

Abstract
Background
Peach [Prunus persica (L.) Batsch] is an economically important fruit crop that has become a genetic-genomic model for all Prunus species in the family Rosaceae. A doubled haploid reference genome sequence length of 227.3 Mb, a narrow genetic base contrasted by a wide phenotypic variability, the generation of cultivars through hybridization with subsequent clonal propagation, and the current accessibility of many founder genotypes, as well as the pedigree of modern commercial cultivars make peach a model for the study of inter-cultivar genomic heterogeneity and its shaping by artificial selection.
Results
The quantitative genomic differences among the three genotypes studied as genomic variants, included small variants (SNPs and InDels) and structural variants (SV) (duplications, inversions and translocations). The heirloom cultivar 'Georgia Belle’ and an almond by peach introgression breeding line 'F8,1-42’ are more heterogeneous than is the modern cultivar 'Dr. Davis’ when compared to the peach reference genome ('Lovell’). A pair-wise comparison of consensus genome sequences with 'Lovell’ showed that 'F8,1-42’ and 'Georgia Belle’ were more divergent than were 'Dr. Davis’ and 'Lovell’.
Conclusions
A novel application of emerging bioinformatics tools to the analysis of ongoing genome sequencing project outputs has led to the identification of a range of genomic variants. Results can be used to delineate the genomic and phenotypic differences among peach genotypes. For crops such as fruit trees, the availability of old cultivars, breeding selections and their pedigrees, make them suitable models for the study of genome shaping by artificial selection. The findings from the study of such genomic variants can then elucidate the control of pomological traits and the characterization of metabolic pathways, thus facilitating the development of protocols for the improvement of Prunus crops.
Electronic supplementary material
The online version of this article (doi: 10.1186/1471-2164-14-750) contains supplementary material, which is available to authorized users.
Background
High-throughput DNA sequencing has made available large quantities of genomic information allowing a more complete characterization of genomes at the chromosome level. This approach, which has been successfully applied to human genomics through The 1000 Genomes Project Consortium project [1], shows similar promise for the genetic analysis and improvement of crop species [2].
Comparative genomics has been used to distinguish intraspecific differences such as among different agronomic cultivars. Recently, determination of the genome sequences of important tree crops promises to advance genomic analysis of these perennial and clonally propagated crops to the genomic analysis levels now routine for agronomic crops such as rice (Oryza sativa L.) and maize (Zea mays L.).
Unlike sexual seed propagation common to agronomic crops, most fruit tree crops, such as Prunus species, are propagated through vegetative methods; this permits the capture of the individual genetic and epigenetic composition, including chromosomal variants, which may play important roles in their genetic improvements and even domestication.
Peach [Prunus persica (L.) Batsch] has become a model species for genetic and genomic studies in the Rosaceae because it has several characteristics facilitating genetic studies, including: important genes described and mapped, a small diploid genome [3], self-compatibility, and a short juvenile period. As a result of the International Peach Genome Initiative (IPGI), a peach reference genome sequence has been obtained [4]. The peach genome size is approximately 227.3 million base pairs (227.3 MB), and its eight main scaffolds align with the eight linkage groups in the reference physical genetic map developed for peach, which was generated from an F2 progeny of an interspecific cross between peach and almond [5–8]. The publically available peach genome sequence shows high correspondence to the previous physical map obtained for peach [9, 10]. The reference genome is based on a doubled-haploid sample of the 'Lovell’ cultivar [9], which was chosen as the preferred model for pursuing several types of genetic and genomic studies since all of the alleles are represented as homozygous. Peach possesses a haploid chromosome set of eight chromosomes [11]. The eight principal scaffolds of the genome sequence are concordant with the eight linkage groups of the peach physical and genetic maps. 'Lovell’ exhibits the typical phenotype of domesticated peach, which has yellow flesh, yellow skin with around 15% blush, detached pit (freestone), and a melting type flesh texture, with some red pigmentation around the pit (Zhebentyayeva, manuscript in preparation).
Peach, a species domesticated over 4000 years ago [12], exhibits high phenotypic variability but restricted genetic diversity. Low genetic diversity is a consequence of the self-compatibility in peach [13], as well as a recent genetic-bottleneck during the development of modern European and American cultivars [14].
Chromosome 1 is the largest and sub-metacentric, chromosomes 2 and 4 to 7 are metacentric, while chromosomes 3 and 8 are acrocentric. Chromosome 8 is the shortest. Chromosomes 6 and 7 are nucleolus-organizers [15, 16]. Techniques such as fluorescence in situ hybridization (FISH) in almond, which has high chromosomal synteny with peach [17], has led to the identification of each chromosome based on the positions of ribosomal DNA genes [18, 19]. Most current cultivars have been developed in the last 100 to 150 years [20]. Because of the low genetic diversity among cultivars [13], the sequence of an individual genome should be representative of the general genic organization in peach.
While several protocols for genetic transformation had been reported for this species [14, 21–23]; an efficient standardized transformation system is not yet available for the species [24]. The consequent limitation on detailed genome annotation further emphasizes the value of genome sequencing as a promising approach for genomic analysis and manipulation.
The genome sequences of three different genotypes of peach were sequenced at the University of California, Davis [25] and aligned to the 'Lovell’ peach reference genome. 'Lovell’ is a double haploid line developed with colchicine by Toyama [26]. The accessions consisted of the heirloom fresh-market cultivar 'Georgia Belle’ (also known as 'Belle of Georgia’), the modern processing cultivar 'Dr. Davis’ and the almond breeding introgression line 'F8,1-42’ from the Processing Peach Breeding Program at UC Davis. These accessions were selected because of their commercial relevance, historic context, diverse phenotypes, and the generation of mapped progenies from these parent cultivars.
The discovery and quantification of genomic variants enables researchers to characterize genomic differences among specific genotypes. For clonally propagated crops, such as peach, individual genotypes or clones can represent a large proportion of the commercial acreage around the world. Genomic variants include both changes in the nucleotides as well as changes in chromosome structure. For trait mapping, nucleotide variants, such as Single Nucleotide Polymorphisms (SNPs, in which one nucleotide is substituted for another) are commonly studied. Insertions and Deletions (InDels, i.e. the addition or loss of a number of nucleotides in a chain no longer than 50) are commonly used to study evolutionary divergence and speciation. Genomic rearrangements (or chromosomal rearrangements) longer than 50 nucleotides are often considered structural variants (SV) [27] since they have a direct impact on the structure and behavior of the chromosomes as well as causing variations in gene dosage. Such structural variants are the result of rearrangements within a chromosome or between chromosomes. While the importance of such variation is recognized in plants, their study remains limited. Typical sources of variation include insertions (longer than 50 bp), inversions, duplications, translocations, and, where they have been characterized, mobile-elements in the target genome, or a combination of such events in balanced or unbalanced signatures [27].
Analysis of SNPs and InDels has become common in genetic and genomic studies such as genetic linkage maps and Quantitative Trait Loci (QTL). In addition to their frequency, they provide information concerning recombination, selection, divergence and genetic structure. In human studies, structural variants have increasingly been considered as a major driving force in evolution [28]. Structural variations are the main source of genomic variation, having been associated with important phenotypic changes, including several rare and complex diseases in humans [27]. The association between structural variants and associated phenotypes in plants has been less thoroughly studied, except for maize [29] with comparisons among inbreed lines [30] and a comparison with teosinte (Zea mays ssp. parviglumis H.H.Iltis & Doebley) [31]. Recent studies have shown this variation to be associated with changes of Copy Number Variation (CNV) in Arabidopsis [32] and intra-cultivar variation in soybean [Glycine max (L.) Merr.] [33, 34]. The discovery and quantification of genomic variants can be used in comparative genomics in order to estimate the genomic heterogeneity among genotypes of the same species, including different cultivars and even different clones of the same cultivar.
Methods of phylogenetic reconstruction which take advantages of powerful statistical approaches and mathematical models, have become indispensable tools in describing the patterns of DNA base substitution, amino acid replacement, and the structural differences among genomes [35]. The use of methods such as the genome conservation matrix [36] enables researchers to make quantitative measurements of comparison among and between genomes, and the application of these measurements to the study of inter-cultivar genome differences is particularly valuable.
The ready availability of genomic and genetic information generated by high-throughput sequencing allows the application of advanced bioinformatic methods to characterize the quantity and distribution of the small and structural variants, and so clarify the effects of such genomic variants.
Genome heterogeneity among three peach genotypes was studied through the discovery and quantification of genomic variants, including small variants, such as SNPs and InDels, and structural variants, such as inversions, duplications and translocations, to better understand the quantitative differences in the genome sequences and their relationship to the number, type and impact of variants. The implications for improved understanding of peach genomics and genetic improvement are discussed. Because desirable genetic and epigenetic genomic variation can be captured in clonally propagated crops such as peach, unique opportunities for clonal crop improvement are possible.
Results
Small variants
Most common small variants (SNPs, InDels) for the three genotypes are summarized in Table 1 and compared with the genome reference sequence. The most common variants were SNPs. Insertions and Deletions were present in similar numbers among the three genotypes, and proportionally, these variants represent approximately 8% of the small variants in 'F8,1-42’, 9% in 'Georgia Belle’ and 10% in 'Dr. Davis’. The distribution and frequency of the variants among the eight scaffolds is shown in Figure 1. The differences in small variants exhibited among the genotypes and among the chromosomes were evident, the most distinct being the high frequency of variants in 'F8,1-42’ at the end of chromosomes 4 and 8, and the particular pattern of variation exhibited at the end of chromosome 5, suggesting possible chromosomal rearrangements in this genotype.
Table 1.
Total number of variants, type and zygosity of variants for each genotype
| Genotype | Total | SNPs | Insertions | Deletions | |||
|---|---|---|---|---|---|---|---|
| Hom | Het | Hom | Het | Hom | Het | ||
| 'Georgia Belle’ | 639,062 | 581,616 | 27,515 | 29,931 | |||
| 2,910 | 578,706 | 7,745 | 19,770 | 7,790 | 22,141 | ||
| 'Dr. Davis’ | 399,649 | 358,648 | 19,148 | 21,853 | |||
| 1,428 | 357,220 | 6,756 | 12,392 | 6,995 | 14,858 | ||
| 'F8,1-42’ | 593,720 | 546,542 | 22,543 | 24,635 | |||
| 3,698 | 542,844 | 8,617 | 13,926 | 8,674 | 16,159 | ||
“Hom” refers to homozygous variants and “Het” to heterozygous variants.
Figure 1.
