

Abstract
Background
Natural selection has molded evolution across all taxa. At an arguable date of around 330,000 years ago there were already at least two different types of cattle that became ancestors of nearly all modern cattle, the Bos taurus taurus more adapted to temperate climates and the tropically adapted Bos taurus indicus. After domestication, human selection exponentially intensified these differences. To better understand the genetic differences between these subspecies and detect genomic regions potentially under divergent selection, animals from the International Bovine HapMap Experiment were genotyped for over 770,000 SNP across the genome and compared using smoothed FST. The taurine sample was represented by ten breeds and the contrasting zebu cohort by three breeds.
Results
Each cattle group evidenced similar numbers of polymorphic markers well distributed across the genome. Principal components analyses and unsupervised clustering confirmed the well-characterized main division of domestic cattle. The top 1% smoothed FST, potentially associated to positive selection, contained 48 genomic regions across 17 chromosomes. Nearly half of the top FST signals (n = 22) were previously detected using a lower density SNP assay. Amongst the strongest signals were the BTA7:~50 Mb and BTA14:~25 Mb; both regions harboring candidate genes and different patterns of linkage disequilibrium that potentially represent intrinsic differences between cattle types. The bottom 1% of the smoothed FST values, potentially associated to balancing selection, included 24 regions across 13 chromosomes. These regions often overlap with copy number variants, including the highly variable region at BTA23:~24 Mb that harbors a large number of MHC genes. Under these regions, 318 unique Ensembl genes are annotated with a significant overrepresentation of immune related pathways.
Conclusions
Genomic regions that are potentially linked to purifying or balancing selection processes in domestic cattle were identified. These regions are of particular interest to understand the natural and human selective pressures to which these subspecies were exposed to and how the genetic background of these populations evolved in response to environmental challenges and human manipulation.
Electronic supplementary material
The online version of this article (doi:10.1186/1471-2164-14-876) contains supplementary material, which is available to authorized users.
Keywords: Bos, Taurus, Indicus, FST, Selection, Speciation
Background
Natural selection has shaped the genome of all living creatures in our planet, including domesticated animals. Nearly all modern cattle can be associated with one of two types or sub-species. This division between the types Bos taurus taurus (taurine cattle) and Bos taurus indicus (zebu cattle) is estimated to have occurred from a common ancestor between 330,000 [1] and 2 million [2] years ago. Since divergence, cattle types have accumulated different genetic variations, which have contributed to highly differentiated phenotypes. It is assumed that the divergence between cattle types was long before domestication, which is estimated to have occurred between 10,000 to 7,000 BC in two separate locations: the Fertile Crescent (taurine cattle) and the Indus Valley (zebu cattle) [3, 4]. After domestication human-oriented selection added further complexity to the evolution of cattle.
For most of the history of human-cattle coexistence the environment was the main force driving changes in the animals’ genome. Shortly after domestication, human breeders preferred traits that enabled easy management; however, breeders also sought production improvement traits as well [5]. The introduction of the concept of breed in the 19th century led to human-oriented selection imposing strong bottlenecks, which created population demes based on phenotypes. Breed formation was followed by breed expansion via the use of artificial insemination, which reduced genetic variability within breeds particularly in the sex chromosomes and mitochondrial DNA [6]. This is due to the fact that only one haplotype is passed on to the following generation, and subjected to stronger selective forces when compared to autosomal chromosomes.
Positive and balancing selections are terms used to characterize different aspects that selection forces might impose on a population. Positive selection, also termed directional or purifying selection, refers to the selection process through which a particular phenotype (or genotype) is favored in a given environment, which leads to an increase of its frequency in a population. In contrast, balancing selection refers to the selective process through which multiple alleles are selected, thus preserving the genetic diversity in a population. Balancing selection is often observed when heterozygous animals have a competitive advantage. Alternatively, these may be regions of convergent selection across groups. Importantly, both positive and balancing selection phenomena can be tracked using SNP genotypes or sequence data from the cattle genome.
SNP genotyping has become widely used in animal genetics and a number of methods have been developed to identify regions under selection. Out of these FST is a widely used statistic to evaluate the diversity of subpopulations of animals or to determine the relative distance between populations. Many variations of the FST concept [7] exist, but all adhere to the core principle of being a metric of allele frequencies and their variance. This metric has also been used to identify loci under selection [8–10].
In this study, we used a pure drift FST model [11] which assumes all animals originated from the same ancestral population. This model was applied to taurine and zebu animals to identify loci under selection. These two groups correspond to the main (and most ancestral) separation of domestic cattle, which in most but not all cases corresponds to animals adapted to tropical and temperate environments. The identification of such loci can aid in the identification of genes and genomic variants that are related to environmental adaptation and/or selection derived from human agro-pastoral activities.
Methods
Statement on the ethical use of animals
No ethics statement was required for the collection of genetic material. The DNA from animals included in this study were either part of previous analyses that obtained specific permissions [12] or were extracted from semen straws collected in accredited AI centers in accordance with the Brazilian legislation on animal welfare.
Cattle samples and SNP genotypes
All individuals were genotyped using the BovineHD BeadChip that includes ~777 K SNP (Illumina, Inc. San Diego, USA) following standard procedures. The SNP set included in this genotyping platform was carefully selected to reduce the potential for ascertainment bias during SNP discovery. Seven different grouping of breeds were used to assess the minor allele frequency of all available SNP, this included Holstein, Angus, Nelore, Bos taurus taurus dairy excluding Holstein, Bos taurus taurus beef ignoring Angus, Bos taurus indicus excluding Nelore, and adapted Bos taurus taurus (e.g. Senepol). This was complemented with sequence data from 30 breeds that were compiled and weighted to minimize ascertainment bias. More information on the BovineHD can be found in the supplier’s website (http://www.illumina.com/documents//products/datasheets/datasheet_bovineHD.pdf).
Only animals with call rates > = 98%, and SNP with more than 95% successful genotypes were kept in the final dataset. Filtering was also based on available pedigree information and the estimated proportion of alleles shared identical-by-descent (PI_HAT > 0.8) ([13]http://pngu.mgh.harvard.edu/~purcell/plink/), animals with high relatedness were excluded. A total of 339 Bos taurus taurus or taurine individuals from the Bovine Hapmap DNA panel [12] were included in the analyses. Breeds represented in this group were: Angus (n = 44), Brown Swiss (n = 24), Charolais (n = 37), Guernsey (n = 21), Hereford (n = 36), Holstein (n = 63), Jersey (n = 39), Limousin (n = 47), Norwegian Red (n = 17), and Red Angus (n = 11). The Bos taurus indicus or Zebu animals (n = 166) were also from the Bovine Hapmap experiment, and they were complemented with additional individuals. Breeds represented in this group were: Nelore (n = 91), Gir (n = 50), and Guzera (n = 25). Even though Brahmans are considered zebu animals, it is known that taurine animals were also used during the breed formation and expansion; therefore they were not included in these analyses.
