

Abstract
Background
It is generally agreed that horizontal gene transfer (HGT) is common in phagotrophic protists. However, the overall scale of HGT and the cumulative impact of acquired genes on the evolution of these organisms remain largely unknown.
Results
Choanoflagellates are phagotrophs and the closest living relatives of animals. In this study, we performed phylogenomic analyses to investigate the scale of HGT and the evolutionary importance of horizontally acquired genes in the choanoflagellate Monosiga brevicollis. Our analyses identified 405 genes that are likely derived from algae and prokaryotes, accounting for approximately 4.4% of the Monosiga nuclear genome. Many of the horizontally acquired genes identified in Monosiga were probably acquired from food sources, rather than by endosymbiotic gene transfer (EGT) from obsolete endosymbionts or plastids. Of 193 genes identified in our analyses with functional information, 84 (43.5%) are involved in carbohydrate or amino acid metabolism, and 45 (23.3%) are transporters and/or involved in response to oxidative, osmotic, antibiotic, or heavy metal stresses. Some identified genes may also participate in biosynthesis of important metabolites such as vitamins C and K12, porphyrins and phospholipids.
Conclusions
Our results suggest that HGT is frequent in Monosiga brevicollis and might have contributed substantially to its adaptation and evolution. This finding also highlights the importance of HGT in the genome and organismal evolution of phagotrophic eukaryotes.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-14-729) contains supplementary material, which is available to authorized users.
Keywords: Genome evolution, Choanoflagellates, HGT frequency, Eukaryotic evolution, Adaptation
Background
While horizontal gene transfer (HGT) in prokaryotes has been extensively studied and its significance in prokaryotic evolution is well known, our knowledge about HGT in eukaryotes is relatively limited [1–4]. In eukaryotes, a large number of genes are of bacterial origin, many of which are derived from mitochondria or plastids through endosymbiotic gene transfer (EGT), whereas some others are from independent HGT events. A gene ratchet mechanism “you are what you eat” has been proposed to explain frequent gene transfer events in protists, especially those of phagotrophic lifestyles [5]. The list of HGT-derived genes in diverse protists becomes increasingly longer thanks to recent studies [6–9].
Monosiga brevicollis is a unicellular member of choanoflagellates, a group of free-living and phagotrophic microbial eukaryotes. Characterized by a central flagellum surrounded by a ring of 30–40 microvilli, choanoflagellates resemble sponge choanocytes morphologically [10]. Molecular phylogenetic analyses show that choanoflagellates form a distinct lineage that is closely related to animals [11, 12]. Because of their unique evolutionary position, choanoflagellates bear great significance in understanding the origin of animals. Genome of M. brevicollis has been sequenced and annotated [13], thus offering a good opportunity for comparative genomic studies to understand the evolution of choanoflagellates.
Monosiga brevicollis has structures to facilitate swimming and feeding. Its flagella can cause water current when in motion, which in turn propel itself to swim freely. Its microvillar collar helps hold bacteria and other detritus from water flow and then engulfs them as foodstuff. Because of their high feeding efficiency, M. brevicollis and other choanoflagellates play a critical ecological role in marine ecosystems, particularly related to global carbon cycle [14]. Previous studies identified over 100 algal genes in M. brevicollis genome, and it has been suggested that many of these genes were likely acquired from food sources and might have benefited M. brevicollis in food digestion and adaptation to environmental stresses [15–18]. Although these studies identified an impressive number of acquired genes in M. brevicollis, the major sources of these genes were all from eukaryotic groups, and those from prokaryotes were not extensively investigated.
Currently, several computational programs, including PhyloGenie [19], DarkHorse [20] and AlienG [21], are available for genome screening of horizontally acquired genes. PhyloGenie predicts acquired genes by extracting generated gene trees that match specific topological constraints [19], and it has often been used in HGT identification [16, 22–25]. DarkHorse is a similarity-based tool for rapid identification of HGT candidates at genome level. This program predicts acquired genes by re-ranking the matches in BLAST search based on their species relationships with the query [20]. This approach alleviates the over-reliance on top-scoring BLAST hits for HGT identification and has been used in several studies [16, 26, 27]. AlienG is a newly developed computational program for HGT identification [21]. Based on an assumption that sequence similarity is correlated to sequence relatedness, AlienG detects candidates of acquired genes by comparing sequence similarities of the query to distantly related organisms versus those to close relatives. This program has recently been used in detecting acquired virulence effector gene homologs in chytrids [28], algae-related genes in animals [29] and HGT-derived genes in the basal land plant Physcomitrella patens[30]. In this study, we performed a comprehensive analysis to identify acquired genes in M. brevicollis based on predictions from these three computational programs. Through this extensive study, we aim to understand the overall scope and role of HGT in the evolution of Monosiga.
Results and discussion
Genome screening for foreign genes in M. brevicollis
Although both PhyloGenie [19] and DarkHorse [20] have been successfully used in some studies [16, 27, 28, 31], their limitations are obvious. Because PhyloGenie samples top hits of BLAST search for phylogenetic tree construction, a large database may lead to biased taxonomic sampling when the top hits are from the same or closely related taxonomic groups. Likewise, DarkHorse only accepts the NCBI non-redundant (nr) database, and genomes absent from nr would be missed in the analysis, thus leading to a large pool of candidates with many false positives. To obtain more reliable prediction results, we created a customized database covering representative species for prediction of foreign genes using PhyloGenie. Additionally, other available eukaryotic genomes were added to the NCBI nr database for AlienG analyses.
Identification of HGT is always complicated by multiple issues, such as differential losses, insufficient taxonomic sampling, and phylogenetic artifacts due to data quality or long-branch attraction [23, 32–34]. For each predicted foreign gene, we performed additional manual inspection for shared indels, conserved amino acid positions, unique gene structure, alignment quality, and potential contamination [16, 31]. The possibility of potential contamination was largely eliminated by checking whether the adjacent genes on genomic scaffolds showed metazoan/fungal affiliation. We also considered phyletic distribution of the gene (e.g., distribution only in choanoflagellates, prokaryotes and/or algae) and performed further manual phylogenetic analyses. A potential HGT event was inferred if the subject choanoflagellate gene forms a monophyletic group with homologs from prokaryotes and/or algae (with 70% or higher bootstrap support), to the exclusion of sequences from fungi/metazoans. Here, the term “algae” is loosely defined to include organisms with primary, secondary or tertiary plastids. Because oomycetes and ciliates are often considered to be of photosynthetic ancestry [35], they were also deemed as algae in this study. These measures would effectively reduce the artifacts associated with the gene tree construction.
