

1. Introduction
Ovarian cancer is the most lethal gynecologic malignancy in women, which causes nearly 15,000 deaths in the United States every year. Its high death rate is a result of the fact that most patients (>75%) are diagnosed at an advanced stage of disease at which point the 5-year survival rate is less than 30% [1, 2]. The vast majority of ovarian cancers are epithelial in origin and characterized by the rapid growth and spread of solid treatment for patients with advanced ovarian cancer is maximal surgical cytoreduction followed by systemic platinum-based chemotherapy [2]. Cytotoxic action of platinum compounds such as cisplatin (cis-dichlorodiamminoplatinum (II), CDDP) is mediated through the formation of platinum-DNA adducts, which in turn inhibits DNA replication and/or transcription and results in apoptosis and necrosis [3, 4]. Despite its success, use of CDDP is greatly limited by severe dose limiting side effects, rapid elimination, and development of acquired drug resistance. No new small molecule platinum drug have gained international marketing approval since 1999 indicating a shift in focus in the last decade away from drug design and towards drug delivery [5]. Among drug delivery systems, those based on self-assembled block copolymer micelles have great potential for delivery of anticancer drugs [6]. The advantages of polymer micelles include their small size, long circulation times, and ability to circumvent renal excretion and extravasate at sites of enhanced vascular permeability. However, disintegration of the polymer micelles is a major drawback for their application in drug delivery. In order to overcome this limitation, we have proposed hydrophilic polyelectrolyte micelles with core composed of a network of cross-linked polyanion chains and PEG shell that can encapsulate CDDP with high capacity (up to 35 w/w%) [7, 8]. We have demonstrated significant improvement of drug safety and drug delivery using such CDDP-loaded cross-linked micelles. However, single drug therapy agents directed to individual targets frequently show limited efficacies. Hence, multi-component therapies and multi-targeted drug combinations are key to most cancer treatments [9]. Combination chemotherapy using sequential administration of paclitaxel (PTX) with a platinum-based regimen is currently the standard first-line therapy for ovarian cancer resulting in increased success rates in eradicating tumors and/or longer remission periods [10]. These clinical benefits of the combination therapy can be possibly maximized by controlled delivery of the desired drug ratio to the in vivo target. Combining drugs in one delivery carrier is well-suited strategy for controlling the pharmacokinetics and co-delivery of the desired drug ratio in vivo. The drug loading and structure of such carriers can be tuned to control the drug release rates, maximize the therapeutic potency and minimize drug-associated toxicities. Nanoscale delivery carriers such as liposomes, nanoparticles, polymeric micelles have been explored for co-delivery of anticancer drugs [11–13]. However, co-incorporation of drug molecules with different physicochemical properties, such as hydrophilic CDDP and hydrophobic PTX, has been challenging. As an attempt to address this challenge, we report a design of multi-compartment cross-linked micelles based on hybrid triblock copolymers, poly(ethylene glycol)-block-poly(L-glutamic acid)-block-poly(L-phenylalanine) (PEG-b-PGlu-b-PPhe) for simultaneous loading and delivery of binary CDDP and PTX combination. Such micelles have 1) a hydrophobic core formed by PPhe chains, which serves as a reservoir for PTX solubilization, 2) an anionic layer, which incorporates CDDP through coordination with the carboxylic groups of PGlu, and 3) an outer PEG shell stabilized, which stabilizes micelles in aqueous dispersion. The crosslinks incorporated into PGlu block layer ensure that the PEG-b-PGlu-b-PPhe micelles remain stable until they encounter proteases, which degrade the micelles by cleaving the polypeptide chains of the block copolymers. This work demonstrates that such biodegradable hybrid micelles carrying CDDP and PTX drug combination exert superior antitumor efficacy in treatment of ovarian cancer in xenograft mouse tumor model.
2. Material and Methods
2.1. Materials
PEG-b-PGlu-b-PPhe block copolymers with different ratios of glutamic acid and phenylalanine units (90:7 and 90:25) were synthesized as described below. α-Amino-ω-methoxy poly(ethylene glycol) (mPEG-NH2, Mw = 5,000 g mol−1, Mw/Mn = 1.05) was purchased from Creative PEGWorks Inc., (NC, USA). CDDP was purchased from Acros Organics. L-glutamic acid γ-benzyl ester (BGlu), L-phenylalanine (Phe), 1,2-ethylenediamine, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC), paclitaxel, and other chemicals were purchased from Sigma-Aldrich (St Louis, MO) and were used without further purification. Fetal bovine serum (FBS), RPMI 1640 medium, penicillin, streptomycin, Trypsin–ethylenediaminetetraacetic acid (EDTA) (0.5% trypsin, 5.3 mM EDTA tetra-sodium) and other chemicals were purchased from Invitrogen (Carlsbad, CA, USA).
2.2. N-carboxyanhydrides (NCA) of BGlu and Phe
BGlu (0.020 mole) or Phe (0.026 mole) and anhydrous tetrahydrofuran (THF) were added into a dried glass reactor to form a suspension. Triphosgen (0.023mole) was separately dissolved in fresh anhydrous THF and injected drop wise into reaction mixture. Nitrogen was bubbled through the mixture during synthesis. The mixture was heated at about 40°C with constant stirring till mixture became transparent. The solution was precipitated in n-hexane and then stored at −20°C overnight in order to allow complete precipitation of γ-benzyl L-glutamate-N-carboxyanhydride (BGlu-NCA) or L-phenulalanine –N-carboxyanhydride (Phe-NCA). The white solids obtained were collected and purified further by repeated precipitation with n-hexane. The resulting NCA monomers were dried under vacuum for 24 h and characterized by the proton nuclear magnetic resonance (1H-NMR).
2.3. PEG-b-PGlu-b-PPhe block copolymer
Monoaminomethoxypoly(ethylene glycol) (mPEG-NH2) (0.02 mmol) was dissolved under stirring in 10 mL of anhydrous dimethylformamide (DMF) in nitrogen atmosphere at 40°C. BGlu-NCA (2 mmol, the feed molar ratio of mPEG-NH2 to BGlu-NCA was 1: 100) dissolved in 5 mL of anhydrous DMF was added dropwise and the solution was stirred for 24 hrs. The aliquot of the reaction mixture was precipitated using excess of diethyl ether, dried under vacuum, and the composition of PEG-PBGlu diblock copolymer was determined by 1H-NMR from the peak intensity ratios of the methylene protons of PEG and the phenyl protons of the γ-benzyl groups (400 MHz in CDCl3: δ = 3.5 (s, 4H, -OCH2CH2-), 5.0 (m, 2H, benzyl), 7.3 (m, 5H, aryl)). The calculated amount of the monomer for the third block, Phe-NCA, dissolved in anhydrous DMF was then injected dropwise and mixture was stirred under a N2 atmosphere at 40 °C for additional 24 hrs. The product (PEG-b-PBGlu-b-PPhe) was precipitated by diethyl ether, purified by repeated precipitation in diethyl ether and dried in a vacuum. The benzyl groups of PEG-b-PBGA-b-PPhe were removed in the presence of NaOH to obtain PEG-b-PGlu-b-PPhe. Polymer sample was dissolved in 10 mL of THF followed by addition of 5 mL of 1N NaOH. After stirring for 10 hrs at 40°C THF was removed at reduced pressure, the residual solution was neutralized by 1M HCl and dialyzed using a dialysis membrane (MWCO 3,500 Da) against distilled water for 24 hrs. After dialysis the product was dried, washed 2–3 times with ethanol followed by lyophilization. By varying the feed molar ratio of mPEG-NH2 to Phe-NCA (1:10 and 1:30), two copolymers with targeted compositions PEG-b-PBGlu(100)-b-PPhe(10) and PEG-b-PBGlu(100)-b-PPhe(30) were synthesized. The concentration of carboxylate groups in the copolymer samples was estimated by potentiometric titration. Self-assembly behavior of PEG-b-PGlu-b-PPhe copolymers was examined using pyrene as a hydrophobic fluorescence probe [14].