Population and linkage disequilibrium (LD) structure
Pairs of markers with high linkage disequilibrium (LD) provide redundant information and impose higher computational demands for population structure analyses. To remove extraneous information, the dataset was pruned based on LD between markers using the PLINK [13] command --indep-pairwise 50 10 0.1, which calculates LD for each pair of marker in a window of 50 SNP. If a pair of SNP had r2 > 0.1, then one of the SNP was removed, the window was moved 10 SNP and the process restarted. The pruned genotypes defined a dataset including 38,681 SNP that were then used to assess the population structure using three methods: 1) unsupervised clustering of individuals based on maximum likelihood as implemented in the program Admixture Version 1.20 [14] with cluster number (K) equal 2; 2) principal components analysis as implemented in GCTA [15]; and 3) estimated genetic relationship matrix [16] visualized as heat map using R [17]. For plots of LD between markers, r2 were calculated using Haploview [18].
Identification of genomic regions under selection
FST statistics were used to characterize the differentiation between taurine and zebu animals by first identifying SNP potentially under selection. Next, genomic regions with a high proportion of such SNP were identified, and then the genic content of regions with extreme signals for positive and balancing selection were further analyzed. The estimation of SNP FST was based on a pure drift model defined by Nicholson et al. [11], following the simplification proposed by Flori et al. [10]. These analyses were performed using R [17] scripts. The SNP FST were smoothed across the Bovine genome reference assembly UMD 3.1 [19] using a local variable bandwidth kernel estimator [20] (R package lokern), where every fifteen SNP FST values generated one smoothed FST value. This bandwidth was used because it covers a region of ~50Kb which is the average extent of LD found in these populations. The genomic regions with predominantly higher FST values usually resulted in high values of smoothed FST and were potentially associated to positive selection. In contrast, regions with mainly low FST values generated low smoothed FST values and were potentially associated to regions under balancing selection. The top and bottom 1% smoothed FST values were identified, translated into genomic position (UMD 3.1) and the genic content of each region was tested for gene ontology overrepresentation. The cattle chromosome X (BTAX) is highly differentiated between taurine and zebu animals. Therefore, the identification of the top and bottom 1% values included only the autosomes, being the BTAX analyzed separately as it contains regions under relatively strong positive selection. Similar analyses were also performed only within-taurine (n = 9 breeds, the Red Angus was excluded due to small sample size) and only within-zebu (n = 3 breeds). These analyses were performed to gather hints as to the origin of the differentially selected regions seen between zebu and taurine cattle.
Regions harboring copy number variants (CNV) might also be under selection and contributing to an observed selection signal, therefore CNV regions that coincide to smoothed FST peaks were further explored. Gene content of cattle CNV regions was assessed using Ensembl (ftp://ftp.ensembl.org/pub/current_fasta/bos_taurus/pep/). It is worthwhile to point out that FST and CNV results did not use the exact same samples. CNV results are based on Bickhart et al. [21] that use a Holstein, a Nelore, a Hereford and 3 Angus samples, also included in the FST analyses. Intersections between balancing selection region coordinates and exon positions were compared using MySQL queries. We obtained a catalog of all bovine peptides from Ensembl. This yielded 22,118 peptides, 345 of which overlap with 24 predicted balancing selection regions, and corresponded to 318 unique Ensembl genes. Using PANTHER version 7 [22], we tested for over representation of biological process, molecular function and pathway terms under the balancing selection regions. Results were Bonferroni [23] adjusted and PANTHER terms with less than five observations were not further analyzed. Similar analyses were performed on the peptides under the 48 positive selection regions. PANTHER results were similar when all peptides under the 24 balancing selection regions and 48 positive selection regions were combined in a single analysis.
Results
SNP genotypes
After quality control, a total of 768,506 SNP were considered. In taurine, most of the autosomes had >90% of markers polymorphic and in zebu slightly less markers were polymorphic (between 80-90%). This distribution was similar across all autosomes; however, the taurine group had a reduced proportion of polymorphic markers when compared to the zebu on BTAX (Figure 1A). Most autosomes had >80% of SNP polymorphic in both groups, with ~10% polymorphic only in taurine and only a reduced number of SNP exclusively polymorphic in zebu. The zebu exclusive SNP group was different again for the BTAX where ~50% of the SNP were polymorphic in both groups and close to 40% polymorphic only in zebu (Figure 1B). Within cattle types, the average heterozygosity was 0.21 and 0.29 for zebu and taurine.
Figure 1.
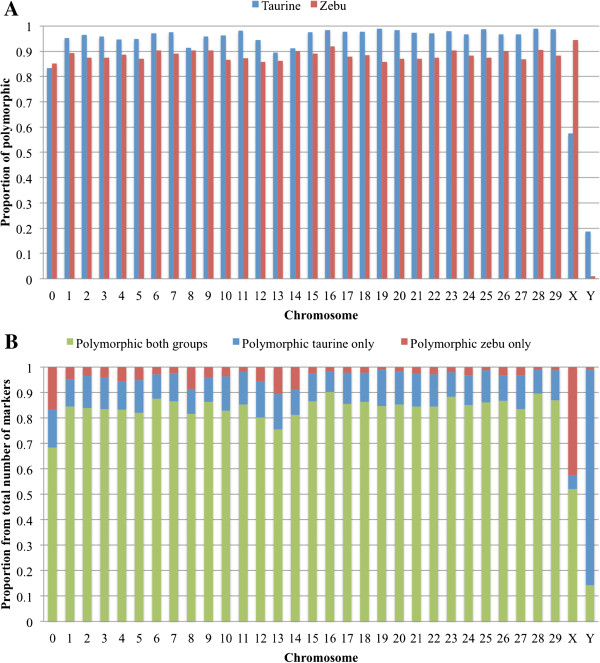
Polymorphic status of the BovineHD (Illumina) markers in zebu and taurine cattle. A) Proportion of polymorphic markers, and B) Proportion of markers by polymorphic status across both cattle types.
Population substructure
The separation between taurine and zebu is the most substantial type-distinction between domestic cattle. Clustering animals based on the genetic relationship matrix clearly demonstrates this division between cattle populations (Figure 2), which is also seen using an unsupervised clustering with selected number of clusters K = 2 (Additional file 1: Figure S1A). This latter analysis evidences the majority of individuals are pure bred within each cattle type assigning an estimated proportion of more than 0.9 for either the zebu or taurine clusters.
Figure 2.
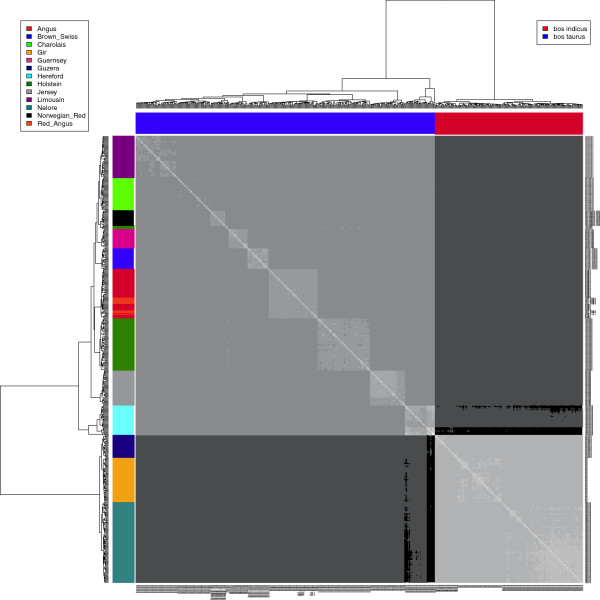
Heatmap of relationship between individuals of 10 taurine and 3 zebu cattle breed (n = 505) based on the genetic relationship matrix calculated using 768,506 SNP genotypes.