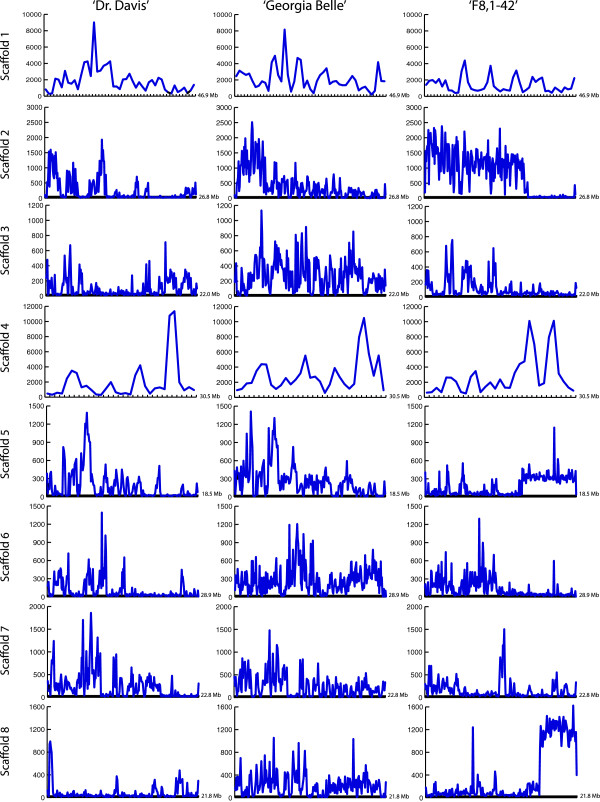
Comparison in the frequency distribution of the variants along each scaffold for 'Dr. Davis’, 'Georgia Belle’, and 'F8,1-42’. The frequency is given in number of variants per 100 Kb for a particular position in the scaffold.
The heirloom cultivar 'Georgia Belle’ exhibited the greatest variation with respect to the 'Lovell’ reference genome, followed by the breeding introgression line 'F8,1-42’ and then the modern cultivar 'Dr. Davis’. A similar pattern was followed for each type of small variants, as well as for zygosity. The genome-wide change rate for 'Georgia Belle’ was 1 change for every 355 bases, 1 for every 382 for 'F8-1.42’ and 1 for every 568 for 'Dr. Davis’.
The output of SnpEff 3.0c (see Additional files 1, 2 and 3) provided detailed information on the number of changes and the change rate per chromosome (scaffolds as denominated by the Peach Genome Initiative). Among the eight scaffolds that comprise the genome of peach, the highest change rate was observed in scaffold 2. This finding was observed for all three genotypes, with one change for every 122 bases for 'F8,1-42’, one change for every 235 bases for 'Georgia Belle’, and one change for every 397 bases for 'Dr. Davis’. Interestingly, scaffold 8 in 'Dr. Davis’ shows the lowest rate of change, with one change for every 1268 bases, followed by scaffold 5 of 'F8,1-42’, which exhibits one change for every 1111 bases. Also, notable is that the change rate for the eight scaffolds of 'Georgia Belle’ ranges from 235 to 462, while for 'F8,1-42’, it is between 122 and 1111 and for 'Dr. Davis’ it is 392 and 1268.
'Georgia Belle’ exhibits the highest proportion of heterozygous versus homozygous variants (97.1%), followed by 'F8,1-42’ (96.5%) and then 'Dr. Davis’ (96.2%). SnpEff also evaluated the impact of the changes based on the known annotation for the peach reference genome. Around 95% of the changes reported by genotype were considered sequence modifiers; the remaining ~5% consisted of moderate impact (~2.68% avg.), low impact (~1.85% avg.) and high impact (~0.28%) changes in the transcript unit. Few high impact variants were reported for each genotype, being greater for 'F8,1-42’ and 'Georgia Belle’, both with over 2000 changes. A total of 2729 changes were considered high impact changes in 'F8,1-42’ (0.281% of the total number of changes), 2277 in 'Georgia Belle’ (0.221%), and 1691 (0.268%) in 'Dr. Davis’.
For the three effects per functional class (missense, nonsense and silent), the three genotypes showed between 57 and 59% missense changes, 38.85 and 40.3% silent changes, and a very small proportion of nonsense changes, ranging between 1.403 and 1.88%. The Missense/Silent ratio for 'Dr. Davis’ is 1.5262, 1.4481 for 'Georgia Belle’ and 1.4347 for 'F8,1-42’.
SnpEff also provided a detailed summary of the occurrence of small variants by type (Table 2) and by genomic region (Table 3, the two tables are complementary). The most common type of change is Non-Synonymous-Coding change, which ranges in each genotype between 2.5 and 3% of the total changes. Synonymous Coding changes were the next most common type of change, ranging between 1.6 and 2%. The remaining types of changes were present in low frequencies, since these do not exceed 0.14%. Changes such as Frame Shift surpass 1000 events in 'Georgia Belle’ (1,134) and in 'F8,1-42’ (1,284), while the lowest frequency change was the Non-Synonymous-Start type, with less than 10 events per genotype.
Table 2.
Count and percentage of changes given by small variants by type of change for each genotype
| Type of change (alphabetical order) | 'Georgia Belle’ | 'Dr. Davis’ | 'F8,1-42’ | |||
|---|---|---|---|---|---|---|
| Count | Percent | Count | Percent | Count | Percent | |
| Codon Change + Codon Deletion | 98 | 0.01% | 64 | 0.01% | 95 | 0.01% |
| Codon Change + Codon Insertion | 125 | 0.012% | 79 | 0.013% | 131 | 0.013% |
| Codon Deletion | 143 | 0.014% | 82 | 0.013% | 135 | 0.014% |
| Codon Insertion | 56 | 0.005% | 35 | 0.006% | 63 | 0.006% |
| Frame Shift | 1,134 | 0.11% | 847 | 0.134% | 1,284 | 0.132% |
| Non-Synonymous-Coding | 25,607 | 2.489% | 15,537 | 2.464% | 28,699 | 2.953% |
| Non-Synonymous-Start | 6 | 0.001% | 2 | 0.0005% | 4 | 0.0001% |
| Start Gained | 258 | 0.025% | 169 | 0.027% | 211 | 0.032% |
| Start Lost | 49 | 0.005% | 35 | 0.006% | 42 | 0.004% |
| Stop Gained | 635 | 0.062% | 499 | 0.079% | 947 | 0.097% |
| Stop Lost | 75 | 0.007% | 45 | 0.007% | 70 | 0.007% |
| Synonymous Coding | 17,743 | 1.725% | 10,217 | 1.62% | 20,046 | 2.062% |
| Synonymous Stop | 25 | 0.002% | 16 | 0.003% | 38 | 0.004% |
Table 3.
Count and number of changes per genomic region in each genotype
| Region (alphabetical order) | 'Georgia Belle’ | 'Dr. Davis’ | 'F8,1-42’ | |||
|---|---|---|---|---|---|---|
| Count | Percent | Count | Percent | Count | Percent | |
| Downstream | 351,984 | 34.216% | 210,781 | 33.431% | 332,654 | 34.226% |
| Exon | 45,696 | 4.442% | 27,458 | 4.355% | 51,554 | 5.304% |
| Intergenic | 162,860 | 15.831% | 108,303 | 17.178% | 147,753 | 15.202% |
| Intron | 79,677 | 7.745% | 47,897 | 7.597% | 82,602 | 8.499% |
| Splice site acceptor | 191 | 0.019% | 121 | 0.019% | 183 | 0.019% |
| Splice site donor | 193 | 0.019% | 144 | 0.023% | 203 | 0.021% |
| Upstream | 382,086 | 37.142% | 231,850 | 36.773% | 349,884 | 35.998% |
| UTR-3′ | 3,863 | 0.376% | 2,430 | 0.385% | 4,602 | 0.473% |
| UTR-5′ | 2,168 | 0.211% | 1,504 | 0.239% | 2,507 | 0.258% |
Most changes were downstream (33-34%) and upstream (36-37%) of the genes included in the annotation of the peach genome reference. The changes in the intergenic regions of the genomes account for 15-17% of the total, while the changes in introns represented between 7.6 and 8.5% of the changes. The portion of changes within the exonic regions ranged between 4.35 and 5.30%; 'F8,1-42’ showed 51,554 changes (5.304%), while 'Georgia Belle’ showed 45,696 (4.442%) and 'Dr. Davis’ 27,458 (4.355%). Changes occurring within the Untranslated Regions (UTR) 3′ and 5′ were present in proportions between 0.211 and 0.473%.
The base change from guanine (G) to adenine (A) was the most common in 'Georgia Belle’ and 'Dr. Davis’, with 96,058 and 59,129 changes, respectively. Most changes were from cytosine (C) to thymine (T) in 'F8,1-42’. In all cases, changes were denominated as transitions. The total number of transitions and transversions per genotype, as well as their respective Transitions/Transvertion (Ti/Tv) ratios, were presented in Table 4. All three genotypes exhibited Ti/Tv ratios above 3, with 'Georgia Belle’ showing a value above 3.6.
Table 4.
Number of transitions and transversions per genotype
| 'Georgia Belle’ | 'Dr. Davis’ | 'F8,1-42’ | |
|---|---|---|---|
| Transitions | 374,886 | 227,722 | 339,879 |
| Transversions | 206,730 | 130,926 | 206,663 |
| Ti/Tv ratio | 3.6268 | 3.4786 | 3.2892 |
Ti/Tv is a ratio of rates, not of observed events. Since transitions are two times more frequent than transversions, the Ti/Tv ratio is twice the ratio of events = 2×(Ti/Tv).
For codon changes (based in SNPs), 'F8,1-42’ exhibited CCG (Proline) to CCA (Proline) as the most common change (325 events), which results in a synonymous change in transcription. The most common non-synonymous codon change was that from GAG (Glutamic Acid) to AAG (Lysine), with 309 events. 'Georgia Belle’ exhibited AAG (Lysine) to AAA (Lysine) as the most common synonymous codon change (306 events), and GGA (Glycine) to AAA (Lysine) as the most common non-synonymous change with 282 events. 'Dr. Davis’ exhibited GGA (Glycine) to AAA (Lysine) as the most common non-synonymous codon change, with 183 events, and AAC (Asparagine) to AAT (Asparagine) as the most common synonymous codon change with 176 events.
The most common amino acid changes per genotype were: Alanine to Valine, 666 times in 'F8,1-42’, followed by 655 Valine to Isoleucine events, and 603 Alanine to Tyrosine events. For 'Georgia Belle’, the change from Alanine to Valine occurs 553 times, followed by the change from Valine to Isoleucine, with 523 events, and 497 changes from Alanine to Tyrosine. Finally, 'Dr. Davis’ exhibits 352 changes from Glutamic acid to Lysine, followed by Alanine to Tyrosine, with 351 changes, and 349 Alanine to Valine changes.
Structural variants
Two hundred and ninety two significant structural variants were identified from the comparisons of the three peach genotypes with the 'Lovell’ reference genome. The longest structural variant was a balanced inversion of a genomic fragment (Bal-Inv-Framt) in 'Georgia Belle’ at 1075 bp (variant ID 69,825 in Table 5).
Table 5.