Determination of HGT direction is not always straightforward. Other than gene tree topologies, we also considered additional lines of evidence when determining the direction of HGT, such as behavioral ecology of transfer partners and phyletic distribution of the transferred genes. For genes that are only distributed in prokaryotes and Monosiga, or only in algae and Monosiga, HGT from prokaryotes or algae to Monosiga was concluded; for genes with algal affiliation and sometimes broad distributions in diverse eukaryotic lineages, HGT from algae to Monosiga was inferred. Such inference of HGT direction can be justified based on: 1) Monosiga is phagotrophic and consumes algae and bacteria as food [36, 37]; 2) bacteria and many algal groups are more ancient than Monosiga; HGT in reverse directions would require ancestors of some major bacterial or algal groups as recipients, or it might entail multiple secondary transfer events among bacteria and algae; both are less likely scenarios. We should note here that some previously defined autotrophic algae are actually mixotrophic [38, 39] and, therefore, the possibility that these mixotrophs acquired genes from Monosiga cannot be excluded. However, given its highly efficient feeding activities, Monosiga may far more frequently be predators than being prey.
In addition to the algal and bacterial affiliations, anomalous relationships among other taxa can be observed in most gene trees in our analyses, where multiple eukaryotic sequences sporadically branch with prokaryotic homologs (Figure 1; Additional file 1). Such anomalous relationships are somewhat expected, given the frequent HGT within and between domains [1, 40], EGT from mitochondria, plastids and other endosymbionts [41], as well as homologous replacements [42]. In theory, differential gene loss can always be invoked as an explanation alternative to HGT. Although we cannot confidently exclude the possibility of differential gene loss, the patchy distribution of most putatively transferred genes in distantly related taxa would otherwise invoke many gene losses in other groups, a less parsimonious scenario. It should be cautioned, however, that this list of putatively acquired genes in Monosiga will likely change when improved phylogenetic methods and larger taxonomic samplings become possible in future.
Figure 1.
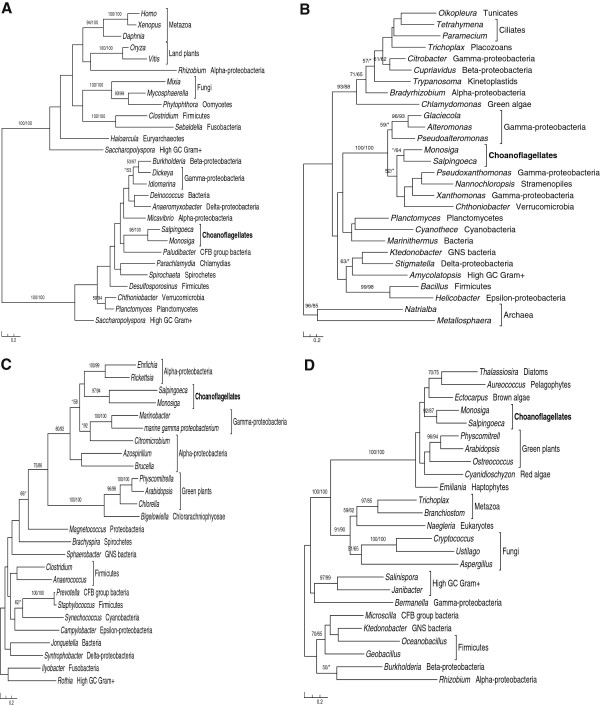
Molecular phylogenies of bacterial or algal genes in M. brevicollis . A. L-threonine 3-dehydrogenase (GenBank accession number: XP_001746273). B. D-beta-hydroxybutyrate dehydrogenase (GenBank accession number: XP_001744068). C. Metallo-beta-lactamase (GenBank accession number: XP_001747251). D. L-galactono-1,4-lactone dehydrogenase (GenBank accession number: XP_001748157). Numbers associated with branches show bootstrap values from maximum likelihood and distance analyses, respectively. Asterisks indicate bootstrap values lower than 50%. Taxonomic affiliations are shown after genus names, with choanoflagellates bolded.
Upon further manual curation, 405 genes in M. brevicollis were found to be more closely related to sequences from prokaryotes and/or algae (Additional file 1), more than 80% of which contain introns (Additional file 1: Table S1). Interestingly, after comparing with our previous studies [31] and unpublished data, we found that 17 genes were absent from the candidate lists predicted by all three programs. Three of these genes were identified when we studied the evolutionary history of the branched aspartate-derived pathway [31]; 14 other genes were identified when we performed analyses on other candidates. Most of these missed genes have an alien index score (bit score ratio between the top hit from distantly related taxa and that from closely related taxa) less than 1.2, which is the default setting of AlienG. Increasing alien index would produce fewer false positives in the prediction, but might miss true positives [21].
Of the 388 remaining genes, 358 (92.3%) were predicted by AlienG, and 345 (88.9%) and 204 (52.6%) by DarkHorse and PhyloGenie, respectively (Figure 2). The positive rate of AlienG in HGT prediction (43%) is also higher than those of PhyloGenie (34%) and DarkHorse (24%) (Figure 2). Other than the algorithmic difference, the better performance of AlienG may be attributed to the larger customized database used in the analyses. Because these three programs are based on different algorithms, analyses using a combination of two or all three programs would increase the total number of acquired genes identified. It is also important to note that some transferred genes could still be missed due to the balance between prediction sensitivity and specificity [21], which is reflected in the parameter settings.
Figure 2.
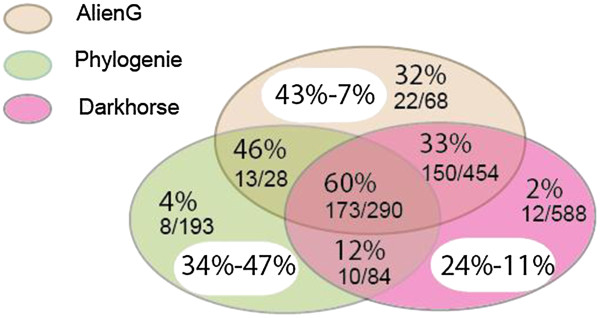
Evaluation of three computational programs on prediction of prokaryotic and algal genes in M. brevicollis .ÉFor AlienG, the alien index threshold was set to 1.2. For PhyloGenie, bootstrap value threshold for interested branches was set to 50%. Prediction results from three programs are shown in three different colors. The percentages in white ovals indicate positive rates (before hyphen) and false negative rates (after hyphen). The percentage in colored background indicates the positive rate for each part and is shown above. The numbers of foreign genes identified by manual curation (before slash) and originally predicted (after slash) are shown below.