The first principal component, which is the axis that explains the most variance, not surprisingly corresponds to the same main division. The second principal component starts to subdivide the taurine animals (Additional file 1: Figure S1B and C). This subdivision of taurine animals was also seen in four independent runs of principal components analyses that used the same number of individuals per breed and different random combinations of taurine breeds in addition to the three zebu breeds (Additional file 2: Figure S2). This agrees with the lower pair-wise FST observed between zebu breeds in comparison to taurine breeds (Additional file 3: Table S1).
Genomic regions under selection
Regions under positive and balancing selection were defined as the regions in the top and bottom 1% of all smoothed FST values, respectively (Figure 3, Tables 1 and 2).
Figure 3.
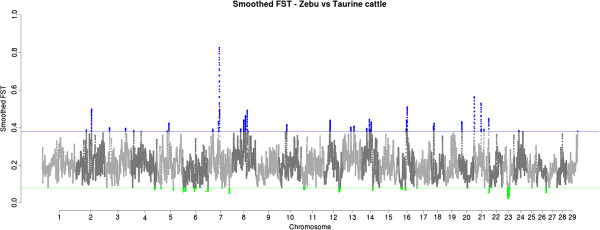
Smoothed FST comparing taurine and zebu animals. Only autosomes are plotted in alternated shades of gray. The top and bottom 1% values are highlighted in blue and green, corresponding to the regions under positive and balancing selections.
Table 1.
Positive selection: regions in the top 1% smoothed F ST values
| Region | BTA | SNP start pos | SNP end pos | Highest sFST | CNV [21] | Within cattle type** | Candidate genes | Cross reference |
|---|---|---|---|---|---|---|---|---|
| P1 | 2 | 47,857,335 | 48,065,161 | 0.387 | 1 | |||
| P2* | 2 | 71,565,086 | 72,885,823 | 0.498 | 1 | Hol | [24] | |
| P3 | 3 | 19,689,648 | 20,166,059 | 0.399 | 0 | Hol, Nor, Bro | CDC42SE1 | |
| P4 | 3 | 94,742,479 | 95,401,345 | 0.396 | 0 | |||
| P5 | 4 | 12,106,878 | 12,361,811 | 0.384 | 0 | |||
| P6 | 4 | 46,670,940 | 46,814,875 | 0.380 | 0 | [25] | ||
| P7 | 5 | 48,229,556 | 48,336,996 | 0.381 | 0 | [9, 12, 24] | ||
| P8 | 5 | 55,881,766 | 56,801,729 | 0.424 | 1 | Hol | STAT6, GLI1 | [24, 25] |
| P9 | 7 | 21,008,805 | 21,606,667 | 0.391 | 0 | Gue | ITGB1BP3 | [24, 25] |
| P10 | 7 | 47,299,497 | 47,859,329 | 0.433 | 3 | Lim | SPOCK, PPP2CA | [12] |
| P11* | 7 | 50,951,861 | 53,757,384 | 0.826 | 5 | Ang, Cha, Gue, Nor, Gir | CD14, CDC23, EGR1, MYOT, TMEM173 | [24] |
| P12 | 8 | 39,288,115 | 39,800,492 | 0.393 | 0 | CD274 | [24] | |
| P13 | 8 | 53,490,845 | 54,592,381 | 0.440 | 1 | Nor | [26] | |
| P14 | 8 | 58,649,674 | 58,727,004 | 0.380 | 0 | |||
| P15 | 8 | 61,543,379 | 62,874,750 | 0.464 | 1 | |||
| P16 | 8 | 69,691,214 | 70,488,061 | 0.493 | 0 | Ang | POLR3D, PPP3CC | |
| P17 | 8 | 73,617,355 | 73,704,634 | 0.382 | 0 | |||
| P18 | 10 | 36,488,829 | 37,051,537 | 0.416 | 0 | Her | CHP | [25] |
| P19 | 12 | 27,935,604 | 29,508,940 | 0.439 | 1 | Hol | [24, 25] | |
| P20 | 13 | 34,119,211 | 35,054,048 | 0.402 | 0 | ZEB1 | ||
| P21 | 13 | 48,893,096 | 49,816,619 | 0.408 | 0 | Guz | [9] | |
| P22 | 14 | 24,603,090 | 25,298,972 | 0.395 | 0 | Nor | PLAG1, XKR4, MOS | [10] |
| P23 | 14 | 36,715,710 | 37,511,658 | 0.444 | 0 | Gue | [24] | |
| P24 | 14 | 38,919,669 | 39,027,008 | 0.383 | 0 | Gue | ||
| P25 | 14 | 42,121,450 | 42,376,970 | 0.389 | 0 | |||
| P26 | 14 | 45,478,315 | 46,437,276 | 0.430 | 0 | [24] | ||
| P27 | 16 | 40,318,965 | 40,656,961 | 0.390 | 0 | |||
| P28 | 16 | 40,886,797 | 41,149,860 | 0.383 | 0 | |||
| P29 | 16 | 41,564,542 | 42,407,997 | 0.440 | 0 | Jer | ||
| P30 | 16 | 43,250,880 | 43,501,100 | 0.390 | 0 | [27] | ||
| P31* | 16 | 44,277,286 | 45,534,177 | 0.510 | 2 | PIK3CD | ||
| P32 | 18 | 11,298,096 | 11,959,392 | 0.409 | 0 | Bro | IRF8 | [24] |
| P33 | 18 | 14,171,624 | 14,702,657 | 0.423 | 0 | ACSF3, SPATA2L | [10] | |
| P34 | 20 | 13,714,109 | 15,135,107 | 0.429 | 0 | [24] | ||
| P35* | 20 | 71,629,018 | 71,967,622 | 0.508 | 0 | |||
| P36* | 21 | 83,766 | 2,416,432 | 0.564 | 0 | Gue | ||
| P37* | 21 | 31,681,776 | 33,273,658 | 0.530 | 1 | Her | ||
| P38 | 21 | 45,793,883 | 45,979,589 | 0.391 | 0 | |||
| P39 | 21 | 68,349,152 | 68,943,249 | 0.448 | 0 | Ang | HSP90AA1, PPP2R5C | |
| P40 | 24 | 24,114,816 | 24,452,344 | 0.383 | 1 | Nor | ||
| P41 | 29 | 51,452,986 | 52,452,986 | 0.382 | 1 | |||
| P42 | 30 | 33,064,381 | 50,329,406 | 0.810 | 0 | IRAK1, BCAP31, CETN2, GAB3, IKBKG, KIR3DL2, MTM1, SRPK3 | ||
| P43 | 30 | 53,439,167 | 57,258,157 | 0.711 | 0 | BTK | ||
| P44 | 30 | 68,834,838 | 71,300,049 | 0.861 | 0 | CYLC1 | [12] | |
| P45 | 30 | 72,360,206 | 79,580,964 | 0.844 | 0 | [12] | ||
| P46 | 30 | 84,352,052 | 85,706,219 | 0.764 | 0 | IL2RG | [12] | |
| P47 | 30 | 96,229,383 | 100,603,158 | 0.699 | 0 | ALAS2, SMC1A, LOC524601, SPIN2, VSIG4 | ||
| P48 | 30 | 130,500,087 | 132,116,040 | 0.670 | 0 | RS1 |
*Regions containing smoothed FST (sFST) in the top 0.1%.
