

Summary
Altered cellular bioenergetics and mitochondrial function are major features of several diseases including cancer, diabetes, and neurodegenerative disorders. Given this important link to human health, we sought to define proteins within mitochondria that are critical for maintaining homeostatic ATP levels. We screened an RNAi library targeting >1,000 nuclear-encoded genes whose protein products localize to the mitochondria in multiple metabolic conditions to examine their effect on cellular ATP levels. We identified a mechanism by which electron transport chain perturbation under glycolytic conditions increased ATP production through enhanced glycolytic flux; thereby highlighting the cellular potential for metabolic plasticity. Additionally, we identified a mitochondrial adenylate kinase (AK4) that regulates cellular ATP levels, AMPK signaling, and whose expression significantly correlates with glioma patient survival. As a result, this study maps the bioenergetic landscape of >1,000 mitochondrial proteins in the context of varied metabolic substrates and begins to link key metabolic genes with clinical outcome.
Introduction
The production of ATP to in order to fuel energy consuming processes is a principal function of both quiescent and proliferative cellular metabolism. Sufficient energy levels must be maintained for cells to thrive (Wallace, 2011), and it is clear that dysregulated bioenergetics plays an important role in many diseases (Raimundo, 2014). In cancer, energy production is increased to support rapid proliferation (Formentini et al., 2010; Vander Heiden et al., 2009; Vander Heiden et al., 2012); while in many neurodegenerative diseases, core energy producing pathways are compromised leading to impaired cellular function and decreased viability (Breuer et al., 2013; Federico et al., 2012; Xun et al., 2012).
The major pathways directly responsible for ATP production in quiescent and proliferative cells are well-described. Mitochondria house much of the core ATP-generating machinery and are recognized as important for maintaining cellular energy homeostasis through integrating cellular environmental and nutritional signals to produce the bulk of cellular ATP. However, the individual contributions to cellular energy homeostasis by each mitochondrial protein and the numerous mitochondrial non-cellular respiration functions have not been comprehensively investigated. Establishing a catalogue of each protein’s impact on the cellular metabolic economy would provide a useful reference for investigating normal and disease bioenergetics (Pagliarini and Rutter, 2013). Because cells respond to different fuel sources by utilizing different bioenergetic programs (Stanley et al., 2014), defining these bioenergetic contributions in the context of multiple fuel sources provides added biological relevance.
Previous studies have identified the contributions of individual metabolic genes to cancer cell survival (Ros et al., 2012) or tumor formation (Possemato et al., 2011), identified drugs that are effective in distinct bioenergetic programs (Gohil et al., 2010), mapped proteomic components of mitochondria (Pagliarini et al., 2008; Rensvold et al., 2013; Rhee et al., 2013), or derived computational models of central carbon metabolism (Greenberg et al., 2011; Noor et al., 2010; Shlomi et al., 2011). In this study, we developed a high throughput method to identify critical components regulating cellular ATP levels in specific metabolic programs and performed a functional RNAi screen to characterize cellular bioenergetics under glycolytic and oxidative phosphorylation (OXPHOS) conditions. We analyzed the entire complement of MitoCarta genes (a catalogue of >1,000 genes whose protein products localize to the mitochondria (Pagliarini et al., 2008)) for global effects on cellular energy levels in response to four fuel sources (glucose, pyruvate, glutamine, galactose). In addition to cataloguing each gene, our study identified specific mitochondrial functions associated with maintaining ATP levels in distinct fuel sources, as cultured cells are able to utilize a variety of carbon sources for bioenergetic requirements (Genzel et al., 2005; Reitzer et al., 1979). We also identified a mechanism of metabolic plasticity wherein genetic or chemical disruption of the electron transport chain (ETC) significantly increased overall ATP levels through enhanced glycolytic flux. Finally, we characterized adenylate kinase 4 (AK4), the gene most significantly associated with increased ATP production in our screen. Adenylate kinases are critical regulators of adenine nucleotide homoeostasis, maintaining proper cellular AMP/ADP/ATP ratios (Dzeja and Terzic, 2009; Noma, 2005). As one of three mitochondrial adenylate kinases, little is known about AK4 function. AK4 has been proposed to play a role in cellular stress responses (Edhager et al., 2014; Kong et al., 2013; Liu et al., 2009) and the invasive potential of lung cancer cell lines (Jan et al., 2012). We found that AK4 regulates ATP levels across multiple cell types, cellular proliferation, and is also associated with glioma patient survival. Remarkably, AK4 knockdown also activated the AMPK-signaling pathway, providing a mechanistic link between mitochondrial adenylate kinase function and important energy sensing pathways.