Active feeding and gene acquisition in Monosiga
Of all 405 genes identified in our analyses, 240 were likely acquired from algae, 139 from bacteria, and 26 from either bacteria or algae. Because gene duplication may occur after HGT, we also estimated the number of HGT events by counting the acquired genes clustering together in the phylogenetic trees as a single event. The results suggested about 210 HGT events from algae, 100 from bacteria, and 20 from either bacteria or algae. Therefore, HGT from algae occurred nearly twice as frequently as those from bacteria. This raises an interesting question whether these algal genes resulted from past plastid (or algal) endosymbioses or from other sources. It is theoretically possible that the large number of algal genes detected in this study might have resulted from a historical plastid in Monosiga or choanoflagellates, even though no plastids or algal endosymbionts have ever been found in them. On the other hand, M. brevicollis is a protozoan species feeding on bacteria and microscopic algae. Based on the hypothesis “you are what you eat” [5], it is also likely that M. brevicollis acquired a large number of foreign genes from food sources.
Circumstantial evidences for the mechanism of gene acquisition may come from the details of HGT events and the lifestyles of recipient organisms. Although both active feeding and historical plastids (or algal endosymbionts) may explain the impressive number of algal genes in M. brevicollis[16], the numbers and sources of acquired genes through these two processes are different. Because any specific endosymbiont (including the plastid) will have a fixed gene pool, the number and sources of genes acquired from this endosymbiont are limited. By contrast, gene acquisition through feeding activities has no such strict limitation. Theoretically, phagotrophic protists could acquire a large number of foreign genes from diverse food sources over time, and their diet may be reflected in the sources (or donors) of acquired genes. The proportion of acquired genes in Monosiga genome (4.4%) is considerably higher than reported in many protozoan eukaryotes [8, 9, 40, 43, 44], but is in line with those reported in some other free-living microbial eukaryotes such as the red alga Galdieria sulphuraria[45] and bdelloid rotifers [46]. The potential donors for these acquired genes include diverse microscopic algal lineages such as green algae (Micromonas and Ostreococcus), diatoms (Thalassiosira and Phaeodactylum), haptophytes (Emiliania and Isochrysis), pelagophytes (Aureococcus), as well as numerous bacterial taxa, all of which are abundant and coexist in the same marine habitat with M. brevicollis. Given these considerations, we reason that many of the algal and bacterial genes identified in Monosiga are likely derived from food sources. However, because of the complication related to HGT identification (see above section), other scenarios cannot be definitely excluded. Such scenarios may include transfer events associated with parasites or other pathogens, viruses, mobile gene elements, phylogenetic artifacts, and misinterpretation due to insufficient taxon sampling.
Acquired genes and the adaptation of Monosiga
HGT in prokaryotes has been extensively studied [1, 47] and its role in eukaryotic evolution has gained increasing appreciation. Like in prokaryotes, HGT in eukaryotes can confer adaptive traits to recipient organisms and allow them to utilize new resources or explore new niches. For instance, it has been suggested that anaerobic diplomonads were derived from an aerobic ancestor, and their adoption of an anaerobic lifestyle was facilitated by the acquisition of anaerobic metabolism-related genes from prokaryotes [8]. Comparative genomic analyses also identified 84 foreign genes in the diplomonad parasite Spironucleus salmonicida, suggesting an important impact of HGT on diplomonad genome evolution [48]. The role of algal genes in the adaptation of M. brevicollis has been discussed in previous studies [15, 16, 49]. A more complete list of acquired genes identified in this study allows better understanding of HGT in the evolution and adaptation of Monosiga.
Of all 405 genes identified in this study, 212 have unknown biological functions, but 89 of them do contain known domains. We categorized the remaining 193 genes according to their putative biological functions (Figure 3). About one third of them (32.1%, 62 genes) are related to carbohydrate metabolism, 28 of which were also identified in earlier analyses [15, 16, 31, 49] and 34 are newly reported in this study (Additional file 1). Because of the importance of carbohydrates as basic energy sources and structural components, carbohydrate metabolism is interwoven with multiple other biochemical processes. Thirteen genes identified in our analyses encode glycoside hydrolases, which are common enzymes and involved in nutrient uptake and plant cell wall degradation. Acquisition of genes encoding glycoside hydrolases has also been reported in other organisms including rumen ciliates and the rumen fungus Orpinomyces, where the acquired genes are critical for the recipient organisms to adapt to an anaerobic, carbohydrate-rich environment [50, 51]. Likewise, acquisition of multiple carbohydrate metabolism-related genes might allow M. brevicollis to digest diverse food sources.
Figure 3.
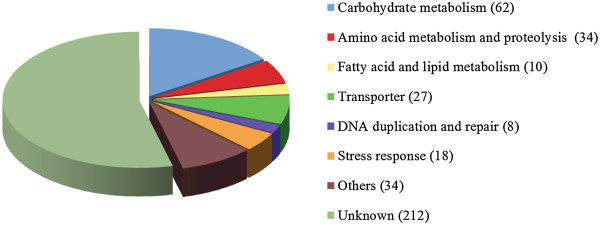
Functional categories for genes acquired from algae and bacteria in M. brevicollis .
The second largest functional category includes genes related to amino acid metabolism and protein degradation (Additional file 1). Among them, 12 acquired genes are related to proteolysis. Twenty-two genes are involved in the metabolism of amino acids, such as the biosynthesis of lysine, glutamate, histidine, and aspartate. In particular, acquired genes in Monosiga contributed greatly to the establishment of the branched aspartate-derived pathway that is responsible for the biosynthesis of methionine, isoleucine, threonine, and lysine [31]. All Monosiga genes specific to the diaminopimelic acid (DAP) pathway of lysine biosynthesis were acquired from either bacteria or algae [31]. By acquiring or improving capabilities of protein degradation and amino acid metabolism, M. brevicollis might ensure sufficient supply of amino acids. Ten other genes identified in our analyses are related to fatty acid and lipid metabolism (Additional file 1). In total, 106 acquired genes are related to metabolism of carbohydrates, proteins, or lipids, indicating foreign genes might have played an important role in basic and essential biological processes of M. brevicollis.
