that these data would be normally distributed. On average, 1.7 compounds (2.2%) fell in the NP-only space (standard deviation 1.1). Because 9 (12%) of the SB compounds exist in this space, we conclude it unlikely to have occurred by chance, ρ = 8 × 10–12.
Activity of Synthetic Biology Compounds
The yeast strain used for this study was equipped with a Brome Mosaic Virus functional assay, such that the yeast only survives in the presence of a small molecule inhibitor of viral replication. Only when the yeast strain was transformed with YACs encoding cDNA libraries and metabolic pathways producing different small molecules were surviving colonies obtained. BMV served as a surrogate for human infectious positive-sense single-stranded (+ve ss) RNA viruses. A secondary BMV assay guided fractionation of supernatant extract from surviving colonies led to fractions with high antiviral activity, which were then further fractionated for structural elucidation and activity determination of the compounds.
Table 2 (and Supporting Information Figure S3) shows activity data (IC50) of selected SB compounds in the secondary BMV assay and against two viruses of therapeutic interest, Dengue (DENV) and human hepatitis C (HCV) virus. (Characterization data for the 6 compounds can be found in the Supporting Information.) Compounds 31 and 32–34 were up-scaled by metabolic engineering or synthetic chemistry, respectively, and activity data were determined on the resynthesized products. Activity data in the BMV assay (Table 2) was recorded for a set of compounds within or above the MW range of fragments. pIC50 values vary between 4.3 (compound 30) and 6.7 (compound 34) (typical, literature-derived value for fragment-like molecules pIC50 = 4.4)16 reflecting, in accordance with Hann’s complexity model,8,9 the slightly larger size of some of the molecules. Compound 34 shows above average activity for its size. Interestingly, μM activity was also found for two therapeutically relevant viruses, HCV and DENV, which, like brome mosaic virus, also belong to Group IV +ve ss RNA viruses (Table 2). Overall, these data show the potential of the BMV model system to detect compounds that might serve as starting points for the development of antiviral drugs against Group IV +ve ss RNA viruses of therapeutic interest.
Table 2. IC50 Values of Synthetic Biology Compounds in a Secondary Brome Mosaic Virus (BMV), Human Hepatitis C Virus (HCV), and Dengue Virus (DENV) Assaya.
| compd | pathway/cDNA combination | MW [Da] | BMV, IC50 [μM](pIC50) | other viruses, IC50 [μM] |
|---|---|---|---|---|
| 25 | type III polyketide/Brachionis plicatilis | 272 | 32 (4.49) | HCV, 17.2 |
| 30 | type III polyketide/Hypericum perforatum | 214 | 50 (4.30) | |
| 31 | tetra- and diterpenoids/virus-infected tobacco | 318 | 1.3 (5.89) | |
| 32b | type III polyketide/Brachionis plicatilis | 283 | 40.3 (4.39) | |
| 33 | lignans/Hypericum perforatum | 259 | 3.8 (5.42) | HCV, 18.2 |
| 34 | flavonoids/Strobilantes ciliates | 259 | 0.2 (6.70) | HCV, 40.6 |
| DENV, 50 | ||||
| Ribavirin (control) | 244 | 113 (3.95) | HCV, 28 | |
| DENV, 120 |
The pathway/cDNA combination refers to the YAC clone from which they were isolated.
Racemic mixture used (resynthesized).
Utility of the Approach
Overall, the activity, low lipophilicity and MW, the low number of aromatic rings, the absence of basic centers, a favorable stereochemical, and high shape complexity make the isolated SB compounds high quality and optimizable start points for drug discovery. On the basis of the molecules identified, we assert that yeast-produced SB products allow the exploration of new areas of biologically relevant chemical space. A significant number of the isolated compounds are novel and diverse according to the tests described in the Results section. Importantly, the novelty is biologically derived and not at the expense of drug-likeness.
Given the attractive features of the SB approach, it is important to also consider some of the practical aspects. Cycle times, consisting of the creation of the libraries, primary and secondary screening, dereplication, isolation, and structure elucidation of active metabolome components, have averaged 6–9 months for screens of 50–60 libraries, resulting in the identification of the hits described in this study. Overall, this is comparable to a typical complete time frame for HTS- or FBS-based hit-finding approaches.