Results
Segregation of cellular bioenergetic programs
To restrict cells to different bioenergetic contexts, we cultured cells in media containing specified carbon nutrient sources which forced reliance on either glycolysis or OXPHOS for ATP production (Guppy et al., 2002; Stanley et al., 2014). We restricted cells to glucose as a model of glycolytic bioenergetics; to either pyruvate or glutamine as different models of common OXPHOS bioenergetics; and to galactose as a model of bioenergetics reliant on both glycolysis and OXPHOS (Figure 1A) (Colombo et al., 2011; Gohil et al., 2010; Hensley et al., 2013; Marroquin et al., 2007; Robinson et al., 1992; Rossignol et al., 2004).
Figure 1. A Sensitized RNAi Screen to Identify Regulators Glycolytic and OXHPOS Bioenergetics.

(A) Depiction of the nutrient source strategy utilized in this study.
(B) Relative ATP/cell measurements from cells after a four hour treatment with DMSO, iodoacetic acid (IAA, 10 μM), or rotenone (500 nM). Error bars represent SD values.
(C) Compliment of metabolic pathways and functions targeted in the RNAi screen.
(D) RNAi screen results. Labeled data points indicate siRNAs that increased or decreased ATP levels 25% or more compared to control siRNAs in all four conditions.
See also Figure S1.
To validate our strategy of restricting cells to glycolytic or OXPHOS bioenergetic programs, we treated cells with inhibitors of glycolysis (iodoacetic acid, IAA, which inhibits GAPDH) or OXPHOS (rotenone, which inhibits Respiratory Complex I). ATP levels (ATP/cell, Figure S1A, see Experimental Procedures) in cells maintained in glucose and galactose media were sensitive to acute glycolytic inhibition with IAA, whereas ATP production in cells maintained in pyruvate, glutamine, or galactose media were sensitive to acute OXPHOS inhibition with rotenone (Figure 1B), as expected based on previous studies (Gohil et al., 2010; Marroquin et al., 2007; Rossignol et al., 2004; Warburg et al., 1967). ATP levels in cells grown in complete (Figure S1B) or glucose only media were insensitive to OXPHOS inhibition, indicating that HeLa cells preferentially rely on glycolysis, even in the presence of OXPHOS-permissive carbon substrates (Warburg, 1956). In the absence of glucose, however, HeLa cells retain the ability to utilize OXPHOS-specific bioenergetics, as they are viable and proliferate in OXPHOS-restricted media conditions. These results of acute metabolic inhibition confirm our approach of utilizing alternative fuel sources to restrict cells to specific bioenergetic programs. The metabolic plasticity of the HeLa cell model revealed here also highlights the challenges of targeting cancer cell bioenergetics as a therapeutic strategy.