2.4. Preparation of cross-linked polymeric micelles
Micelles were prepared by directly dissolving PEG-b-PGlu-b-PPhe copolymer in aqueous media at a concentration of about 1 mg/mL. EDC in water (0.2 eq with respect to the amount of carboxylate groups) was added dropwise to this solution and allowed to stir for additional 10 min at r.t.. An aqueous solution of cross-linker, 1,2-ethylenediamine (0.1 eq) was then added to the dispersion of micelles to achieve 20% of cross-linking degree. This degree represents the maximum theoretical amount of cross-linking that could take place, rather than the precise extent of amidation. The reaction mixture was allowed to stir overnight at r.t. Byproducts of the cross-linking reaction were removed by exhaustive dialysis of the reaction mixtures against distilled water.
2.5. Physicochemical methods of characterization
The 1H NMR spectra for the monomers and copolymers were acquired in CDCl3 or D2O at 25 °C using a Bruker 400 MHz spectrometer. Effective hydrodynamic diameters (Deff) and ζ-potential of micelles were determined by dynamic light scattering (DLS) using a Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, UK). All measurements were performed in automatic mode at 25°C. Software provided by the manufacturer was used to calculate the size, polydispersity indices (PDI), and ζ-potential of micelles. All measurements were performed at least in triplicate to calculate mean values ± SD. Morphology of the micelles was characterized by transmission electron microscopy (TEM) and atomic force microscopy (AFM) as previously described [24].
2.6. Drug loading
A solution of PTX in ethanol (2 mg/mL) was added dropwise into the aqueous dispersion of cl-micelles (1 mg/mL) and the mixture was stirred at r.t. overnight in an open-air system to allow slow evaporation of ethanol [15]. The residual ethanol was then removed at reduced pressure. The precipitate of unbound PTX was removed by centrifugation. The aqueous dispersion of PTX-loaded cl-micelles was mixed with an aqueous solution of CDDP (1 mg/mL) at pH 9.0 at 0.5 molar ratio of CDDP to carboxylate groups followed by incubation at 37°C for 48 h. Unbound CDDP was removed by Ultracon filter units (MWCO 10,000 Da, Millipore). Pt content in cl-micelles (Pt194/Pt195) was assayed on Nexion ion coupled plasma-mass spectrometer (NexION 300Q, PerkinElmer) calibrated with Pt (2–100 ng/ml) and Holmium as the internal standard. Samples were diluted in 0.1 N HCl. PTX levels were determined by high-performance liquid chromatography (HPLC) analysis under isocratic conditions using an Agilent 1200 HPLC system a diode array detector set at 227 nm. As stationary phase a Nucleosil C18 column was used (250 mm × 4.6 mm), a mobile phase of acetonitrile/water mixture (55/45, v/v) was applied at a flow rate of 1 mL/min.
2.7. Release studies
Drug release from the cl-micelles was examined in PBS (pH 7.4), acetate buffered saline (ABS, pH 5.5, 0.14 M NaCl), and ABS in presence of cathepsin B (10 units/mL) by dialysis method using a membrane with 3,500 Da cutoff. The concentrations of PTX and Pt(II) released were determined by HPLC and inductively coupled plasma mass spectrometry (ICP-MS), respectively, and expressed as a percentage of the total PTX or Pt(II) available vs. time.
2.8. Cell culture and cytotoxicity assay
A2780 human ovarian carcinoma cells were provided by Dr. P. Rogers (Institute of Cancer Research, University of Bristol, UK). Cells were maintained in RPMI 1640 medium with 2 mM glutamine supplemented with 10% (v/v) FBS in the presence of penicillin and streptomycin (100 U/ml and 0.1 mg/ml, respectively) at 37°C in a humidified atmosphere containing 5% CO2. Cells were harvested with trypsin-EDTA (Life Technologies) after 80% confluence. Cells seeded in 96-well plates (5,000 cells/well) 24 h prior the experiment were exposed to various doses (0–10 μg/mL on CDDP or PTX basis) of CDDP alone, polymeric micelles alone, CDDP-loaded cl-micelles, PTX-loaded cl-micelles and (CDDP+PTX)-loaded cl-micelles for 72 h at 37 °C, followed by washing with PBS, and maintaining in RPMI 1640 medium with 10% FBS for additional 24 h. Cytotoxicity of drug-loaded cl-micelles was assessed by a standard MTT assay [16] and the IC50 values were calculated using GraphPad Prism software. Combination index (CI) analysis based on Chou and Talaly method [17] was performed using CompuSyn software for CDDP and PTX combinations, determining synergistic, additive, or antagonistic cytotoxic effects against A2780 breast cancer cells. Values of CI < 1 demonstrate synergism while CI = 1 and CI > 1 values represent additive and antagonistic effects of drug combination, respectively.
2.9. Animal studies
Upon arrival, animals were placed in a facility accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Food and reverse osmosis water were available ad libitum throughout the study. Euthanasia was performed by CO2 asphyxiation. Treatments were administered by tail vein injection. Drug amount was calculated based on the average animal body weight. The University of Nebraska Medical Center Institutional Animal Care and Use Committee approved all animal protocols. A xenograft human ovarian carcinoma model was used as previously described [8, 18]. Briefly, A2780 cells (5 × 106 cells/site) were subcutaneously transplanted into the flanks, one above each hind limb, of four-week-old female athymic (Ncr-nu/nu) mice (National Cancer Institute). When the tumors reached a size of about 200–400 mm3 (12–15 days after transplantation) animals were randomized (5 treatment groups, n = 8) and treated with free CDDP or CDDP/cl-micelles or PTX/cl-micelles or (CDDP+PTX)/cl-micelles at an equivalent dose of 4 mg/kg CDDP, or 1mg/kg PTX or (4 mg/kg CDDP+1 mg/kg PTX) or 5% dextrose solution. Treatments were administered via tail vein injections at 4-day intervals. Animal body weight and tumor volume were monitored every second day. Tumor volume (V = 0.5 × L × W2) was estimated by measuring two orthogonal diameters (longer dimension: L, and smaller dimension: W) of the tumor using electronic calipers. Animals were sacrificed when tumor volume exceeded 3000 mm3, greatest tumor dimension exceeded 20 mm, tumor became necrotic, or animal exhibited a body weight loss of more than 20%. All other animals were sacrificed by day 45.
2.10. Apoptosis and proliferation
Tumors from mice that received different treatments were excised at day 14 (2–3 mice per group). The tumors were dissected and fixed in 10% neutral buffered formalin. Then, the tissues were processed routinely into paraffin, sectioned at a thickness of 4 μm. Proliferating and apoptotic cells were detected using an antibody against Ki-67 and caspase-3, respectively. Visualization was done by incubation with DAB+ (brown, for Ki-67) and Permanent red (for caspase-3) (DAKO) for 2 min. After being rinsed with distilled water, the sections were counterstained with hematoxylin. For quantification of Ki-67 and caspase-3 expression, the area of positive cells was determined (Image J) in 5 random high power fields (20X magnification) and divided by the total area of cells for each field of slice.