Exclusive Structural Variants per genotype, their length, their type and the genomic region in which they occurred
| 'Dr. Davis’ | ||||||
|---|---|---|---|---|---|---|
| ID | Scaffold | Coordinates | SV Type | Length | Sequence | Gene or Repeat |
| 1495 | 1 | 13799591..13800210 | UnBal-Inv-Dup | 619 | Gene | ppb020139m.g |
| 16911 | 2 | 10443723..10444191 | UnBal-Inv-Dup | 468 | - | - |
| 17043 | 2 | 10707357..10707900 | UnBal-Inv-Dup | 543 | Repeat | Repeat_45491 |
| 19815 | 2 | 17082238..17082630 | Bal-Inv-Trans | 392 | Repeat | Repeat_50409, Repeat_50410, Repeat_50411 |
| 19815 | 3 | 5906870..5907047 | Bal-Inv-Trans | 177 | Repeat | Repeat_61206 |
| 20201 | 2 | 1815521..1816145 | UnBal-Inv-Dup | 624 | Repeat | Repeat_39494 |
| 23151 | 2 | 2648789..2649409 | UnBal-Inv-Dup | 620 | Repeat | Repeat_40108 |
| 23712 | 2 | 383807..384764 | Bal-Inv-Framt | 957 | Repeat | Repeat_38367, Repeat_38368 |
| 24146 | 2 | 4837884..4838548 | UnBal-Inv-Dup | 664 | Repeat | Repeat_41631 |
| 26318 | 3 | 1013398..1014058 | UnBal-Inv-Dup | 660 | Repeat | Repeat_57671 |
| 29142 | 3 | 18696965..18697347 | UnBal-Inv-Dup | 382 | Repeat | Repeat_70838, Repeat_70839 |
| 29263 | 3 | 19066495..19066675 | UnBal-Large-Dup | 180 | Repeat | Repeat_71125 |
| 29263 | 3 | 19068151..19068360 | UnBal-Large-Dup | 209 | Repeat | Repeat_71125 |
| 32395 | 3 | 8050690..8051662 | Bal-Inv-Framt | 972 | Repeat | Repeat_62915 |
| 43139 | 5 | 128216..128814 | UnBal-Inv-Dup | 598 | Repeat | Repeat_94279 |
| 46422 | 5 | 6900639..6900801 | UnBal-Trans | 162 | Repeat | Repeat_99387, Repeat_99388 |
| 46422 | 8 | 11283205..11283711 | UnBal-Trans | 506 | Repeat | Repeat_151873, Repeat_151874 |
| 52028 | 6 | 2620470..2620776 | UnBal-Trans | 306 | Repeat | Repeat_108508, Repeat_108509 |
| 52028 | 8 | 11283214..11283719 | UnBal-Trans | 505 | Repeat | Repeat_151873, Repeat_151874 |
| 58484 | 7 | 4749087..4750073 | Bal-Inv-Framt | 986 | Repeat | Repeat_130958 |
| 58485 | 7 | 4749430..4750258 | Bal-Inv-Framt | 828 | Repeat | Repeat_130958 |
| 63963 | 8 | 7122023..7122827 | Bal-Inv-Framt | 804 | mRNA | ppa026667m |
| 64422 | 8 | 9086244..9087200 | Bal-Inv-Framt | 956 | Repeat | Repeat_150549 |
| 'F8,1-42’ | ||||||
| ID | Scaffold | Coordinates | SV Type | Length | Sequence | Gen or Repeat |
| 20993 | 2 | 10443560..10444206 | Bal-Inv-Framt | 646 | - | - |
| 21986 | 2 | 12156442..12157007 | UnBal-Inv-Dup | 565 | Repeat | Repeat_46629, Repeat_46630 |
| 24536 | 2 | 16606936..16607425 | UnBal-Inv-Dup | 489 | - | - |
| 29055 | 2 | 2650281..2650575 | UnBal-Inv-Trans | 294 | Repeat | Repeat_49991, Repeat_49992 |
| 29055 | 3 | 15046335..15046888 | UnBal-Inv-Trans | 553 | Gene | ppa020237m.g |
| 30173 | 2 | 4307001..4307685 | UnBal-Inv-Dup | 684 | Repeat | Repeat_41315, Repeat_41316, Repeat_41317 |
| 33929 | 3 | 10480044..10480270 | UnBal-Inv-Dup | 226 | Gene | ppa011613m.g |
| 37571 | 3 | 19066494..19066675 | UnBal-Large-Dup | 181 | - | - |
| 37571 | 3 | 19068151..19068359 | UnBal-Large-Dup | 208 | Repeat | Repeat_71125 |
| 46467 | 4 | 19153499..19153637 | UnBal-Inv-Dup | 138 | Repeat | Repeat_86571 |
| 55460 | 5 | 10569336..10569979 | UnBal-Inv-Dup | 643 | EST | EST217 [GenBank ID: FE969391.1] |
| 55461 | 5 | 10569391..10570047 | UnBal-Inv-Dup | 656 | EST | EST217 [GenBank ID: FE969391.1] |
| 65545 | 6 | 19832212..19832895 | UnBal-Inv-Dup | 683 | Repeat | Repeat_121473, Repeat_121474, Repeat_121475 |
| 77074 | 7 | 4761867..4762779 | Bal-Inv-Framt | 912 | Repeat | Repeat_130964 |
| 77412 | 7 | 5482889..5483887 | Bal-Inv-Framt | 998 | EST | HPL-01-A08 [GenBank: DN552811.1] |
| 84240 | 8 | 5353089..5353931 | Bal-Inv-Framt | 842 | Repeat | Repeat_147771 |
| 'Georgia Belle’ | ||||||
| ID | Scaffold | Coordinates | SV Type | Length | Sequence | Gen or Repeat |
| 2525 | 1 | 1390693..1391565 | Bal-Inv-Framt | 872 | EST | PP_LEc0006H18f [GenBank ID: DW341826.1] |
| 32059 | 2 | 191135..192115 | Bal-Inv-Framt | 980 | EST | PP_LEc0012I17f [GenBank ID: DW342898.1] |
| 33996 | 2 | 22282633..22282891 | UnBal-Inv-Dup | 258 | Repeat | Repeat_53962 |
| 34581 | 2 | 23312824..23313409 | UnBal-Inv-Dup | 585 | Repeat | Repeat_54614, Repeat_54615, Repeat_54616 |
| 37966 | 2 | 4837563..4838555 | Bal-Inv-Framt | 992 | Repeat | Repeat_41631 |
| 49338 | 3 | 4508991..4509132 | UnBal-Inv-Trans | 141 | Repeat | Repeat_60164 |
| 49338 | 7 | 1525434..1525564 | UnBal-Inv-Trans | 130 | Repeat | Repeat_128579 |
| 57742 | 4 | 19154182..19154816 | UnBal-Inv-Dup | 634 | EST | AJ873513 [GenBank ID: AJ873513.1] |
| 69825 | 5 | 10568959..10570034 | Bal-Inv-Framt | 1075 | EST | EST217 [GenBank ID: FE969391.1] |
| 69826 | 5 | 10569191..10570123 | Bal-Inv-Framt | 932 | EST | EST217 [GenBank ID: FE969391.1] |
| 76451 | 5 | 6900036..6900768 | UnBal-Inv-Dup | 732 | Repeat | Repeat_99387, Repeat_99388 |
| 95603 | 7 | 22382739..22383456 | UnBal-Inv-Dup | 717 | Repeat | Repeat_143336 |
| 95633 | 7 | 22436698..22437437 | Bal-Inv-Framt | 739 | Repeat | Repeat_143367 |
| 96867 | 7 | 4749469..4750167 | UnBal-Inv-Dup | 698 | Repeat | Repeat_130958 |
ID identification number for each structural variant, SV Type Structural variant type, which includes UnBal-Inv-Dup Unbalanced Inverted Duplication, Bal-Inv-Trans Balanced Inverted Translocation, Bal-Inv-Framt Inversion of a genomic fragment, defined by balanced signatures, UnBal-Large-Dup Unbalanced large Duplication, UnBal-Trans Unbalanced Translocation, Sequence type of functional sequence, Length number of nucleotides rearranged in the sequence.
Structural Variants (SV) exhibit a different pattern than the small variants. A global comparison of SV showed that 258 structural variations with respect to the 'Lovell’ sequence were shared by the three genotypes. Among these genotypes, 329 structural variations occur with respect to the peach reference genome sequence, of which 292 are inter-chromosomal and 37 are intra-chromosomal. Inverted translocations (172) are the most frequent variation, followed by inversions and duplications.
The number of exclusive SV in 'Dr. Davis’ was 285, 169 in 'F8,1-42’, and 151 in 'Georgia Belle’ (Figure 2). The number of exclusive SV with a high significance score per genotype longer than 100 nucleotides was 19 for 'Dr. Davis’ (detected by SVDetect release 0.8a). 'F8,1-42’ exhibited 14 structural variations, while 'Georgia Belle’ exhibited 13 (Figure 2, lower panel). Among the three genotypes, the most common types of SV were the unbalanced inverted duplications, or balanced inversions of genomic fragments. 'Dr. Davis’ exhibited one balanced inverted translocation and two unbalanced translocations, which occurred from the first third of chromosomes 5 and 6 to the middle part of chromosome 8. 'F8,1-42’ exhibited one unbalanced inverted translocation occurring between the first third of chromosome 2 and going to the middle part of chromosome 3, and one large unbalanced duplication in the terminal part of chromosome 3. 'Georgia Belle’ exhibited one unbalanced inverted translocation (details in Table 5) between the first fourth of chromosome 3 to the top of chromosome 7.
Figure 2.
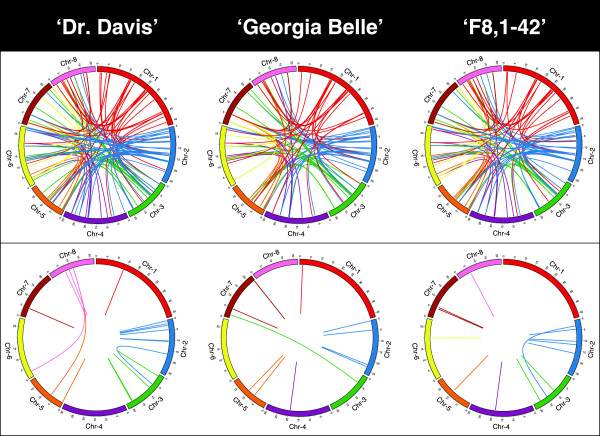
Visual comparison of the structural variants for three peach cultivars using Circos graphs. The variants were obtained through comparisons with the 'Lovell’ Peach Genome Reference Sequence ('Lovell’, upper row) and with the exclusive structural variants per genotype (lower row). Non-connected lines correspond to intra-chromosomal variations. Color of lines corresponds to the source chromosome as defined by the 'Lovell’ reference.
A search for genes within SV regions showed that, in 'Dr. Davis’, just two SV fell in regions with annotated transcripts in the genome annotation of the peach genome sequence reference: the gen ppb020139m.g and the mRNA ppa026667. The remaining SV fell in regions annotated with sequence repeats. A balanced inversion of a genomic fragment (Bal-Inv-Framt) with ID 63,963 in scaffold 8 is located at the gene ppa026667m. It is an mRNA, without a functional annotation. 'F8,1-42’ exhibits two SV within genic regions; a reciprocal translocation that affects the region of the Repeat_49992 in scaffold 2 and the region of the gen ppa020237m.g in scaffold 3, in addition to an inversion within the gen ppa011614m.g in scaffold 3. Three SV (two in scaffold 5 and one in scaffold 7) overlap with Expressed Sequence Tags (ESTs).
'Georgia Belle’ had no SV overlap with a genic region, and five SV (in scaffolds 1, 2, 4 and 5) overlapped with the PP_LEc0006H18f [GenBank ID: DW341826.1], PP_LEc0012I17f [GenBank ID: DW342898.1], ESTs AJ873513 [GenBank ID: AJ873513.1] and EST217 [GenBank ID: FE969391.1] (Additional details in Table 5).