Identification of mitochondrial genes that regulate cellular ATP levels
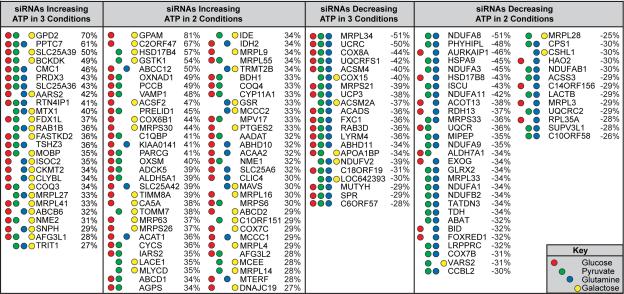
Next, we used this strategy to interrogate a set of nuclear-encoded genes that comprises >1,000 known mitochondria-localized proteins (Figure 1C, Figure S1C, Table S1). We performed duplicate RNAi screens targeting these genes in each of the four carbon sources (glucose, pyruvate, glutamine, galactose). Our results identified genes whose knockdown altered ATP levels across multiple conditions in addition to unique sets of bioenergetic regulators for each condition (Figure 1D, Table S2). Knockdown of five genes (AK4, ADCK2, CASP8, MECR, TMEM70) increased ATP levels in all conditions, while knockdown of six genes (DNAJC11, VDAC2, FAM65C, C12ORF62, HSCB, APEX2) decreased ATP levels in all conditions. Because these genes altered ATP levels in all four nutrient conditions, the respective functions of their protein products likely affect mechanisms which commonly impact homeostatic ATP levels. Knockdown of additional genes altered ATP levels by ≥ 25% in at least two or three of the four conditions examined (Table 1). Importantly, each metabolic condition contained unique genes whose knockdown impacted ATP levels in only that nutrient source (Table S3). Taken together, these results identify mitochondrial proteins that maintain cellular ATP levels across multiple metabolic conditions, as well as mitochondrial proteins that uniquely regulate ATP levels in specific metabolic conditions.
Table 1. Mitochondrial Genes Impacting Cellular Bioenergetics.
Genes altering ATP levels by ≥ 25% in multiple metabolic conditions. Red, green, blue, and yellow circles represent ≥ 25% ATP/cell changes in glucose, pyruvate, glutamine, and galactose conditions, respectively. Percentages are the combined averages of ATP/cell changes in the indicated metabolic conditions.
|
We next defined mitochondrial functions that impact ATP levels by grouping genes whose knockdown altered ATP levels into common mitochondrial functions (Figure 2, Table S3). In each of the four conditions, knockdown of genes associated with the electron transport chain (ETC), fatty acid metabolism, amino acid metabolism, membrane transporters, and ribosomal proteins altered ATP levels. However, a comparison of the overall effect on each function revealed opposing effects in glucose and galactose compared to pyruvate and glutamine conditions (Figure S2A). This may reflect the common routes of carbon flux into mitochondria in response to these fuels. Glucose and galactose carbons must flux through the glycolytic pathway prior to delivery into mitochondria, while pyruvate and glutamine can be directly imported into the mitochondria and incorporated into the TCA cycle after only two enzymatic modifications. Interestingly, as opposed to a comparison between functions, gene-by-gene comparisons revealed that galactose and pyruvate conditions to be more similar than galactose and glucose conditions (Figure S2B), which is expected based on the results of previous studies (Gohil et al., 2010; Marroquin et al., 2007; Rossignol et al., 2004; Warburg et al., 1967). In addition to these mitochondrial functions commonly affecting ATP levels across all metabolic conditions, each condition also contains unique mitochondrial functions. Redox maintenance genes are associated with pyruvate-specific bioenergetics, translocases are associated with galactose-specific bioenergetics, mitochondrial signaling proteins are associated with glutamine-specific bioenergetics, and lipid metabolism, nucleotide metabolism, and aminoacyl tRNA synthetases are associated with glucose-specific bioenergetics. Of note, many proteins with unknown functions are associated with maintaining homeostatic ATP levels in all conditions. Seventeen percent of the MitoCarta remains uncharacterized, and these results reveal that many of these uncharacterized proteins have a measurable impact on at least one aspect of cell biology, providing an impetus for further characterization of these proteins.
Figure 2. Mitochondrial Functions Impacting Cellular Bioenergetics.

Cytoscape (see Methods section) was used to define mitochondrial functions affecting ATP levels in response to each metabolic condition. Central colored nodes represent individual nutrient sources. Gray nodes radiating from the central node represent all mitochondrial functions associated with siRNAs altering ATP by ≥25%. The distance between each functional node and the central node is proportional to the number of genes associated with that function that increased or decreased ATP by ≥ 25% in that condition. Functions with five or more associated genes altering ATP by ≥ 25% are labeled and include the associated genes, represented as nodes with colored edges. Solid colored edges represent increased ATP levels; dotted edges represent decreased ATP levels. The distance between each gene node and the associated functional node is proportional to the magnitude of ATP level change. See also Table S3 and Figure S2.
