

Abstract
Hydrogen bonds are ubiquitous in enzyme active sites, providing binding interactions and stabilizing charge rearrangements on substrate groups over the course of a reaction. But understanding the origin and magnitude of their catalytic contributions relative to hydrogen bonds made in aqueous solution remains difficult, in part because of complexities encountered in energetic interpretation of traditional site-directed mutagenesis experiments. It has been proposed for ketosteroid isomerase and other enzymes that active site hydrogen bonding groups provide energetic stabilization via “short, strong” or “low-barrier” hydrogen bonds that are formed due to matching of their pKa or proton affinity to that of the transition state. It has also been proposed that the ketosteroid isomerase and other enzyme active sites provide electrostatic environments that result in larger energetic responses (i.e., greater “sensitivity”) to ground-state to transition-state charge rearrangement, relative to aqueous solution, thereby providing catalysis relative to the corresponding reaction in water. To test these models, we substituted tyrosine with fluorotyrosines (F-Tyr’s) in the ketosteroid isomerase (KSI) oxyanion hole to systematically vary the proton affinity of an active site hydrogen bond donor while minimizing steric or structural effects. We found that a 40-fold increase in intrinsic F-Tyr acidity caused no significant change in activity for reactions with three different substrates. F-Tyr substitution did not change the solvent or primary kinetic isotope effect for proton abstraction, consistent with no change in mechanism arising from these substitutions. The observed shallow dependence of activity on the pKa of the substituted Tyr residues suggests that the KSI oxyanion hole does not provide catalysis by forming an energetically exceptional pKa-matched hydrogen bond. In addition, the shallow dependence provides no indication of an active site electrostatic environment that greatly enhances the energetic response to charge accumulation, consistent with prior experimental results.
Introduction
Hydrogen bonds are a common feature of enzyme active sites, and their energetic contribution to catalysis has been the subject of much discussion. Most basically, when hydrogen bonds are made to functional groups that undergo charge rearrangement over the course of a reaction, their strength is expected to change in going from the ground state to the transition state. The classic example in enzyme catalysis is that of the serine protease oxyanion hole, in which hydrogen bonds are donated to the oxygen atom of a carbonyl group that accumulates charge as the reaction proceeds to the transition state and the oxyanion intermediate.1,2 These hydrogen bonds can contribute energetically to catalysis if their net strengthening in going from the ground state to the transition state exceeds that for the hydrogen bonds formed to water in the corresponding nonenzymatic reaction in aqueous solution.
The importance of active site hydrogen bonds is highlighted by the plethora of site-directed mutagenesis experiments that have demonstrated deleterious effects upon the removal of active site hydrogen bond donors and acceptors.3 However, traditional mutagenesis replaces hydrogen-bonding groups with hydrophobic side chains and/or “holes” and can further be accompanied by structural rearrangements and the introduction of water molecules within an idiosyncratic environment that differs from bulk solution and may also be altered relative to the environment of the wild type enzyme.3 Comparisons between wild type and mutant enzymes are therefore complex, and the rate effects from mutations cannot simply be interpreted as an energetic contribution to wild type catalysis from the removed group.3,4
Another level of complexity in understanding the contribution of hydrogen bonds to enzymatic catalysis arises because their free energies of formation are highly dependent on the surrounding environment.5−7 For example, while the free energy change upon hydrogen bond formation between fluoride ion and hydrogen fluoride is −0.8 kcal/mol in water, in the gas phase the formation energy is estimated to be between −32 to −39 kcal/mol.8−10 The surrounding solvent can also affect the sensitivity of hydrogen bond energetics to changes in the electrostatic properties of the hydrogen bond donor and acceptor. For example, the dependence of the free energy of hydrogen bond formation on the difference in pKa (ΔpKa) between the donor and acceptor for a series of substituted salicylates is shallow in water but is 15-fold steeper in DMSO;11,12 the ΔpKa series provides a surrogate for charge accumulation.11
An analogous increased sensitivity due to the active site could contribute to catalysis. According to this model, as charge accumulates in the course of a reaction there would be a larger increase in hydrogen bond energy in the active site relative to the increase in water.7 However, the energetics of active site hydrogen bonds cannot be inferred from model compound studies in any other solvent, because enzyme active sites, with polar and nonpolar residues that are dynamically restrained and are surrounded by an aqueous phase, are not analogous to or authentically modeled by any homogeneous solvent or the gas phase.13,14 Thus, experimental tests within active site environments are needed.
Another model for enhanced hydrogen bond contributions proposes that the enzyme active site environment promotes the formation of “short, strong” or “low-barrier” hydrogen bonds by matching the pKa of hydrogen bonding groups in the active site to those in the transition state.15−19 Indeed, hydrogen bonds with unusual physical properties, including short donor–acceptor distances, large 1H NMR chemical shifts, and low deuterium fractionation factors have been observed in several proteins.20−28 However, experimental tests of the energetic significance of such hydrogen bonds have been limited by an inability to assess the catalytic consequences of systematically varying the pKa of hydrogen bonding groups while minimizing accompanying steric or structural changes.29
Computational studies can and have been used to develop insightful models,30−32 but all calculations employ simplifying approximations, and theoretical predictions still require rigorous and independent experimental tests. As noted above, traditional site-directed mutagenesis studies also do not cleanly assay the hydrogen bond energetic properties predicted from the above proposals.
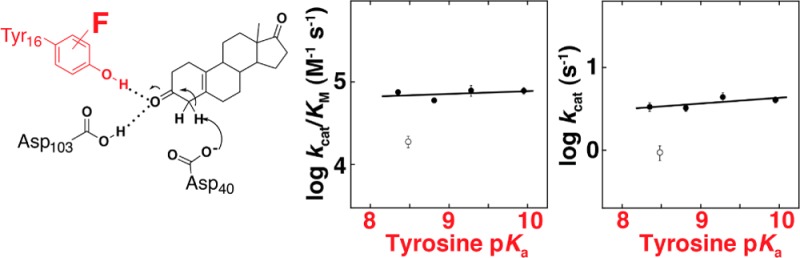
Unnatural amino acids allow maintenance of a hydrogen bond while perturbing its electrostatic properties and thereby provide, in principle, a powerful means to probe the energetics of intact hydrogen bonds within enzyme active sites. Unnatural amino acids have been incorporated and used in several protein systems to incisively probe cation–pi interactions, radical transfer reactions, general acid–base catalysis, hydrogen bonds, and other molecular properties and interactions.33−39 Here we use unnatural amino acids to probe the energetics of hydrogen bonds in the oxyanion hole of bacterial ketosteroid isomerase (KSI; Figure 1).
Figure 1.

Mechanism of KSI-catalyzed isomerization. KSI uses a general base Asp40 to isomerize double bonds adjacent to carbonyl groups through a dienolate intermediate that is stabilized by the oxyanion hole formed from the Tyr16 and Asp103 side chains. Residues are numbered according to their sequence position in KSI from Pseudomonas putida, the enzyme used in this study.
KSI, unlike serine proteases, has side chains contributing the hydrogen bonds within its oxyanion hole and thus allows systematic perturbation. Prior studies have identified short hydrogen bonds in the KSI oxyanion hole24,28,29,40−45 and have proposed that these hydrogen bonds have the energetic properties described above.16−19,24,40−44,46−49 We report the catalytic consequences of substituting fluorotyrosines (hereafter abbreviated as F-Tyr’s; Figure 2A) in place of Tyr16 to systematically vary the electrostatic properties of this oxyanion hole hydrogen bond donor. The results provide a test of whether pKa matching is important for maximal catalysis and a measure of the sensitivity of catalytic power to the electrostatic properties of an active site hydrogen bond donor.
Figure 2.

Semisynthesis of KSI with substituted tyrosines in the oxyanion hole. (A) Structures, abbreviations, and side chain pKa values of inserted tyrosines. The pKa values are for the corresponding phenols.86,87 (B) Substituted tyrosines were protected and incorporated into a 23-mer synthetic peptide with a C-terminal thioester. The remainder of the protein was expressed as a SUMO fusion, followed by removal of the N-terminal SUMO tag and ligation to the synthetic peptide to generate full-length protein. (C) Electrospray mass spectra of semisynthetic (black) and recombinant (gray) WT enzyme (i), and semisynthetic enzymes with substituted tyrosines (ii–v). The expected masses for KSI with Tyr, 2-F- or 3-F-Tyr, 2,6-F2-Tyr, and 3-Cl-Tyr are shown as brown, red, blue, and green dashed lines.
Experimental Procedures
Materials
All reagents were of the highest purity commercially available (≥97%). Ultralow total organic carbon (TOC) biological grade type I deionized water (18.2 MΩ·cm) was generated using an Aqua Solutions 2121BL system (Jasper, GA, USA) and used to prepare all buffers and aqueous solutions. Any materials not mentioned below were purchased from Sigma-Aldrich (St. Louis, MO, USA), Fisher (Pittsburgh, PA, USA), or Mallinckrodt Baker (Phillipsburg, NJ, USA).
Pyruvic acid (≥98%), p-dioxane (anhydrous, >99.8%) and Dowex 50WX8 cation exchange resin (100–200 mesh) were purchased from Acros Organics (Geel, Belgium); 3-chloro-l-tyrosine (97%) was purchased from Alfa Aesar (Ward Hill, MA, USA); di-tert-butyl dicarbonate (≥99%), trifluoroacetic acid (≥99.9%), O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (≥99%), N2-(tert-butoxycarbonyl)-N6-(((2-chlorobenzyl)oxy)carbonyl)-l-lysine (99%), O-benzyl-N-(tert-butoxycarbonyl)-l-threonine (99%) and N-(tert-butoxycarbonyl)-l-leucine hydrate (99%) were purchased from Chem-Impex International (Wood Dale, IL, USA); N2-(tert-butoxycarbonyl)-N4-trityl-l-asparagine was purchased from AAPPTec (Louisville, KY, USA); S-trityl-β-mercaptopropionic acid was purchased from Peptides International (Louisville, KY, USA); and Boc-Leu-phenylacetamidomethyl resin and 1,4-dithio-dl-threitol (DTT) were purchased from Bachem (Torrance, CA, USA). All other N-Boc protected amino acids were purchased from Novabiochem (EMD Millipore, Billerica, MA, USA).