Some other HGT-derived genes are related to the biosynthesis of important metabolites. For example, L-galactono-1, 4-lactone dehydrogenase (Figure 1D) and 1, 4-dihydroxy-2-naphthoate octaprenyl-transferase are involved in the biosynthesis of vitamins C and K12, respectively. Given the antioxidant activities of vitamin C, acquisition of genes related to vitamin C biosynthesis might allow M. brevicollis to tolerate oxidative stress. Five other acquired genes are involved in oxidative stress response, two of which encode ascorbate peroxidase and have been reported previously [15] (Additional file 1). Because oxidative stress may damage cellular contents such as DNA, lipids and proteins, organisms developed various antioxidant defense mechanisms [52, 53]. Of the above six antioxidant-related genes, the osmotically inducible protein C (OsmC) and alkyl hydroperoxide reductase/thiol specific antioxidant (AhpC/TSA) protein families encode antioxidant enzymes as part of the enzymatic defense systems [54, 55], while the remaining four genes are involved in the biosynthesis of ascorbate, the ionized form of ascorbic acid (vitamin C), and belong to the non-enzymatic defense systems [56–58]. Additionally, several other identified genes are functionally related to resistance to heavy metal toxicity, osmotic stress, and pathogen infection (Additional file 1). For example, mercuric reductase might allow M. brevicollis to reduce mercury to nontoxic forms, and enterotoxin may be important in defense against pathogen infection. Acquisition of genes related to stress response would potentially facilitate M. brevicollis to adapt to various habitats, which might partly explain the wide distribution of Monosiga in marine ecosystems.
For protists engaging phagocytosis such as ciliates, food particles are firstly digested in phagolysosomes, and nutrients are then released and transported to the cytosol to be utilized in other metabolic processes [59]. Consequently, a complex transporter system is important for phagotrophic protists to shuffle metabolic products (e.g., amino acids, nucleotides, phosphates and sugars) and release nutrients from the phagolysosomes to the cytosol. For instance, genes encoding UDP-galactose translocator identified in our analyses are responsible for nucleotide and sugar transport [60, 61]. Thirteen of the 27 acquired transporter genes in Monosiga are responsible for ion transfer, such as the Ca2+/cation antiporter (CaCA) family participating in Ca2+ homeostasis and signaling [62] and the potassium inwardly-rectifying channel for maintenance of K+ homeostasis [63]. Intriguingly, a gene encoding multidrug efflux transporter, which confers resistance to toxins in bacteria and plants [64], was also found in Monosiga and may allow Monosiga to pump out toxic compounds. These transporter-related genes might represent an adaptation of Monosiga to a phagotrophic lifestyle and marine environments, where variable ion concentrations and toxic substances may be common.
Acquired genes may either introduce novel functions or replace pre-existing homologs. Introduction of novel functions or phenotypes may potentially aid the adaptation of recipient organisms to their environments [15]. Of the 405 identified genes, 192 have no identifiable homologs in another choanoflagellate Salpingoeca rosetta, representing HGT events after the divergence of Monosiga and Salpingoeca, or alternatively, HGT events prior to the divergence of the two organisms followed by gene loss in the latter. The remaining 213 genes in M. brevicollis are also present in S. rosetta (Figure 1A-D; Additional file 1), indicating that most genes identified in our analyses were acquired prior to the divergence of Monosiga and Salpingoeca. Many of these acquired genes fall into different categories discussed above, suggesting a possibly profound impact of HGT on the evolution of M. brevicollis and other choanoflagellates.
The scale of HGT in Monosiga
Prokaryotic genomes are usually fluid as a result of pervasive and dynamic HGT events [65]. Such fluid genomes are often linked to the widespread distribution and tremendous metabolic variation of individual species. It has been suggested that individual prokaryotic organisms sample genes from a large global gene pool or pan-genome in response to shift in niches and resources [66, 67]. In eukaryotes, although acquired genes have been reported in many studies [7–9, 16, 44, 51, 68, 69], the overall scale of HGT in eukaryotes remains elusive. Because the evolutionary impact of HGT is largely correlated to the number of acquired genes, such a scale is critical for understanding genome evolution and speciation of recipient organisms.
To date, numerous cases of HGT have been reported in microbial eukaryotes, particularly phagotrophic microbes [3, 5, 70]. For example, about 20% of genes encoding plastid-targeted proteins in the chlorarachniophyte Bigelowiella natans were likely acquired through HGT events [7]. Fifteen HGT-derived genes were identified in diplomonad parasites [8] and 96 genes of prokaryotic origin in the parasite Entamoeba histolytica[9]. About 4.1% of ESTs from rumen ciliates were interpreted as derived from prokaryotes, most of which are related to the degradation of plant cell wall [51]. Several recent studies also indicate that up to 3.34% of protein-coding genes in the root-knot nematode Meloidogyne incognita[61], at least 5% in the red alga G. sulphuraria[45] and 8-9% in the bdelloid rotifer Adineta ricciae were acquired from other organisms [46]. Although the methods and criteria used in above analyses might be different, available data indicate that the rate of HGT may vary among eukaryotic lineages.
Our analyses identified 405 putatively HGT-derived genes, which account for approximately 4.4% (405/9,200) of the Monosiga genome. This number is among the highest HGT frequencies reported for protozoan eukaryotes, but still substantially lower than that reported in bdelloid rotifers. It should be noted here that our analyses are largely based on initial genome screening using three computational programs, none of which predicts all the identified genes. This indicates that available computational programs may not be able to identify all acquired genes in a genome. Several other factors may lead to possible underestimation of the HGT scale in this study. For instance, many genes of patchy distribution, which is frequently associated with gene transfer [44], are not considered in our analyses. Additionally, anciently acquired genes, such as those acquired by the common ancestor of choanoflagellates and animals, and genes acquired from many other eukaryotic lineages are also not included in our data. In fact, the very dynamic nature of HGT can be evidenced by the ultimately bacterial origin of many algal genes in Monosiga, which suggests recurrent HGT among different lineages (i.e. HGT from bacteria to algae and then to Monosiga) [16]. This mirrors the suggestion that the patchy distribution of many genes may be attributed to frequent HGT and gene losses [44]. Therefore, we expect that the overall scale of HGT in Monosiga would be higher than our current finding, even though the evolutionary histories depicted for some identified genes may be different with more data becoming available.
Conclusions
Based on the performance comparison of three common computational programs (i.e., PhyloGenie, DarkHorse, and AlienG) in HGT prediction, we recommend that a combination of two or all three programs be used to identify acquired genes. HGT contributes approximately 4.4% of the Monosiga genome. Many of the acquired genes in Monosiga are probably derived from food sources. Acquired genes are involved in different metabolic processes and stress responses, and they might have played a significant role in the adaptation of M. brevicollis to its environments.