**Regions at sFST top 1% of within taurine and within zebu breeds. Full table of within cattle type results for candidate regions under positive selection is on Additional file 6: Table S3. Ang – Angus; Bro – Brown Swiss, Cha – Charolais, Gue – Guernsey, Jer – Jersey, Her – Hereford, Hol – Holstein, Lim – Limousin, Nor – Norwegian Red, Guz – Guzera, Nel – Nelore.
Table 2.
Balancing selection: regions in the bottom 1% smoothed F ST values
| Region | BTA | SNP start pos | SNP end pos | Lowest | CNV [21] | Within cattle type** | Candidate genes | Cross reference |
|---|---|---|---|---|---|---|---|---|
| B1 | 4 | 110,295,764 | 111,378,106 | 0.070 | 2 | |||
| B2 | 4 | 111,742,866 | 112,562,902 | 0.076 | 2 | CNTNAP2 | ||
| B3 | 5 | 19,457,756 | 19,898,237 | 0.075 | 0 | ATP2B1 | ||
| B4 | 5 | 76,719,327 | 77,207,435 | 0.067 | 0 | PKP2 | ||
| B5 | 6 | 2,883,313 | 4,231,143 | 0.059 | 6 | CCNA2, ANXA5 | ||
| B6 | 6 | 12,490,545 | 13,266,473 | 0.062 | 1 | CAMK2D | ||
| B7 | 6 | 54,759,464 | 55,199,755 | 0.062 | 4 | |||
| B8 | 6 | 61,590,746 | 61,892,976 | 0.076 | 0 | APBB2 | ||
| B9 | 6 | 118,252,961 | 118,649,364 | 0.061 | 0 | Guz | ||
| B10 | 7 | 65,205,183 | 65,242,121 | 0.079 | 0 | GLRA1 | ||
| B11 | 7 | 98,598,188 | 99,371,157 | 0.050 | 3 | ERAP2, LNPEP | ||
| B12 | 11 | 11,993,676 | 13,090,823 | 0.072 | 0 | DYSF | ||
| B13 | 11 | 16,914,701 | 17,716,204 | 0.074 | 2 | |||
| B14 | 12 | 70,094,561 | 76,785,743 | 0.059 | 6 | ABCC4 | ||
| B15 | 14 | 53,550,213 | 54,231,380 | 0.066 | 2 | Nel, Guz, Jer | ||
| B16 | 16 | 16,039,261 | 17,069,240 | 0.076 | 1 | FAM5C | ||
| B17 | 16 | 19,740,336 | 20,450,779 | 0.073 | 0 | ESRRG | ||
| B18 | 16 | 36,476,830 | 37,151,556 | 0.068 | 1 | XCL2 | ||
| B19 | 17 | 8,512,165 | 8,575,700 | 0.079 | 0 | |||
| B20 | 21 | 69,852,429 | 70,269,531 | 0.054 | 0 | Guz | ||
| B21 | 22 | 1,504,583 | 1,623,884 | 0.078 | 1 | SEC61G, NEK10 | ||
| B22* | 23 | 24,242,547 | 31,194,961 | 0.025 | 30 | Ang, Cha, Her, Lim | BOLA (MHC) genes, TNF, AGER, NCR3, C2, CFB, LY6G6F, BTNL2, IL17A, IL17F, CLIC1, CSNK2B, MOG | [12, 24] |
| B23 | 23 | 32,608,468 | 33,237,258 | 0.069 | 1 | Cha | ALDH5A1, TDP2, GMNN | |
| B24 | 26 | 46,663,802 | 47,234,109 | 0.055 | 0 | Cha |
*Regions containing smoothed FST (sFST) in the bottom 0.1%.
**Regions at sFST bottom 1% of within taurine and within zebu breeds. Full table of within cattle type results for candidate regions under balancing selection is on Additional file 7: Table S4. Ang – Angus; Bro – Brown Swiss, Cha – Charolais, Gue – Guernsey, Jer – Jersey, Her – Hereford, Hol – Holstein, Lim – Limousin, Nor – Norwegian Red, Guz – Guzera, Nel – Nelore.
Regions under positive selection
The top 1% smoothed FST values were distributed across 48 regions in 17 chromosomes (Table 1) including the BTAX (not shown in Figure 3). Of those, 12 regions were known to harbor copy number variations, and 22 regions had been described as under positive selection in previous studies (Table 1). Twenty of them also overlapped on one or more breed specific peaks in the within cattle type analyses. Among the previously described peaks, 10 of them overlapped to taurine breed signals, and 1 to a zebu breed peak.
The search for overrepresentation of gene ontology terms was not conclusive. Nevertheless, some regions can be highlighted because of their genic content and/or results from previous studies identifying them as being under selection. The BTA7:47.2-53.7 Mb region (Table 1: regions P10 and P11) harbors two closely linked regions that are potentially under selection. These regions contain a number of immune-related and imprinted genes (CD14, HSPA9 and PCDH family) previously identified to be under selection, and associated with cattle fertility (SPOCK). Moreover, a number of CNV are located in the same region and linkage disequilibrium (LD) blocks larger than the average genomic LD are present in both taurine and zebu animals with LD blocks varying in length (Additional file 4: Figure S3A). Another interesting region is the BTA14:24.6-25.2 Mb region (Table 1: region P22), which confirmed previous results [10] and was recently associated with cattle production-related traits. Interestingly, the zebu and taurine LD patterns also markedly vary within this region (Additional file 4: Figure S3B). The BTAX is the final region to be highlighted, as almost the entire chromosome was shown to be highly differentiated between taurine and zebu.
Regions under balancing selection
The bottom 1% smoothed FST values consisted of 24 genomic regions across 13 chromosomes (Table 2). Of those, only a region on BTA23 had been previously described as a candidate for balancing selection. This region also overlapped taurine breed signals from the within-taurine analysis. In total, 6 regions overlapped within cattle type analyses, three to zebu breed peaks and four to taurine breeds.
Fourteen of these regions have been described as having CNV. These included the large region (Table 2: B22) on BTA23:24.2-31.1 Mb comprising the BOLA gene family (MHC – II molecules) which harbors 30 described CNV. This region has also been previously associated with balancing selection [12, 24] in cattle (Table 2).