For the first time, novel compounds produced by SB are reported. In view of their activity and physicochemical and structural properties, these compounds represent good start points for medicinal chemistry optimization and can be considered qualified hits or early leads for drug discovery. Furthermore, the SB approach complements current hit-finding practices and can be easily integrated in the drug discovery process.
Methods
Yeast Strains
The haploid yeast strains used for constructing the primary screening assay strains have a S288C genetic background with the following genotypes MATa LYS2 ade8Δ his3Δ ura3Δ leu2Δ trp1Δ arg4Δ and MATα lys2Δ ADE8 his3Δ ura3Δ leu2Δ trp1Δ arg4Δ. The haploid yeast strain used for constructing the secondary screening assay has a S288C genetic background with the following genotype MATa met15Δ his3Δ ura3Δ leu2Δ pdr1Δ pdr3Δ erg6Δ
Primary BMV Assay Screening
Evolva has developed a functional BMV replication assay in yeast on the basis of the publications of Ahlquist and co-workers.33−35 The assay has been applied to Evolva’s proprietary SB platform for creating small molecule libraries in yeast14 (see Supporting Information, Table S4).
The primary BMV screening strain contains a BMV replication system consisting of expression cassettes for BMV RNA1, BMV RNA2, BMV RNA3-URA3, and BMV RNA3-Renilla that are integrated into the yeast genome under the control of inducible GAL1 promoters, as described by Ahlquist and co-workers.33,34 Galactose-media-induced expression of the BMV cassettes leads to expression of URA3 and Renilla reporter genes in a BMV replication-specific manner. The URA3 gene encodes orotidine 5-phosphate decarboxylase that can convert 5-fluoroorotic acid (5-FOA) to 5-fluorouracil, which causes cell death. The Renilla gene encodes Renilla luciferase that catalyzes a light-emitting reaction, which can be quantitatively detected by a chemiluminescence reader. The primary BMV strain also has an integrated Firefly expression cassette under the control of a constitutive ADH1 promoter. This gene is expressed independently of BMV replication and is used for normalizing the signal from the Renilla luciferase reaction.
Screening libraries were made by mating haploid primary BMV strains that had been transformed with YAC pools. The YAC pools were constructed and introduced in the haploid primary BMV strains essentially as described previously.14 Each YAC consists of multiple expression cassettes under the control of inducible methionine dependent promoters.
Haploid MATα yeast libraries containing pathway YACs (pathway libraries, LEU2 marker) and haploid MATa yeast libraries containing cDNA YACs (cDNA libraries, HIS3 marker) were prepared. Fifty different combinations of pathway and cDNA libraries were mated to create diploid screening libraries. In addition, diploid “pathway only” screening libraries were created by mating the haploid MATα pathway libraries with the haploid MATa yeast containing an empty HIS3 plasmid. Furthermore, the haploid yeast strains with HIS3 and LEU2 plasmids were mated to make a diploid screening strain control without YACs.
To start the screening process, an overnight culture of the diploid library was grown in glucose media without methionine. The next day, 2 × 50 million diploid cells were spread on two screening plates (24 × 24 cm) containing solid synthetic complete (SC) media without leucine, histidine, or methionine and containing 1% galactose, 1% raffinose, 0.12% FOA, and 80 mg/L uracil. The galactose induces the BMV replication, whereas the absence of methionine induces expression of the YAC genes.
The number of surviving clones varied with the pathway and cDNA combination and was typically in the range of 200–2000 surviving clones per 50 million cells. Survivor clones (176) per diploid library were picked from the FOA plates and transferred to two 96-well master plates with SC media with 2% glucose, without histidine and leucine, and with 2 mM methionine, to shut down BMV replication and expression of the YAC genes. In total, 58 diploid libraries × 176 survivors = 10208 clones were picked from the primary screening.