5(10)-Estrene-3,17-dione (5(10)-EST) and 4-androstene-3,17-dione (4-AND) were purchased from Steraloids (Newport, RI, USA). 5-Androstene-3,17-dione (5-AND) was synthesized from 4-AND as described by Malhotra and Ringold and purified by silica gel flash chromatography, eluting with dichloromethane as described by Pollack et al.50,51 3-Cyclohexen-1-one was synthesized as previously described.52
KSI Semisynthesis
I. Synthesis of N-Boc, O-Silylated Chloro- and Fluorotyrosines
Fluorotyrosine Synthesis
Fluorotyrosines were synthesized on a 20 mmol scale from pyruvic acid, ammonium acetate, and the corresponding fluorophenol using tyrosine phenol lyase as previously described.53 Minor modifications described below were made to the procedure for isolating tyrosine from the reaction mixture following reaction completion.
After complete conversion of substituted phenol to tyrosine, as monitored by HPLC, enzyme was precipitated by lowering the solution pH to 3 by addition of glacial acetic acid (AcOH) and removed by vacuum filtration through a Celite pad and a 0.2 μm membrane. Dowex 50WX8 cation exchange resin (100–200 mesh, 100 mL) was washed sequentially with 6 M HCl, 6 M NaOH and water and then packed into a column and equilibrated with 15% AcOH in water (v/v). All 2 L of pale yellow filtrate was passed over this resin using a siphon under gravity, and the resin was washed with 500 mL of 15% AcOH in water (v/v) and with 500 mL of water.
Aqueous ammonia (10% v/v) was used for elution, and fractions staining positive with ninhydrin were pooled. The resin was washed with water and then with 15% AcOH in water, and the filtrate was passed over the resin two more times to maximize amino acid recovery. Ammonia was removed from the pooled fractions by rotary evaporation, and any remaining water and other volatile components were removed by lyophilization to isolate a brown powder. Typical yields of crude amino acid were 75–90%. This crude product was used directly for synthesis of the N-Boc protected amino acid without further purification or characterization.
General Procedure for N-Boc Protection of 3-Chlorotyrosine and Fluorotyrosines
A 250 mL round-bottom flask equipped with a magnetic stirbar was charged with 10 mmol of substituted tyrosine (2.0–2.2 g depending on amino acid molecular weight), 100 mL of 1:1 p-dioxane:water (v/v) and 2.1 mL of triethylamine (15 mmol, 1.5 equiv). The solution was cooled to 0 °C on ice while stirring, and 2.2 g of di-tert-butyl dicarbonate (11 mmol, 1.1 equiv) was added in one portion. After stirring on ice for 30 min, the solution was allowed to warm to room temperature and stirred overnight. Monitoring reaction progress by thin layer chromatography on silica plates (10% methanol, 0.2% AcOH (v/v) in dichloromethane) revealed the disappearance of starting material (Rf 0) and the appearance of a spot migrating at Rf 0.4.
Dioxane was removed by rotary evaporation, and the remaining aqueous mixture was cooled on ice. The solution pH was lowered to 2 (measured by pH paper) by the slow, dropwise addition of cold 1 M HCl while stirring on ice. The acidified mixture was extracted with ethyl acetate (3 × 20 mL), and the combined organic layers were washed with brine and dried over anhydrous sodium sulfate. Ethyl acetate was removed by rotary evaporation to yield a pale yellow oil.
The crude product was dissolved in a minimal volume of dichloromethane and purified by silica gel flash chromatography using a gradient of 3–5% methanol in dichloromethane (with 0.2% AcOH added to lessen product streaking). Solvent was removed by rotary evaporation and high vacuum to yield a sticky off-white foam that was characterized by ESI-MS and by 1H, 13C, and 19F NMR. Typical isolated yields were 50–80%.
General Procedure for O-Silylation of 3-Chlorotyrosine and Fluorotyrosines with tert-Butyldimethylsilyl Chloride or tert-Butyldiphenylsilyl Chloride
This synthesis is based on a previously published procedure.54 A 25 mL round-bottom flask, oven-dried, flushed with argon and equipped with a magnetic stirbar, was charged with 10 mL of dry N,N-dimethylformamide (DMF), 5 mmol of N-Boc protected modified tyrosine (1.5–1.6 g depending on amino acid molecular weight, 1 equiv) and imidazole. Syntheses using tert-butyldimethylsilyl chloride (TBDMS-Cl) used 3.4 g of imidazole (50 mmol, 10 equiv), while 1.36 g of imidazole (20 mmol, 4 equiv) was added for syntheses using tert-butyldiphenylsilyl chloride (TBDPS-Cl).
The reaction mixture was cooled to 0 °C on ice, and either TBDMS-Cl (3.1 g, 20 mmol, 4 equiv) or TBDPS-Cl (1.65 g, 6 mmol, 1.2 equiv) was added in one portion. The solution was stirred at room temperature under argon for 16 h for reactions using TBDMS-Cl, or for 36 h for reactions using TBDPS-Cl. Reaction progress was monitored by TLC on silica gel plates (10% methanol, 0.2% AcOH (v/v) in dichloromethane), which showed the gradual disappearance of starting material with Rf 0.4 and the appearance of an intense dark spot at Rf 0.5.
DMF was removed by rotary evaporation, and the resulting slurry was dissolved in diethyl ether and washed with 10% citric acid in water (w/v). The aqueous layers were extracted with diethyl ether (3 × 20 mL), and the combined organic layers were washed with saturated lithium chloride in water to remove dissolved DMF and dried over anhydrous sodium sulfate. Diethyl ether was then removed by rotary evaporation to yield a pale yellow oil.
The crude product was dissolved in a minimal amount of dichloromethane and purified by silica gel flash chromatography using a gradient of 2–4% methanol in dichloromethane (with 0.2% AcOH). Solvent was removed by rotary evaporation and high vacuum to yield a white foam that was crushed to a white powder, characterized by ESI-MS and by 1H, 13C, and 19F NMR, and stored at −20 °C until use in peptide synthesis. Typical isolated yields were 40–60%.
II. Synthesis and Purification of N-Terminal KSI Peptide with a Thioester for Native Chemical Ligation
N-Terminal Peptide Synthesis and Cleavage
Peptides containing the first 23 amino acids of pKSI with the R15K and D21N mutations and with a C-terminal β-mercaptopropionyl-Leu thioester for ligation were synthesized on 100 μmol scale using Boc-Leu-PAM resin (0.61 mmol/g) in DMF using manual in situ neutralization protocols as described elsewhere.55
Tyrosine and its unnatural variants were incorporated at position 16. Tyrosine was incorporated using N-Boc-Tyr-(2-Br-Z)-OH, and 2-fluorotyrosine was incorporated using N-Boc-O-(tert-butyldimethylsilyl)-2-fluorotyrosine, synthesized as described above. 3-Fluorotyrosine, 2,6-difluorotyrosine, and 3-chlorotyrosine were incorporated using the corresponding N-Boc-O-(tert-butyldiphenylsilyl) amino acids.
Following incorporation of unnatural tyrosine variants into the peptides, N-Boc deprotection conditions were changed from incubating the resin in neat trifluoroacetic acid (TFA) twice for 1 min, to incubating in 1:1 TFA:dichloromethane (v/v) twice for 4 min, with a dichloromethane flow wash before and after N-Boc deprotection. These conditions were used to minimize premature removal of the silyl protecting group on the tyrosine hydroxyl before peptide synthesis was complete.
Peptides were cleaved from the resin by first adding 750 μL of thioanisole and 5 mL of TFA to dried resin in a 20 mL glass vial while stirring on ice for 10 min, followed by addition of 500 μL of trifluoromethanesulfonic acid dropwise and letting the solution warm to room temperature. After 1 h, the resin was separated from the solution by filtration through a Kimwipe plug in a glass Pasteur pipet, and washed with ∼3–4 mL of TFA. The volume of TFA in the filtered solution was reduced from ∼10 to ∼3–4 mL by evaporation under a stream of nitrogen. Cold diethyl ether was added slowly while the vial was vigorously agitated on ice to precipitate crude peptide. The precipitate was triturated with cold diethyl ether (3 × 15 mL) to remove any soluble impurities, divided into four portions, dried under a stream of nitrogen, and stored at −80 °C until purification.
N-Terminal Peptide Purification
Each portion of crude peptide from above was resuspended in 5 mL of 55% formic acid in water (v/v), filtered through a Kimwipe plug in a glass Pasteur pipet, and loaded onto a semipreparative HPLC C18 column (Vydac 218TP510, 5 μm beads, 250 mm length, 10 mm internal diameter; Grace, Deerfield, IL, USA) pre-equilibrated with 95% buffer A (water, 0.1% TFA) and 5% buffer B (9:1 acetonitrile:water v/v, 0.1% TFA) at a flow rate of 1 mL/min. Gradient elution was then applied at a 5 mL/min flow rate from 95 to 65% buffer A over 20 min, and 65 to 50% buffer A over 30 min. The identity and purity of the peptide, eluting at a retention time of 46–50 min, was confirmed by ESI-MS. Peptide-containing fractions were pooled, lyophilized and stored at −80 °C until use. Isolated yields ranged from 5 to 40 mg (2–15%, theoretical yield = 272–276 mg).