Genome-wide comparison
A conservation matrix was obtained (Table 6) from the genome-wide comparison through the pairwise alignment of 'Lovell’ reference genome sequence and the three genotypes studied. Values of zero indicate complete genome conservation between a pair of genome sequences, while values greater than zero imply some degree of divergence between genome sequences (negative values are not expected), with the value of one denoting complete divergence between a pair of sequences.
Table 6.
Genome conservation matrix among the three genotypes and the peach genome reference sequence
| 'Lovell’ | 'Georgia Belle’ | 'Dr. Davis’ | 'F8,1-42’ | |
|---|---|---|---|---|
| 'Lovell’ | 0 | 0.0264 | 0.0167 | 0.0430 |
| 'Georgia Belle’ | - | 0 | 0.0268 | 0.0429 |
| 'Dr. Davis’ | - | - | 0 | 0.0405 |
| 'F8,1-42’ | - | - | - | 0 |
The analysis, performed using Mauve 2.3.1, identified 'F8,1-42’ as the most divergent genotype with respect to the 'Lovell’ reference (0.0430). 'Georgia Belle’ was intermediate (0.0264), while the least divergent was 'Dr. Davis’ (0.0167). The divergence between 'F8,1-42’ and 'Georgia Belle’ (0.0429) was comparable to that between 'Lovell’ and 'F8,1-42’, and similar to that exhibited between 'F8,1-42’ and 'Dr. Davis’ (0.0405). The divergence between the two peach cultivars was 0.0268, which was comparable to divergence between 'Lovell’ and 'Georgia Belle’. The analysis also determined that the three genotypes exhibit a GC-content of 37.6%.
Discussion
Small variants and structural variants represent different types of genomic variation. While natural selection acts on both types, crop breeding targets primarily small variants, as their inheritance patterns are better understood and therefore, more efficiently manipulated, and because small variants code for single functional changes (amino acid and protein changes). Most crop breeding programs target small incremental changes, while structural variation is manifested as large disruptive changes, including possible sterility as result of genome mismatch. An improved understanding of the process through which structural variants occur, their locations, and their effects on phenotype expression, is now possible through advanced genomic methods.
Small variants
SNP ratios (SNP/bp) observed in this study, differ from previous results observed in other crop plants, which typically occur in a range between 1/100 and 1/300 bp [37]. The SNP/bp ratio also differs among genotypes with respect to the clonal age of the peach cultivars. The heirloom melting flesh cultivar 'Georgia Belle’ (originating before 1870) presented the largest SNP/bp ratio (1/391), agreeing with results of Aranzana et al. [38] showing the highest heterozygozity for this type of cultivar. In contrast, 'Dr. Davis’, which was selected in 1979 and patented in 1982 [39, 40], exhibited a ratio of 1/633, suggesting that modern cultivars tend towards a more homogeneous genomic state, with its associated higher homozygosity. This trend would be an expected consequence of the self-fruitfulness of this species combined with its narrow genetic base, since most important European and North American cultivars have been derived from as few as six Chinese founder genotypes [41]. Both factors promote inbreeding, which leads to homozygosity.
'Georgia Belle’, which is a progeny of 'Chinese Cling’, one of the founder genotypes for modern cultivated peaches, is a melting flesh cultivar, whereas 'Dr. Davis’ is non-melting. Aranzana et al. [38] divided peach cultivars into three main groups based on fruit type rather than geographical distribution [42]. They found that melting flesh cultivars tend to be more heterozygous and probably represent the predominant first domesticated peach types.
'F8,1-42’ exhibited a SNP ratio of 1/415. Selection 'F8,1-42’ represents a more exotic genotype, since the related species Prunus dulcis (Mill.) D.A.Webb (almond) was used as the seed parent in one cross in its lineage (see Additional file 4) [43]. The SNP variant event ratio was closer to that for 'Georgia Belle’ than for 'Dr. Davis’. The genome conservation distance matrix among the four sequences suggests that the almond background in 'F8,1-42’ influences the zygosity of this selection as well as the divergence of the genome sequence relative to 'Lovell’, 'Georgia Belle’, and 'Dr. Davis’.
Earlier studies of the introgression of almond to peach have shown that the rate of recombination between genomes is reduced [44]. Hence, long donor chromosome segments were maintained, resulting in linkage drag. This may be responsible for the wide range in the variants, as well as the change ratios (variant/bp) per scaffold in 'F8,1.42’ (from 1 change every 122 bases to 1 in 1111 bases). Consequently, further backcrossing to peach is desirable to add and fix desired combinations into breeding selections. Interestingly, 'F8,1-42’ exhibits a unique non-melting, freestone phenotype which has not been previously reported in peaches [45], suggesting that the expression of this unique phenotype is a result of unique recombinations of almond and peach genetic material [46].
The differences in the change rates among chromosomes and within chromosomes or scaffolds is, in part, a result of the pattern of crossovers along chromosomes, which is influenced by the length of the chromosome [47] and position on the chromosome [48], as well as genome compatibility in interspecific crosses. Scaffold 2 in all three genotypes exhibited the highest change rate, even though it is not the largest chromosome. The ranking from longest to shortest based on sequencing in the peach reference genome sequence is: scaffold 1, scaffold 4, scaffold 6, scaffold 2, scaffold 7, scaffold 3, scaffold 8 and scaffold 5.
The high rate of variation for chromosome 2 may be a result of the higher number of recombination hotspots, as has been reported by Nachman in the case of humans [49]. Scaffold 2 has been reported to carry important quantitative trait loci (QTL) for fruit, including ripening time, skin color, soluble solids content, and diameter [50], which are important targets of selection. More recombination does not necessarily represent a source of new alleles, since recombination hotspots often occur in intergenic regions in plants [51, 52], and their distribution along the chromosome is influenced by several factors, including proximity to the centromere, gene density, and GC content [53]. A better understanding of the distribution of these hotspots will lead to better modeling of the inheritance and conformation of linkage blocks. Relatively large linkage blocks are anticipated in peach because of the low linkage disequilibrium decay in the species, which ranges from ~6 cM (2524–2644 Kb) in Chinese landraces [42] to 13 to 15 cM (5460–6600 Kb) in commercial cultivars [38].
Scaffold 4 has been reported to carry QTLs for blooming time, ripening time, and glucose/fructose content, as well as the major genes for flesh adhesion (F) (clingstone/freestone) and flesh texture (M) (melting/non-melting) [17], which are discriminator traits for the three genotypes studied here, as well as important targets of selection in the Processing Peach Breeding Program at UC Davis. Also, scaffold 4 is the third longest scaffold in peach, and has exhibited one change every 330 bp in 'Dr. Davis’, one for every 352 bp in 'F8,1-42’, and one for every 505 bp in 'Georgia Belle’ (Figure 1). High rates of variation were exhibited in the terminal sections of the scaffold in the three genotypes, which coincide with identified QTLs for freestone-melting flesh, mealiness, and flesh bleeding in two mapping populations obtained through two crosses using the three genotypes studied here ('Dr. Davis’ used as seed parent in both crosses) [54]. The variations in the genome-wide change rate and scaffold change rate in the three genotypes studied here do not represent some systematic change, but such variations are likely to be due to random variation. However, if different chromosomes have different distributions of non-coding DNA, such difference in non-coding DNA distribution could imply some rate change bias.
Most of the genomic variations would be expected to occur within non-coding regions, thus avoiding changes to transcribed proteins [55]. A relatively low numbers of high impact variants (splice site acceptors, splice site donors, start lost codons, frame shifts, stop gained codons, and stop lost codons) were observed. These variants can alter the amino acid transcript or the length of the ORF and directly impact the structure of the protein. These results were expected since one of the DNA functions is to prevent disruptive changes, which can compromise the integrity of the organism.
The proportion of silent changes (around 39%) and missense modifications (around 58%) among the three genotypes is relevant since the former are considered as evolutionarily neutral (however, these silent changes can affect the structure and function of the resultant protein, see [56]) and the latter are not. Our results support that, from an evolutionary perspective, the proportion of missense and silent modifications, as well as the ratio between these modifications, indicate a strong effect of artificial selection on the peach genome over the last 100 years of cultivar breeding.
The observed genome-wide missense/silent modifications ratios are consistent with the theory that loci under the action of selection present higher ratios of missense/silent modifications than do those under less or nil selection pressure. Thus, if the whole genome is considered as a whole transcribe-able locus, the heirloom cultivar 'Georgia Belle’ exhibited a value of 1.4481, while the modern 'Dr. Davis’ exhibited a value of 1.5262. Selection 'F8,1-42’, with its introgression of genetic material from almond, exhibited a value of 1.4347, which was more similar to the more diverse heirloom cultivar. While these analyses ultimately have to be performed on specific loci (genes or candidate genes, preferably those with agronomic value) they provide initial insights into the ways that artificial selection has configured the peach genome including targets of selection, methods of selection and timing, as has been suggested by Aranzana et al. [41] and Verde et al. [4].
The transition-transversion ratio (Ts:Tv) is around 3.0, which is consistent with the Ts:Tv ratio of 3.0988 from SNPs mapped in closely related peach genotypes reported by Martinez-Garcia et al. [57]. Ts:TV ratios in Non-long Terminal Repeat (Non-LTR) retrotransposon sequences have been estimated as 3.9, 3.6, 1.9, 1.6, and 2.5 for plants such as maize, alfalfa (Medicago sativa L.), eikorn wheat (Triticum monococcum L.), barley (Hordeum vulgare L.) and plants from the genus Lotus, respectively [58]. Information about Ts:Tv ratios in whole genome sequences from other peach relatives, or even other crops, is scarce. The transition-transversion ratio is commonly used for phylogenetic tree reconstruction, divergence time estimation, as well as a better understanding of the mechanisms of molecular evolution [59, 60]. It is a theoretical estimator of mutation rates and evolutionary divergence, which is not directly related to observed rates of change at the phenotypic level [61].
'F8,1-42’ and 'Georgia Belle’ exhibited the same most common amino acid substitutions, Alanine to Valine, Valine to Isoleucine and Alanine to Tyrosine. Nucleotide and amino acid substitutions have been shown to affect important agronomic traits. Barry et al. [62] identified two mutations involved in the degradation of green color in tomato, which can be traced to two specific amino acid substitutions. Previous studies in peach have shown a Quantitative Trait Nucleotide (QTN) located on chromosome 4 to be involved in chilling injury, in particular mealiness [57]. The understanding of nucleotide and amino acid substitutions can therefore facilitate the characterization of metabolic pathways and improvements in phenotyping through the identification of the relevant biochemical changes affecting structure or the availability of substrates.
Structural variants
The peach genome is approximately 227.3 Mb long, and has approximately 62.3Mb (27.4%) of repeats (see [63]); so the effective coding sequence of peach is approximately 165 Mb in length. With 27,852 genes annotated ([4] and see [64]), the average length of a gene in peach is approximately 5924 bp. Thus, if a balanced inversion of a genomic fragment occurred in a genic region, it would constitute a sizable structural change, which could compromise the function of associated genes or prevent recombination in that region. In this particular case, the structural variant with ID 69,825 occurs in scaffold 5, within a reported EST (GenBank ID FE969391.1) described as a protein of unknown function [65].