Confirmation of Primary Hit Clones
A confirmatory process was carried out to rank the survivor clones according to antiviral potency of each clone. The master plate was used to inoculate a 96-well plate with SC media with 2% glucose + 2 mM methionine (YAC expression off) or without methionine (YAC expression on) and incubated overnight. Next day, each plate was used to inoculate two 96 well plates containing SC media with 2% galactose (BMV replication turned on) with either 2 mM methionine (+methionine, YAC expression off) or without methionine (−methionine, YAC expression on) and incubated overnight. The following day, these plates were used for the luciferase assay using the Dual-Glo Luciferase Assay System (Promega). A Microlab Star pipetting robot (Hamilton) was used for handling of plate cultures, lysis buffers, and luciferase substrates. A 7 μL culture from each well was transferred in duplicate to 384-well plates, in which cell lysis, Firefly, and Renilla measurements were carried out. The Firefly and Renilla signals were measured in a Pherastar FS plate reader (BMG Labtech) by first adding 7 μL of lysis buffer containing Firefly substrate. After a 1 h incubation, the Firefly signal was measured. Next, 7 μL of StopGlo buffer containing Renilla substrate was added to quench the Firefly signal and start the Renilla reaction. After 45 min of incubation time, the Renilla signal was measured.
The luciferase readings were used to calculate the YAC-dependent inhibition of BMV viral replication for each clone (% inhibition = (Renilla/Firefly (+methionine = YAC off) – Renilla/Firefly (−methionine = YAC on))/(Renilla/Firefly (+methionine = YAC off)) × 100).
The most potent clones with inhibition ≥60% were picked and used for extraction and secondary assay testing. The number of clones selected based on this cut off value varied from 0–35 clones/library depending on the screening library. Approximately 300 clones from 58 screening libraries were selected on the basis of this confirmation.
Secondary BMV Assay Screening
The secondary BMV strain was used for screening of metabolite fractions to identify the fractions/compounds that are active against BMV. It contains the same integrated expression cassettes as the primary BMV screening assay described above, except for the BMV RNA3-URA3 cassette. In addition, the strain has been deleted in the PDR1, PDR3, and ERG6 genes to enhance the uptake of exogenously added compounds. The PDR1 and PDR3 genes regulate ABC transporters that remove xenobiotic substances from the yeast cytoplasm.36 ERG6 deletion is known to give an increased “permeability” of the yeast membrane to exogenously added compounds.37 The Renilla and Firefly activity is measured by the Dual-Glo kit (Promega) as described above
A culture of secondary assay yeast strain was grown overnight in SC containing 2% glucose. The next day, the culture was washed and diluted to OD600 = 0.3 in SC media containing 2% galactose and distributed in two 96-deep-well plates with 400 μL of cell culture in each well. A 2 μL aliquot of each metabolite fraction resuspended in 20 μL of DMSO was transferred to the 96-deep-well plates with the Microlab Star pipetting robot (Hamilton). The plates were incubated overnight at 30 °C, 300 rpm to let the fraction compounds enter the yeast and interfere with the BMV viral replication system. Negative control (no compound, used as baseline) and positive controls (40 and 200 μM Ribavirin) were included. The following day, the luciferase assay was carried out as described above for the confirmatory process. The inhibition of BMV replication was calculated according to the formula: (% inhibition = (Renilla/Firefly (baseline) – Renilla/Firefly (fraction))/(Renilla/Firefly (baseline)) × 100). Fractions that show activity by inhibiting the viral replication system were identified (see Supporting Information Table S5 and Figure S3 for additional data).
The inhibition data for the CEY fractions were grouped and aligned in a pathway specific manner to identify pathway-specific clusters of fractions, pathway + cDNA specific clusters of fractions, and clone-specific fractions (singlets). A cluster arises when the same active fraction is present in more than one CEY. The clustering analysis is used to identify redundancy of molecules that arise when the same combinations of YAC genes appear in more than one yeast clone.
In total, 457 fractions gave a positive dose response that justified further dereplication work against corresponding, nonactive fractions of the control strain using MSXelerator in order to find YAC-dependent fractions active against BMV. Finally, 28 compounds were isolated (≈10% hit rate based on the number of “clone hits”).
Purification of Compounds for MS and NMR Characterization
Yeast culture batches (4 L) were grown and extracted using the same conditions as those described for 0.5 L batch preparation. The reconstituted supernatant was injected in a preparative HPLC column (XBridge C18, 19 × 250 mm, 5 μm, Waters). Mobile phases used were A (0.1% TFA in water) and B (0.1% TFA in acetonitrile). The separation of the target compounds was achieved by a gradient from 1% B to 100% B in 27 min. The collected fractions were dried in a Genevac HT12 evaporation system, and a final quality control (QC) was completed in the LC-MS using the LC conditions described above.