III. Expression and Purification of C-Terminal KSI Polypeptide
A peptide containing amino acids 24 to 131 of pKSI and incorporating the mutations D24C/Y32F/C69S/C81S/C97S was isolated and purified as described in the following subsections. The procedure is based on one previously described with a few modifications.56 The full protocol is given below for completeness. All affinity chromatography, ion exchange and desalting columns used were from GE Healthcare Life Sciences (Piscataway, NJ, USA) unless otherwise indicated.
Generation of His6-Tagged SUMO–D24C-131/Y32F/C69S/C81S/C97S KSI Plasmid
QuikChange site-directed mutagenesis (Stratagene, La Jolla, CA, USA) was used to incorporate the Y32F/C69S/C81S/C97S mutations into the His6-tagged SUMO–D24C-131 KSI plasmid.56 Miniprep DNA from DH5α cells was sequenced on an ABI3100 capillary sequencer (Stanford Protein and Nucleic Acid sequencing facility).
Expression and Purification of His6-Tagged SUMO-(D24C-131)/Y32F/C69S/C81S/C97S KSI Fusion Protein
Using the construct generated above, a fusion protein containing an N-terminal His6 tag, a small ubiquitin-like modifier protein (SUMO) tag, and the amino acids 24 to 131 of pKSI with the mutations D24C/Y32F/C69S/C81S/C97S was expressed in E. coli BL21(DE3) cells grown in 6 L of lysogeny broth (LB). Cells were grown at 37 °C while shaking at 220 rpm to an OD600 of 0.6–0.8 and then induced with 0.5 mM isopropyl-β-d-1-thiogalactopyranoside (IPTG) and incubated at 25 °C overnight. Cells were harvested by centrifugation, resuspended in 80 mL of cold 20 mM sodium phosphate, pH 7.2, μ = 150 mM NaCl (lysis buffer), and lysed by passage through an Emulsiflex (Avestin, Ottawa, ON, Canada). All subsequent steps were performed at 4 °C unless indicated otherwise.
Inclusion bodies containing fusion protein were isolated by centrifuging cell lysate at 48400g for 20 min. Inclusion bodies were then resuspended in 50 mL of lysis buffer containing 1% Triton X-100 using a dounce to remove membrane lipids, centrifuged at 17400g for 20 min at 4 °C, and then washed several times by resuspension in lysis buffer and centrifugation to remove detergent. Inclusion bodies were solubilized in 25 mL of 50 mM Tris-HCl, 150 mM NaCl, 20 mM imidazole, pH 8.0, 7 M urea (binding buffer) using a dounce, followed by centrifugation at 48400g to remove insoluble material and clarification by passage through a 0.2 μm syringe filter.
The supernatant was loaded on a 5 mL HisTrap HP column pre-equilibrated with binding buffer. The column was washed with binding buffer until no further change was observed in A280. Elution was carried out using 50 mM Tris-HCl, 150 mM NaCl, 250 mM imidazole, pH 8.0, 7 M urea (elution buffer). The column was then re-equilibrated with binding buffer, and the flowthrough from the previous run was passed over the column 5–6 more times with fusion protein being eluted each time to maximize recovery. All steps were performed at a flow rate of 4 mL/min.
The eluted material was buffer exchanged into 50 mM Tris-HCl, 150 mM NaCl, pH 8.0, 7 M urea using 10 kDa cutoff Amicon centrifugal filter units (EMD Millipore, Billerica, MA, USA) to remove imidazole from the previous step. The eluted material was diluted while stirring into 50 mM Tris-HCl, 150 mM NaCl, pH 8.0 to a final urea concentration of 2 M to allow the SUMO protein tag to fold. The purity of the fusion protein was >95% as assessed by Coomassie-stained SDS-PAGE.
Cleavage of His6-Tagged SUMO-(D24C-131)/Y32F/C69S/C81S/C97S KSI Fusion Protein and Purification of D24C-131/Y32F/C69S/C81S/C97S KSI Polypeptide
The SUMO tag was cleaved by adding SUMO protease (1 mg protease per 100 mg fusion protein) and DTT (2 mM final concentration), and stirring overnight at 4 °C. The reaction volume was approximately 300 mL, and the concentration of fusion protein during the cleavage reaction was ∼12 mg/mL on the basis of a Bradford assay using a KSI-derived standard curve that was obtained with expressed KSI with its concentration independently determined by absorbance under denaturing conditions.57 Cleavage efficiency was >90% as assessed by Coomassie-stained SDS-PAGE.
Solid urea was added to a final concentration of 7 M to solubilize a small amount of precipitate that was observed at the end of the cleavage reaction, and imidazole and DTT were added to final concentrations of 20 mM and 2 mM, respectively. The solution was clarified by passage through a 0.2 μm membrane under a vacuum. SUMO protease, SUMO protein tag, and any uncleaved SUMO KSI fusion protein were then removed by subtractive nickel affinity chromatography as follows.
The cleavage reaction mixture was passed over a 5 mL HisTrap HP column pre-equilibrated with 50 mM Tris-HCl, 20 mM imidazole, 150 mM NaCl, 2 mM DTT, pH 8.0, 7 M urea (DTT binding buffer). The column was washed with DTT binding buffer until no further decrease in A280 was observed. Elution of bound SUMO tag, protease, and uncleaved fusion protein was achieved with 50 mM Tris-HCl, 250 mM imidazole, 150 mM NaCl, 2 mM DTT, pH 8.0, 7 M urea (DTT elution buffer), and the column was re-equilibrated with DTT binding buffer. The flowthrough was passed over the column 3 more times to maximize removal of SUMO tag, protease and uncleaved fusion. Purity of the cleaved D24C-131/Y32F/C69S/C81S/C97S peptide was typically >95% as assessed by Coomassie-stained SDS-PAGE.
The cleaved peptide solution was concentrated using 3 kDa cutoff Amicon centrifugal filter units to a volume of ∼30 mL, buffer exchanged into 50 mM ammonium bicarbonate by passage through a HiPrep 26/10 desalting column, and lyophilized to an off-white powder. Typical yields were ∼150 mg. The lyophilized peptide was stored at −80 °C until ligation.
IV. Native Chemical Ligation, Folding, and Purification to Generate Semisynthetic KSI
The synthesized peptide containing tyrosine variants at position 16 and a C-terminal thioester was ligated to the recombinant peptide containing an N-terminal cysteine by native chemical ligation. Lyophilized peptides were added to a final concentration of ∼2 mM (synthesized peptide) and ∼4–6 mM (recombinant peptide) to 500 μL of freshly prepared ligation buffer (100 mM sodium phosphate, 50 mM 4-mercaptophenylacetic acid, 20 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP·HCl), and 6 M guanidine hydrochloride, pH 7.1).
The reaction mixture was incubated at 25 °C for 4 h and then diluted 200-fold into 40 mM potassium phosphate, 1 mM Na·EDTA, 500 mM l-arginine·HCl, pH 7.2 and stirred at 25 °C for 30 min to fold ligated material. Folded protein was purified by deoxycholate affinity chromatography as previously described for other KSI mutants.3 Eluted protein was passed through a 30 mL Superose 12 size exclusion column pre-equilibrated with 40 mM potassium phosphate, 1 mM EDTA, 2 mM DTT, for buffer exchange and then concentrated in a 3 kDa cutoff Amicon centrifugal filter unit. Final protein purity was typically >99% as assessed by Coomassie-stained SDS-PAGE, and ESI-MS was used to confirm the masses of all semisynthetic proteins. Typical purified yields relative to the limiting synthesized peptide were 2–5%.
The concentrations of expressed and semisynthetic enzymes containing Tyr at position 16 were determined using a calculated molar extinction coefficient at 280 nm (15 700 M–1 cm–1).57 The concentrations of semisynthetic enzymes containing unnatural tyrosines were determined by modifying this calculated extinction coefficient with the extinction coefficient of the unnatural tyrosine (data not shown), which was assumed to be the same as that for the corresponding phenol. Calculating protein concentrations without this correction (i.e., assuming that the extinction coefficients of tyrosine and unnatural tyrosines are the same) gives protein concentration values that are within 8% of the corrected values and has no effect on the conclusions of this work.
Kinetics
All reactions were conducted at 25 °C in buffer containing 2% DMSO (v/v) added as a cosolvent for substrate solubility and monitored continuously at 248 nm (5(10)-EST and 5-AND) or at 240 nm (3-cyclohexen-1-one) in a Lambda 25 spectrophotometer (PerkinElmer, Waltham, MA, USA). Initial rates of product formation with 5(10)-EST, 5-AND and 3-cyclohexen-1-one were calculated using previously determined molar extinction coefficients for the products 4-estrene-3,17-dione (4-EST), 4-androstene-3,17-dione (4-AND), and 2-cyclohexen-1-one of 14 800, 14 750, and 7680 M–1 cm–1, respectively.52,58 Substrate concentrations used in the experiments below are summarized in Table S1 (Supporting Information).
Michaelis–Menten Kinetics
Reactions with 5(10)-EST and 5-AND were conducted in 40 mM potassium phosphate and 1 mM sodium·EDTA at pH 7.2 as previously described.52 The values of kcat, KM, and kcat/KM were determined by fitting the initial rates of product formation as a function of substrate concentration to the Michaelis–Menten equation. At least three determinations from independent experiments using different enzyme concentrations varied over 6- to 12-fold were averaged.