OXPHOS inhibition enhances glycolytic ATP production
We chose to pursue our observation that knockdown of ETC genes produces opposite effects in glucose and pyruvate conditions (Figure 2, Table S3). We mapped the effect of silencing each ETC complex subunit on ATP levels. As expected, knockdown of core ETC components negatively impacted ATP levels in OXPHOS (pyruvate) conditions; surprisingly, however, we found that knockdown of many core ETC components increased glucose-dependent ATP levels (Figure 3A, B, Table S2). To test whether acute perturbation of the ETC also elicited this response, we treated cells with various chemical inhibitors of the OXPHOS machinery for four hours. Chemical inhibition of the OXPHOS machinery also increased glucose-dependent ATP levels (Figure 3C, Figure 1B), indicating that both acute (4-hour chemical inhibition) and prolonged (72-hour knockdown) perturbation of the OXPHOS machinery enhances glucose-dependent ATP levels. Because OXPHOS-dependent ATP production is blocked in this context, we predicted that the increased ATP levels could be due to decreased ATP consumption or increased glycolytic flux. To investigate this, we labeled cells with D-[1,6-13C]-glucose and concurrently measured ATP levels and lactate production. We found that knockdown of Respiratory Complex I subunits as well as treatment with rotenone concomitantly increased ATP levels and lactate production (Figure 3D, E), verifying an increase in glycolytic flux and associated ATP production in this context. Interestingly, prolonged perturbation (72-hour knockdown) resulted in higher ATP and lactate levels than acute perturbation (4-hour rotenone), indicating that this effect may lead to the accumulation of metabolic products over time. It is well-known that chemical perturbation of the ETC increases glucose uptake and glycolytic flux in order to counteract ATP depletion (Hardie et al., 2012; Kurth-Kraczek et al., 1999; Marsin et al., 2000). However, in this context, increased glycolytic flux has been observed, but not necessarily increased ATP levels (Hao et al., 2010; Wu et al., 2007).
Figure 3. Suppression of OXPHOS in Glycolytic Conditions Increases Energy Production.

(A) Electron transport chain schematic. Each subunit is colored according to the results of the RNAi screen in ATP/cell standard deviations from control siRNA.
(B) Results of the RNAi screen presented as percent change in ATP/cell for each subunit.
(C) Percent change in relative ATP/cell was determined compared to DMSO treatment. Measurements were obtained after 4 hours of treatment with ETC complex inhibitors (n=5 for each dose). Error bars represent SD values. *p < 0.05.
(D) Relative ATP/cell was measured in cells transfected with control or selected RC I subunit siRNAs and in cells treated for 500 nM rotenone for 4 hours (n=3 for each condition). Error bars represent SD values. **p < 0.01; ***p < 0.001.
(E) Glucose-to-lactate conversion in control and selected RC I subunit siRNA transfected HeLa cells. Cells were cultured with medium containing 1,6 13C-labeled glucose, and enrichment of 13C-lactate (m+3) in the extracellular medium was measured after 6 hours (n=3 for each condition). Error bars represent SD values. **p < 0.01; ***p < 0.001.
Impact of the mitochondrial kinome and phosphatome on cellular ATP levels
We noted that silencing multiple mitochondrial kinases and phosphatases affected ATP levels (Table S2), and it is known that central metabolism can be regulated by phosphorylation (Oliveira et al., 2012). (Hornbeck et al., 2004) To investigate this further, we re-screened the compliment of mitochondrial kinases and phosphatases in quadruplicate in glucose and pyruvate conditions, and generated a volcano plot comparing ATP/cell fold change to p-values. Similar to the MitoCarta analysis, the adenylate kinase, AK4, and the uncharacterized phosphatase, PPTC7, ranked highest in both conditions (Figure 4A), increasing cellular ATP levels from 30 to 50% (Figure 4B). It is possible that the mitochondrial kinome and phosphatome may regulate ETC-mediated ATP production via phosphorylation (Acin-Perez et al., 2011; Hopper et al., 2006; Pagliarini and Dixon, 2006). Indeed, by mining publically available databases for evidence of phosphorylation of ETC complex subunits (Hornbeck et al., 2004), we identified 284 reported phosphorylation events on the ETC machinery (Table S4). Because adenylate kinases catalyze the transfer of a phosphate group between ATP, ADP, and AMP (Dzeja and Terzic, 2009; Noma, 2005; Noma et al., 2001), it is unlikely that AK4 directly phosphorylates the ETC machinery. However, we found that knockdown of PPTC7 increased oxygen consumption rate while having no effect on extracellular acidification rate (Figure 4C), raising the possibility that PPTC7 negatively regulates ETC activity. Future studies examining whether PPTC7 regulates ETC activity and ATP levels by modifying ETC machinery phosphorylation status are warranted.