NMR Analysis of Compounds
All NMR experiments were performed in DMSO-d6 at 25 °C using a Bruker Avance III 600 MHz NMR spectrometer equipped with a 1.7 mm cryogenic TCI probe.
The structures were solved by means of standard homo- and heteronuclear multipulse NMR experiments, namely, 1H,1H–COSY, 1H,1H-ROESY, 1H,13C-HSQC, 1H,13C-HMBC, and 1H,15N-HMBC experiments.
Calculation of Physicochemical Properties
Physicochemical properties were calculated using web-based molinspiration (http://www.molinspiration.com/cgi-bin/properties), OSIRIS Property Explorer (http://www.organic-chemistry.org/prog/peo/), and ChemAxon chemicalize (http://www.chemicalize.org/) software.
Shape and stereochemical complexity were calculated using a custom script written in support vector language within the MOE environment (CCG, Inc., www.chemcomp.com). Cores were defined by removing exocyclic groups with more than one rotatable bond. The drug-like compounds used for these calculations contained a set of 870 compounds which were extracted as a subset from the DrugBank database, selected on the basis of the criteria that molecular weight was less than 500.38
Diversity Analysis Using MACCS Keys
MACCS key fingerprints were generated for the SB compounds using the MOE software package (CCG, Inc.). Cluster composition was then also calculated using the Fingerprint Cluster method in MOE with a 0.85 similarity threshold and Tanimoto similarity metric. The same methodology was applied to the other fragment databases which were compared in the study.
ChemGPS Analysis
Construction of a randomized 20 000 compound set extracted from the Asinex Platinum screening library was undertaken using a script written in support vector language within the MOE operating system. The relevant sets of compounds were uploaded to the ChemGPS-NP webserver. The maximum size of a submitted database was 8000 compounds; therefore, the screening library compounds were submitted as several jobs. The resulting flat result files were imported into MOE databases before 3D graphs were generated in the same environment.
Acknowledgments
The authors gratefully acknowledge H. Waldmann and K.-H. Altmann for fruitful discussions and their constructive comments on the manuscript. We would also like to thank the following employees of Evolva S.A. for their assistance in this work: G. Salerno, F. Brianza, C. Folly, D. Williams, C. Nielsen, and S. Schmid.
Supporting Information Available
Contains a pathway and cDNA combination summary of the YAC clones used, graphs on Ro5 and mol wt. distributions for the SB compounds, structures and analytical data of all 74 SB compounds, diversity (cluster) analysis comparing the SB compounds with other compound libraries, the identity of genes used in the YACs, and luciferase assay data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
∥ Novartis Pharma AG, WSJ.145.10.61, CH-4002 Basel, Switzerland
Author Contributions
J.K. and J.H. led the project. J.K. analyzed yeast metabolomes and carried out all structure elucidation. J.K., J.R.H., and W.D.O.H. calculated all physicochemical, structural, and shape-based descriptors and carried out novelty searches and diversity calculations. J.K., J.R.H., and W.D.O.H. wrote the manuscript. T.B. isolated the compounds. T.O and F.D. performed primary and secondary BMV assays. G.J. made synthetic batches of compounds 32–34 for additional biological tests. A.H. was involved with yeast metabolome analysis and compound isolation.
The authors declare the following competing financial interest(s): J.K., T.O., F.D., and A.H. are employees of Evolva SA. J.H. and T.B have financial interest in Evolva. J.R.H. and W.D.O.H. are employees of and have a financial interest in Prosarix Ltd. T.B. is an employee of Novartis Pharma AG. G.J. is an employee of F. Hoffmann-La Roche Ltd.