Determining kcat/KM values for 3-Cyclohexen-1-one
Reactions with 3-cyclohexen-1-one were conducted in 4 mM potassium phosphate, 0.1 mM sodium·EDTA, pH 7.2 buffer as previously described.52 The substrate concentrations used were chosen on the basis of prior determinations of KM between 100 and 500 mM for several pKSI mutants.52 A lower buffer concentration was used with this substrate than with 5(10)-EST and 5-AND to decrease the background rate of product formation. Values of kcat/KM were determined from initial rates. The maintenance of a subsaturating level of 3-cyclohexen-1-one was tested by ensuring that initial rates increased by 5-fold upon a 5-fold increase in substrate concentration.
pH-Dependent Activity Measurements
Reactions were conducted with 5(10)-EST in 50 mM buffer with a constant ionic strength maintained at 100 mM using NaCl. Buffers with overlapping pH ranges were used:59 sodium acetate (pH 4.1–5.4), sodium succinate (pH 4.8–6.4), sodium phosphate (pH 6.2–7.6), sodium N-(tri(hydroxymethyl) methyl)glycine (tricine) (pH 7.4–9.1), sodium glycine (pH 8.9–10.4), and sodium carbonate (pH 9.7–10.8). Activity measurements were made using initial rates of product formation. Enzyme concentrations were varied over 3- to 4-fold at each pH to ensure that reactions were first order in enzyme concentration. Subsaturating and saturating concentrations of 5(10)-EST were initially chosen on the basis of prior measurements of KM between 20 and 50 μM for several pKSI mutants,52,60 and similar KM values were determined in this work from full Michaelis–Menten curves at pH 7.2.
The observed activity as a function of pH was fit to a model containing an inactivating deprotonation and protonation (Figure S3A and S3B, Supporting Information) in Kaleidagraph 4.04 (Synergy Software, Reading, PA, USA) using eqs 1 and 2 for subsaturating and saturating activity, respectively.
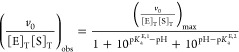 |
1 |
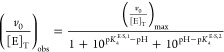 |
2 |
where v0 is the observed initial rate of product formation, [E]T and [S]T are the total initial concentrations of enzyme and substrate, pKaE,1 and pKaE·S,1 are the pKa values for inactivation at low pH under subsaturating and saturating conditions, and pKaE,2 and pKaE·S,2 are the pKa values for inactivation at high pH under subsaturating and saturating conditions, respectively.
Solvent Isotope Effect Determinations
Reactions were conducted in 40 mM potassium phosphate, 1 mM sodium·EDTA, pH 7.2 (H2O) or pD 7.6 (D2O). The pD of deuterated buffer was calculated by adding 0.4 units to the reading displayed by a pH meter connected to a glass electrode.61 A deuterated buffer with a pD 0.4 units higher than the pH of protiated buffer was used to ensure that the same fraction of titratable groups would be ionized in both H2O and D2O reactions, as the pKa values of most ionizable groups are elevated by 0.4 units in D2O relative to H2O.61 The reactions in each independent experiment comparing activity in H2O and D2O for a particular KSI variant were initiated by adding the same volume of the same concentrated enzyme stock (prepared in 40 mM potassium phosphate, 1 mM sodium·EDTA, pH 7.2 (H2O)), to H2O or D2O buffer containing 5(10)-EST. The final mole fraction of deuterium in the water used in all reactions conducted in D2O was greater than 99 atom % D.
Solvent isotope effects were determined by dividing the initial rate of product formation in H2O by the rate observed in D2O (initial rates were used as deuterium is incorporated into substrate over time; see Supporting Information Text II). The ratios observed in 6–12 (subsaturating) or 2–3 (saturating) independent experiments were averaged to obtain a mean and standard deviation for each KSI variant. Enzyme concentrations were varied over a 3-fold range with 2.3, 4.7, and 300 μM substrate.
Primary Kinetic Isotope Effect Measurements
Reactions were conducted in 40 mM potassium phosphate, 1 mM sodium·EDTA, pH 7.2 (H2O) or pD 7.6 (D2O). The pD of deuterated buffer was determined as described in the section above. Values of kcat/KM were determined by fitting the change in absorbance with subsaturating substrate monitored nearly to completion to a single exponential and plotting the observed rate constants as a function of enzyme concentration varied over a 3-fold range. Kinetic isotope effects for the semisynthetic enzymes under subsaturating conditions were determined by dividing kcat/KM for protiated 5(10)-EST in H2O by kcat/KM for 4,4-dideuterated 5(10)-EST in D2O. Saturating kinetic isotope effects were determined by dividing v0/[E] for protiated 5(10)-EST in H2O by v0/[E] for 4,4-dideuterated 5(10)-EST in D2O, where v0 is the initial rate of product formation observed with 300 μM substrate.
4,4-Dideuterated 5(10)-EST was generated by incubating 150, 300, or 7500 μM protiated 5(10)-EST with 1 mM sodium deuteroxide in D2O for 18 min, with either 2% DMSO (150 or 300 μM substrate) or 50% DMSO (7500 μM substrate). Aliquots of these reactions were diluted 25-fold into deuterated buffer containing semisynthetic enzyme to start the enzymatic reactions. The activity of semisynthetic enzymes with protiated 5(10)-EST in H2O was determined using substrate that had been incubated in H2O instead of 1 mM NaOD/D2O and diluting into protiated rather than deuterated buffer.
KSI Equilenin Affinity Determinations and Active Site Titrations
All fluorescence measurements were made at 20 °C on a FluoroLog-3 spectrofluorometer (Horiba Jobin Yvon, Edison, NJ, USA) using 45 μL microcuvettes (Starna Cells, Atascadero, CA, USA).
The affinity of D40N-containing KSI variants for EqA488–1, a fluorescent analogue of equilenin, was determined by fitting the observed fluorescence of EqA488–1 measured over a range of enzyme concentrations to a quadratic binding isotherm as described previously (Table S6, Supporting Information).29 All experiments used 0.1 nM EqA488–1, and fluorescence measurements were made with no enzyme and at enzyme concentrations varying in 2-fold increments from 0.8 to 200 nM. All measurements were conducted in pH 7.2 buffer containing 10 mM potassium phosphate, 0.1 mM sodium·EDTA and ionic strength maintained at 100 mM using KCl, with excitation at 480 nm, emission at 515 nm, and excitation and emission bandpass values of 10 and 14.7 nm, respectively.
Active site titrations were conducted in pH 7.2 buffer containing 10 mM potassium phosphate, 0.1 mM sodium·EDTA, 5% DMSO (v/v) and ionic strength maintained at 100 mM using KCl, with excitation at 335 nm, emission at 365 nm, and excitation and emission bandpass values of 2 and 10 nm, respectively. Equilenin fluorescence was monitored over a range of enzyme concentrations varied up to twice the concentration of equilenin (900 nM), and the observed data were fit to the following quadratic binding isotherm using Kaleidagraph 4.04 (Synergy Software, PA):
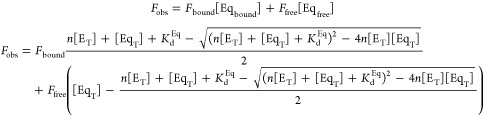 |
3 |
where n is the fraction of enzyme that is active or can bind equilenin, KdEq is the affinity for equilenin determined using EqA488–1 in an independent experiment and held constant, [ET] and [EqT] are the total concentrations of enzyme and equilenin, respectively, and Fbound and Ffree are the fluorescence values of the bound and free states.
Results
KSI catalyzes the migration of double bonds adjacent to carbonyl groups on cyclic substrates (Figure 1). In an initial enolization step, the general base Asp40 abstracts an α-proton from bound substrate, generating a dienolate intermediate stabilized by hydrogen bonding interactions with the side chains of Tyr16 and Asp103 in an oxyanion hole. In a subsequent ketonization step, the intermediate is reprotonated at a different position to generate conjugated product.62
We investigated the catalytic consequences of systematically varying the electrostatic properties of an oxyanion hole hydrogen bond donor by substituting F-Tyr’s (Figure 2A) in place of Tyr16 in the KSI oxyanion hole. F-Tyr substituted KSI mutants were prepared using a recently published semisynthesis procedure with modifications described in Experimental Procedures (Figure 2B).56 F-Tyr’s were protected and incorporated into a synthesized peptide fragment containing the first 23 amino acids of KSI and a C-terminal thioester. The rest of the protein was expressed as a SUMO fusion, followed by cleavage and removal of the SUMO tag. Full-length KSI was produced by native chemical ligation of the two peptides and folding the ligated material.
Preparation and Initial Characterization of KSI Variants with Unnatural Tyrosines in the Oxyanion Hole
Mutants were prepared in the Y32F background, as Y32F eliminates potentially complicating active site ionizations that could obscure comparisons of different F-Tyr mutants;56 this mutant has activity within 2-fold of wild type pKSI (Table S2, Supporting Information). Mutations R15K and D21N were also incorporated, as they facilitate peptide synthesis using Boc chemistry with triflic acid cleavage, and a D24C mutation was incorporated to facilitate ligation. The surface cysteine residues C69, C81, and C97 were mutated to serine, as we observed by ESI-MS that the presence of cysteine residues at those positions led to the formation of 4-mercaptophenylacetic acid (MPAA) adducts that could not be disrupted by denaturation or treatment with DTT (data not shown). Bacterially expressed enzyme containing all of these mutations had activity that matched that of Y32F KSI within 20%, indicating the absence of substantial functional effects from these substitutions (Table S2, Supporting Information). For simplicity, this enzyme is referred to as WT hereafter.