The majority of the exclusive variants in our analysis were found within repeats. Thus, 'Dr. Davis’ exhibited an unbalanced inverted duplication (UnBal-Inv-Dup) within the gen ppb020139m.g in scaffold 1 (variant ID 1495, Table 5), which is associated with the cytochrome C assembly protein family, in homologous Arabidopsis thaliana L. and rice sequences.
Construction of a complete reliable functional annotation for peach has not been completed [9]. An initial annotation was done several years ago (see [66]); however, there are gaps and inconsistencies such as the unbalanced inverted translocation (UnBal-Inv-Trans) occurring between scaffolds 2 and 3, associated with a non-plant functional annotation for the human Fanconi anemia pathway. The, Kegg Orthology (entry K10891) for this annotation is “a rare genetic disorder characterized by aplastic anemia, greater susceptibility to cancer/leukemia as well as cellular hypersensitivity to DNA crosslinking agents, such as cisplatin” [67].
An UnBal-Inv-Dup (ID 33,929) was present in the first exon of gene ppa011613m.g, which appears related to Ribosomal protein L13, controlling the structural constituents of the ribosome. Two UnBal-Inv-Dup and one Bal-Inv-Framt overlapping within two ESTs, (one of them being the same EST described above in 'Georgia Belle’,) occurred twice in 'F8,1-42’. The Bal-Inv-Framt (ID 77,412) overlapped with the EST HPL-01-A08 (GenBank: DN552811.1 from a Plum Pox Virus (PPV) study [68], in which this particular EST was obtained from non-infected 'Baby Gold #5’ cultivar leaf tissue).
The distribution of variants observed in chromosomes 4 and 8 of 'F8,1-42’ (Figure 1) suggested that SV has occurred at the terminal portions of the chromosome. Thus, on chromosome 4, seven translocations (Trans) and inverted translocations (Inv-Trans) between the nucleotides 19,153,501 and 27,502,845, in addition to four inverted duplications (Inv-Dup) have occurred (details in Additional file 5, sheet F8_Exclusive). Chromosome 8 in 'F8,1-42’ exhibited seven translocation and inverted translocations events between the nucleotides 11,283,140 and 17,453,927. It has been reported that QTLs for chilling and heat requirement are located within the middle and terminal portion of chromosome 8 [69]; therefore, the SV reported in 'F8,1.42’ for this chromosome would have implication in altering characteristics such as blooming date (BD) or maturation time (MT). For the three genotypes studied, the number of Julian days for BD and MT are different among genotypes by 10 to 15 days, being the earliest for 'Georgia Belle’, followed by 'Dr. Davis’, and 'F8,1-42’ (latest flowering). These SV are not exclusive to 'F8,1-42’, since some are shared with least one other genotype (mostly 'Dr. Davis’).
A set of 62 SV (of 292), on chromosome 8, was shared by the three genotypes, and those SV were different from that of 'Lovell’, which suggests that this specific chromosome has undergone a severe rearrangement. In the case of 'F8,1-42’ rearrangement effects may be magnified as a result of almond genetic material introgression. However, this restructuring had also taken place (to a limited extent) in the other genotypes, as seen in by Jauregui et al. [70] in F2 progeny between an almond and peach with introgression of Prunus davidiana (Carrière) Franch in upstream generations, indicating that this chromosome is under constant restructuring in peaches. Restructuring may be occurring as a result of the mode of evolution shaping the Prunus genome, as it is hypothesized that the ancestral genome of Rosaceae had nine chromosomes [71], and that chromosome 8 in Prunus may have resulted from a fission event in the Rosaceae ancestral chromosome A1, when the shortest portion formed chromosome 8, and the fusion of the largest portion of A1 and the whole A2 formed chromosome 1 [72]. Similarly, chromosome 4 was formed from the larger portion of an A9 fission event, while the smaller A9 portion fused with A8 to form chromosome 6 [72]. Interestingly, chromosome 4 carries genes relevant to the fruit phenotypic differences among the three genotypes in this study (particularly genes F and M mentioned above, which are located within the range of high frequency of variation); but chromosome 8 in Prunus is recognized as a chromosome with little evidence for the maintenance of simply inherited (and critical) genes [73] or QTLs [74] responsible for the anthropocentric discrimination of useful agronomic traits used for subsequent selection of peaches during domestication and current breeding.
'Georgia Belle’, in addition to the EST mentioned above, displayed exclusive structural variants (inversions) overlapping with ESTs: PP_LEc0006H18f (GenBank ID: DW341826.1) and PP_LEc0012I17f (GenBank ID: DW342898.1) [75]. The EST AJ873513.1 (GenBank ID: AJ873513.1) has been identified in mesocarp with epidermis tissues at 30 days after bloom in studies of the early stages of fruit development in the peach cultivar 'Fantasia’ (unpublished data [76]).
An estimation of divergence among genotypes provides an overview of whole genome differences. Thus, the divergence between a complete homozygous genome ('Lovell’) and an heirloom cultivar ('Georgia Belle’) is comparable to that exhibited by a genotype of peach with introgressed material from almond ('F8,1-42’). This finding suggests that introgression from almond and subsequent backcrosses with conventional peach genotypes promotes genome heterogeneity similar to that exhibited by the direct progeny of the peach founder genotype 'Chinese Cling’. The divergence between 'Lovell’ and 'Dr. Davis’ supports the assertion that modern cultivars of peach tend to be genomically homogeneous and, thus, tend to be more homozygous. The genomic divergence between 'Georgia Belle’ and 'Dr. Davis’ possesses relevance in terms of fruit characteristics, since the genotypes are completely opposite. 'Georgia Belle’ is a cultivar selected for fresh consumption of the fruit, since the fruits are freestone, melting and white, while 'Dr. Davis’ is a cultivar for the processing industry (e.g. canning and baby food production), with fruits that are clingstone, non-melting and yellow. However, 'Dr. Davis’ and 'Lovell’ fruits are phenotypically distinct only with respect to the detachment of the endocarp from the mesocarp, since the fruits are clingstone and freestone, respectively, and they exhibit the least divergence among the four genotypes.
Our results were consistent with previous discoveries from other crops. In watermelon [Citrullus lanatus (Thunb.) Matsum. & Nakai, Cucurbitaceae], genome heterogeneity has been observed in genomic regions affected by the domestication process, such as disease-resistance genes [77]. In the case of soybean (Fabaceae), a comparison between wild and cultivated soybeans showed long Linkage Disequilibrium blocks in cultivated soybeans, which may result from a combination of the lower genetic diversity given by the domestication bottleneck, low frequency of genetic recombination, and self-fertilization [78]. Similar processes can also be occurring in peach [13, 14].
Several resequencing projects of genomes at the intra-specific level (cultivar founders, breeding lines, cultivars, hybrids) have been carried out to understand genomic heterogeneity [33, 77–82]. In tomato (Solanum lycopersicum L.), the model species for the evolution of species possessing fleshy fruits [83], more than 150 genotypes are being resequenced in the largest resequencing project until now for a crop species [84], The discoveries from this kind of project will have significant relevance for their application in various biological systems of several agricultural crop species. However, researchers should be cautious when extrapolating results, since differences in biology, life history, crop production systems, etc. may result in comparisons/correlations that are not appropriate. For example, peach is a vegetatively propagated species (cloned) and intra-cultivar genome heterogeneity is not an issue, while for soybeans, a sexually generated crop, it is a consideration [33]. The extrapolation of results from closely related species should be done cautiously. For example, although apple (Malus × domestica Borkh., Rosaceae) is a closely related species to peach and vegetatively propagated, apple’s domestication history is totally different [85]. Hence, the context in which each biological system has evolved is relevant when making decisions about which discoveries can be extrapolated.
Our findings suggest that identification of genomic variants may be particularly important in breeding programs incorporating interspecies germplasm to expand the genetic base. A more accurate characterization of the structural variants identified could facilitate “smart breeding”, as suggested by McCouch et al. [86], thus facilitating the recycling of genes that domestication and associated artificial selection had left behind. A useful tool is the genome conservation matrix, which estimates the extent of the genetic-genomic difference between one genotype and another through measurement of their divergence-conservation distance. Thus, the genome conservation matrix “expresses the conservation of both sequence and gene content between two genomes” [36].
This study, to the authors’ knowledge, is the first to use the measurement of conservation-divergence to compare three phenotypically distinct peach genotypes, two commercial peaches, and a peach with almond in its pedigree. Although this measurement may be biased as a result of the assumption of same gene content (an unbiased assessment would require a de novo genome sequence, structural and functional annotations per genotype) and the absence of a comparison with the almond genome sequence (not yet completed). However, given the current status of and the trends for high-throughput sequencing and the comparison of individual genomes [87], future reports with enhanced accuracy and specific trait targets will likely be published.
Conclusions
We combined Illumina/Solexa and Roche 454 sequences to evaluate the genome heterogeneity in three peach genotypes using the doubled haploid cultivar 'Lovell’ as reference sequence. We counted the number of small variants and structural variants among these genotypes and we also estimated the divergence between each genome with the peach reference genome. The main objective was to try to understand the quantitative differences in peach genome sequences and improve the knowledge about the relationship of phenotype and genome features through the application of bioinformatic procedures.
The heterogeneity among the genomes of three peach genotypes was analyzed to characterize and quantify genomic variants. Further analysis showed that the heirloom cultivar 'Georgia Belle’ and the almond by peach introgression breeding line F8,1-42’ are more heterogeneous than is the modern cultivar 'Dr. Davis’, when compared with the 'Lovell’ peach reference genome. The differences in heterogeneity per peach genotype are reflected in the number of variants, the types of variants, and the impacts of those variants on the transcribe-table and non-transcribe-table portions of each genotype analyzed.
The pair-wise comparison of consensus genome sequences with 'Lovell’ showed that 'F8,1-42’ and 'Georgia Belle’ are more divergent compared to 'Dr. Davis’ and 'Lovell’. The results suggest that progenies close to peach founder genotypes conserve more heterogeneity than modern cultivars do, and that the introgression of genetic material from related species can promote genomic heterogeneity in modern breeding lines.
The study of genomic variants is useful for the elucidation of genetic control of pomological traits, the characterization of metabolic pathways and the modeling of the inheritance of complex traits, and thus can lead to improved protocols for phenotyping in research and breeding.
Methods
Plant materials
'Georgia Belle’ (also called 'Belle of Georgia’ [88]) is a freestone peach (the endocarp detaches freely from the mesocarp) with white flesh obtained no later than 1870 on the East Coast of the US. It exhibits melting flesh (losing of firmness and structure, for an accurate description see [89]), a high acid/sugar ratio, and is prone to flesh mealiness and significant browning. This cultivar is a progeny from an open pollination of a tree of the cultivar 'Chinese Cling’; however, other studies suggest the cultivar 'Late Crawford’ is the male parent [88].
'Dr. Davis’ is a clingstone peach (the endocarp does not detach freely from the mesocarp) with yellow flesh, exhibiting non-melting flesh and bland-flavor, with a non-mealy flesh showing only slight oxidative-browning. It is considered a quality reference for canning peach cultivars [39]. It was patented in 1982 (PP4861) and is the result of a cross between the selections D25-9E and G40-5E in the UC Davis breeding program.