Supplementary Material
References
- Macarron R.; Banks M. N.; Bojanic D.; Burns D. J.; Cirovic D. A.; Garyantes T.; Green D. V.; Hertzberg R. P.; Janzen W. P.; Paslay J. W.; Schopfer U.; Sittampalam G. S. (2011) Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discovery 10, 188–195. [DOI] [PubMed] [Google Scholar]
- Barker A.; Kettle J. G.; Nowak T.; Pease J. E. (2013) Expanding medicinal chemistry space. Drug Discovery Today 18, 298–304. [DOI] [PubMed] [Google Scholar]
- Siegel M. G.; Vieth M. (2007) Drugs in other drugs: a new look at drugs as fragments. Drug Discovery Today 12, 71–79. [DOI] [PubMed] [Google Scholar]
- Babaoglu K.; Shoichet B. K. (2006) Deconstructing fragment-based inhibitor discovery. Nat. Chem. Biol. 2, 720–723. [DOI] [PubMed] [Google Scholar]
- Kerserü G. M.; Makara G. M. (2009) The influence of lead discovery strategies on the properties of drug candidates. Nat. Rev. Drug Discovery 8, 203–212. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Kerserü G. M. (2012) Finding the sweet spot: the role of nature and nurture in medicinal chemistry. Nat. Rev. Drug Discovery 11, 355–365. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; St-Gallay S. A. (2011) The influence of the ’organizational factor’ on compound quality in drug discovery. Nat. Rev. Drug Discovery 10, 749–765. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Leach A. R.; Harper G. (2001) Molecular complexity and its impact on the probability of finding leads for drug discovery. J. Chem. Inf. Comput. Sci. 41, 856–864. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Hann M. M. (2011) Molecular complexity and fragment-based drug discovery: ten years on. Curr. Opin. Chem. Biol. 15, 489–496. [DOI] [PubMed] [Google Scholar]
- Evans B. E.; Rittle K. E.; Bock M. G.; DiPardo R. M.; Freidinger R. M.; Whitter W. L.; Lundell G. F.; Veber D. F.; Anderson P. S. (1988) Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J. Med. Chem. 31, 2235–2246. [DOI] [PubMed] [Google Scholar]
- Breinbauer R.; Manger M.; Scheck M.; Waldmann H. (2002) Natural product guided compound library development. Curr. Med. Chem. 9, 2129–2145. [DOI] [PubMed] [Google Scholar]
- Wetzel S.; Schuffenhauer A.; Roggo S.; Ertl P.; Waldmann H. (2007) Cheminformatic analysis of natural products and their chemical space. Chimia Int. J. Chem. 61, 355–360. [Google Scholar]
- Over B.; Wetzel S.; Grütter C.; Nakai Y.; Renner S.; Rauh D.; Waldmann H. (2013) Natural-product-derived fragments for fragment-based ligand discovery. Nat. Chem. 5, 21–28. [DOI] [PubMed] [Google Scholar]
- Naesby M.; Nielsen S. V.; Nielsen C. A.; Green T.; Tange T. O.; Simón E.; Knechtle P.; Hansson A.; Schwab M. S.; Titiz O.; Folly C.; Archila R. E.; Maver M.; van Sint Fiet S.; Boussemghoune T.; Janes M.; Kumar A. S.; Sonkar S. P.; Mitra P. P.; Benjamin V. A.; Korrapati N.; Suman I.; Hansen E. H.; Thybo T.; Goldsmith N.; Sorensen A. S. (2009) Yeast artificial chromosomes employed for random assembly of biosynthetic pathways and production of diverse compounds in Saccharomyces cerevisiae. Microb. Cell Fact. 8, 45–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. (2003) A ’rule of three’ for fragment-based lead discovery?. Drug Discovery Today 8, 876–877. [DOI] [PubMed] [Google Scholar]
- Schultes S.; de Graaf C.; Haaksma1 E. E. J.; de Esch I. J. P.; Leurs R.; Krämer O. (2010) Ligand efficiency as a guide in fragment hit selection and optimization. Drug Discovery Today: Technol. 7, 157–162. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. (2001) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Grabowski K.; Baringhaus K.-H.; Schneider G. (2008) Scaffold diversity of natural products: inspiration for combinatorial library design. Nat. Prod. Rep. 25, 892–904. [DOI] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. (2009) Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- Clemons P. A.; Bodycombe N. E.; Carrinski H. A.; Wilson J. A.; Shamji A. F.; Wagner B. K.; Koehler A. N.; Schreiber S. L. (2010) Small molecules of different origins have distinct distributions of structural complexity that correlate with protein-binding profiles. Proc. Natl. Acad. Sci. U.S.A. 107, 18787–18792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons P. A.; Wilson J. A.; Dančík V.; Muller S.; Carrinski H. A.; Wagner B. K.; Koehler A. N.; Schreiber S. L. (2011) Quantifying structure and performance diversity for sets of small molecules comprising small-molecule screening collections. Proc. Natl. Acad. Sci. U.S.A. 108, 6817–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray C. W.; Verdonk M. L.; Rees D. C. (2012) Experiences in fragment-based drug discovery. Trends Pharmacol. Sci. 33, 224–232. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H. Y.; Smith B. R.; Ward K. W.; Kopple K. D. (2002) Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 45, 2615–2623. [DOI] [PubMed] [Google Scholar]
- Tanimoto T.An Elementary Mathematical Theory of Classification and Prediction. Internal IBM Technical Report, November 17, 1957.