Semisynthetic WT, 2-F-Tyr, 3-F-Tyr, 2,6-F2-Tyr, and 3-Cl-Tyr substituted enzymes were folded, purified, and found by ESI-MS to have the expected masses (Figure 2C). The activity of semisynthetic WT was 60–80% of that of recombinant WT enzyme (Figure S1, Supporting Information). The activities of semisynthetic WT and 2-F-Tyr-substituted enzymes were the same within 25% across independent preparations (Figures S1 and S2, Supporting Information), suggesting that the activity of semisynthetic KSI is reproducible between preparations and that any small systematic differential in activity between expressed and semisynthetic KSI was also reproducible. Experiments with the tight-binding inhibitor equilenin, using semisynthetic KSI also containing a D40N mutation to allow very strong binding and thus active site titration, gave stoichiometries near to 1:1 for binding to enzymes with Tyr, F-Tyr’s, or 3-Cl-Tyr at position 16 (see Supporting Information Text I, Table S7). These results are consistent with all or nearly all of each semisynthetic protein being in a fully active conformation and provide no indication of differential enzyme misfolding with different unnatural amino acid substitutions.
Semisynthetic KSI pH Dependencies
To compare the activities of semisynthetic KSIs under reaction conditions where all variants had the same ionization state, we first determined pH dependencies of activity under subsaturating and saturating conditions. We used 5(10)-estrene-3,17-dione [5(10)-EST] as prior work showed that a chemical step is rate-limiting for this substrate.63 In all cases, activity was constant between pH 5 and 9, consistent with all variants having the same ionization state across this plateau region (Figure S3, Supporting Information). The measured activities with overlapping buffers fall on the same best-fit line, consistent with the prior observed absence of buffer-specific effects (Figure S3, Supporting Information).59 Further experiments were carried out at pH 7.2, in the center of this region. In all variants, an inactivating protonation, presumably corresponding to protonation of the general base Asp40,64 was observed at low pH and with the same pKa of 4.3 ± 0.1 (Table 1). This observation is consistent with an absence of structural or environmental changes around the general base upon Tyr substitution.
Table 1. pKa Values for Inactivation at Low pH for Semisynthetic KSI Tyr16 Variants from pH Dependencies of Activity with Subsaturating (pKaE,1) and Saturating (pKaE·S,1) 5(10)-ESTa.
| residue at position 16 | side chain solution pKab | pKaE,1 (subsaturating) | pKaE·S,1 (saturating) |
|---|---|---|---|
| Tyr (WT) | 9.95 | 4.31 ± 0.05 | 4.28 ± 0.11 |
| 2-F-Tyr | 9.28 | 4.35 ± 0.10 | 4.24 ± 0.07 |
| 3-F-Tyr | 8.81 | 4.30 ± 0.07 | 4.52 ± 0.09 |
| 2,6-F2-Tyr | 8.35 | 4.29 ± 0.05 | 4.18 ± 0.06 |
| 3-Cl-Tyr | 8.48 | 4.17 ± 0.12 | 4.35 ± 0.06 |
Values are from fits to data in Figure S3 (Supporting Information) and are shown together with standard errors from regression analysis.
In WT enzyme, an inactivating deprotonation is observed at high pH with a pKa of 10.00 ± 0.04 and 10.06 ± 0.12 under subsaturating and saturating conditions, respectively (Figure S3, Supporting Information, Table 2). The numerical agreement between this value and the Tyr side chain solution pKa 9.95 raised the possibility that this deprotonation corresponds to ionization of Tyr16. However, similar inactivating pKa values near 10 were observed for the F-Tyr and 3-Cl-Tyr substituted KSI variants, despite side chain pKa values in solution that are considerably lower than 10 (Figure 2A, Figure S3 and S4, Supporting Information, Table 2). Further, a change in rate-limiting step is unlikely, given that the same inactivating deprotonation pKa was observed with a different substrate 5-androstene-3,17-dione (5-AND) (data not shown). Thus, the observed constant pKa of 10 likely corresponds to ionization of a residue other than Tyr16.
Table 2. pKa Values for Inactivation at High pH for Semisynthetic KSI Tyr16 Variants from pH Dependencies of Activity with Subsaturating (pKaE,2) and Saturating (pKaE·S,2) 5(10)-ESTa.
| residue at position 16 | side chain solution pKab | pKaE,2 (subsaturating) | pKaE·S,2 (saturating) |
|---|---|---|---|
| Tyr (WT) | 9.95 | 10.00 ± 0.04 | 10.06 ± 0.12 |
| 2-F-Tyr | 9.28 | 9.58 ± 0.08 | 10.38 ± 0.07 |
| 3-F-Tyr | 8.81 | 9.84 ± 0.07 | 10.09 ± 0.10 |
| 2,6-F2-Tyr | 8.35 | 9.58 ± 0.04 | 10.28 ± 0.06 |
| 3-Cl-Tyr | 8.48 | 9.71 ± 0.12 | 9.60 ± 0.08 |
Values are from fits to data in Figure S3 (Supporting Information) and are shown together with standard errors from regression analysis.
Our observations also provide evidence for elevated pKa values for substituted tyrosines in this position. The solution pKa values of 9.28, 8.81, and 8.35 for the 2-F-, 3-F-, and 2,6-F2-Tyr side chains are lower than the observed inactivating pKa values of 9.6–10.4 (Table 2). For 2,6-F2-Tyr, the most acidic tyrosine examined in this work, the enzymatic pKa is perturbed at least 1.23 units relative to its solution pKa value (Table 2). The unmodified Tyr16 presumably experiences a similar perturbation (i.e., to ∼11.2 or higher) that is masked by a different group that titrates with a pKa of around 10 and inactivates the enzyme.
Dependence of KSI Activity on Tyrosine pKa
To carefully compare the activities of the F-Tyr-substituted KSI variants, we determined kcat/KM, kcat and KM at pH 7.2 for 5(10)-EST, a substrate for which prior work has shown that a chemical step is rate-limiting,63 using nine different substrate concentrations with 3–4 different enzyme concentrations (Figure S5, Table S3, Supporting Information). The values of kcat/KM and kcat showed no significant dependence on tyrosine acidity, with kcat/KM decreasing by 1.1-fold and kcat by 1.2-fold over a 40-fold increase in F-Tyr acidity (Figure 3A,B, closed circles). No change in KM was observed (Figure 3C).
Figure 3.

Dependence of (A) kcat/KM, (B) kcat, and (C) KM for isomerization of 5(10)-EST on the side chain pKa of substituted tyrosine. Tyr and F-Tyr containing variants are shown as closed circles; 3-Cl-Tyr is shown as open circles. Error bars correspond to standard deviations from 3 to 4 independent experiments using different enzyme concentrations. The slopes in parts A, B, and C are 0.03 ± 0.05, 0.07 ± 0.04, and 0.04 ± 0.04, respectively.
To control for possible complexities with rate-limiting steps or other idiosyncrasies that might arise with a particular substrate, we determined the activity of the F-Tyr substituted KSI variants with two other substrates, 5-androstene-3,17-dione (5-AND) (Figure S7, Supporting Information, closed circles) and 3-cyclohexen-1-one, a single-ring substrate (Figure S8, Supporting Information, closed circles). We found similar shallow dependencies of activity on F-Tyr pKa. These results suggest that the previously observed shallow dependence of the 5-AND reaction on F-Tyr pKa with metabolic F-Tyr incorporation39 did not arise either from masking of the chemical step by (partially) rate-limiting binding and release of 5-AND and its product or by modest levels of Tyr contamination in KSI preparations expressed in the presence of F-Tyr’s.
Isotope Effects on Semisynthetic KSI Reactions
To test whether F-Tyr substitutions result in a change in the mechanism of the KSI-catalyzed reaction, we determined whether F-Tyr substitutions change the solvent kinetic isotope effect or the primary or secondary kinetic isotope effects on C–H abstraction. We observed the same initial rates of product formation in H2O and D2O with 5(10)-EST (Table 3), indicating no significant change in the solvent kinetic isotope effect. We also determined the activity of Tyr and F-Tyr containing semisynthetic enzymes with protiated 5(10)-EST in H2O and 4,4-dideuterated 5(10)-EST in D2O (Figures S13 and S14, Supporting Information; Tables 4 and 5; see Supporting Information Text II for discussion on the generation of 4,4-dideuterated 5(10)-EST). Again, there was no significant change in the ratio of reaction rates of protiated 5(10)-EST in H2O versus 4,4-dideuterated 5(10)-EST in D2O for the different enzymes (Tables 4 and 5), suggesting that the primary and secondary kinetic isotope effects for C–H proton transfer are also the same for each of these enzymes. These results suggest that the KSI reaction is unaffected in both rate and mechanism by F-Tyr substitution. These results also provide no indication of coupling of proton transfer, which involves Asp40 as a general base, to the electrostatic properties of the oxyanion hole hydrogen bond donor.65
Table 3. Solvent Isotope Effects on the Initial Rates of Product Formation from 5(10)-EST with Semisynthetic KSIs under Subsaturating and Saturating Conditionsa.
| residue at position 16 | vH2O/vD2O (subsaturating) | vH2O/vD2O (saturating) |
|---|---|---|
| Tyr (WT) | 0.99 ± 0.12 | 0.94 ± 0.05 |
| 2-F-Tyr | 1.04 ± 0.13 | 1.00 ± 0.03 |
| 3-F-Tyr | 1.03 ± 0.09 | 1.10 ± 0.05 |
| 2,6-F2-Tyr | 1.09 ± 0.12 | 1.03 ± 0.01 |
Conditions: 40 mM potassium phosphate, 1 mM EDTA, 2% DMSO (v/v), pH 7.2 (H2O) or pD 7.6 (D2O). Standard deviations are from 6 to 12 (subsaturating) or 2 to 3 (saturating) independent experiments. Subsaturating data were collected with 2.3, 4.7, 9.4, 18.8, or 37.5 μM substrate, and saturating data were collected with 300 μM substrate. Enzyme concentrations were varied over a 3-fold range with 2.3, 4.7, and 300 μM substrate.