2.11. Sample preparation and drug content measurement in tissue
Known weights of thawed tissue, collected from the animals sacrificed at different days during treatment, were decomposed by wet-ashing in screw-capped vials with six volumes of concentrated nitric acid, overnight heating, and stirring at 65°C. An iridium internal standard was added prior to digestion. Total platinum concentrations were determined by ICP-MS using iridium correction. Calibration range for the assay was platinum 2–100 ng/mL with extrapolation to platinum 1000 ng/mL. Necessary dilutions were made when the platinum concentration exceeded the calibration range. Assay sensitivity was 0.8 ng of Pt/mL, with inter- and intraday assay variability not exceeding 5%. For PTX measurements, tissue sample (200 mg) was spiked with 50 uL diazepam (internal standard, I.S.) to achieve a final concentration about 25 ug/mL, then 2mL of tert-butyl methyl ether (TBME) was added and tissue was homogenized. Homogenized samples were centrifuged at 3000g for 10 min, 1mL of supernatant TBME layer was collected and dried under air. Residue was reconstituted using 1mL of mobile phase and 20uL of it was injected into HPLC system. Samples of PTX was prepared by directly dissolving PTX and I.S. in mobile phase and measured in HPLC system to calculate recovery rate.
2.12. Blood chemistry and histopathology
Blood from the sacrificed animals was collected in EDTA tubes and analyzed for blood cell count and liver enzymes using Vetscan VS (Abaxis). Fixed tissues were processed, sectioned, inserted into tissue cassettes, dehydrated in 70% ethanol overnight, and paraffin embedded (UNMC Tissue Sciences Facility, Omaha, NE). Serial 5 μm sections were stained with either hematoxylin and eosin (H&E) or by immunohistochemistry (IHC). For histopathological diagnosis, H&E-stained slides were examined by light microscopy and photomicrographs were taken using a Nikon camera mounted on a Nikon Eclipse 600 microscope (both Nikon Instruments, Melville, NY) with Adobe Elements 3.0 software (Adobe Systems, San Jose, CA). For IHC detection of cl-micelles in tissues, rabbit monoclonal antibody to PEG (anti-PEG methoxy group; Epitomics, Burlingame, CA) was used. Goat anti-rabbit secondary antibody conjugated with fluorescence label AF 488 was used for the detection followed by counterstaining with Mayer’s hematoxylin. Stained slides were visualized using confocal imaging (Carl Zeiss LSM 510).
2.13. Statistical analysis
Statistical comparisons except animal studies were carried out using Student t-test. For the antitumor study and toxicity studies, group means for tumor volume and body weights were evaluated using repeated measures analysis of variance with the Bonferroni post test. Survival was estimated using Kaplan–Meier analysis and compared using log-rank test. P values less than 0.05 were considered significant. Analysis of variance with Bonferroni test and Kaplan–Meier analysis with log-rank test were performed using GraphPad Prism 5 (GraphPad Software, Inc.).
3. Results and Discussion
3.1. Synthesis and characterization of PEG-b-PGlu-b-PPhe
The synthesis of the amphiphilic hybrid polypeptide-based triblock copolymers PEG-b-PGlu-b-PPhe is illustrated in Scheme 1.
The copolymers were synthesized via sequential ring-opening polymerization of BGlu-NCA and Phe-NCA monomers using amino-terminated PEG (Mw = 5,000 g mol−1) as macroinitiator in three stages. At first, PEG-b-PBGlu diblock copolymer was prepared. The length of PBGlu block was set constant by using feed molar ratio of mPEG-NH2 to BGlu-NCA at 1 : 100. The second step was polymerization of Phe-NCA initiated by PEG-b-PBGlu copolymer. The length of PPhe block was varied by adjusting the feed molar ratio of NCA monomers, BGlu-NCA : Phe-NCA (10:1 and 10:3). At the final step, the deprotection of the glutamate residues was carried out by alkali hydrolysis to obtain PEG-b-PGlu-b-PPhe. The chemical composition of two resulting triblock copolymers was determined by 1H-NMR analysis (Supporting Information, Fig. S1) and further confirmed by Fourier transform infrared spectroscopy (FTIR) (data not shown). The polymerization degrees of PGlu and PPhe blocks are summarized in Table 1.
Table 1.
Characteristics of the PEG-PGlu-PPhe triblock copolymer.
| Polymer | Feed molar ratio (mmol) | Repeating units ratio* | Mw* (g/mol) | ||||
|---|---|---|---|---|---|---|---|
| PEG | BGlu-NCA | Phe-NCA | PEG | PGlu | Phe | ||
| PEG-PGlu92-PPhe7 | 0.02 | 2 | 0.2 | 114 | 92 | 7 | 19,700 |
| PEG-PGlu90-PPhe25 | 0.02 | 2 | 0.6 | 114 | 90 | 25 | 22,400 |
Calculated from 1H-NMR spectra.
Notably, PEG-b-PGlu-b-PPhe maintained the same degree of polymerization after complete removal of the benzyl protecting groups. These copolymers had a constant PEG block (114 repeating units), practically identical length of anionic PGlu block (about 90 repeating units), and differed in the length of PPhe blocks (7 and 25 repeating units). They are further denoted as PEG-PGlu90-PPhe7 and PEG-PGlu90-PPhe25, respectively.
3.2. Synthesis and characterization of cl-micelles
We anticipated that incorporation of hydrophobic PPhe block into triblock copolymer confers amphiphilic properties and facilitate the formation of micellar aggregates in aqueous medium. Indeed, formation of small (intensity-average diameter of approximately 90 nm) particles with relatively narrow particle size distribution (PDI = 0.16) and net negative charge (ξ-potential = −22 mV) was detected in aqueous solutions of PEG-PGlu90-PPhe25 copolymer. In contrast, formation of large aggregates with a broad size distribution was observed only in relatively concentrated PEG-PGlu90-PPhe7 solutions. Differences in association behavior of PEG-PGlu-PPhe block copolymers were further confirmed by a fluorescence measurements using pyrene as a probe (Fig. S2). Using this approach, we determined that the critical micelle concentration (cmc) for PEG-PGlu90-PPhe25 was low (62.5 mg/L). In contrast the onset of aggregation of PEG-PGlu90-PPhe7 chains was detected at concentrations higher than 1000 mg/L. These data indicate that increase in the length of the hydrophobic PPhe block from 7 to 25 units is accompanied by an order of magnitude increase in the cmc value. A similar trend in the dependency of cmc on the length of core-forming block was observed earlier for other amphiphilic block copolymers [19]. In dilute solution, hydrophobic and π-π stacking interactions of the phenylalanine residues play a major role in driving self-assembly of Phe-based peptides like amyloid β peptide [20] or synthetic polypeptides [21]. A red shift of the emission peak from the normal phenylalanine fluorescence maximum (around 280 nm) has been previously observed upon assembly in amyloid system [20]. In the present case, the fluorescence spectra obtained from 0.01% solutions of PEG-PGlu-PPhe copolymers displayed a peak at 320 nm that is attributable to π-π stacking interactions of phenyl units; though the intensity of the fluorescence was lower for PEG-PGlu90-PPhe7 copolymer (Fig. S3). Notably, the fluorescence spectrum for PEG-PGlu90-PPhe7 was recorded below the cmc. It is possible that some loose pre-micelle aggregates of copolymer chains with short PPhe blocks might exist below experimentally determined cmc and the observed fluorescence characteristics may reflect the local association of aromatic units in such pre-aggregates. Interestingly, it was previously reported that peptide-PEG amphiphiles containing the hydrophobic sequence of four Phe residues conjugated to PEG of molar mass 5,000 g mol−1 (PEG-PPhe4 using our nomenclature) exhibit self-assembly behavior despite its relatively short peptide sequence with cmc value of 950 mg/L in water. It seems that insertion of ionic PGlu block into PEG-b-PGlu-b-PPhe triblock copolymer provides for additional negative contribution to micellization and increasing cmc. This may be explained, on the one hand, by stabilization of the single polymer chains in solution, and, on the other hand, by destabilization of multimolecular micelles due to the strong electrostatic repulsion of PGlu chains in the micelle corona. Overall, the optimal hydrophilic-lipophilic balance (HLB) in terms of numbers of Phe groups with respect to PEG and PGlu chains is essential for the formation of multilayer micelles with well-defined and distinct domains: hydrophobic PPhe core, the intermediate ionic PGlu layer, and hydrophilic nonionic PEG outer shell.