The 24 balancing selection regions overlap with 345 Ensembl peptides, corresponding to 318 unique Ensembl genes (Table 2). Additionally ~83% (20/24) of the balancing selection regions completely or partially span cattle Ensembl genes. We assigned PANTHER accessions to a total of 332 overlapping peptides. Statistically significant over represented peptides were observed for multiple categories. Five pathways were found significantly overrepresented (adjusted p-value <0.05): the olfactory transduction, systemic lupus erythematosus, type I diabetes mellitus, antigen processing and presentation, graft-versus-host disease and allograft rejection pathways; all of which could be linked to immune response systems (a biological process also overrepresented).
The average FST for each chromosome in each analysis can be found in the Additional file 5: Table S2. Also in the supplementary material all top and bottom FST peaks for all analyses are presented (Additional file 6: Table S3 and Additional file 7: Table S4).
Discussion
In all, 505 animals derived from 10 taurine and 3 zebu cattle breeds were genotyped across more than 770,000 SNP markers to investigate the genomic changes subsequent to the separation between taurine and zebu cattle, which occurred at a date between 330 thousand and 2 million years ago [1, 2]. Evaluation of the SNP genotyping platform suggested there was minimal bias in properly characterizing both subspecies of animals, except possibly on the sex chromosomes. As expected, most of the chromosomes had a higher proportion of polymorphic markers in taurine animals, also resulting in higher heterozygosity, when compared to zebu (Figure 1A). This is due to the fact that most of the SNP described for cattle were identified using the reference sequence of a taurine animal [19, 28], but this should not overly impact population diversity metrics [29]. Nevertheless, all chromosomes have >80% SNP polymorphic in both cattle types, exception made for BTA1, 13, X and Y (Figure 1B), providing a large number of informative markers.
Clustering animals based on the genetic relationship matrix, not surprisingly, split the animals into two groups (taurine and zebu) in agreement to the division along the first principal component and the magnitude of pair-wise FST between breeds. The split along the second principal component between taurine breeds suggests that there is more variation within this cattle type than there is within zebu. Since it is known that unbalanced principal components analyses could mislead interpretations of population structures [30], four randomized evenly sampled analyses were run (Additional file 2: Figure S2). These additional analyses supported the previous results. This could be partly due to more intensive selection and reproductive isolation in taurine breeds than among zebu cattle. However, even though the BovineHD BeadChip was developed to minimize potential ascertainment bias, one cannot entirely reject the possibility that the subdivision seen on principal component 2 was due to this potential bias carried over by the genotyping platform. In the near future when whole genome sequences from a number of breeds and cattle types become available a definitive conclusion about this aspect will be drawn.
The BTAX and Y carry a great number of SNP with high difference in allelic frequencies between groups. These chromosomes have probably undergone much stronger selection or, more parsimoniously, higher genetic drift, due to their unique inheritance [6], and the history of domestication, selection, breed formation. Furthermore, the intensive use of artificial insemination techniques have likely contributed to the reduction of genetic variability within breeds (or cattle types) in these chromosomes. It is understood that in the case of the SNP that are polymorphic in both cattle types, the alternative allele likely arose within the cattle population before the split between taurine and zebu, and remained in both populations at variable frequencies. Alleles that are fixed in one subspecies and variable in the other possibly arose after the split. However, this understanding does not take into account that alleles that were fixed in one population also might have arisen before the split, but were fixed due to different selection processes or as a result of different bottlenecks on the populations. The identification of the ancestral allele of these SNP, ideally using whole genome sequences of other Bovids, would contribute to understand the evolutionary processes behind these monomorphic sites.
The use of metrics based on variance of allelic frequencies in order to identify genomic regions that are potentially under selection, such as FST, have already been explored in a number of studies using cattle [10, 28, 31], sheep [9] and dogs [8]. In this study a relatively high density of markers (average gap between markers 4Kb) was applied to detect genomic differences between zebu and taurine using FST, identifying regions that were potentially associated with different types of selection. Due to their original geographic distribution, taurine cattle are more adapted to temperate climate, while zebu cattle are better adapted than most taurine cattle to tropical environments. Therefore, differences between these two cattle could be linked to adaptation to the environment; however, it is likely that selection imposed by humans in different geographical locations and livestock-product production goals may have also produced regions that were under differing selective pressures. This study, the most comprehensive to date for cattle, identified 48 regions under potential positive and 24 under balancing selection.
A number of these positive selection candidates have been identified to be under selection in previous studies (22 out of 48, Table 1). These previous studies cannot strictly be considered independent analyses since a subset of markers included in the analyses presented here were already used in those. However, in this work more than a 10 fold increase in marker density was used, thus reducing the overlap of SNP across experiments to less than 10%. Further, different cattle samples and populations were used. Thereafter, even though not absolutely independent, from previous studies, our results lend support to the findings from previous articles provide new insights on ancient differentiation between zebu and taurine cattle. These regions may be genomic segments that were under natural selection or drift, but in fact, might for instance represent zebu fragments that were introgressed in taurine breed potentially defining low-level admixed populations [24, 25]. A parallel could also be drawn to described QTLs that overlap these highly divergent genomic regions, e.g. on BTA14:~25 Mb which harbors quantitative loci for stature [32], fertility [33–35] and subcutaneous fat [36]. The different LD structure in these regions supports the concept of introgressed segments as a way of sharing recent polymorphisms between the cattle types [37], and defines quantitative loci and signatures of selection.
The highest differentiation peak was found in BTA7:~50 Mb. This region had previously been identified as a site containing a signature of selection [12, 24]. A number of features were also identified in this region, including different LD structure between zebu and taurine cattle, the presence of imprinted genes, and potential association to fertility traits. This region is among the very few regions for positive selection that also contain CNVs; which may seem antagonistic to purifying selection. It is not clear at this point how CNV are being kept in the population at this site and at the same time there is a differential signal for zebu and taurine cattle. It could be in consequence that these CNV being less likely in LD with neighbouring SNPs, because similar CNVs can occur on different haplotype backgrounds. Another possibility is that duplications can initiate gene conversion events, which can then decrease the LD surrounding such variants. Interestingly, CNVs were often observed at most candidate sites for balancing selection, where variation is expected.
Fourteen out of 24 balancing selection regions overlap identified CNVs, including the highly variable region on BTA23: ~24 Mb with 30 described CNV (Table 2). This set of balance selection-derived genes possess a wide spectrum of molecular functions and provide a rich resource for testing hypotheses on the genetic basis of phenotypic variation within and among breeds. Consistent with similar analyses in other mammals (human, mouse and dog), several of these genes, which are important in drug detoxification, defense/innate and adaptive immunity, are also highlighted by these analyses in cattle. These gene families include the bovine MHC (BoLA), ATP-binding cassette (ABC) transporters, Glutathione S-transferases, Complement factors, Interleukin-17A (IL17A), Heat shock 70 kDa protein 1A (HSPA1A), Chloride intracellular channel protein 1 (CLIC1), and Casein kinase II subunit beta (CSNK2B), which support the shared GO terms among mammals. Conservation of these genes across mammals suggests that selective pressure may drive acquisition or retention of species-specific gene functions.