Table 4. Comparison of kcat/KM Values for Isomerization of Protiated 5(10)-EST in H2O and 4,4-Dideuterated 5(10)-EST in D2O by Semisynthetic KSIsa.
| residue at position 16 | kcat/KM for protiated 5(10)-EST in H2O (M–1 s–1) | kcat/KM for 4,4-dideuterated 5(10)-EST in D2O (M–1 s–1) | ratio of kcat/KM values for protiated and 4,4-dideuterated 5(10)-EST |
|---|---|---|---|
| Tyr (WT) | (8.2 ± 1.6) × 104 | (1.2 ± 0.3) × 104 | 6.8 ± 2.1 |
| 2-F-Tyr | (8.3 ± 0.3) × 104 | (1.3 ± 0.2) × 104 | 6.3 ± 1.0 |
| 3-F-Tyr | (6.4 ± 1.2) × 104 | (7.4 ± 1.2) × 103 | 8.7 ± 2.2 |
| 2,6-F2-Tyr | (6.5 ± 0.8) × 104 | (9.5 ± 1.6) × 103 | 6.8 ± 1.4 |
Conditions: 40 mM potassium phosphate, 1 mM EDTA, 2% DMSO (v/v), pH 7.2 (H2O) or pD 7.6 (D2O). Values of kcat/KM are from best fits to data in Figure S13 (Supporting Information) and are shown together with standard deviations from 3 to 4 independent experiments using enzyme concentrations varied over a 3-fold range, using either 6 or 12 μM substrate. Uncertainties in the ratios of kcat/KM values were obtained by error propagation.
Table 5. Comparison of kcat Values for Isomerization of Protiated 5(10)-EST in H2O and 4,4-Dideuterated 5(10)-EST in D2O by Semisynthetic KSIsa.
| residue at position 16 | kcat for protiated 5(10)-EST (in H2O) (s–1) | kcat for 4,4-dideuterated 5(10)-EST (in D2O) (s–1) | ratio of kcat values |
|---|---|---|---|
| Tyr (WT) | 3.2 ± 0.5 | (3.9 ± 0.7) × 10–1 | 8.1 ± 2.0 |
| 2-F-Tyr | 3.1 ± 0.4 | (4.0 ± 0.4) × 10–1 | 7.7 ± 1.2 |
| 3-F-Tyr | 2.9 ± 0.6 | (3.5 ± 0.5) × 10–1 | 8.2 ± 2.0 |
| 2,6-F2-Tyr | 3.4 ± 0.3 | (3.9 ± 0.1) × 10–1 | 8.9 ± 0.8 |
Conditions: 40 mM potassium phosphate, 1 mM EDTA, 2% DMSO (v/v), pH 7.2 (H2O) or pD 7.6 (D2O). Values of kcat are from best fits to data in Figure S14 (Supporting Information) collected at 300 μM substrate, a concentration shown to be saturating in independent experiments (Figure S5, Table S3, Supporting Information). Values are shown together with standard deviations from 2 to 3 independent experiments using enzyme concentrations varied over a 4-fold range. Uncertainties in the ratios of kcat values were obtained by error propagation.
Assessing Potential Steric Effects
To explore the potential impact of steric differences between substituted tyrosines, we measured the activity of KSI with 3-Cl-Tyr. This side chain has a solution pKa of 8.48, within the pKa range of the F-Tyr’s examined in this work, but chlorine is larger than fluorine (covalent radii: Cl = 100 pm, F = 60 pm) and the C–Cl bond is longer (C–Cl = 174 pm, C–F = 136 pm).66 We found that kcat and kcat/KM for 5(10)-EST were 4-fold below the nearly flat line defined by Tyr and the F-Tyr’s (Figure 3, open circles). Similar 4- to 7-fold effects were observed with the two other substrates 5-AND and 3-cyclohexen-1-one (Figure S7 and Figure S8, Supporting Information, open circles). The modest but reproducible rate effects for 3-Cl-Tyr and not for F-Tyr’s suggest that substituents on the tyrosyl ring can affect the rate of reaction in ways other than perturbing the electrostatic properties of the hydrogen bond donor.67
Discussion
This study provides tests of proposed models for large catalytic contributions from active site hydrogen bonds. We first address the model invoking matched pKa values and then discuss the model in which the enzyme provides an environment that substantially enhances the energetic response to charge accumulation.
Several researchers have proposed that the pKa values or proton affinities of hydrogen bonding groups in enzyme active sites are matched to those of the transition state, promoting the formation of “short, strong” or “low-barrier” hydrogen bonds that may provide substantial selective transition state stabilization.15−19 KSI is among the enzymes for which this proposal has been made,16−19,24,40−44,47 on the basis of 1H NMR chemical shifts indicative of short oxyanion hole hydrogen bonds to oxyanionic transition state analogues and large energetic effects of oxyanion hole mutants.24,41−43,47,68,69
Recently, Childs and Boxer showed that substituted naphthols bind to KSI in both protonated (neutral) and deprotonated (anionic) states and observed that the fraction of bound anionic ligand as a function of napthol ligand solution pKa fit a titration curve with an equivalence point of 10, similar to the estimated pKa of the reaction intermediate (Figure 1).70 This observation was regarded as support for catalysis via short, strong hydrogen bonds that form in the transition state as a result of matching of proton affinities between the transition state’s incipient oxyanion and an oxyanion hole hydrogen bond donor.70
Our observation that perturbation of the Tyr proton affinity by nearly 2 orders of magnitude (nearly two pKa units, Figure 2A) has <20% effect on the reaction rate, leaving the more than billion-fold catalysis by KSI virtually intact,52 suggests that there is no substantial stabilization in this system from the formation of pKa-matched hydrogen bonds. This conclusion is bolstered by the expectation that steric effects from the substitutions, if present, would likely be deleterious, giving a rate decrease in addition to any from breaking pKa matching. The ability here to perturb the intrinsic Tyr proton affinity via subtle fluorine substitutions allows stronger conclusions to be made about the absence of an energetic contribution from pKa matching, relative to traditional site-directed mutagenesis in KSI and other systems.27,71−73
The oxyanion hole Tyr side chain substitutions also test, more generally, the sensitivity of hydrogen bond energetics to charge accumulation within the active site environment. Studies with model hydrogen-bonded complexes have revealed that, in nonaqueous solvents, the free energy of hydrogen bond formation is much more sensitive to the electrostatic properties of the donor and acceptor than in water.7,11 This observation could be highly relevant to enzymatic catalysis, as the electrostatic properties of substrates also change as they morph into transition states and the active site is a “solvent” environment distinct from aqueous solution.74
Given the electrostatic changes along the KSI reaction coordinate, and as generally occurs in reactions, if there were substantially greater sensitivity of hydrogen bond energetics to charge accumulation within the active site than in aqueous solution, substantial catalysis could accrue via this mechanism. But determining whether or not there is an enhanced energetic sensitivity in active site hydrogen bonds, relative to water, i.e., determining whether or not this mechanism holds, requires tests with the enzyme itself, because the enzyme interior cannot be confidently modeled by the structural or energetic behavior of any other solvent,13,14 and because its energetic properties cannot be considered accurately quantitated by current computational models in the absence of nontrivial predictions that can be tested by experiment.
The KSI F-Tyr substitutions provide a rare window into this sensitivity, as the strength of a hydrogen bond donor in the active site is varied systematically; i.e., among acids of similar structure, lower pKa acids are stronger hydrogen bond donors.75 Our observations of nearly flat dependences of activity on Tyr pKa (Figure 3, Figures S7 and S8, Supporting Information) suggest that the sensitivity of hydrogen bond energetics to charge within the KSI oxyanion hole is not as steep as it is in some organic solvents and is not substantially steeper than in aqueous solution.76 These results are consistent with the conclusions of a previous study where little to no change was observed in the binding affinity of KSI across a series of fluorophenolate ligands (oxyanion intermediate analogues) varying in pKa and thereby mimicking charge accumulation over the course of the reaction.29
The interpretation above assumes that the −F substitutions affect the hydrogen bond via inductive effects on the tyrosyl hydroxyl group. But, given the larger size of fluorine relative to hydrogen, fluorine substitutions on the tyrosyl ring could have introduced complicating steric effects or structural rearrangements. Nevertheless, the data provide no indication of such steric effects. All of the F-Tyr’s fall on a single line, along with the parent Tyr; for the high sensitivity model to hold, coincidentally larger steric effects for lower pKa F-Tyr’s would have to precisely compensate for enhanced intrinsic hydrogen bond strength for each F-Tyr such that the observed straight line was obtained. Further, although there clearly are steric effects from the larger chloro-substitution, its deleterious effect is a modest 4- to 7-fold.