The PEG-PGlu90-PPhe25 micelles were further utilized as “core–shell” templates for synthesis of cl-micelles. The cross-linking was achieved via condensation reactions between the carboxylic groups of PGlu chains in intermediate layer and the amine groups of ethylenediamine in the presence of a water-soluble carbodiimide, EDC. The targeted extent of cross-linking (20%) was controlled by the molar ratio of cross-linker to carboxylic acid groups of the Glu residues. The cl-micelles displayed an effective diameter of about 90 nm (ξ-potential = −20 mV) and were uniform (monomodal, narrow size distribution) as determined by DLS. It is important to note that cl-micelles had comparable sizes with the precursor PEG-PGlu90-PPhe25 micelles (91 ± 1 nm) indicating that the micelles retained their integrity and formation of cross-links was limited to intramicellar reactions. It is likely that the outer PEG shell of the micelles prevented the undesirable intermicellar reactions. The dimensions and morphology of cl-micelles were further characterized by TEM and tapping-mode AFM in air. Based on AFM and TEM images PEG-PGlu90-PPhe25 cl-micelles had a spherical morphology. As expected the number-average particle height (Hav) and diameter (Dav) values of dehydrated cl-micelles were reduced compared to the hydrodynamic diameters Deff determined by DLS (Table 2). Upon adsorption on mica surface cl-micelles exhibited a high diameter versus height aspect ratios (Dav/Hav) suggesting substantial flattering of the particles. This observation was in agreement with the expected flexible character of the PEG-PGlu shell of the cl-micelles. The electrostatic interactions of the negatively charged cl-micelles with the positively charged amino-modified mica surface may induce additional flattering of cl-micelles.
Table 2.
Summary of characterization data for cl-micelles by TEM, AFM, and DLS.
| Sample | TEMa | AFMb | DLSc | |
|---|---|---|---|---|
| Dav, nm | Dav, nm | Hav, nm | Deff, nm | |
| cl-micelles | 45 ± 3.3 | 47 ± 6.8 | 8.7 ± 3.7 | 90 ± 1.2 |
Number-average diameter (Dav), n = 27. A drop of the sample solution was allowed to settle on a Formvar precoated copper grid and the sample was allowed to air-dry.
Number-average height (Hav) and diameter (Dav), n = 55. cl-micelles were deposited from aqueous solutions at pH 6.5 onto APS mica surface and allowed to dry in vacuum.
Intensity-average hydrodynamic diameter(Deff) for DLS was determined at pH 7.4, n= 5.
Data are expressed as mean ± SD.
Interestingly, the hybrid PEG-PGlu90-PPhe25 cl-micelles were characterized by a significantly lower aspect ratio (ca. 6) compared to PEG-poly(methacrylic acid) (PEG-PMA) cl-micelles containing entirely hydrophilic cross-linked PMA cores [22]. For example, PEG-PMA133 and PEG-PMA75 cl-micelles were characterized by dimension aspect ratios of 13.3 and 15.0, respectively. The observed topological differences can be explained by the presence of relatively rigid hydrophobic PPhe cores in the PEG-PGlu90-PPhe25 cl-micelles that rendered them more stiff and deceased their deformation at the substrate surface.
The pH-induced dimensional changes (nanogel-like behavior) of the cl-micelles were evident from an increase in the micelle size and net negative charge (Fig. 2A).
Figure 2.
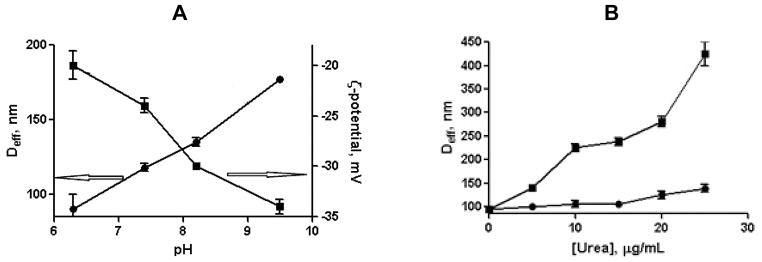
Swelling behavior (A) and stability (B) of cl-micelles. (A) The effective diameter, Deff (●) and ζ-potential (■) of cl-micelles as a function of pH. (B) Changes in Deff of cl-micelles (●) and non-cl-micelles (■) upon treatment with different concentrations of 8M urea.
Such behavior was indicative of ionization and swelling of the intermediate layer formed by the cross-linked PGlu chains in the shell of the micelles. The kinetic stability of cl-micelles was further analyzed in the presence of urea (Fig. 2B). Urea, a small hydrophilic molecule, is widely used as protein denaturant due to its ability to form stronger attractive dispersion interactions with the protein side chains and backbone than does water. Therefore, it was expected that urea is able to dissolve the PPhe core hydrophobic region. Indeed, for non-cross-linked micelles addition of 8 M urea led to significant increase in the size and PDI of the aggregates. In the meantime, only little changes in the average size of cl-micelles were detected in the presence of urea (Fig. 2B). The observed limited swelling of PEG-PGlu90-PPhe25 cl-micelles can be thus directly related to a restricted mobility of the PGlu chains in the shell and provides additional evidence that the physical stability of micelles can be greatly enhanced via covalent crosslinking. Importantly, the cross-linking did not prevent the degradation of cl-micelles by proteolytic enzymes. The enzymatic biodegradability of PEG-PGlu90-PPhe25 cl-micelles was determined by incubating the micelles with cathepsin B followed by analysis of the reaction mixture using size exclusion chromatography (SEC) (Fig. S4). A gradual decrease in the UV absorption of the micelle peak was observed in SEC chromatograms over the incubation time, which almost disappeared after 24 hrs. Furthermore, decrease in light scattering intensity along with the drastic increase of PDI was detected by DLS of the cl-micelles dispersions already after 12 hrs of incubation with cathepsin B suggesting structural disintegration of such micelles (Table S1). Overall, these results imply that the reinforcing cross-links within the shell of PEG-PGlu90-PPhe25 micelles can permanently suppress their dissociation. Resulting cl-micelles can withstand environmental challenges and sink conditions encountered after systemic administration and, consequently, provide means for temporal control over drug disposition. On the other hand, enzymatic degradation of the polypeptide-based building blocks of cl-micelles reduces the risk of polymer accumulation inside cells and can also facilitate specific intracellular drug release.