On the other hand, lineage-specific selection events were detected in mammals, especially in mice and rats. In this regard, it is intriguing to note that mammary gland development genes, such as Butyrophilin-like protein 2 (BTNL2) and Myelin-oligodendrocyte glycoprotein (MOG) were enriched in GO Biological process on the PANTHER analyses. We also detected marked variation between individuals and across diverse cattle breeds, which indicates that these selection events may have occurred within the artiodactyla and/or Bos lineages contributing to cattle speciation and domestication.
Genome-wide, most CNVs evolved under neutral evolutionary pressures. Their frequency and sequence context were shaped by demographic events, mutation rate and genetic drift. However, most CNVs in potentially functional regions, especially those overlapping genes, are under purifying selection and there are only a few examples of CNVs on these positive selection sites. Regions that differ in copy number between subspecies can be informative about ancient adaptations that may have led to species-specific phenotypes [38]. Recent copy number changes can inform about human selection that may have led to genetic and phenotypic differences between breeds.
Similar to selection for variability seen in balancing regions that result in low FST values, it is worth noting that low values could also represent purifying selection forces that are simultaneously applied in both populations in the same direction, imposing high similarity between the compared groups which would result in low differentiation (low FST). In this case, a potential deleterious mutation affecting both populations would be selected against in both groups. This can partially explain the high frequency of genes associated to Mendelian diseases within those potential balancing selection regions. Highlighting a few examples, Dysferlin (DYSF) is associated to muscular dystrophy [39], ATPase, Ca (2+)-transporting, plasma membrane, 1 (ATP2B1), where mouse knockouts have identified variation underlying embryonic lethality, and has a critical role in male fertility [40], Plakophilin 2 (PKP2), which is linked to circulatory system conditions [41, 42], and Cyclin A (CCNA2) that is an essential regulatory molecule for the cell’s cycle [43]. It is not clear at this point, and it will require further investigation to define if the selection signals seen in these regions are due to the presence of those candidate genes or not.
It is not completely clear at this point how the observed signals of selection originated. The within-taurine and within-zebu FST complement the taurine-zebu contrast analysis providing hints on the breed driving each signal. From the autossomal regions previously described as candidate regions under positive selection, around half of them overlap to signals of one or more breeds in the within-taurine analysis (10 out of 19), which is consistent with one’s expectation, since the majority of previous work was done using mostly taurine breeds, and in a few cases also composite cattle. There was only one region previously described as a candidate for balancing selection, in BTA23, and this also overlaps with within-breed type signals. A number of peaks were characterized with more than one breed specific peak in the within-breed analyses, supporting a commonality of selective pressure in at least a few regions in some breeds. However, not all observed signals from the comparison taurine-zebu could be attributed to a specific breed (s), and these suggest that they represent a deeper degree of separation and, possibly, adaptation between cattle types.
In summary, genomic regions that are linked to positive and balancing selection were detected within taurine and zebu cattle, which represent the major sub-division of domestic cattle. A number of previously described regions containing positive selection were confirmed. Novel selection regions were likely discovered due to the higher resolution of informative SNPs available in this study compared to previous analyses. Some of these regions overlap with production QTL, and e.g. immune-related genes, suggesting that favorable variations to adaptation and production are present in the general cattle population, however the application of these results into breeding programs to accelerate creation of synthetic breeds with high production value in tropical environments remains elusive until subsequent investigations confirm the underlying effect of the variants underlying the signatures. This information is needed to define breeding systems able to efficiently introgress specific genomic fragments of zebu in taurine cattle and vice-versa.
Conclusions
Genomic regions that are potentially linked to purifying or balancing selection processes in domestic cattle were identified genome-wide. The genetic variants imposing such selective pressure are not known, even though for some regions candidate genes could be assigned, and could serve as resource for new hypothesis testing in the future. These regions are of particular interest to understand the natural and human selective pressures to which these subspecies were exposed and how the genetic background of these populations evolved in response to environmental challenges and human manipulation.
Availability of supporting data
Supporting information is available in the additional files and further supporting data is available from the authors on request.
Electronic supplementary material
Additional file 1: Figure S1: Population substructure, the main division in domestic cattle (based on 505 individuals, 38,681 SNP). A) Unsupervised clustering result (inferred number of clusters K = 2). The two clusters represent the main division in ancestry of domestic cattle, the zebu (red) and taurine (blue). The estimated proportion of each cluster (y) is given for each individual. #1-91 Nelore, #92-141 Gir, #142-166 Guzera, #167-187 Guernsey, #188-226 Jersey, #227-270 – Angus, #271-281 Red Angus, #282-317 Hereford, #318-364 Limousin, #365-401 Charolais, #402-425 Brown Swiss, #426-488 Holstein, #489-505 Norwegian Red. B-C) Principal components analysis (PCA1 vs PCA2), taurine and zebu animals are plotted B) by cattle type zebu (blue) and taurine (red), and C) by breed. (TIFF 911 KB)
Additional file 2: Figure S2: “Balanced” principal components analyses (PCA). In order to investigate if the distribution of the breeds within the principal components factorial plan was due to the uneven number of individuals in each breed, four independent evenly balanced PCA were run. (TIFF 354 KB)
Additional file 3: Table S1: Wright’s F-statistics FIS and pair-wise FST between cattle breeds based on 768,506 SNP genotypes. (PDF 72 KB)
Additional file 4: Figure S3: Linkage Disequilibrium (r2) of selected regions potentially under positive selection. a) BTA7:47 – 54 Mb. b) BTA14: 24 – 26 Mb. (TIFF 1 MB)
Additional file 5: Table S2: Average FST per chromosome for each analysis. (PDF 61 KB)
Additional file 6: Table S3: Candidate region for positive selection: top 1% smoothed FST values for all breeds in all analyses. (PDF 2 MB)
Additional file 7: Table S4: Candidate regions for balancing selection: bottom 1% smoothed FST values for all breeds in all analyses. (PDF 769 KB)
Acknowledgements
The authors would like to thank the Bovine Hapmap Consortium for providing access to their genotypes and CRV Lagoa and Alta Genetics do Brasil ltda for donation of semen doses from Guzera bulls. LRPN and CG were supported by a grant from the Next-Generation BioGreen 21 Program (No. PJ008196), Rural Development Administration, Republic of Korea. This project was also partially supported by FAPESP (2010/52030-2) and CNPq (475914/2010-4), Brazil.
This project was supported by projects 1265-31000-104-00D (BFGL) from the USDA Agricultural Research Service. The SNP data were supported by National Research Initiative Competitive Grant no. 2008-35205-18846 from the USDA National Institute of Food and Agriculture. Mention of trade names or commercial products in this article is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the U.S. Department of Agriculture.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
LRPN, TSS and CPVT planned the experiment; LRPN, GEL, DMB and CG analyzed data; TSS, CPVT, MVBS, MAM, JFG and YTU provided material or technical support; LRPN, TSS, GEL, DMB wrote the manuscript. All authors have read and approved the final manuscript.