'F8,1-42’ is an advanced breeding line with an exotic genetic background including an almond introgression ('Nonpareil’) and several processing peach cultivars (e.g. 'Jungerman’ and 'Everts’) in its lineage. Therefore, it is considered to be an exotic breeding accession, although it is distinctly peach for all fruit and tree phenotypes. It has an unusual phenotype combination, as it has non-melting flesh at maturity, comparable to the standard canning clingstone peach cultivars. Unlike standard canning clingstone peach cultivars; however, it is a freestone, non-melting cultivar. Consequently, F8,1-42 is the breeding line closest to the much desired Non-melting-Freestone cultivar, even though it appears to possesses the standard Non-melting-Clingstone endopolygalacturonase (endoPG) f1 allelic genotype [46].
Methods
For this study, the binary alignment mapped (BAM) files generated from the study of Ahmad et al. [25] were used to generate Simple Alignment Map (SAM) and, subsequently, Variant Filter Calling (VCF) files through the use the routine mpileup in the software SAMtools [90]. The alignment files were developed from the combined Illumina/Solexa and Roche 454 sequences for 'Dr. Davis’ and 'F8,1-42’, and exclusively Illumina/Solexa for 'Georgia Belle’. The alignments were performed with the Burrows-Wheeler Aligner (BWA) tool [91, 92] against the peach reference genome 'Lovell’ (available at [64]). As given by Ahmad et al., aligned positions for 'Dr. Davis’, 'F8,1-42’ and 'Georgia Belle’ were calculated to be 94.7%, 92.0% and 93.7%, respectively. Additionally, consensus genome sequences were generated per genotype through the application of the routine: samtools mpileup -uf ref.fa aln.bam | bcftools view -cg - | vcfutils.pl vcf2fq > cns.fq to each BAM file, resulting in three files in FASTA format of 230.1 MB each.
The quantification, estimation of general statistics, distribution, and prediction of effects on the genomic variants were performed with the software SnpEff 3.0c [93], and are available at the developer’s web page [94]. This software is a bioinformatics tool that annotates the variants (SNPs, insertions, deletions, and multiple nucleotide polymorphisms) and calculates the effects they produce on known genes present in the annotation of the reference genome sequence through an algorithm based on interval trees, which is implemented in the Java programming language.
A SnpEff predictor database file in binary format (.bin) was created to locate each SNP within annotated transcripts or intronic regions. This predictor database is available through SnpEff, and it is based on the 'peach v1.0 genome’ sequence. Annotation of the peach v1.0 is available at Genome Database for Rosaceae (GDR) [64], which was generated by gene models based on homology prediction using information publically available from several organisms. The default parameters of SnpEff ver 3.0c were used to generate the predictor database and perform the Variant Effect Analysis of the three genotypes of peach in annotated transcripts within the 5000 bases of the upstream and downstream portions of the Open Reading Frames (ORF). Both HTML and text output files were generated from SnpEff. The output included the position of the SNP on the scaffold, the reference nucleotide, the changed nucleotide, whether it was a transition or a transversion, the transitions/tranversions ratio (Ts/Tv), warnings, the gene ID, the gene name, the biotype, the transcript ID, the exon ID, the exon rank effect, the amino acid change (old aa/new aa), old codon/new codon, the number of effects, the effects by functional class, the missense/silent ratio, the codon number [based on the coding sequence (CDS)], and the CDS size.
SVDetect release 0.8a [95] was used for the detection of structural variants. This program is specifically designed to identify genomic structural variations through sliding-window and clustering strategies by processing sorted BAM or SAM files resulting from the alignment of the whole sequences for 'Dr. Davis’, 'F8,1-42’ and 'Georgia Belle’ against 'Lovell’. Each alignment file was processed, using a read length of 84, window size of 832 in 'Dr. Davis’, 840 for 'F8,1-42’, and 915 for 'Georgia Belle’. The step length values were 208, 210, and 229, respectively. The values for window size and step size were calculated by running the script BAM_preprocessingPairs.pl (included in SVDetect) per genotype. The script outputs the values for mu_length and sigma_length parameters. Once the values were set for each genotype, all the structural variants (inter and intra chromosomal, as well as balanced and unbalanced) were identified and quantified to convert the output to a graphical form through the visualization tool Circos 0.6.2 [96].
Mauve 2.3.1 [97] [progressiveMauve (multiple genome alignment) using the default settings and the assumption of collinear genomes for the four sequences] was used for the pair-wise comparison among the three consensus genome sequences of the three genotypes previously generated through SAMtools and the peach genome reference genome 'Lovell’.
Authors’ information
JFR is a PhD Candidate in the field of plant genetics and breeding. Currently working on the development and application of genomic resources for the breeding of peach and almond. Areas of interest are plant genetic resources, applied bioinformatics, quantitative genetics and the breeding of fruit tree crops.
PJMG Postdoctoral Associate at UC Davis, Department of Plant Sciences, in David Neale’s lab. His research focuses on genetic and comparative mapping, marker-assisted selection, breeding, population genetics and genome evolution in forest trees.
DEP Lecturer and Pomologist in the College of Agricultural and Environmental Sciences (AES). He is a plant geneticist, breeder with a research focus on fruit and nut germplasm diversity, genetic relationships, and tree breeding.
CHC Specialist and Pomologist, his research and extension program is focus on the postharvest biology and technology of fruits through the application of genomic techniques to identify gene(s) responsible for fruit sensory attributes (both desirable and undesirable), and investigating physiological disorders such as chilling injury.
TMG Professor and Breeder, his research focuses on the development of improved breeding lines and varieties of almond and processing peach through introgression of genetic material from other Prunus relatives to solve problems such as brown rot of clingstone peach, aflatoxin contamination of almond, and pollination efficacy in almond.
Electronic supplementary material
Additional file 1: Summary file of SnpEff output for 'Dr. Davis’. SnpEff_DD.pdf: Summary of statistics of the output of SnpEff 3.0c for the variants present in the peach genotype 'Dr. Davis’ in portable document format (PDF). (PDF 757 KB)
Additional file 2: Summary file of SnpEff output for 'F8,1-42’. SnpEff_F8.pdf: Summary of statistics of the output of SnpEff 3.0c for the variants present in the peach genotype 'F8,1-42’ in portable document format (PDF). (PDF 806 KB)
Additional file 3: Summary file of SnpEff output for 'Georgia Belle’. SnpEff_GB.pdf: Summary of statistics of the output of SnpEff 3.0c for the variants present in the peach genotype 'Georgia Belle’ in portable document format (PDF). (PDF 796 KB)
Additional file 4: Pedigree of the advanced breeding line 'F8,1-42’. F8,1-42_Ped.pdf: 'F8,1-42’ has an exotic genetic background, including introgression of almond (P. dulcis) from the cultivar 'Nonpareil’ (pink box in the center) and several peach cultivars. This figure was generated through PediMap® version 1.2 [98]. (EPS 1 MB)
Additional file 5: Summary of SV identified in the three peach genotypes. SV_DD_F8_GB.xls: Summary of the intra and inter-chromosomal SV identified in exclusive or shared among the peach genotypes 'Dr. Davis’, 'F8,1-42’ and 'Georgia Belle’ in Microsoft Excel format (XLS). (XLS 268 KB)
Acknowledgements
We gratefully acknowledge the support of the National Research Initiative of USDA’s National Institute of Food and Agriculture (NIFA) grant # 2008-35300-04432, UC Davis, UC Agricultural Experiment Station, USDA-CREES (Hatch Experiment Station funding), Henry A. Jastro Graduate Research Award and CONACYT-UCMEXUS, which provides a PhD fellowship to Jonathan Fresnedo-Ramírez. We would especially like to thank Dr. Pablo Cingolani, developer of SnpEff at McGill University, for his useful help comments and clarifications, to Dr. Tatyana Zhebentyayeva at Clemson University, for the communication of the phenotypic characteristics of 'Lovell’, to Dr. Jill L. Wegrzyn at UC Davis for her comments during the correction of the manuscript, and last but not least to Palma Lower, writing specialist at UC Davis, for her valuable comments and corrections during the redaction of this article.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
JFR conceived the study, carried out the bioinformatics, and drafted the manuscript. PJMG participated in the small variant analysis and in the design of the study and helped to draft the manuscript. DEP and CHC participated in the design of the study and helped to draft and edit the manuscript. TMG coordinated the study and elaborated on manuscript. All authors read and approved the final manuscript.
Contributor Information
Jonathan Fresnedo-Ramírez, Email: jfresnedoramirez@ucdavis.edu.
Pedro J Martínez-García, Email: pjmartinezgarcia@ucdavis.edu.
Dan E Parfitt, Email: deparfitt@ucdavis.edu.
Carlos H Crisosto, Email: chcrisosto@ucdavis.edu.
Thomas M Gradziel, Email: tmgradziel@ucdavis.edu.