3.3. Drug loading and release from cl-micelles
PEG-PGlu90-PPhe25 cl-micelles have a well-defined structure with a central hydrophobic core formed by PPhe chains, swollen intermediate layer of a cross-linked PGlu network and external PEG shell surrounding the core and intermediate layer. Each of these three nanodomains contributes to the utility of cl-micelles as drug nanocarriers. A PPhe core can serve as a cargo for water-insoluble drugs. An ionic intermediate layer can incorporate hydrophilic drug molecules through electrostatic or covalent bonding while PEG shell in addition to stabilizing micelles in dispersion also allows minimizing interactions of the micelles with blood components. Therefore, such cl-micelles would provide unique opportunity to simultaneously incorporate and deliver multiple chemotherapeutic drugs with very different physical properties and modes of action inside a single polymeric carrier in order to achieve a synergistic effect and enhance the efficacy of treatment. In the present study cl-micelles with binary drug combination of hydrophilic CDDP and hydrophobic PTX were prepared. At first, PTX was solubilized into the PPhe cores of the cl-micelles using micelle extraction method. According to this method, PTX dissolved in ethanol was added to the aqueous dispersion of cl-micelles upon agitation followed by evaporation of organic solvent and removal of unbound PTX. Under these conditions PTX loading capacity (the net amount of drug loaded into a carrier) was about 9 w/w% weight as was quantified by HPLC. The size and ζ-potential of the PTX-loaded cl-micelles were comparable to those of empty cl-micelles indicating that the PTX incorporation into PPhe cores did not affect the macroscopic characteristics of the pre-formed cl-micelles. As the next step, CDDP was incubated with the aqueous dispersions of PTX-loaded cl-micelles for 48 h at pH 9.0 [7]. Such conditions were chosen to maximize the swelling of PGlu cross-linked layer (Fig. 2A) and increase the accessibility of the bulk of the carboxylate groups to CDDP. As expected, the net negative charge and particle size of cl-micelles decreased upon CDDP loading consistent with neutralization and condensation of PGlu segments by CDDP (Table 3).
Table 3.
Physicochemical characteristics of cl-micelles before and after drug loading.
| Sample | Deff (nm) | PDI | ζ-potential (mV) | LC (w/w%)b |
|---|---|---|---|---|
| cl-micelles | 90 ± 1.2 | 0.14 | −20.0 ± 1.2 | - |
| PTX/cl-micelles | 105 ± 2.8 | 0.23 | −19.1 ± 1.5 | 8.4 |
| CDDP/cl-micelles | 71 ± 3.3 | 0.19 | −3.7 ± 1.0 | 22.4 |
| (CDDP+PTX)/cl-micelles | 76 ± 4.0 | 0.24 | −5.7 ± 0.47 | 24.8c |
Effective diameter (Deff), polydispersity index (PDI) and ζ-potential were determined in water (pH 6.5).
Pt and PTX content were deternined by ICP-MS and HPLC, respectively. Loading capacity (LC) is expressed as mass of incorporated drug per mass of drug loaded cl-micelles (w/w).
Total LC for combination of CDDP (15.4%) and PTX (9.4%).
Data are expressed as mean ± SD (n = 3).
15 w/w% loading was achieved with respect to CDDP while total loading values for binary drug combination was about 25% and corresponded to 10 : 1 molar ratio (CDDP : PTX) of drugs in the micelles. (CDDP+PTX)/cl-micelles maintained their spherical morphology as was confirmed by TEM (Fig. S5). Notably, (CDDP+PTX)-loaded cl-micelles were stable in aqueous dispersions, exhibiting no aggregation or precipitation for a prolonged period of time (weeks). Interestingly, the loading capacity of cl-micelles with respect to CDDP was decreased after loading of PTX as compared to single CDDP/cl-micelle formulation (15% vs. 23%). This apparent difference in drug loading is not yet understood and needs to be investigated further. We hypothesize that the presence of hydrophobic solute in the core of the micelle affects the conformation of the polypeptide chains at the PPhe core - PGlu shell interface. Specifically, in more hydrophobic environment PGlu chains can transition from random coil to α-helical conformation leading to structural condensation in this region which may restrict space for the loading CDDP molecules. Validation of this hypothesis is of considerable interest and is ongoing in our laboratory.
The release of the drugs from cl-micelles was studied by equilibrium dialysis at 37 °C at either pH 7.4 (PBS) or pH 5.5 (ABS), which reflect conditions encountered in plasma and in intracellular compartments (lysosomes), respectively. The kinetic profiles of drug release from (CDDP+PTX)/cl-micelles are presented in Fig. 3. As seen from these data sustained but temporally distinct release of Pt(II) species and PTX was observed. Notably, PTX release was much faster than that of Pt(II), which is expected since PTX is physically entrapped into the PPhe core. In contrast, CDDP binds with PGlu chains through electrostatic and coordination interactions and its release usually proceeds via ligand exchange reactions with chloride or other biologically abundant anions, thus delaying its liberation from the cl-micelles. Pt(II) release from the cl-micelles was also a pH-dependent process. Indeed, Pt(II) species were liberated from the micelles faster at pH 5.5 than at pH 7.4 (Fig. 3B), probably, due to protonation of carboxylic groups of PGlu, which weakens the drug and micelle electrostatic coupling. For example, during 24 hrs (CDDP+PTX)/cl-micelles released 16.7 ± 5.5% of loaded Pt(II) at pH 5.5 and only 8.8 ± 3.6% at pH 7.4, respectively.
Figure 3.
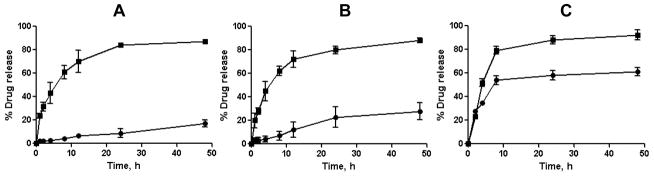
In vitro drug release profiles for Pt (●) and PTX (■) from cl-micelles in (A) PBS, pH 7.4; (B) ABS, pH 5.5; and (C) ABS in presence of cathepsin B (10 units/mL) at 37°C. Data are expressed as mean ± SD (n=3).
Notably, pH changes practically did not affect the release of PTX. Also, noteworthy that Pt (II) release from (CDDP+PTX)/cl-micelles at the acidic pH was further accelerated in presence of cathepsin B in the media (Fig. 3C). Cathepsin B is a lysosomal thiol-dependent protease [23] and is also extracellularly present in pathological tissues such as tumors and sites of inflammation [24, 25]. These results indicate that degradation of cl-micelles can further facilitate the drug release once located within targeted tissue and cells.
3.4. In vitro cytotoxicity of the drug-loaded cl-micelles
The cytotoxicity of drug combination loaded into cl-micelles was determined in human ovarian carcinoma A2780 cells using MTT assay. Calculated IC50 values are summarized in Table 4.
Table 4.
Comparison of IC50 values for various drug-loaded cl-micelles and free CDDP against A2780 cells as determined by the MTT assay.
| IC50 (nM) | |
|---|---|
| Free CDDP | 1.5 |
| CDDP-loaded cl-micelles | 3.9 |
| PTX-loaded cl-micelles | 0.17 |
| (CDDP+PTX)-loaded cl-micelles | 0.14* |
Calculated with respect to CDDP.