In the absence of “short, strong” pKa-matched hydrogen bonds or a substantially greater active site sensitivity of hydrogen bond energetics to charge accumulation in KSI and, potentially, in other enzymes as well, how can we account for the enormous rate enhancements attained by KSI and other enzymes? For KSI, which provides 1012 fold catalysis relative to a corresponding solution reaction,52 conservative mutation of the oxyanion hole side chains, Tyr to Phe and Asp to Ala or Leu give enormous rate decreases of 105–108 fold.68,69 These observations led to suggestions of distinct structural and energetic properties of the native hydrogen bonds.19,68,69 However, these mutations create a hydrophobic environment that is likely destabilizing to the incipient oxyanion relative to an aqueous environment.3,4
Indeed, a different mutational approach by Schwans et al. led to a much smaller estimate of the contribution from the oxyanion hole relative to the corresponding reaction in aqueous solution.60 Schwans et al. replaced the active site hydrogen bond donors (and surrounding side chains) with a water-filled cavity and observed a rate decrease of ∼103-fold; several similar KSI mutants maintained ∼109-fold catalysis relative to the corresponding solution reaction.60,77 Thus, the KSI oxyanion hole hydrogen bonds appear to contribute ∼103-fold to catalysis relative to an aqueous environment.
As noted above, our observation of a shallow dependence of reaction rate over a ∼102-fold change in proton affinity of the Tyr hydrogen bond donor, and prior observations of little to no change in the binding affinity of KSI across a series of oxyanion intermediate analogues varying in proton affinity by ∼102-fold, strongly suggest that the enzyme environment does not have energetic properties that substantially enhance the sensitivity of hydrogen bonds to charge rearrangement relative to that observed in aqueous solution. Nevertheless, given the large increase in proton affinity over the course of the reaction, estimated to be 1013-fold going from the substrate carbonyl to the intermediate oxyanion,78 the ∼103-fold catalytic contribution from the oxyanion hole could arise from a slightly higher energetic sensitivity to charge accumulation within the active site than in aqueous solution that would not have been detected in this or the previous study.
However, there are multiple differences between hydrogen bonds made to water in aqueous solution and those made to residues in an active site, so that it would be unwarranted to ascribe the oxyanion hole catalytic contribution solely to a slightly higher sensitivity to charge. Most basically, positioning of the oxyanion hole hydrogen bond donors would be expected to play a role in this catalytic contribution, consistent with classical descriptions of enzymatic catalysis.81−85 Additional differences that remain to be dissected and quantitatively understood include the number of hydrogen bonds formed in the active site versus water, differences in the identities of the hydrogen bond donors, the interactions with second solvation shell water molecules or surrounding enzyme residues, and differential rearrangements and conformational entropy of solution versus the enzyme as charge accumulates in going to the transition state. Such understanding will require systematic and multifaceted experimental approaches and computation that provides quantitative and nontrivial predictions that can be independently tested. The remaining identified catalytic features, substrate binding and the positioned active site general base, will also require careful investigation in order to understand how these and perhaps unidentified catalytic features provide the remaining 109-fold catalysis.
Acknowledgments
We would like to thank Tom Muir for suggesting SUMO fusions as a method to express and purify the C-terminal KSI fragment, and members of the Herschlag lab for commenting on the manuscript and for stimulating discussions. This work was funded by National Science Foundation Grant MCB-1121778 (to D.H.). A.N. was supported in part by a Howard Hughes Medical Institute International Student Research Fellowship, a Natural Sciences and Engineering Research Council of Canada Postgraduate Scholarship, and a William and Sara Hart Kimball Stanford Graduate Fellowship. J.P.S. was supported in part by a National Institutes of Health Postdoctoral Fellowship.
Supporting Information Available
Description of active site titrations of semisynthetic enzymes (Text I); description of the generation of 4,4-dideuterated 5(10)-EST for kinetic isotope effect determinations (Text II); comparison of activity of recombinant and semisynthetic WT KSI with 5(10)-EST (Figure S1); comparison of activity of independent preparations of semisynthetic 2-F-Tyr KSI with 5(10)-EST (Figure S2); pH dependencies of activity of semisynthetic KSI variants with 5(10)-EST (Figure S3); dependence of inactivating deprotonation pKa values observed in pH dependencies of activity with 5(10)-EST on side chain pKa of substituted tyrosine (Figure S4); initial rates of product formation with 5(10)-EST and semisynthetic KSI variants (Figure S5); initial rates of product formation with 5-AND and semisynthetic KSI variants (Figure S6); dependence of kcat/KM, kcat, and KM for isomerization of 5-AND on side chain pKa of substituted tyrosine (Figure S7); dependence of kcat/KM for isomerization of 3-cyclohexen-1-one on side chain pKa of substituted tyrosine (Figure S8); determination of equilenin affinity for KSI variants containing a D40N mutation (Figure S9); active site titrations of semisynthetic KSI variants (Figure S10); 1H NMR spectra of protiated 5(10)-EST incubated with NaOD/D2O monitored over 18 min, and of 4-EST with no added base in D2O (Figure S11); product formation from 4,4-dideuterated 5(10)-EST with semisynthetic WT KSI (Figure S12); subsaturating activity of semisynthetic KSIs with 4,4-dideuterated 5(10)-EST in D2O and protiated 5(10)-EST in H2O (Figure S13); saturating activity of semisynthetic KSIs with 4,4-dideuterated 5(10)-EST in D2O and protiated 5(10)-EST in H2O (Figure S14); substrate concentrations used in kinetic characterizations of KSI variants (Table S1); Michaelis–Menten parameters for KSI-catalyzed isomerization of 5(10)-EST by expressed proteins (Table S2); Michaelis–Menten parameters for isomerization of 5(10)-EST by semisynthetic KSIs (Table S3); Michaelis–Menten parameters for isomerization of 5-AND by semisynthetic KSIs (Table S4); kcat/KM values for isomerization of 3-cyclohexen-1-one by semisynthetic KSIs (Table S5); affinities of expressed and semisynthetic KSI variants for a fluorescent analogue of equilenin EqA488–1 (Table S6); fractions of expressed and semisynthetic KSI variants that bind equilenin (Table S7); 1H, 13C, and 19F NMR spectra and masses of protected tyrosines. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Department of Chemistry and Biochemistry, California State University Long Beach, Long Beach, California 90840, United States.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Robertus J. D.; Kraut J.; Alden R. A.; Birktoft J. J. Biochemistry 1972, 11, 4293–4303. [DOI] [PubMed] [Google Scholar]
- Hedstrom L. Chem. Rev. 2002, 102, 4501–4524. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Sigala P. A.; Fenn T. D.; Herschlag D. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1960–1965and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschlag D.; Natarajan A. Biochemistry 2013, 52, 2050–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hine J. J. Am. Chem. Soc. 1972, 94, 5766–5771. [Google Scholar]
- Stahl N.; Jencks W. P. J. Am. Chem. Soc. 1986, 108, 4196–4205. [Google Scholar]
- Shan S.-O.; Herschlag D. Methods Enzymol. 1999, 308, 246–276and references therein. [DOI] [PubMed] [Google Scholar]
- Kresge A. J.; Chiang Y. J. Phys. Chem. 1973, 77, 822–825. [Google Scholar]
- Larson J. W.; McMahon T. B. J. Am. Chem. Soc. 1982, 104, 5848–5849. [Google Scholar]
- Wenthold P. G.; Squires R. R. J. Phys. Chem. 1995, 99, 2002–2005. [Google Scholar]
- Shan S.-O.; Herschlag D. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 14474–14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The enhanced sensitivity in DMSO relative to water can also be seen in a plot of the equilibrium constants for hydrogen bond formation in DMSO against those for the same series of compounds in water. Such a plot has a slope of 15 (see Figure 2 in Shan and Herschlag11), in contrast to a slope of 1 expected if there were no dependence on the solvent.