Methods
Database selection
Predicted protein sequences of the choanoflagellate M. brevicollis were downloaded from the Joint Genome Institute (http://genome.jgi-psf.org/Monbr1/Monbr1.download.ftp.html). The NCBI nr protein sequence database was used in DarkHorse analyses, and two customized databases were constructed for PhyloGenie and AlienG analyses, respectively. The database for PhyloGenie analyses contained genomic or EST sequences of 260 representative taxa from all three domains of life, of which 15 were from archaea, 126 from bacteria, and 119 from eukaryotes. For AlienG analyses, the NCBI nr database was combined with genomic or EST sequences of 59 eukaryotic representative taxa that are absent from nr. Complete genome sequences of heterokont Aureococcus anophagefferens, haptophyte Emiliania huxleyi, and heterolobosean Naegleria gruberi were downloaded from the Joint Genome Institute. Annotated protein sequences of red algal Cyanidioschyzon merolae were downloaded from its genome project (http://merolae.biol.s.u-tokyo.ac.jp). ESTs were downloaded from the Taxonomically Broad EST Database (TBestDB) [71] and the NCBI dbEST database, and then translated into amino acid sequences over six frames using transeq in EMBOSS package after removing redundancy using miraEST [72].
Parameter settings for PhyloGenie, DarkHorse, and AlienG
Parameter settings for each of the three analyses were determined after testing with multiple sample datasets. For analyses using PhyloGenie, BLAST search was carried out against the customized database. The expectation value (E-value) cutoff and the number for alignment display were set to 10-10 and 250, respectively. Phylogenetic trees were constructed using a maximum of 150 sequences, with sequence length coverage over 60% of the query. All trees showing a clade of choanoflagellates, prokaryotes (bacteria and archaea) or/and algae (green plants, glaucophytes, red algae, alveolates, cryptophytes, euglenids, haptophytes, chlorarachniophytes, and stramenopiles) were retrieved using the program phat included in the PhyloGenie package. Analyses using DarkHorse were performed with BLAST results against nr database as the input file; the filter threshold was set to 1% and the self-definition to choanoflagellates. For analyses using AlienG, BLAST search was performed against the comprehensive database described above. The default parameters were used except that E-value cutoff and the number for alignment display were set to 10-5 and 1,000 respectively. The following three types of hits were excluded from further analyses: 1) sequences from choanoflagellates, which were used to exclude self-sequences; 2) sequences with length coverage below 10%; 3) pseudo-sequences annotated as “artificial sequences”, “synthetic construct”, or “plasmids”.
Phylogenetic analyses
Each HGT candidate predicted by the three computational programs was subject to further manual phylogenetic analyses. Homologous sequences were sampled from representative groups of three domains of life (bacteria, archaea, and eukaryotes). The comprehensive database built for AlienG analyses was used for sequence sampling. Protein sequence alignments were performed using both MUSCLE [73] and ClustalX [74], followed by cross-comparison and manual refinement. Gaps and ambiguously aligned regions were removed manually. The alignment data are available upon request. The optimal model of protein sequence substitution and rate heterogeneity for each dataset were chosen using ModelGenerator based on the AIC1 criterion [75]. Phylogenetic analyses were performed with a maximum likelihood method using PHYML 3.0 [76] and a distance method using neighbor of PHYLIP version 3.69 [77], with maximum likelihood distance calculated using TREE-PUZZLE [78]. Bootstrap analyses used 100 pseudo-replicates.
Identification of acquired genes homologs in the choanoflagellate S. rosetta
The genome of the choanoflagellate S. rosetta was not available to the public when we initiated our analyses of M. brevicollis. To investigate whether the genes identified in M. brevicollis were also acquired by S. rosetta, we downloaded a total of 11,731 predicted protein sequences of S. rosetta from the Origins of Multicellularity Sequencing Project (Broad Institute of Harvard and MIT, http://www.broadinstitute.org) [79] and then identified the homologs based on sequence similarity comparison. The acquired genes in M. brevicollis were used as queries to search against the genome of S. rosetta with E-value cutoff set to 1e-40. The genes shared by M. brevicollis and S. rosetta were considered to be acquired prior to the split of S. rosetta and M. brevicollis.
Electronic supplementary material
Additional file 1: Table S1: Algal and prokaryotic genes (405) identified in M. brevicollis. Figure S1-S109. Maximum likelihood trees for the algal and bacterial genes identified in M. brevicollis. Genes identified in our previous studies and some of those uniquely distributed in prokaryotes and/or algae besides choanoflagellates are not included. (PDF 5 MB)
Acknowledgments
This work is supported by a NSF Assembling the Tree of Life grant (DEB 0830024), an NSFC Oversea, Hong Kong, Macao collaborative grant (31328003), and the CAS/SAFEA International Partnership Program for Creative Research Teams. GS is partly supported by a startup grant from Kunming Institute of Botany, CAS, to the Group of Chemical and Molecular Ecology.
Abbreviations
- HGT
Horizontal gene transfer
- EGT
Endosymbiotic gene transfer.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JH conceived and designed the study and wrote manuscript. GS generated the data. GS, JY and XH performed the analyses and wrote the manuscript. All authors read and approved the final manuscript.
Contributor Information
Jipei Yue, Email: yuej@ecu.edu.
Guiling Sun, Email: sung@mail.kib.ac.cn.
Xiangyang Hu, Email: huxiangyang@mail.kib.ac.cn.
Jinling Huang, Email: huangj@ecu.edu.