Importantly, cl-micelles alone were nontoxic to the cells up to the concentration of 5 mg/mL. CDDP/cl-micelles displayed lower cytotoxic activity than free CDDP. Since unloaded cl-micelles did not affect the cell survival in the entire range of concentrations the cytotoxic activity of CDDP/cl-micelles was mediated by the Pt(II) species slowly released from the micelles. However, we found that (CDDP+PTX)/cl-micelles were significantly more effective in killing A2780 ovarian cancer cells than CDDP/cl-micelles. The dose of CDDP in cl-micelles was indeed reduced nearly by 30-fold when used in combination with PTX (at 10 : 1 CDDP/PTX molar ratio) to achieve the same cytotoxic effect produced by the CDDP loaded cl-micelles. The CI value at IC50 for this (CDDP+PTX)/cl-micelles combination was 0.078, indicating strong synergistic cytotoxicity against A2780 ovarian cancer cells.
3.5. Antitumor activity
Motivated by the enhanced in vitro efficacy of (CDDP+PTX)/cl-micelle formulation, we evaluated its antitumor efficacy in vivo in mice bearing A2780 human ovarian cancer xenografts. Free CDDP and CDDP/cl-micelles were injected 4 times at 4-day intervals at an equivalent dose of 4 mg-CDDP/kg determined as the maximum tolerated dose upon this treatment schedule [18]. Animals injected with (CDDP+PTX)/cl-micelles received 4mg/kg CDDP and 1 mg/kg PTX equivalents per dose. The changes in the relative tumor volume, body weight and animal lifespan are shown in Fig. 4.
Figure 4.
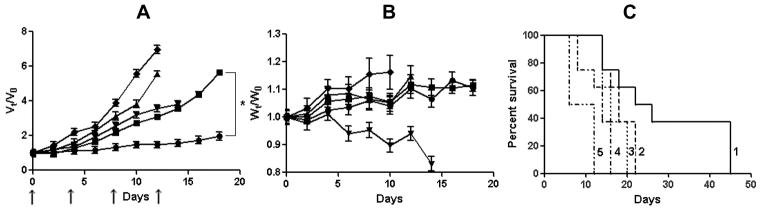
In vivo antitumor efficacy of (CDDP+PTX)/cl-micelles in A2780 human ovarian cancer xenograft-bearing female nude mice. Relative changes in (A) tumor volume and (B) body weight were measured following intravenous administration of (CDDP+PTX)/cl-micelles(●) or CDDP/cl-micelles (■) or PTX/cl-micelles (▲) or free CDDP (▼) or 5% dextrose (◆). Drug formulations were injected in100 μL at a dose of 4 mg CDDP or 1mg PTX equivalents/kg body weight 4 times at 4-day intervals as indicated by the arrows. Values indicated are means ± SEM (n = 8). (C) Kaplan–Meier analysis of overall survival in (CDDP+PTX)/cl-micelles group (1) or CDDP/cl-micelles group (2) or PTX/cl-micelles group (4) or free CDDP group (3) or control group (5). Tumor volume and body weight are normalized with respect to tumor volume or body weight at day 0. * P<0.05.
Tumor burden was significantly decreased by both free CDDP (P<0.05) and CDDP/cl-micelle (P<0.05) treatments compared to control (Fig. 4A). However, tumors in the animals treated with CDDP/cl-micelles remained relatively smaller than in animals treated with free CDDP between days 0 and 18. As a result, increased survival of the animals was observed in mice treated with CDDP/cl-micelle compared to CDDP alone (Fig. 4C). Notably, treatment with PTX-loaded cl-micelles did not have any substantial effect on the tumor growth and only minor increase in survival was observed over controls. In contrast, nearly complete inhibition of tumor growth was observed for the mice treated with (CDDP+PTX)/cl-micelles from day 0 to 14, which translated into significantly increased overall survival of the animals treated with (CDDP+PTX)/cl-micelle compared to either CDDP/cl-micelles (P<0.05) or CDDP alone (P<0.05) (Fig. 4A and 4C). On the second day post treatment animals injected with either (CDDP+PTX)/cl-micelles or CDDP/cl-micelles displayed significantly higher levels of Pt in tumors compared to free CDDP treatment group (P<0.05) which may be attributed to the enhanced permeability and retention (EPR) effect (Fig. 5A) [26].
Figure 5.
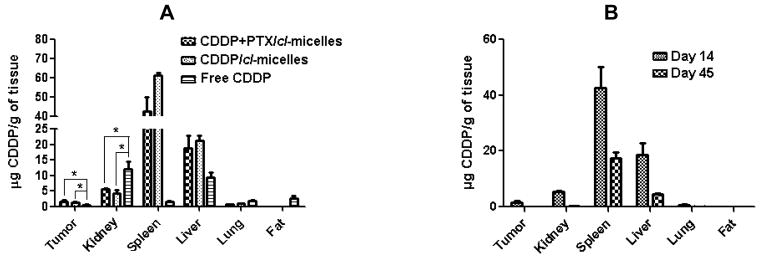
Tissue distribution of platinum in different treatment groups as determined ICP- MS. (A) Mice were sacrificed at day 14 of the treatment with (CDDP+PTX)/cl-micelles or CDDP/cl-micelles or free CDDP. (B) Significant reduction in the Pt levels in various organs of mice treated with (CDDP+PTX)/cl-micelles was confirmed by comparing Pt levels at day 14 and day 45. Values indicated are means ± SEM (n = 3), * P<0.05.
To elucidate the mechanism underlying the enhanced antitumor activity of (CDDP+PTX)/cl-micelles, the tumors were excised post-treatment (on 2nd day after last injection) and processed for Ki-67-Caspase-3 assay to examine the effect of each treatment on induction of apoptosis (Fig. 6).
Figure 6.
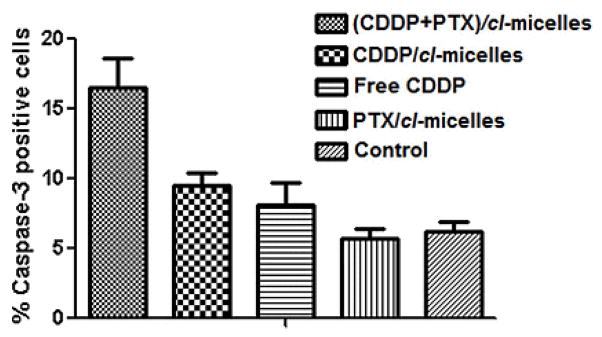
Ki-67-caspase-3 apoptosis assay. Quantification of caspase-3 positive cells in tumor tissue from mice from various groups. Data are presented as mean ± SD (n = 5 random microscopic fields for each tumor slice).