Contributor Information
Laercio R Porto-Neto, Email: laercio.portoneto@csiro.au.
Tad S Sonstegard, Email: Tad.Sonstegard@ars.usda.gov.
George E Liu, Email: George.Liu@ars.usda.gov.
Derek M Bickhart, Email: derek.bickhart@ars.usda.gov.
Marcos VB Da Silva, Email: marcos@cnpgl.embrapa.br.
Marco A Machado, Email: machado@cnpgl.embrapa.br.
Yuri T Utsunomiya, Email: ytutsunomiya@gmail.com.
Jose F Garcia, Email: jfgarcia@fmva.unesp.br.
Cedric Gondro, Email: cgondro2@une.edu.au.
Curtis P Van Tassell, Email: Curt.VanTassell@ars.usda.gov.
References
- 1.Achilli A, Olivieri A, Pellecchia M, Uboldi C, Colli L, Al-Zahery N, Accetturo M, Pala M, Kashani BH, Perego UA, et al. Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr Biol. 2008;18(4):R157–R158. doi: 10.1016/j.cub.2008.01.019. [DOI] [PubMed] [Google Scholar]
- 2.Hiendleder S, Lewalski H, Janke A. Complete mitochondrial genomes of Bos taurus and Bos indicus provide new insights into intraspecies variation, taxonomy and domestication. Cytogenet Genome Res. 2008;120(1–2):150–156. doi: 10.1159/000118756. [DOI] [PubMed] [Google Scholar]
- 3.Bradley DG, Loftus RT, Cunningham P, MacHugh DE. Genetics and domestic cattle origins. Evol Anthropol. 1998;6(3):79–86. doi: 10.1002/(SICI)1520-6505(1998)6:3<79::AID-EVAN2>3.0.CO;2-R. [DOI] [Google Scholar]
- 4.Helmer D, Gourichon L, Monchot H, Peters J, Segui M. Identifying early domestic cattle from pre-pottery Neolithic sites on the Middle Euphrates using sexual dimorphism. In: Vigne JD PJ, Helmer D, editors. The first steps of animal domestication: new archeological approaches. Oxford: Oxbow Books; 2005. [Google Scholar]
- 5.Ajmone-Marsan P, Garcia JF, Lenstra JA, Globaldiv C. On the origin of cattle: how aurochs became cattle and colonized the world. Evol Anthropol. 2010;19(4):148–157. doi: 10.1002/evan.20267. [DOI] [Google Scholar]
- 6.Schaffner SF. The X chromosome in population genetics. Nat Rev Genet. 2004;5(1):43–51. doi: 10.1038/nrg1247. [DOI] [PubMed] [Google Scholar]
- 7.Wright S. The genetical structure of populations. Ann Eugen. 1951;15(4):323–354. doi: 10.1111/j.1469-1809.1949.tb02451.x. [DOI] [PubMed] [Google Scholar]
- 8.Akey JM, Ruhe AL, Akey DT, Wong AK, Connelly CF, Madeoy J, Nicholas TJ, Neff MW. Tracking footprints of artificial selection in the dog genome. Proc Natl Acad Sci U S A. 2010;107(3):1160–1165. doi: 10.1073/pnas.0909918107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kijas J, Lenstra JA, Hayes B, Boitard S, Porto Neto LR, San Cristobal M, Servin B, McCulloch R, Whan V, Gietzen K, et al. Genome wide analysis of the world’s sheep breeds reveals high levels of historic mixture and strong recent selection. PLoS Biol. 2012;10(2):e1001258. doi: 10.1371/journal.pbio.1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flori L, Fritz S, Jaffrezic F, Boussaha M, Gut I, Heath S, Foulley JL, Gautier M. The genome response to artificial selection: a case study in dairy cattle. PLoS ONE. 2009;4(8):e6595. doi: 10.1371/journal.pone.0006595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson G, Smith AV, Jonsson F, Gustafsson O, Stefansson K, Donnelly P. Assessing population differentiation and isolation from single-nucleotide polymorphism data. J R Stat Society Ser B-Stat Method. 2002;64:695–715. doi: 10.1111/1467-9868.00357. [DOI] [Google Scholar]
- 12.Gibbs RA, Taylor JF, Van Tassell CP, Barendse W, Eversole KA, Gill CA, Green RD, Hamernik DL, Kappes SM, Lien S, et al. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science. 2009;324(5926):528–532. doi: 10.1126/science.1167936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, De Bakker PIW, Daly MJ, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19(9):1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang JA, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.VanRaden PM. Efficient methods to compute genomic predictions. J Dairy Sci. 2008;91(11):4414–4423. doi: 10.3168/jds.2007-0980. [DOI] [PubMed] [Google Scholar]
- 17.R: A Language and Environment for Statistical Computinghttp://www.R-project.org
- 18.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 19.Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, Hanrahan F, Pertea G, Van Tassell CP, Sonstegard TS, et al. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10(4):R42. doi: 10.1186/gb-2009-10-4-r42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gasser T, Kneip A, Kohler W. A flexible and fast method for automatic smoothing. J Am Stat Assoc. 1991;86(415):643–652. doi: 10.1080/01621459.1991.10475090. [DOI] [Google Scholar]
- 21.Bickhart DM, Hou YL, Schroeder SG, Alkan C, Cardone MF, Matukumalli LK, Song JZ, Schnabe RD, Ventura M, Taylor JF, et al. Copy number variation of individual cattle genomes using next-generation sequencing. Genome Res. 2012;22(4):778–790. doi: 10.1101/gr.133967.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mi H, Dong Q, Muruganujan A, Gaudet P, Lewis S, Thomas PD. PANTHER version 7: improved phylogenetic trees, orthologs and collaboration with the Gene Ontology Consortium. Nucleic Acids Res. 2010;38(Database issue):D204–210. doi: 10.1093/nar/gkp1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu GE, Hou YL, Zhu B, Cardone MF, Jiang L, Cellamare A, Mitra A, Alexander LJ, Coutinho LL, Dell’Aquila ME, et al. Analysis of copy number variations among diverse cattle breeds. Genome Research. 2010;20(5):693–703. doi: 10.1101/gr.105403.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gautier M, Flori L, Riebler A, Jaffrezic F, Laloe D, Gut I, Moazami-Goudarzi K, Foulley JL. A whole genome Bayesian scan for adaptive genetic divergence in West African cattle. BMC Genomics. 2009;10:550. doi: 10.1186/1471-2164-10-550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gautier M, Naves M. Footprints of selection in the ancestral admixture of a New World Creole cattle breed. Mol Ecol. 2011;20(15):3128–3143. doi: 10.1111/j.1365-294X.2011.05163.x. [DOI] [PubMed] [Google Scholar]