- Luethi E.; Nguyen K. T.; Bürzle M.; Blum L. C.; Suzuki Y.; Hediger M.; Reymond J. L. (2010) Identification of selective norbornane-type aspartate analogue inhibitors of the glutamate transporter 1 (GLT-1) from the chemical universe generated database (GDB). J. Med. Chem. 53, 7236–7250. [DOI] [PubMed] [Google Scholar]
- Reymond J.-L.; Blum L. C.; van Deursen R. (2011) Exploring the chemical space of known and unknown organic small molecules at www.gdb.unibe.ch. Chimia 65, 863–867. [DOI] [PubMed] [Google Scholar]
- Ruddigkeit L.; van Deursen R.; Blum L. C.; Reymond J.-L. (2012) Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17. J. Chem. Inf. Model. 52, 2864–2875. [DOI] [PubMed] [Google Scholar]
- Ruddigkeit L.; Blum L. C.; Reymond J.-L. (2013) Visualization and virtual screening of the chemical universe database GDB-17. J. Chem. Inf. Model. 53, 56–65. [DOI] [PubMed] [Google Scholar]
- Larsson J.; Gottfries J.; Muresan S.; Backlund A. (2007) ChemGPS-NP: tuned for navigation in biologically relevant chemical space. J. Nat. Prod. 70, 789–794. [DOI] [PubMed] [Google Scholar]
- Rosén J.; Lövgren A.; Kogej T.; Muresan S.; Gottfries J.; Backlund A. (2009) ChemGPS-NP(Web): chemical space navigation online. J. Comput.-Aided Mol. Des. 23, 253–259. [DOI] [PubMed] [Google Scholar]
- Asinex Ltd. http://www.asinex.com/download-zone.html Platinum 01/13
- AnalytiCon Discovery GmbH. http://www.ac-discovery.com/content/Products%26Technologies/MEGAbolite.php. Purified Natural Products of microbial and plant origin Release_MEGx_R130101.zip
- Janda M.; Ahlquist P. (1993) RNA-dependent replication, transcription, and persistence of brome mosaic virus RNA replicons in S. cerevisiae. Cell 26, 961–970. [DOI] [PubMed] [Google Scholar]
- Ishikawa M.; Janda M.; Krol M. A.; Ahlquist P. (1997) In vivo DNA expression of functional brome mosaic virus RNA replicons in Saccharomyces cerevisiae. J. Virol. 71, 7781–7790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner D. B.; Lindenbach B. D.; Grdzelishvili V. Z.; Noueiry A. O.; Paul S. M.; Ahlquist P. (2003) Systematic, genome-wide identification of host genes affecting replication of a positive-strand RNA virus 2003. Proc. Natl. Acad. Sci. U.S.A. 100, 15764–15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfger H.; Mamnun Y. M.; Kuchler K. (2001) Fungal ABC proteins: pleiotropic drug resistance, stress response and cellular detoxification. Res. Microbiol. 152, 375–389. [DOI] [PubMed] [Google Scholar]
- Gaber R. F.; Copple D. M.; Kennedy B. K.; Vidal M.; Bard M. (1989) The yeast gene ERG6 is required for normal membrane function but is not essential for biosynthesis of the cell-cycle-sparking sterol. Mol. Cell. Biol. 9, 3447–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Shrivastava S.; Hassanali M.; Stothard P.; Chang Z.; Woolsey J. (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 34, D668–D672. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.