The number of caspase-3 positive cells were significantly higher in tumors from mice that received (CDDP+PTX)/cl-micelles compared to tumors in CDDP/cl-micelles (P<0.05) or PTX/cl-micelles (P<0.05) or free CDDP (P<0.05) (Fig. 6 and Fig. S6). This assay further corroborates the superior antitumor efficacy of binary drug combination cl-micelle formulation over single drug-loaded micelles or CDDP alone. Since the levels of Pt accumulation in tumors were comparable for the treatments with both single or binary drug formulations, we rationalize that an increase in therapeutic efficacy of (CDDP+PTX)/cl-micelles can be related to highly synergistic interactions between CDDP and PTX simultaneously delivered to the tumors. Indeed, the average weight ratio of the CDDP and PTX in the tumors of the animals treated with (CDDP+PTX)/cl-micelles was determined to be 4 : 1, the same as the drug loading ratio in cl-micelle, indicating the spatial-temporal synchronization of drug exposure. CDDP and PTX are highly suited for combination chemotherapy because they have distinct mechanisms of action. CDDP binds to DNA base pairs creating adducts, cross-links, and strand breaks that inhibit DNA replication. Unrepairable DNA damage often results in activation of the apoptotic pathway. Paclitaxel acts by binding to intracellular β-tubulin, which leads to microtubule stabilization, G2–M arrest and apoptosis [27]. CDDP is thought to be relatively non-cell cycle specific in terms of its cell-killing effects and tends to synergize or have additive cytotoxicity with agents that alter mitosis (PTX) [28, 29]. Apart from the mechanism basis, synergistic effect greatly depends on drug dosages, combination ratios, cell lines and intervention schedules. Indeed, synergy between CDDP and PTX was reported to be highly schedule dependent [30, 31]. Profound synergistic effects were observed when PTX was administered prior to CDDP [28, 30] while the sequence of CDDP before PTX had less antitumor activity in vitro [31], and induced more profound neutropenia in vivo [32]. One can speculate that the ability of (CDDP+PTX)/cl-micelles to supply drugs in a temporally controlled fashion by releasing PTX significantly faster than CDDP may contribute into observed improvement of antitumor efficacy of drug combination.
It should be emphasized that PTX and CDDP may also interact synergistically with respect to toxicity to the normal tissues as well. However, this was not the case in our studies. Fig. 4B shows that either single or binary drug loaded cl-micelles did not induce body weight loss while the same dose of the free CDDP produced a considerable body weight loss (P < 0.05), which indicated systemic toxicity of free CDDP. In the kidney, the primary target organ of CDDP toxicity [33, 34], single or binary drug loaded cl-micelles showed significantly lower Pt levels than the free CDDP (Fig. 5A). Reduced Pt accumulation in kidney after (CDDP+PTX)/cl-micelles or CDDP/cl-micelle treatment ameliorated CDDP-induced nephrotoxicity as confirmed by tissue histopathology analysis.
Light microscopic examination of H&E stained kidney sections from sacrificed animals indicated focal tubular basophilia with regeneration in kidneys from all animals treated with free CDDP, while no histopathological changes were observed in kidney from animals in the (CDDP+PTX)/cl-micelle-treated or CDDP/cl-micelles-treated groups compared with the control (Fig. 7). Reduced nephrotoxicity of (CDDP+PTX)/cl-micelles or CDDP/cl-micelles is likely due to its macromolecular size coupled with slow release of the drug, which leads to a lower renal tubular drug exposure. This is a significant result considering renal toxicity is the most severe, dose-limiting toxicity with CDDP treatment [35]. (CDDP+PTX)/cl-micelles showed very high Pt accumulation in spleen and liver (Fig. 5A), but this amount reduced significantly (P<0.01) with time (Fig. 5B). Despite much higher exposure of liver and spleen to CDDP in the (CDDP+PTX)/cl-micelle or CDDP/cl-micelles treatment, there was no significant change in hepatic toxicity markers of blood chemistry (Table S2), indicating no obvious toxicity to the liver. There was no evidence of liver or splenic toxicity by histopathology (data not shown). In spleen, the micelles (either free or localized inside macrophages) were detected predominantly in the red pulp with little to no staining in the lymphoid region (Fig. S7). Free CDDP treatment has previously been reported to have a remarkable effect on immunological function due to its ability to reduce lymphocytes in the thymus and the spleen with changes in splenic white pulp being the most significant [36]. Absence of (CDDP+PTX)/cl-micelles in lymphoid regions of white pulp may thus limit free-drug exposure to these follicles. Collectively, these results demonstrate that biodegradable PEG-polypeptide hybrid cl-micelles carrying CDDP and PTX drug combination exerted superior antitumor efficacy, both in terms of tumor inhibition and survival, which could be attributed to the preferential simultaneous accumulation, and increased potency.
Figure 7.

Light microscopy images (original magnification 100×) of hematoxylin and eosin-stained kidney sections. of kidney. Tissue samples were collected at day 14.
4. Conclusions
In the present investigation, micelles based on biodegradable and biocompatible amphiphilic PEG-b-PGlu-b-PPhe triblock copolymers were evaluated as potential carriers for co-delivery of chemotherapeutic drugs with various physicochemical properties and modes of action. These nanostructures were designed to have multicompartment morphology for drug loading and were cross-linked at an intermediate layer within the polymer micelles to ensure their prolong stability upon systemic administration. Hydrophilic CDDP and hydrophobic PTX were loaded in such micelles with high efficiency, exhibited differential release profiles and synergistic cytotoxic effect against ovarian cancer cells. The binary drug combination simultaneously delivered using cl-micelles exhibited superior antitumor efficacy over single drug cl-micelle analogues in vivo in synergistic manner. Altogether, this study demonstrates a fundamental possibility for simultaneous delivery of chemotherapeutics and molecular targeting agents via single well-defined and structurally tunable polymeric nanocarrier.
Figure 1.

Scheme for the synthesis of PEG-b-PGlu-b-PPhe polymer via ring-opening NCA-based polymerization
Acknowledgments
This work was supported by National Institute of Health grant CA116590 (T.K.B.). We acknowledge the assistance of the Nanomaterials Core facility of the Center for Biomedical Research Excellence (CoBRE) Nebraska Center for Nanomedicine supported by the by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health under grant number P20GM103480. We also thank NMR, Electron Microscopy and Nanoimaging Cores (University of Nebraska Medical Center). The authors are grateful to Dr. Larisa Poluektova for assistance with IHC studies and Dr. Hardeep Oberoi for help with animal studies. We acknowledge assistance of Jinjin Zhang and Zhengyuan Zhou for the help in preparation of illustration for this paper.