- 26.Stella A, Ajmone-Marsan P, Lazzari B, Boettcher P. Identification of Selection Signatures in Cattle Breeds Selected for Dairy Production. Genetics. 2010;185(4):1451–U1498. doi: 10.1534/genetics.110.116111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qanbari S, Gianola D, Hayes B, Schenkel F, Miller S, Moore S, Thaller G, Simianer H. Application of site and haplotype-frequency based approaches for detecting selection signatures in cattle. BMC Genomics. 2011;12:318. doi: 10.1186/1471-2164-12-318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Elsik CG, Tellam RL, Worley KC, Gibbs RA, Muzny DM, Weinstock GM, Adelson DL, Eichler EE, Elnitski L, Guigo R, et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science. 2009;324(5926):522–528. doi: 10.1126/science.1169588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Porto Neto LR, Barendse W. Effect of SNP origin on analyses of genetic diversity in cattle. Anim Prod Sci. 2010;50:792–800. doi: 10.1071/AN10073. [DOI] [Google Scholar]
- 30.McVean G: A Genealogical Interpretation of Principal Components Analysis.PLoS Genet 2009.,5(10): [DOI] [PMC free article] [PubMed]
- 31.Barendse W, Harrison BE, Bunch RJ, Thomas MB, Turner LB. Genome wide signatures of positive selection: the comparison of independent samples and the identification of regions associated to traits. BMC Genomics. 2009;10:178. doi: 10.1186/1471-2164-10-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karim L, Takeda H, Lin L, Druet T, Arias JAC, Baurain D, Cambisano N, Davis SR, Farnir F, Grisart B, et al. Variants modulating the expression of a chromosome domain encompassing PLAG1 influence bovine stature. Nat Genet. 2011;43(5):405. doi: 10.1038/ng.814. [DOI] [PubMed] [Google Scholar]
- 33.Fortes MR, Reverter A, Hawken RJ, Bolormaa S, Lehnert SA. Candidate genes associated with testicular development, sperm quality, and hormone levels of inhibin, luteinizing hormone, and insulin-like growth factor 1 in brahman bulls. Biol Reprod. 2012;87(3):58. doi: 10.1095/biolreprod.112.101089. [DOI] [PubMed] [Google Scholar]
- 34.Fortes MRS, Lehnert SA, Bolormaa S, Reich C, Fordyce G, Corbet NJ, Whan V, Hawken RJ, Reverter A. Finding genes for economically important traits: Brahman cattle puberty. Anim Prod Sci. 2012;52(2–3):143–150. doi: 10.1071/AN11165. [DOI] [Google Scholar]
- 35.Hawken RJ, Zhang YD, Fortes MRS, Collis E, Barris WC, Corbet NJ, Williams PJ, Fordyce G, Holroyd RG, Walkley JRW, et al. Genome-wide association studies of female reproduction in tropically adapted beef cattle. J Anim Sci. 2012;90(5):1398–1410. doi: 10.2527/jas.2011-4410. [DOI] [PubMed] [Google Scholar]
- 36.Bolormaa S, Porto Neto LR, Zhang YD, Bunch RJ, Harrison BE, Goddard ME, Barendse W. A genome wide association study of meat and carcass traits in Australian cattle. J Anim Sci. 2011;89:2297–2309. doi: 10.2527/jas.2010-3138. [DOI] [PubMed] [Google Scholar]
- 37.Fortes MR, Kemper K, Sasazaki S, Reverter A, Pryce J, Barendse W, Bunch R, McCulloch R, Harrison BE, Bolormaa S, et al. Evidence for pleiotropism and recent selection in the PLAG1 region in Australian Beef cattle. Anim Genet. 2013;44(6):636–647. doi: 10.1111/age.12075. [DOI] [PubMed] [Google Scholar]
- 38.Iskow RC, Gokcumen O, Lee C. Exploring the role of copy number variants in human adaptation. Trends Genet. 2012;28(6):245–257. doi: 10.1016/j.tig.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ibelli AMG, Ribeiro ARB, Giglioti R, Regitano LCA, Alencar MM, Chagas ACS, Paco AL, Oliveira HN, Duarte JMS, Oliveira MCS. Resistance of cattle of various genetic groups to the tick Rhipicephalus microplus and the relationship with coat traits. Vet Parasitol. 2012;186(3–4):425–430. doi: 10.1016/j.vetpar.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 40.Frisch JE, O’Neill CJ. Comparative evaluation of beef cattle breeds of African, European and Indian origins. 2. Resistance to cattle ticks and gastrointestinal nematodes. Anim Sci. 1998;67:39–48. doi: 10.1017/S1357729800009772. [DOI] [Google Scholar]
- 41.Save and grow: a policy-maker’s guide to the sustainable intensification of smallholder crop production. Rome: FAO; 2011. p. 182. [Google Scholar]
- 42.Frkonja A, Gredler B, Schnyder U, Curik I, Solkner J. Prediction of breed composition in an admixed cattle population. Anim Genet. 2012;43(6):696–703. doi: 10.1111/j.1365-2052.2012.02345.x. [DOI] [PubMed] [Google Scholar]
- 43.Khatkar MS, Nicholas FW, Collins AR, Zenger KR, Al Cavanagh J, Barris W, Schnabel RD, Taylor JF, Raadsma HW. Extent of genome-wide linkage disequilibrium in Australian Holstein-Friesian cattle based on a high-density SNP panel. BMC Genomics. 2008;9:187. doi: 10.1186/1471-2164-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1: Population substructure, the main division in domestic cattle (based on 505 individuals, 38,681 SNP). A) Unsupervised clustering result (inferred number of clusters K = 2). The two clusters represent the main division in ancestry of domestic cattle, the zebu (red) and taurine (blue). The estimated proportion of each cluster (y) is given for each individual. #1-91 Nelore, #92-141 Gir, #142-166 Guzera, #167-187 Guernsey, #188-226 Jersey, #227-270 – Angus, #271-281 Red Angus, #282-317 Hereford, #318-364 Limousin, #365-401 Charolais, #402-425 Brown Swiss, #426-488 Holstein, #489-505 Norwegian Red. B-C) Principal components analysis (PCA1 vs PCA2), taurine and zebu animals are plotted B) by cattle type zebu (blue) and taurine (red), and C) by breed. (TIFF 911 KB)
Additional file 2: Figure S2: “Balanced” principal components analyses (PCA). In order to investigate if the distribution of the breeds within the principal components factorial plan was due to the uneven number of individuals in each breed, four independent evenly balanced PCA were run. (TIFF 354 KB)
Additional file 3: Table S1: Wright’s F-statistics FIS and pair-wise FST between cattle breeds based on 768,506 SNP genotypes. (PDF 72 KB)
Additional file 4: Figure S3: Linkage Disequilibrium (r2) of selected regions potentially under positive selection. a) BTA7:47 – 54 Mb. b) BTA14: 24 – 26 Mb. (TIFF 1 MB)
Additional file 5: Table S2: Average FST per chromosome for each analysis. (PDF 61 KB)
Additional file 6: Table S3: Candidate region for positive selection: top 1% smoothed FST values for all breeds in all analyses. (PDF 2 MB)
Additional file 7: Table S4: Candidate regions for balancing selection: bottom 1% smoothed FST values for all breeds in all analyses. (PDF 769 KB)