References
- 1.Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, Chakravarti A, Clark AG, Donnelly P, Eichler EE, Flicek P, Gabriel SB, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Edwards D, Imelfort M. De novo sequencing of plant genomes using second-generation technologies. Brief Bioinform. 2009;10(6):609–618. doi: 10.1093/bib/bbp039. [DOI] [PubMed] [Google Scholar]
- 3.Shulaev V, Korban SS, Sosinski B, Abbott AG, Aldwinckle HS, Folta KM, Iezzoni A, Main D, Arus P, Dandekar AM, et al. Multiple models for rosaceae genomics. Plant Physiol. 2008;147(3):985–1003. doi: 10.1104/pp.107.115618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.International Peach Genome I, Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, et al. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45(5):487–494. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 5.Bliss FA, Arulsekar S, Foolad MR, Becerra V, Gillen AM, Warburton ML, Dandekar AM, Kocsisne GM, Mydin KK. An expanded genetic linkage map of Prunus based on an interspecific cross between almond and peach. Genome. 2002;45(3):520–529. doi: 10.1139/g02-011. [DOI] [PubMed] [Google Scholar]
- 6.Dirlewanger E, Cosson P, Boudehri K, Renaud C, Capdeville G, Tauzin Y, Laigret F, Moing A. Development of a second-generation genetic linkage map for peach [Prunus persica (L.) Batsch] and characterization of morphological traits affecting flower and fruit. Tree Genet Genomes. 2007;3(1):1–13. doi: 10.1007/s11295-006-0053-1. [DOI] [Google Scholar]
- 7.Dirlewanger E, Pronier V, Parvery C, Rothan C, Guye A, Monet R. Genetic linkage map of peach [Prunus persica (L.) Batsch] using morphological and molecular markers. Theor Appl Genet. 1998;97(5–6):888–895. doi: 10.1007/s001220050969. [DOI] [Google Scholar]
- 8.Foolad MR, Arulsekar S, Becerra V, Bliss FA. A genetic-map of Prunus based on an interspecific cross between peach and almond. Theor Appl Genet. 1995;91(2):262–269. doi: 10.1007/BF00220887. [DOI] [PubMed] [Google Scholar]
- 9.Genome Database for Rosaceae: Prunus persicaWhole Genome v1.0 Assembly & Annotation. [http://www.rosaceae.org/species/prunus_persica/genome_v1.0]
- 10.Zhebentyayeva TN, Swire-Clark G, Georgi LL, Garay L, Jung S, Forrest S, Blenda AV, Blackmon B, Mook J, Horn R, et al. A framework physical map for peach, a model rosaceae species. Tree Genet Genomes. 2008;4(4):745–756. doi: 10.1007/s11295-008-0147-z. [DOI] [Google Scholar]
- 11.Pozzi C, Vecchietti A. Peach structural genomics. In: Folta KM, Gardiner SE, editors. Genetics and genomics of rosaceae. New York: Springer; 2009. pp. 235–257. [Google Scholar]
- 12.Huang H, Cheng Z, Zhang Z, Wang Y: History of cultivation and trends in China. In . In: The peach: botany, production and uses. Layne D, Bassi D, editors. Wallinford: CABI; 2008. pp. 37–60. [Google Scholar]
- 13.Font i Forcada C, Oraguzie N, Igartua E, Moreno MÁ, Gogorcena Y. Population structure and marker–trait associations for pomological traits in peach and nectarine cultivars. Tree Genet Genomes. 2012;9(2):331–349. doi: 10.1007/s11295-012-0553-0. [DOI] [Google Scholar]
- 14.Scorza R, Mehlenbacher SA, Lightner GW. Inbreeding and coancestry of freestone peach cultivars of the eastern United States and implications for peach germplasm improvement. J Am Soc Hortic Sci. 1985;110(4):547–552. [Google Scholar]
- 15.Abbott AG, Arus P, Scorza R. Genetic engineering and genomics. In: Layne D, Bassi D, editors. The peach: botany, production and uses. Wallinford: CABI; 2008. pp. 85–105. [Google Scholar]
- 16.Jelenkovic G, Harrington E. Morphology of the pachytene chromosomes in Prunus persica. Can J Genet Cytol. 1972;14(2):317–324. [Google Scholar]
- 17.Abbott AG, Arús P, Scorza R. Peach. In: Kole C, editor. Fruits and nuts. Berlin Heidelberg: Springer; 2007. pp. 137–156. [Google Scholar]
- 18.Corredor E, Roman M, Garcia E, Perera E, Arus P, Naranjo T. Physical mapping of rDNA genes establishes the karyotype of almond. Ann Appl Biol. 2004;144(2):219–222. doi: 10.1111/j.1744-7348.2004.tb00336.x. [DOI] [Google Scholar]
- 19.Yamamoto M, Haji T, Yamaguchi M, Yaegaki H, Sanada T, Kudo K, Mase N. Fluorescent banding pattern of peach Prunus persica (L.) Batsch chromosomes. J Jpn Soc Hortic Sci. 1999;68(3):471–475. doi: 10.2503/jjshs.68.471. [DOI] [Google Scholar]
- 20.Okie WR. Five eastern peach breeders. HortSci. 2006;41(1):11–13. [Google Scholar]
- 21.Callahan A, Scorza R, Mante S, Cordts J, Cohen R, Walton E, Morgens P. Searching for peach genes affecting fruit-quality and progress in regeneration transformation of peach. HortSci. 1988;23(3):793–793. [Google Scholar]
- 22.Hammerschlag FA, Owens LD, Smigocki AC. Agrobacterium-mediated transformation of peach cells derived from mature plants that were propagated in vitro. J Am Soc Hortic Sci. 1989;114(3):508–510. [Google Scholar]
- 23.Ye XJ, Brown SK, Scorza R, Cordts J, Sanford JC. Genetic-transformation of peach tissues by particle bombardment. J Am Soc Hortic Sci. 1994;119(2):367–373. [Google Scholar]
- 24.Padilla IMG, Golis A, Gentile A, Damiano C, Scorza R. Evaluation of transformation in peach Prunus persica explants using green fluorescent protein (GFP) and beta-glucuronidase (GUS) reporter genes. Plant Cell Tiss Org. 2006;84(3):309–314. doi: 10.1007/s11240-005-9039-1. [DOI] [Google Scholar]
- 25.Ahmad R, Parfitt DE, Fass J, Ogundiwin E, Dhingra A, Gradziel TM, Lin D, Joshi NA, Martinez-Garcia PJ, Crisosto CH. Whole genome sequencing of peach (Prunus persica L.) for SNP identification and selection. BMC Genomics. 2011;12:569. doi: 10.1186/1471-2164-12-569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toyama TK. Haploidy in peach. HortSci. 1974;9:187–188. [Google Scholar]
- 27.Alkan C, Coe BP, Eichler EE. Applications of next-generation sequencing: genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–375. doi: 10.1038/nrg2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8(8):639–646. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 29.Goettel W, Messing J. Divergence of gene regulation through chromosomal rearrangements. BMC Genomics. 2010;11:678. doi: 10.1186/1471-2164-11-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Springer NM, Ying K, Fu Y, Ji T, Yeh C-T, Jia Y, Wu W, Richmond T, Kitzman J, Rosenbaum H, et al. Maize inbreds exhibit high levels of copy number variation (CNV) and presence/absence variation (PAV) in genome content. PLoS Genet. 2009;5(11):e1000734. doi: 10.1371/journal.pgen.1000734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Swanson-Wagner RA, Eichten SR, Kumari S, Tiffin P, Stein JC, Ware D, Springer NM. Pervasive gene content variation and copy number variation in maize and its undomesticated progenitor. Genome Res. 2010;20(12):1689–1699. doi: 10.1101/gr.109165.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeBolt S. Copy number variation shapes genome diversity in Arabidopsis over immediate family generational scales. Genome Biol Evol. 2010;2:441–453. doi: 10.1093/gbe/evq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haun WJ, Hyten DL, Xu WW, Gerhardt DJ, Albert TJ, Richmond T, Jeddeloh JA, Jia GF, Springer NM, Vance CP, et al. The composition and origins of genomic variation among individuals of the soybean reference cultivar Williams 82. Plant Physiol. 2011;155(2):645–655. doi: 10.1104/pp.110.166736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McHale LK, Haun WJ, Xu WW, Bhaskar PB, Anderson JE, Hyten DL, Gerhardt DJ, Jeddeloh JA, Stupar RM. Structural variants in the soybean genome localize to clusters of biotic stress-response genes. Plant Physiol. 2012;159(4):1295–1308. doi: 10.1104/pp.112.194605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liò P, Goldman N. Models of molecular evolution and phylogeny. Genome Res. 1998;8(12):1233–1244. doi: 10.1101/gr.8.12.1233. [DOI] [PubMed] [Google Scholar]
- 36.Kunin V, Ahren D, Goldovsky L, Janssen P, Ouzounis CA. Measuring genome conservation across taxa: divided strains and United Kingdoms. Nucleic Acids Res. 2005;33(2):616–621. doi: 10.1093/nar/gki181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Appleby N, Edwards D, Batley J. New technologies for ultra-high throughput genotyping in plants. Methods Mol Biol. 2009;513:19–39. doi: 10.1007/978-1-59745-427-8_2. [DOI] [PubMed] [Google Scholar]
- 38.Aranzana MJ, Abbassi EK, Howad W, Arus P. Genetic variation, population structure and linkage disequilibrium in peach commercial varieties. BMC Genet. 2010;11:69. doi: 10.1186/1471-2156-11-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cummins JN. Register of New fruit and Nut varieties brooks and olmo list 36. HortSci. 1994;29(9):942–969. [Google Scholar]
- 40.Davis LD, Brooks DR 1982. [Peach tree (7–7-52): United States Patent Office, Plant Patent Number: PP4861. Assigned to: The Regents of the University of California; United States of America]
- 41.Aranzana M, Illa E, Howad W, Arús P. A first insight into peach [Prunus persica (L.) Batsch] SNP variability. Tree Genet Genomes. 2012;8(6):1359–1369. doi: 10.1007/s11295-012-0523-6. [DOI] [Google Scholar]
- 42.Cao K, Wang L, Zhu G, Fang W, Chen C, Luo J. Genetic diversity, linkage disequilibrium, and association mapping analyses of peach (Prunus persica) landraces in China. Tree Genet Genomes. 2012;8(5):975–990. doi: 10.1007/s11295-012-0477-8. [DOI] [Google Scholar]
- 43.Gradziel TM. Almond (Prunus dulcis) breeding. In: Priyadarshan M, Jain SM, editors. Breeding plantation tree crops: temperate species. New York: Springer; 2009. pp. 1–31. [Google Scholar]
- 44.Martinez-Gomez P, Arulsekar S, Potter D, Gradziel TM. Relationships among peach, almond, and related species as detected by simple sequence repeat markers. J Am Soc Hortic Sci. 2003;128(5):667–671. [Google Scholar]
- 45.Van der Heyden CR, Holford P, Richards GD. A new source of peach germplasm containing semi-freestone nonmelting flesh types. HortSci. 1997;32(2):288–289. [Google Scholar]
- 46.Peace CP, Crisosto CH, Gradziel TM. Endopolygalacturonase: a candidate gene for freestone and melting flesh in peach. Mol Breed. 2005;16(1):21–31. doi: 10.1007/s11032-005-0828-3. [DOI] [Google Scholar]
- 47.Fledel-Alon A, Wilson DJ, Broman K, Wen X, Ober C, Coop G, Przeworski M. Broad-scale recombination patterns underlying proper disjunction in humans. PLoS Genet. 2009;5(9):e1000658. doi: 10.1371/journal.pgen.1000658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McMahan S, Kohl KP, Sekelsky J. Variation in meiotic recombination frequencies between allelic transgenes inserted at different sites in the drosophila melanogaster genome. G3-Genes Genom Genet. 2013;3(8):1419–1427. doi: 10.1534/g3.113.006411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nachman MW. Single nucleotide polymorphisms and recombination rate in humans. Trends Genet. 2001;17(9):481–485. doi: 10.1016/S0168-9525(01)02409-X. [DOI] [PubMed] [Google Scholar]
- 50.Hancock JF, Scorza R, Lobos GA. Peaches. In: Hancock J, editor. Temperate fruit crop breeding. Netherlands: Springer; 2008. pp. 265–298. [Google Scholar]
- 51.Mezard C. Meiotic recombination hotspots in plants. Biochem Soc Trans. 2006;34:531–534. doi: 10.1042/BST0340531. [DOI] [PubMed] [Google Scholar]
- 52.Schnable PS, Hsia A-P, Nikolau BJ. Genetic recombination in plants. Curr Opin Plant Biol. 1998;1(2):123–129. doi: 10.1016/S1369-5266(98)80013-7. [DOI] [PubMed] [Google Scholar]