References
- 1.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405(6784):299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 2.Gogarten JP, Doolittle WF, Lawrence JG. Prokaryotic evolution in light of gene transfer. Mol Biol Evol. 2002;19(12):2226–2238. doi: 10.1093/oxfordjournals.molbev.a004046. [DOI] [PubMed] [Google Scholar]
- 3.Andersson JO. Lateral gene transfer in eukaryotes. Cell Mol Life Sci. 2005;62(11):1182–1197. doi: 10.1007/s00018-005-4539-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008;9(8):605–618. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- 5.Doolittle WF. You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 1998;14(8):307–311. doi: 10.1016/S0168-9525(98)01494-2. [DOI] [PubMed] [Google Scholar]
- 6.Nixon JE, Wang A, Field J, Morrison HG, McArthur AG, Sogin ML, Loftus BJ, Samuelson J. Evidence for lateral transfer of genes encoding ferredoxins, nitroreductases, NADH oxidase, and alcohol dehydrogenase 3 from anaerobic prokaryotes to Giardia lamblia and Entamoeba histolytica. Eukaryot Cell. 2002;1(2):181–190. doi: 10.1128/EC.1.2.181-190.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Archibald JM, Rogers MB, Toop M, Ishida K, Keeling PJ. Lateral gene transfer and the evolution of plastid-targeted proteins in the secondary plastid-containing alga Bigelowiella natans. Proc Natl Acad Sci USA. 2003;100(13):7678–7683. doi: 10.1073/pnas.1230951100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andersson JO, Sjogren AM, Davis LA, Embley TM, Roger AJ. Phylogenetic analyses of diplomonad genes reveal frequent lateral gene transfers affecting eukaryotes. Curr Biol. 2003;13(2):94–104. doi: 10.1016/S0960-9822(03)00003-4. [DOI] [PubMed] [Google Scholar]
- 9.Loftus B, Anderson I, Davies R, Alsmark UC, Samuelson J, Amedeo P, Roncaglia P, Berriman M, Hirt RP, Mann BJ, et al. The genome of the protist parasite Entamoeba histolytica. Nature. 2005;433(7028):865–868. doi: 10.1038/nature03291. [DOI] [PubMed] [Google Scholar]
- 10.Leadbeater BSC, Kelly M. Evolution of animals’ choanoflagellates and sponges. Water Atmosph Online. 2001;9(2):9–11. [Google Scholar]
- 11.Carr M, Leadbeater BS, Hassan R, Nelson M, Baldauf SL. Molecular phylogeny of choanoflagellates, the sister group to Metazoa. Proc Natl Acad Sci USA. 2008;105(43):16641–16646. doi: 10.1073/pnas.0801667105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavrov DV, Forget L, Kelly M, Lang BF. Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol Biol Evol. 2005;22(5):1231–1239. doi: 10.1093/molbev/msi108. [DOI] [PubMed] [Google Scholar]
- 13.King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, Fairclough S, Hellsten U, Isogai Y, Letunic I, et al. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008;451(7180):783–788. doi: 10.1038/nature06617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leakey RJG, Leadbeater BSC, Mitchell E, McCready SMM, Murray AWA. The abundance and biomass of choanoflagellates and other nanoflagellates in waters of contrasting temperature to the north-west of South Georgia in the Southern Ocean. Eur J Protistol. 2002;38(4):333–350. doi: 10.1078/0932-4739-00860. [DOI] [Google Scholar]
- 15.Nedelcu AM, Miles IH, Fagir AM, Karol K. Adaptive eukaryote-to-eukaryote lateral gene transfer: stress-related genes of algal origin in the closest unicellular relatives of animals. J Evol Biol. 2008;21(6):1852–1860. doi: 10.1111/j.1420-9101.2008.01605.x. [DOI] [PubMed] [Google Scholar]
- 16.Sun G, Yang Z, Ishwar A, Huang J. Algal genes in the closest relatives of animals. Mol Biol Evol. 2010;27(12):2879–2889. doi: 10.1093/molbev/msq175. [DOI] [PubMed] [Google Scholar]
- 17.Tucker RP, Beckmann J, Leachman NT, Scholer J, Chiquet-Ehrismann R. Phylogenetic analysis of the teneurins: conserved features and premetazoan ancestry. Mol Biol Evol. 2012;29(3):1019–1029. doi: 10.1093/molbev/msr271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Foerstner KU, Doerks T, Muller J, Raes J, Bork P. A nitrile hydratase in the eukaryote Monosiga brevicollis. PloS one. 2008;3(12):e3976. doi: 10.1371/journal.pone.0003976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frickey T, Lupas AN. PhyloGenie: automated phylome generation and analysis. Nucleic Acids Res. 2004;32(17):5231–5238. doi: 10.1093/nar/gkh867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Podell S, Gaasterland T. DarkHorse: a method for genome-wide prediction of horizontal gene transfer. Genome Biol. 2007;8(2):R16. doi: 10.1186/gb-2007-8-2-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tian J, Sun G, Ding Q, Huang J, Oruganti S, Xie B. AlienG: an effective computational tool for phylogenetic identification of horizontally transferred genes. New Orleans, Louisiana: The third International Conference on Bioinformatics and Computational Biology (BICoB); 2011. [Google Scholar]
- 22.Hackett JD, Yoon HS, Li S, Reyes-Prieto A, Rummele SE, Bhattacharya D. Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of rhizaria with chromalveolates. Mol Biol Evol. 2007;24(8):1702–1713. doi: 10.1093/molbev/msm089. [DOI] [PubMed] [Google Scholar]
- 23.Huang J, Gogarten JP. Ancient horizontal gene transfer can benefit phylogenetic reconstruction. Trends Genet. 2006;22(7):361–366. doi: 10.1016/j.tig.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 24.Huang J, Gogarten JP. Did an ancient chlamydial endosymbiosis facilitate the establishment of primary plastids? Genome Biol. 2007;8(6):R99. doi: 10.1186/gb-2007-8-6-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Nosenko T, Hackett JD, Bhattacharya D. Phylogenomic analysis identifies red algal genes of endosymbiotic origin in the chromalveolates. Mol Biol Evol. 2006;23(3):663–674. doi: 10.1093/molbev/msj075. [DOI] [PubMed] [Google Scholar]