- Gilson M. K.; Rashin A.; Fine R.; Honig B. J. Mol. Biol. 1985, 184, 503–516. [DOI] [PubMed] [Google Scholar]
- Warshel A. Nature 1987, 330, 15–16. [DOI] [PubMed] [Google Scholar]
- Cleland W. W. Biochemistry 1992, 31, 317–319. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Gassman P. G. Biochemistry 1993, 32, 11943–11952. [DOI] [PubMed] [Google Scholar]
- Cleland W. W.; Kreevoy M. M. Science 1994, 264, 1887–1890. [DOI] [PubMed] [Google Scholar]
- Gerlt J. A.; Kreevoy M. M.; Cleland W. W.; Frey P. A. Chem. Biol. 1997, 4, 259–267. [DOI] [PubMed] [Google Scholar]
- Cleland W. W.; Frey P. A.; Gerlt J. A. J. Biol. Chem. 1998, 273, 25529–25532. [DOI] [PubMed] [Google Scholar]
- Robillard G.; Shulman R. G. J. Mol. Biol. 1974, 86, 519–540. [DOI] [PubMed] [Google Scholar]
- Loh S. N.; Markley J. L. Biochemistry 1994, 33, 1029–1036. [DOI] [PubMed] [Google Scholar]
- Frey P. A.; Whitt S. A.; Tobin J. B. Science 1994, 264, 1927–1930. [DOI] [PubMed] [Google Scholar]
- Tong H.; Davis L. Biochemistry 1995, 34, 3362–3367. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Abeygunawardana C.; Talalay P.; Mildvan A. S. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 8220–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers P. M.; Klevit R. E. Nat. Struct. Biol. 1996, 3, 522–531. [DOI] [PubMed] [Google Scholar]
- Hur O.; Leja C.; Dunn M. F. Biochemistry 1996, 35, 7378–7386. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Luecke H.; Yao N.; Quiocho F. A. Nat. Struct. Biol. 1997, 4, 519–522. [DOI] [PubMed] [Google Scholar]
- Petrounia I. P.; Pollack R. M. Biochemistry 1998, 37, 700–705. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Sigala P. A.; Pybus B.; Liu C. W.; Ringe D.; Petsko G. A.; Herschlag D. PLoS Biol. 2006, 4, 501–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feierberg I.; Aqvist A. Theor. Chem. Acc. 2002, 108, 71–84. [Google Scholar]
- Gao J.; Ma S.; Major D. T.; Nam K.; Pu J.; Truhlar D. G. Chem. Rev. 2006, 106, 3188–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshel A.; Sharma P. K.; Kato M.; Xiang Y.; Liu H.; Olsson M. H. M. Chem. Rev. 2006, 106, 3210–3235. [DOI] [PubMed] [Google Scholar]
- Zhong W.; Gallivan J. P.; Zhang Y.; Li L.; Lester H. A.; Dougherty D. A. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 12088–12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyedsayamdost M.; Reece S.; Nocera D.; Stubbe J. J. Am. Chem. Soc. 2006, 128, 1569–1579. [DOI] [PubMed] [Google Scholar]
- Messmore J. M.; Fuchs D. N.; Raines R. T. J. Am. Chem. Soc. 1995, 117, 8057–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson M. A.; Falke J. J. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 163–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L.; Chin J. W. Nat. Rev. Mol. Cell Biol. 2012, 13, 168–182. [DOI] [PubMed] [Google Scholar]
- Thorson J. S.; Chapman E.; Schultz P. G. J. Am. Chem. Soc. 1995, 117, 9361–9362. [Google Scholar]
- Brooks B.; Phillips R. S.; Benisek W. F. Biochemistry 1998, 37, 9738–9742. [DOI] [PubMed] [Google Scholar]
- Kim S. W.; Cha S.-S.; Cho H.-S.; Kim J.-S.; Ha N.-C.; Cho M.-J.; Jou S.; Kim K. K.; Choi K. Y.; Oh B.-H. Biochemistry 1997, 36, 14030–14036. [DOI] [PubMed] [Google Scholar]
- Zhao Q.; Abeygunawardana C.; Gittis A. G.; Mildvan A. S. Biochemistry 1997, 36, 14616–14626. [DOI] [PubMed] [Google Scholar]
- Cho H. S.; Ha N. C.; Choi G.; Kim H. J.; Lee D.; Oh K. S.; Kim K. S.; Lee W.; Choi K. Y.; Oh B. H. J. Biol. Chem. 1999, 274, 32863–32868. [DOI] [PubMed] [Google Scholar]
- Ha N.-C.; Kim M.-S.; Lee W.; Choi K. Y.; Oh B.-H. J. Biol. Chem. 2000, 275, 41100–41106. [DOI] [PubMed] [Google Scholar]
- Oh K. S.; Cha S.-S.; Kim D.-H.; Cho H.-S.; Ha N.-C.; Choi G.; Lee J. Y.; Tarakeshwar P.; Son H. S.; Choi K. Y.; Oh B.-H.; Kim K. S. Biochemistry 2000, 39, 13891–13896. [DOI] [PubMed] [Google Scholar]
- Hanoian P.; Sigala P. A.; Herschlag D.; Hammes-Schiffer S. Biochemistry 2010, 49, 10339–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack R. M.; Thornburg L. D.; Wu Z. R.; Summers M. F. Arch. Biochem. Biophys. 1999, 370, 9–15. [DOI] [PubMed] [Google Scholar]
- Ha N. C.; Choi G.; Choi K. Y.; Oh B. H. Curr. Opin. Struct. Biol. 2001, 11, 674–678. [DOI] [PubMed] [Google Scholar]
- Feierberg I.; Aqvist J. Biochemistry 2002, 41, 15728–15735. [DOI] [PubMed] [Google Scholar]
- Chakraborty D. K.; Soudackov A. V.; Hammes-Schiffer S. Biochemistry 2009, 48, 10608–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringold H. J.; Malhotra S. K. Tetrahedron Lett. 1962, 3, 669–672. [Google Scholar]
- Pollack R. M.; Zeng B.; Mack J. P.; Eldin S. J. Am. Chem. Soc. 1989, 111, 6419–6423. [Google Scholar]
- Schwans J. P.; Kraut D. A.; Herschlag D. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 14271–14275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.; Cole P. A. J. Am. Chem. Soc. 1998, 120, 6851–6858. [Google Scholar]
- Corey E. J.; Venkateswarlu A. J. Am. Chem. Soc. 1972, 94, 6190–6191. [Google Scholar]
- Schnölzer M.; Alewood P.; Jones A.; Alewood D.; Kent S. B. Int. J. Pept. Protein Res. 1992, 40, 180–193. [DOI] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Gonzalez A.; Tsai Y.; Herschlag D. Biochemistry 2013, 52, 7840–7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill S. C.; von Hippel P. H. Anal. Biochem. 1989, 182, 319–326. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Churchill M. J.; Dawson P. E.; Herschlag D. ACS Chem. Biol. 2009, 4, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruben E. A.; Schwans J. P.; Sonnett M.; Natarajan A.; Gonzalez A.; Tsai Y.; Herschlag D. Biochemistry 2013, 52, 1074–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwans J. P.; Sunden F.; Gonzalez A.; Tsai Y.; Herschlag D. J. Am. Chem. Soc. 2011, 133, 20052–20055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schowen K. B.; Schowen R. L. Methods Enzymol. 1982, 87, 551–606. [PubMed] [Google Scholar]
- Pollack R. M. Bioorg. Chem. 2004, 32, 341–353. [DOI] [PubMed] [Google Scholar]
- Pollack R. M.; Bantia S.; Bounds P. L.; Koffman B. M. Biochemistry 1986, 25, 1905–1911. [DOI] [PubMed] [Google Scholar]
- Yun Y. S.; Lee T.-H.; Nam G. H.; Jang D. S.; Shin S.; Oh B.-H.; Choi K. Y. J. Biol. Chem. 2003, 278, 28229–28236. [DOI] [PubMed] [Google Scholar]
- Wilde T. C.; Blotny G.; Pollack R. M. J. Am. Chem. Soc. 2008, 130, 6577–6585. [DOI] [PubMed] [Google Scholar]
- CRC Handbook of Chemistry and Physics, 94th ed.; CRC Press: Boca Raton, FL, 2013–2014; pp 9–6, 9–49, 9–50. [Google Scholar]
- Sigala P. A.; Kraut D. A.; Caaveiro J. M. M.; Pybus B.; Ruben E. A.; Ringe D.; Petsko G. A.; Herschlag D. J. Am. Chem. Soc. 2008, 130, 13696–13708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi G.; Ha N.-C.; Kim S. W.; Kim D.-H.; Park S.; Oh B.-H.; Choi K. Y. Biochemistry 2000, 39, 903–909. [DOI] [PubMed] [Google Scholar]
- Thornburg L. D.; Goldfeder Y. R.; Wilde T. C.; Pollack R. M. J. Am. Chem. Soc. 2001, 123, 9912–9913. [DOI] [PubMed] [Google Scholar]
- Childs W.; Boxer S. G. Biochemistry 2010, 49, 2725–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton J. R.; Pelton J. G.; Kirsch J. F. Biochemistry 2001, 40, 10411–10416. [DOI] [PubMed] [Google Scholar]
- Usher K. C.; Remington S. J.; Martin D. P.; Drueckhammer D. G. Biochemistry 1994, 33, 7753–7759. [DOI] [PubMed] [Google Scholar]
- Mitra B.; Kallarakal A. T.; Kozarich J. W.; Gerlt J. A.; Clifton J. R.; Petsko G. A.; Kenyon G. L. Biochemistry 1995, 34, 2777–2787. [DOI] [PubMed] [Google Scholar]
- Kraut D. A.; Carroll K. S.; Herschlag D. Annu. Rev. Biochem. 2003, 72, 517–571. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Grellier P. L.; Prior D. V.; Duce P. P.; Morris J. J.; Taylor P. J. J. Chem. Soc., Perkin Trans. 2 1989, 6, 699–711. [Google Scholar]
- We have varied the electrostatic properties of the hydrogen bond donor, whereas in the catalyzed reaction the hydrogen bond acceptor varies in its charge density. ‘Commutative’ behavior of hydrogen bond energetics has been observed, i.e., equal sensitivity to variation in the hydrogen bond acceptor or donor.6 This equivalence is also predicted from simple hydrogen bond models.5−7
- Additional deleterious effects from the mutations to create this aqueous cavity would render the 103 fold effect from the oxyanion hole an overestimate, not an underestimate, of the total effect.
- A protonated aliphatic ketone has pKa −3,79 while the dienolate reaction intermediate has pKa 10,80 corresponding to a 1013-fold change in proton affinity over the course of the reaction.
- Bagno A.; Lucchini V.; Scorrano G. J. Phys. Chem. 1991, 95, 345–352. [Google Scholar]
- Zeng B.; Pollack R. M. J. Am. Chem. Soc. 1991, 113, 3838–3842. [Google Scholar]
- Page M. I.; Jencks W. P. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 1678–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blow D. Structure 2000, 8, R77–81. [DOI] [PubMed] [Google Scholar]
- Pauling L. Chem. Eng. News 1946, 24, 1375–1377. [Google Scholar]
- Jencks W. P.; Page M. I. Proc. FEBS Meet. 1972, 29, 45–58. [Google Scholar]
- Jencks W. P. Adv. Enzymol. Relat. Areas Mol. Biol. 1975, 43, 219–410. [DOI] [PubMed] [Google Scholar]
- Phenol pKa values were used in this work to avoid potential ionic effects from the amine and carboxylate groups in the free amino acids that can alter pKa values. Nevertheless, the observed pKa values for the side chains of free tyrosine and fluorotyrosines are within 0.3 units of the values for the corresponding phenols,53,87 and their use would not alter any conclusions herein.
- Jencks W. P.; Regenstein J. In Handbook of Biochemistry and Molecular Biology; Fasman G. D., Ed.; CRC Press: Cleveland, OH, 1976; pp 305–351. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.