- 53.Paape T, Zhou P, Branca A, Briskine R, Young N, Tiffin P. Fine-scale population recombination rates, hotspots, and correlates of recombination in the medicago truncatula genome. Genome Biol Evol. 2012;4(5):726–737. doi: 10.1093/gbe/evs046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martinez-Garcia PJ, Parfitt DE, Ogundiwin EA, Fass J, Chan HM, Ahmad R, Lurie S, Dandekar A, Gradziel TM, Crisosto CH. High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.) Tree Genet Genomes. 2013;9(1):19–36. doi: 10.1007/s11295-012-0522-7. [DOI] [Google Scholar]
- 55.Goode DL, Cooper GM, Schmutz J, Dickson M, Gonzales E, Tsai M, Karra K, Davydov E, Batzoglou S, Myers RM, et al. Evolutionary constraint facilitates interpretation of genetic variation in resequenced human genomes. Genome Res. 2010;20(3):301–310. doi: 10.1101/gr.102210.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Komar AA. SNPs, silent but Not invisible. Science. 2007;315(5811):466–467. doi: 10.1126/science.1138239. [DOI] [PubMed] [Google Scholar]
- 57.Martínez-García P, Fresnedo-Ramírez J, Parfitt D, Gradziel T, Crisosto C. Effect prediction of identified SNPs linked to fruit quality and chilling injury in peach [Prunus persica (L.) Batsch] Plant Mol Biol. 2013;81(1–2):161–174. doi: 10.1007/s11103-012-9989-8. [DOI] [PubMed] [Google Scholar]
- 58.Vitte C, Bennetzen JL. Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci USA. 2006;103(47):17638–17643. doi: 10.1073/pnas.0605618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ina Y. Estimation of the transition/transversion ratio. J Mol Evol. 1998;46(5):521–533. doi: 10.1007/PL00006333. [DOI] [PubMed] [Google Scholar]
- 60.Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide-sequences. J Mol Evol. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- 61.Yang Z, Yoder AD. Estimation of the transition/transversion rate bias and species sampling. J Mol Evol. 1999;48(3):274–283. doi: 10.1007/PL00006470. [DOI] [PubMed] [Google Scholar]
- 62.Barry CS, McQuinn RP, Chung M-Y, Besuden A, Giovannoni JJ. Amino acid substitutions in homologs of the STAY-GREEN protein Are responsible for the green-flesh and chlorophyll retainer mutations of tomato and pepper. Plant Physiol. 2008;147(1):179–187. doi: 10.1104/pp.108.118430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peach genome v1.0. http://services.appliedgenomics.org/projects/drupomics/
- 64.Prunus persica whole genome v1.0 Assembly & annotation, Gene functions [http://www.rosaceae.org/sites/default/files/peach_genome/Prunus_persica_v1.0_gene_function.xls; ftp://ftp.bioinfo.wsu.edu/species/Prunus_persica/Prunus_persica-genome.v1.0/]
- 65.Bassett CL, Wisniewski ME, Artlip TS, Norelli JL, Renaut J, Farrell RE. Global analysis of genes regulated by low temperature and photoperiod in peach bark. J Am Soc Hortic Sci. 2006;131(4):551–563. [Google Scholar]
- 66.Horn R, Lecouls AC, Callahan A, Dandekar A, Garay L, McCord P, Howad W, Chan H, Verde I, Main D, et al. Candidate gene database and transcript map for peach, a model species for fruit trees. Theor Appl Genet. 2005;110(8):1419–1428. doi: 10.1007/s00122-005-1968-x. [DOI] [PubMed] [Google Scholar]
- 67.Jacquemont C, Taniguchi T. The Fanconi anemia pathway and ubiquitin. BMC Biochem. 2007;8 Suppl 1:S10. doi: 10.1186/1471-2091-8-S1-S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang A, Chapman P, Chen L, Stobbs LW, Brown DCW, Brandle JE. A comparative survey, by expressed sequence tag analysis, of genes expressed in peach leaves infected with plum pox virus (PPV) and free from PPV. Can J Psychiatry. 2005;27(3):410–419. [Google Scholar]
- 69.Fan S, Bielenberg DG, Zhebentyayeva TN, Reighard GL, Okie WR, Holland D, Abbott AG. Mapping quantitative trait loci associated with chilling requirement, heat requirement and bloom date in peach (Prunus persica) New Phytol. 2010;185(4):917–930. doi: 10.1111/j.1469-8137.2009.03119.x. [DOI] [PubMed] [Google Scholar]
- 70.Jauregui B, de Vicente MC, Messeguer R, Felipe A, Bonnet A, Salesses G, Arus P. A reciprocal translocation between 'Garfi’ almond and 'Nemared’ peach. Theor Appl Genet. 2001;102(8):1169–1176. doi: 10.1007/s001220000511. [DOI] [Google Scholar]
- 71.Jung S, Cestaro A, Troggio M, Main D, Zheng P, Cho I, Folta KM, Sosinski B, Abbott A, Celton JM, et al. Whole genome comparisons of Fragaria, Prunus and Malus reveal different modes of evolution between Rosaceous subfamilies. BMC Genomics. 2012;13:129. doi: 10.1186/1471-2164-13-129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vilanova S, Sargent DJ, Arus P, Monfort A. Synteny conservation between two distantly-related Rosaceae genomes: Prunus (the stone fruits) and Fragaria (the strawberry) BMC Plant Biol. 2008;8:67. doi: 10.1186/1471-2229-8-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Soriano JM, Badenes ML. Mapping and tagging of simply inhereted traits. In: Kole C, Abbott AG, editors. Genetics, genomics and breeding of stone fruits. Boca Raton: CRC Press; 2012. pp. 105–125. [Google Scholar]
- 74.Olukolu BA, Chittaranjan K. Molecular mapping of complex traits. In: Kole C, Abbott AG, editors. Genetics, genomics and breeding of stone fruits. Boca Raton: CRC Press; 2012. pp. 126–157. [Google Scholar]
- 75.Completion of the peach genome database: a reference genome for rosaceae. http://www.rosaceae.org/node/174
- 76.AJ873513 Prunus persica fruit mesocarp plus epidermis 30 days after bloom Prunus persica cDNA clone PR0114G01, mRNA sequence. http://www.ncbi.nlm.nih.gov/nucest/AJ873513
- 77.Guo SG, Zhang JG, Sun HH, Salse J, Lucas WJ, Zhang HY, Zheng Y, Mao LY, Ren Y, Wang ZW, et al. The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat Genet. 2013;45(1):51–U82. doi: 10.1038/ng.2470. [DOI] [PubMed] [Google Scholar]
- 78.Lam HM, Xu X, Liu X, Chen WB, Yang GH, Wong FL, Li MW, He WM, Qin N, Wang B, et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection (vol 42, pg 1053, 2010) Nat Genet. 2011;43(4):387–387. doi: 10.1038/ng0411-387. [DOI] [PubMed] [Google Scholar]
- 79.Gao ZY, Zhao SC, He WM, Guo LB, Peng YL, Wang JJ, Guo XS, Zhang XM, Rao YC, Zhang C, et al. Dissecting yield-associated loci in super hybrid rice by resequencing recombinant inbred lines and improving parental genome sequences. Proc Natl Acad Sci USA. 2013;110(35):14492–14497. doi: 10.1073/pnas.1306579110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lai JS, Li RQ, Xu X, Jin WW, Xu ML, Zhao HN, Xiang ZK, Song WB, Ying K, Zhang M, et al. Genome-wide patterns of genetic variation among elite maize inbred lines. Nat Genet. 2010;42(11):1027–U1158. doi: 10.1038/ng.684. [DOI] [PubMed] [Google Scholar]
- 81.Xu X, Liu X, Ge S, Jensen JD, Hu FY, Li X, Dong Y, Gutenkunst RN, Fang L, Huang L, et al. Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat Biotechnol. 2012;30(1):105–U157. doi: 10.1038/nbt.2050. [DOI] [PubMed] [Google Scholar]
- 82.Zheng LY, Guo XS, He B, Sun LJ, Peng Y, Dong SS, Liu TF, Jiang SY, Ramachandran S, Liu CM, et al. Genome-wide patterns of genetic variation in sweet and grain sorghum (Sorghum bicolor) Genome biology. 2011;12(11):R114. doi: 10.1186/gb-2011-12-11-r114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Consortium TTG. The tomato genome sequence provides insights into fleshy fruit evolution. Nature. 2012;485(7400):635–641. doi: 10.1038/nature11119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.150 Tomato genome ReSequencing project. http://www.tomatogenome.net/index.html
- 85.Cornille A, Gladieux P, Smulders MJM, Roldan-Ruiz I, Laurens F, Le Cam B, Nersesyan A, Clavel J, Olonova M, Feugey L, et al. New Insight into the History of Domesticated Apple: Secondary Contribution of the European Wild Apple to the Genome of Cultivated Varieties. Plos Genetics. 2012;8(5):e1002703. doi: 10.1371/journal.pgen.1002703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.McCouch S. Diversifying selection in plant breeding. PLoS Biol. 2004;2(10):e347. doi: 10.1371/journal.pbio.0020347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morrell PL, Buckler ES, Ross-Ibarra J. Crop genomics: advances and applications. Nat Rev Genet. 2012;13(2):85–96. doi: 10.1038/nrg3097. [DOI] [PubMed] [Google Scholar]
- 88.Miller E. The natural origins of some popular varieties of fruit. Econ Bot. 1954;8(4):337–348. doi: 10.1007/BF02908606. [DOI] [Google Scholar]
- 89.Citterio S, Ghiani A, Onelli E, Aina R, Cocucci M. A comparative study of melting and non-melting flesh peach cultivars reveals that during fruit ripening endo-polygalacturonase (endo-PG) is mainly involved in pericarp textural changes, not in firmness reduction. J Exp Bot. 2011;62(11):4043–4054. doi: 10.1093/jxb/err109. [DOI] [PubMed] [Google Scholar]
- 90.Durbin R, Li H. Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Durbin R, Li H. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010;26(5):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Durbin R, Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Proc GPD. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25(16):2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cingolani P, Platts A, le Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w (1118); iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.SnpEff. http://snpeff.sourceforge.net
- 95.Zeitouni B, Boeva V, Janoueix-Lerosey I, Loeillet S, Legoix-ne P, Nicolas A, Delattre O, Barillot E. SVDetect: a tool to identify genomic structural variations from paired-end and mate-pair sequencing data. Bioinformatics. 2010;26(15):1895–1896. doi: 10.1093/bioinformatics/btq293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Darling AE, Mau B. Perna NT: progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE. 2010;5(6):e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Voorrips RE, Bink MCAM, van de Weg WE. Pedimap: software for the visualization of genetic and phenotypic data in pedigrees. J Hered. 2012;103(6):903–907. doi: 10.1093/jhered/ess060. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Summary file of SnpEff output for 'Dr. Davis’. SnpEff_DD.pdf: Summary of statistics of the output of SnpEff 3.0c for the variants present in the peach genotype 'Dr. Davis’ in portable document format (PDF). (PDF 757 KB)
Additional file 2: Summary file of SnpEff output for 'F8,1-42’. SnpEff_F8.pdf: Summary of statistics of the output of SnpEff 3.0c for the variants present in the peach genotype 'F8,1-42’ in portable document format (PDF). (PDF 806 KB)
Additional file 3: Summary file of SnpEff output for 'Georgia Belle’. SnpEff_GB.pdf: Summary of statistics of the output of SnpEff 3.0c for the variants present in the peach genotype 'Georgia Belle’ in portable document format (PDF). (PDF 796 KB)
Additional file 4: Pedigree of the advanced breeding line 'F8,1-42’. F8,1-42_Ped.pdf: 'F8,1-42’ has an exotic genetic background, including introgression of almond (P. dulcis) from the cultivar 'Nonpareil’ (pink box in the center) and several peach cultivars. This figure was generated through PediMap® version 1.2 [98]. (EPS 1 MB)
Additional file 5: Summary of SV identified in the three peach genotypes. SV_DD_F8_GB.xls: Summary of the intra and inter-chromosomal SV identified in exclusive or shared among the peach genotypes 'Dr. Davis’, 'F8,1-42’ and 'Georgia Belle’ in Microsoft Excel format (XLS). (XLS 268 KB)