- 26.Podell S, Gaasterland T, Allen EE. A database of phylogenetically atypical genes in archaeal and bacterial genomes, identified using the DarkHorse algorithm. BMC Bioinform. 2008;9:419. doi: 10.1186/1471-2105-9-419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhaxybayeva O, Swithers KS, Lapierre P, Fournier GP, Bickhart DM, DeBoy RT, Nelson KE, Nesbo CL, Doolittle WF, Gogarten JP, et al. On the chimeric nature, thermophilic origin, and phylogenetic placement of the Thermotogales. Proc Natl Acad Sci USA. 2009;106(14):5865–5870. doi: 10.1073/pnas.0901260106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun G, Yang Z, Kosch T, Summers K, Huang J. Evidence for acquisition of virulence effectors in pathogenic chytrids. BMC Evol Biol. 2011;11:195. doi: 10.1186/1471-2148-11-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ni T, Yue J, Sun G, Zou Y, Wen J, Huang J. Ancient gene transfer from algae to animals: Mechanisms and evolutionary significance. BMC Evol Biol. 2012;12(1):83. doi: 10.1186/1471-2148-12-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yue J, Hu X, Sun H, Yang Y, Huang J. Widespread impact of horizontal gene transfer on plant colonization of land. Nat Commun. 2012;3:1152. doi: 10.1038/ncomms2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun G, Huang J. Horizontally acquired DAP pathway as a unit of self-regulation. J Evol Biol. 2011;24(3):587–595. doi: 10.1111/j.1420-9101.2010.02192.x. [DOI] [PubMed] [Google Scholar]
- 32.Kurland CG, Canback B, Berg OG. Horizontal gene transfer: a critical view. P Natl Acad Sci USA. 2003;100(17):9658–9662. doi: 10.1073/pnas.1632870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stiller JW. Experimental design and statistical rigor in phylogenomics of horizontal and endosymbiotic gene transfer. BMC Evol Biol. 2011;11:259. doi: 10.1186/1471-2148-11-259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stiller JW, Huang J, Ding Q, Tian J, Goodwillie C. Are algal genes in nonphotosynthetic protists evidence of historical plastid endosymbioses? BMC genomics. 2009;10:484. doi: 10.1186/1471-2164-10-484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Keeling PJ. Chromalveolates and the evolution of plastids by secondary endosymbiosis. J Eukaryot Microbiol. 2009;56(1):1–8. doi: 10.1111/j.1550-7408.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 36.Buck KR, Chavez FP, Thomsen HA. Choanoflagellates of the central California waters: abundance and distribution. Ophelia. 1991;33(3):179–186. doi: 10.1080/00785326.1991.10429708. [DOI] [Google Scholar]
- 37.Marchant H, Scott F. Uptake of sub-micrometre particles and dissolved organic material by Antarctic choanoflagellates. Mar Ecol Prog Ser. 1993;92:59–64. doi: 10.3354/meps092059. [DOI] [Google Scholar]
- 38.Flynn KJ, Stoecker DK, Mitra A, Raven JA, Glibert PM, Hansen PJ, Granéli E, Burkholder JM. Misuse of the phytoplankton–zooplankton dichotomy: the need to assign organisms as mixotrophs within plankton functional types. J Plankton Res. 2013;35(1):3–11. doi: 10.1093/plankt/fbs062. [DOI] [Google Scholar]
- 39.Raven JA, Beardall J, Flynn KJ, Maberly SC. Phagotrophy in the origins of photosynthesis in eukaryotes and as a complementary mode of nutrition in phototrophs: relation to Darwin’s insectivorous plants. J Exp Bot. 2009;60(14):3975–3987. doi: 10.1093/jxb/erp282. [DOI] [PubMed] [Google Scholar]
- 40.Huang JL, Mullapudi N, Sicheritz-Ponten T, Kissinger JC. A first glimpse into the pattern and scale of gene transfer in the Apicomplexa. Int J Parasitol. 2004;34(3):265–274. doi: 10.1016/j.ijpara.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 41.Hackett JD, Yoon HS, Soares MB, Bonaldo MF, Casavant TL, Scheetz TE, Nosenko T, Bhattacharya D. Migration of the plastid genome to the nucleus in a peridinin dinoflagellate. Curr Biol. 2004;14(3):213–218. doi: 10.1016/j.cub.2004.01.032. [DOI] [PubMed] [Google Scholar]
- 42.Dagan T, Martin W. Ancestral genome sizes specify the minimum rate of lateral gene transfer during prokaryote evolution. Proc Natl Acad Sci USA. 2007;104(3):870–875. doi: 10.1073/pnas.0606318104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alsmark UC, Sicheritz-Ponten T, Foster PG, Hirt RP, Embley TM. Horizontal gene transfer in eukaryotic parasites: a case study of Entamoeba histolytica and Trichomonas vaginalis. Methods Mol Biol (Clifton, NJ) 2009;532:489–500. doi: 10.1007/978-1-60327-853-9_28. [DOI] [PubMed] [Google Scholar]
- 44.Andersson JO. Evolution of patchily distributed proteins shared between eukaryotes and prokaryotes: Dictyostelium as a case study. J Mol Microbiol Biotechnol. 2011;20(2):83–95. doi: 10.1159/000324505. [DOI] [PubMed] [Google Scholar]
- 45.Schonknecht G, Chen WH, Ternes CM, Barbier GG, Shrestha RP, Stanke M, Brautigam A, Baker BJ, Banfield JF, Garavito RM, et al. Gene transfer from bacteria and archaea facilitated evolution of an extremophilic eukaryote. Science (New York, NY) 2013;339(6124):1207–1210. doi: 10.1126/science.1231707. [DOI] [PubMed] [Google Scholar]
- 46.Boschetti C, Carr A, Crisp A, Eyres I, Wang-Koh Y, Lubzens E, Barraclough TG, Micklem G, Tunnacliffe A. Biochemical diversification through foreign gene expression in Bdelloid rotifers. PLoS Gen. 2012;8(11):e1003035. doi: 10.1371/journal.pgen.1003035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gogarten JP, Townsend JP. Horizontal gene transfer, genome innovation and evolution. Nat Rev Microbiol. 2005;3(9):679–687. doi: 10.1038/nrmicro1204. [DOI] [PubMed] [Google Scholar]
- 48.Andersson JO, Sjogren AM, Horner DS, Murphy CA, Dyal PL, Svard SG, Logsdon JM, Jr, Ragan MA, Hirt RP, Roger AJ. A genomic survey of the fish parasite Spironucleus salmonicida indicates genomic plasticity among diplomonads and significant lateral gene transfer in eukaryote genome evolution. BMC genomics. 2007;8:51. doi: 10.1186/1471-2164-8-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nedelcu AM, Blakney AJ, Logue KD. Functional replacement of a primary metabolic pathway via multiple independent eukaryote-to-eukaryote gene transfers and selective retention. J Evol Biol. 2009;22(9):1882–1894. doi: 10.1111/j.1420-9101.2009.01797.x. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Vallve S, Romeu A, Palau J. Horizontal gene transfer of glycosyl hydrolases of the rumen fungi. Mol Biol Evol. 2000;17(3):352–361. doi: 10.1093/oxfordjournals.molbev.a026315. [DOI] [PubMed] [Google Scholar]
- 51.Ricard G, McEwan NR, Dutilh BE, Jouany JP, Macheboeuf D, Mitsumori M, McIntosh FM, Michalowski T, Nagamine T, Nelson N, et al. Horizontal gene transfer from Bacteria to rumen Ciliates indicates adaptation to their anaerobic, carbohydrates-rich environment. BMC genomics. 2006;7:22. doi: 10.1186/1471-2164-7-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sies H. Oxidative stress: oxidants and antioxidants. Exp Physiol. 1997;82(2):291–295. doi: 10.1113/expphysiol.1997.sp004024. [DOI] [PubMed] [Google Scholar]