References
- 1.Bast RC, Hennessy B, Mills GB. The biology of ovarian cancer: new opportunities for translation. Nat Rev Cancer. 2009;9:415–428. doi: 10.1038/nrc2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim A, Ueda Y, Naka T, Enomoto T. Therapeutic strategies in epithelial ovarian cancer. J Exp Clin Cancer Res: CR. 2012;31:14. doi: 10.1186/1756-9966-31-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lippert B. Cisplatin: chemistry and biochemistry of a leading anticancer drug. Wiley-VCH; 1999. [Google Scholar]
- 4.Go RS, Adjei AA. Review of the comparative pharmacology and clinical activity of cisplatin and carboplatin. J Clin Oncol. 1999;17:409–409. doi: 10.1200/JCO.1999.17.1.409. [DOI] [PubMed] [Google Scholar]
- 5.Wheate NJ, Walker S, Craig GE, Oun R. The status of platinum anticancer drugs in the clinic and in clinical trials. Dalton Trans. 2010;39:8113–8127. doi: 10.1039/c0dt00292e. [DOI] [PubMed] [Google Scholar]
- 6.Matsumura Y, Kataoka K. Preclinical and clinical studies of anticancer agent-incorporating polymer micelles. Cancer Sci. 2009;100:572–579. doi: 10.1111/j.1349-7006.2009.01103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberoi HS, Laquer FC, Marky LA, Kabanov AV, Bronich TK. Core cross-linked block ionomer micelles as pH-responsive carriers for cis-diamminedichloroplatinum (II) J Control Release. 2011;153:64–72. doi: 10.1016/j.jconrel.2011.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nukolova NV, Oberoi HS, Cohen SM, Kabanov AV, Bronich TK. Folate-decorated nanogels for targeted therapy of ovarian cancer. Biomaterials. 2011;32:5417–5426. doi: 10.1016/j.biomaterials.2011.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han Y, He Z, Schulz A, Bronich TK, Jordan R, Luxenhofer R, Kabanov AV. Synergistic combinations of multiple chemotherapeutic agents in high capacity poly (2-oxazoline) micelles. Mol Pharm. 2012;9:2302–2313. doi: 10.1021/mp300159u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sandercock J, Parmar M, Torri V, Qian W. First-line treatment for advanced ovarian cancer: paclitaxel, platinum and the evidence. Br J Cancer. 2002;87:815–824. doi: 10.1038/sj.bjc.6600567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Batist G, Gelmon KA, Chi KN, Miller WH, Chia SK, Mayer LD, Swenson CE, Janoff AS, Louie AC. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin Cancer Res. 2009;15:692–700. doi: 10.1158/1078-0432.CCR-08-0515. [DOI] [PubMed] [Google Scholar]
- 12.Shin HC, Alani AW, Cho H, Bae Y, Kolesar JM, Kwon GS. A 3-in-1 polymeric micelle nanocontainer for poorly water-soluble drugs. Mol Pharm. 2011;8:1257–1265. doi: 10.1021/mp2000549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci. 2010;107:17939–17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ananthapadmanabhan K, Goddard E, Turro N, Kuo P. Fluorescence probes for critical micelle concentration. Langmuir. 1985;1:352–355. doi: 10.1021/la00063a015. [DOI] [PubMed] [Google Scholar]
- 15.Yang D, Van S, Jiang X, Yu L. Novel free paclitaxel-loaded poly(L-gamma-glutamylglutamine)-paclitaxel nanoparticles. Int J Nanomedicine. 2011;6:85–91. doi: 10.2147/IJN.S15839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ferrari M, Fornasiero MC, Isetta AM. MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J Immunol Methods. 1990;131:165–172. doi: 10.1016/0022-1759(90)90187-z. [DOI] [PubMed] [Google Scholar]
- 17.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 18.Oberoi HS, Nukolova NV, Laquer FC, Poluektova LY, Huang J, Alnouti Y, Yokohira M, Arnold LL, Kabanov AV, Cohen SM. Cisplatin-loaded core cross-linked micelles: comparative pharmacokinetics, antitumor activity, and toxicity in mice. Int J Nanomedicine. 2012;7:2557. doi: 10.2147/IJN.S29145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Astafieva I, Khougaz K, Eisenberg A. Micellization in block polyelectrolyte solutions. 2. fluorescence study of the critical micelle concentration as a function of soluble block length and salt concentration. Macromolecules. 1995;28:7127–7134. [Google Scholar]
- 20.Krysmann MJ, Castelletto V, Kelarakis A, Hamley IW, Hule RA, Pochan DJ. Self-assembly and hydrogelation of an amyloid peptide fragment. Biochemistry. 2008;47:4597–4605. doi: 10.1021/bi8000616. [DOI] [PubMed] [Google Scholar]
- 21.Sun J, Chen X, Deng C, Yu H, Xie Z, Jing X. Direct formation of giant vesicles from synthetic polypeptides. Langmuir. 2007;23:8308–8315. doi: 10.1021/la701038f. [DOI] [PubMed] [Google Scholar]
- 22.Nukolova NV, Yang Z, Kim JO, Kabanov AV, Bronich TK. Polyelectrolyte nanogels decorated with monoclonal antibody for targeted drug delivery. React Funct Polym. 2011;71:315–323. doi: 10.1016/j.reactfunctpolym.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Otto HH, Schirmeister T. Cysteine proteases and their inhibitors. Chem Rev. 1997;97:133–172. doi: 10.1021/cr950025u. [DOI] [PubMed] [Google Scholar]
- 24.Koblinski JE, Ahram M, Sloane BF. Unraveling the role of proteases in cancer. Clin Chim Acta. 2000;291:113–135. doi: 10.1016/s0009-8981(99)00224-7. [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto Y, Kakegawa H, Narita Y, Hachiya Y, Hayakawa T, Kos J, Turk V, Katunuma N. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem Biophys Res Commun. 2001;283:334–339. doi: 10.1006/bbrc.2001.4787. [DOI] [PubMed] [Google Scholar]
- 26.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/s0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 27.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3:502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 28.Jekunen A, Christen R, Shalinsky D, Howell S. Synergistic interaction between cisplatin and taxol in human ovarian carcinoma cells in vitro. Br J Cancer. 1994;69:299. doi: 10.1038/bjc.1994.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engblom P, Rantanen V, Kulmala J, Helenius H, Grenman S. Additive and supra-additive cytotoxicity of cisplatin-taxane combinations in ovarian carcinoma cell lines. Br J Cancer. 1999;79:286. doi: 10.1038/sj.bjc.6690046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milross CG, Peters LJ, Hunter NR, Mason KA, Milas L. Sequence-dependent antitumor activity of paclitaxel (taxol) and cisplatin in vivo. Int J Cancer. 1995;62:599–604. doi: 10.1002/ijc.2910620518. [DOI] [PubMed] [Google Scholar]
- 31.Vanhoefer U, Harstrick A, Wilke H, Schleucher N, Walles H, Schröder J, Seeber S. Schedule-dependent antagonism of paclitaxel and cisplatin in human gastric and ovarian carcinoma cell Lines in vitro. Eur J Cancer. 1995;31:92–97. doi: 10.1016/0959-8049(94)00440-g. [DOI] [PubMed] [Google Scholar]
- 32.Rowinsky EK, Gilbert M, McGuire W, Noe D, Grochow L, Forastiere A, Ettinger D, Lubejko B, Clark B, Sartorius S. Sequences of taxol and cisplatin: a phase I and pharmacologic study. J Clin Oncol. 1991;9:1692–1703. doi: 10.1200/JCO.1991.9.9.1692. [DOI] [PubMed] [Google Scholar]
- 33.Uchino H, Matsumura Y, Negishi T, Koizumi F, Hayashi T, Honda T, Nishiyama N, Kataoka K, Naito S, Kakizoe T. Cisplatin-incorporating polymeric micelles (NC-6004) can reduce nephrotoxicity and neurotoxicity of cisplatin in rats. Br J Cancer. 2005;93:678–687. doi: 10.1038/sj.bjc.6602772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mizumura Y, Matsumura Y, Hamaguchi T, Nishiyama N, Kataoka K, Kawaguchi T, Hrushesky WJM, Moriyasu F, Kakizoe T. Cisplatin-incorporated polymeric micelles eliminate nephrotoxicity, while maintaining antitumor activity. Cancer Sci. 2001;92:328–336. doi: 10.1111/j.1349-7006.2001.tb01099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol. 2003:460. doi: 10.1016/s0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 36.Milićević Ž, Slepčević V, Nikolić D, Živanović V, Milićević N. Effects of cis-Diamminedichloroplatinum II (Cisplatin) on the Splenic Tissue of Rats: A Histoquantitative Study. Exp Mol Pathol. 1994;61:77–81. doi: 10.1006/exmp.1994.1027. [DOI] [PubMed] [Google Scholar]