- 53.Vertuani S, Angusti A, Manfredini S. The antioxidants and pro-antioxidants network: an overview. Curr Pharm Des. 2004;10(14):1677–1694. doi: 10.2174/1381612043384655. [DOI] [PubMed] [Google Scholar]
- 54.Conter A, Gangneux C, Suzanne M, Gutierrez C. Survival of Escherichia coli during long-term starvation: effects of aeration, NaCl, and the rpoS and osmC gene products. Res Microbiol. 2001;152(1):17–26. doi: 10.1016/S0923-2508(00)01164-5. [DOI] [PubMed] [Google Scholar]
- 55.Lee J, Spector D, Godon C, Labarre J, Toledano MB. A new antioxidant with alkyl hydroperoxide defense properties in yeast. J Biol Chem. 1999;274(8):4537–4544. doi: 10.1074/jbc.274.8.4537. [DOI] [PubMed] [Google Scholar]
- 56.Leterrier M, Corpas FJ, Barroso JB, Sandalio LM, del Rio LA. Peroxisomal monodehydroascorbate reductase. Genomic clone characterization and functional analysis under environmental stress conditions. Plant Physiol. 2005;138(4):2111–2123. doi: 10.1104/pp.105.066225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pineau B, Layoune O, Danon A, De Paepe R. L-galactono-1,4-lactone dehydrogenase is required for the accumulation of plant respiratory complex I. J Biol Chem. 2008;283(47):32500–32505. doi: 10.1074/jbc.M805320200. [DOI] [PubMed] [Google Scholar]
- 58.Teixeira FK, Menezes-Benavente L, Margis R, Margis-Pinheiro M. Analysis of the molecular evolutionary history of the ascorbate peroxidase gene family: inferences from the rice genome. J Mol Evol. 2004;59(6):761–770. doi: 10.1007/s00239-004-2666-z. [DOI] [PubMed] [Google Scholar]
- 59.Gronlien HK, Berg T, Lovlie AM. In the polymorphic ciliate Tetrahymena vorax, the non-selective phagocytosis seen in microstomes changes to a highly selective process in macrostomes. J Exp Biol. 2002;205(Pt 14):2089–2097. doi: 10.1242/jeb.205.14.2089. [DOI] [PubMed] [Google Scholar]
- 60.Miura N, Ishida N, Hoshino M, Yamauchi M, Hara T, Ayusawa D, Kawakita M. Human UDP-galactose translocator: molecular cloning of a complementary DNA that complements the genetic defect of a mutant cell line deficient in UDP-galactose translocator. J Biochem. 1996;120(2):236–241. doi: 10.1093/oxfordjournals.jbchem.a021404. [DOI] [PubMed] [Google Scholar]
- 61.Norambuena L, Marchant L, Berninsone P, Hirschberg CB, Silva H, Orellana A. Transport of UDP-galactose in plants. Identification and functional characterization of AtUTr1, an Arabidopsis thaliana UDP-galactos/UDP-glucose transporter. J Biol Chem. 2002;277(36):32923–32929. doi: 10.1074/jbc.M204081200. [DOI] [PubMed] [Google Scholar]
- 62.Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406(3):365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- 63.Abraham MR, Jahangir A, Alekseev AE, Terzic A. Channelopathies of inwardly rectifying potassium channels. FASEB J. 1999;13(14):1901–1910. doi: 10.1096/fasebj.13.14.1901. [DOI] [PubMed] [Google Scholar]
- 64.Diener AC, Gaxiola RA, Fink GR. Arabidopsis ALF5, a multidrug efflux transporter gene family member, confers resistance to toxins. Plant Cell. 2001;13(7):1625–1637. doi: 10.1105/TPC.010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dagan T, Artzy-Randrup Y, Martin W. Modular networks and cumulative impact of lateral transfer in prokaryote genome evolution. Proc Natl Acad Sci U S A. 2008;105(29):10039–10044. doi: 10.1073/pnas.0800679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lapierre P, Gogarten JP. Estimating the size of the bacterial pan-genome. Trends Genet. 2009;25(3):107–110. doi: 10.1016/j.tig.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 67.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc Natl Acad Sci USA. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bowler C, Allen AE, Badger JH, Grimwood J, Jabbari K, Kuo A, Maheswari U, Martens C, Maumus F, Otillar RP, et al. The Phaeodactylum genome reveals the evolutionary history of diatom genomes. Nature. 2008;456(7219):239–244. doi: 10.1038/nature07410. [DOI] [PubMed] [Google Scholar]
- 69.Huang J, Mullapudi N, Lancto CA, Scott M, Abrahamsen MS, Kissinger JC. Phylogenomic evidence supports past endosymbiosis, intracellular and horizontal gene transfer in Cryptosporidium parvum. Genome Biol. 2004;5(11):R88. doi: 10.1186/gb-2004-5-11-r88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Doolittle WF, Boucher Y, Nesbo CL, Douady CJ, Andersson JO, Roger AJ. How big is the iceberg of which organellar genes in nuclear genomes are but the tip? Philos Transac Royal Soc Lond Series B Biol Sci. 2003;358(1429):39–57. doi: 10.1098/rstb.2002.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.O’Brien EA, Koski LB, Zhang Y, Yang L, Wang E, Gray MW, Burger G, Lang BF. TBestDB: a taxonomically broad database of expressed sequence tags (ESTs) Nucleic Acids Res. 2007;35(Database issue):D445–D451. doi: 10.1093/nar/gkl770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Muller WE, Wetter T, Suhai S. Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004;14(6):1147–1159. doi: 10.1101/gr.1917404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25(24):4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006;6:29. doi: 10.1186/1471-2148-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 2003;52(5):696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 77.Felsenstein J. PHYLIP (Phylogeny Inference Package) version 3.65. Distributed by the author. Seattle: Department of Genome Sciences, University of Washington; 2005. [Google Scholar]
- 78.Schmidt HA, Strimmer K, Vingron M, von Haeseler A. TREE-PUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics. 2002;18(3):502–504. doi: 10.1093/bioinformatics/18.3.502. [DOI] [PubMed] [Google Scholar]
- 79.Ruiz-Trillo I, Burger G, Holland PW, King N, Lang BF, Roger AJ, Gray MW. The origins of multicellularity: a multi-taxon genome initiative. Trends Genet. 2007;23(3):113–118. doi: 10.1016/j.tig.2007.01.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1: Algal and prokaryotic genes (405) identified in M. brevicollis. Figure S1-S109. Maximum likelihood trees for the algal and bacterial genes identified in M. brevicollis. Genes identified in our previous studies and some of those uniquely distributed in prokaryotes and/or algae besides choanoflagellates are not included. (PDF 5 MB)