Figure 4. Mitochondrial Kinome and Phosphatome Impact on ATP Levels.

(A) Volcano plot depicting the results of a second RNAi screen targeting the mitochondrial kinome and phosphatome in glucose and pyruvate conditions.
(B) Relative ATP/cell was measured in cells transfected with control, AK4, or PPTC7 siRNAs. Error bars represent SD values (n=4 for each siRNA). **p < 0.01.
(C) Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using a Seahorse Biosciences XFe96 Extracellular Flux Analyzer. Values were normalized to cell number. Cells were cultured in 10mM glucose, 1mM pyruvate, 1mM glutamine. Error bars represent SD values (n=10 for each siRNA).
Adenylate Kinase 4 regulates ATP levels and energy sensor signaling pathways
Adenylate kinases are known to maintain homeostatic ATP, ADP, and AMP levels, and because AK4 was the top gene in both the MitoCarta (Figure 1) and the mitochondrial kinome (Figure 4) screens, we chose to further characterize its effects on ATP levels and cell fate. Silencing AK4 increased ATP levels in HeLa cells (Figure 5A) as well as several other cell lines, including U20S osteosarcoma cells, U251 glioma cells, and HK2 immortalized normal kidney cells (Figure 5B). In addition to increasing ATP levels, AK4 knockdown increased cellular proliferation (Figure 5C). To determine whether AK4 is associated with clinical significance, we interrogated publically available databases for AK4, and found AK4 expression and copy number to be negatively correlated with glioma patient survival (Figure 5D). Low expression and copy number correlate with improved patient survival while high copy number correlates with decreased probability of survival. These results confirm the biological relevance of genes identified in our screen and implicate AK4 as a potentially important gene for normal and diseased cell biology.
Figure 5. AK4 Controls ATP Levels, Proliferation, and Expression Levels Correlate with Glioma Patient Survival.

(A), (B) Relative ATP/cell was measured in cells transfected with control or AK4 siRNAs (n=3 for each siRNA). Error bars represent SD values. **p < 0.01. Western blot depicting level of AK4 knockdown.
(C) Cell proliferation was measured by xCELLigence RTCA. Cell density measurements were taken every hour for 96 hours (n=6 for each siRNA). Error bars represent SD values.
(D) The Rembrandt database was used to perform a Kaplan-Meier survival analysis of glioma patients with differential AK4 expression and copy number.
An increase in cellular ATP levels would be expected to impact cellular energy charge (Hardie and Hawley, 2001; Oakhill et al., 2012)) as well as AMPK activation ((Hardie, 2011; Mihaylova and Shaw, 2011). To evaluate the effect of AK4 silencing on these parameters, we first measured ATP levels, the ADP/ATP ratio as a measure of energy charge, and AMPK activation in different metabolic conditions. As expected, cells maintained in glucose and pyruvate specific conditions for 72 hours exhibited decreased ATP levels, increased ADP/ATP ratios and enhanced AMPK activation (Figure 6A). Because AK4 knockdown increases ATP levels, we predicted that AK4 knockdown would mitigate these effects of decreased energy in glucose and pyruvate specific conditions. However, AK4 knockdown increased the ADP/ATP ratio in all conditions (Figure 6B). Accordingly, AK4 knockdown also induced AMPK phosphorylation of AMPK at T172 in all conditions examined (Figure 2C). AMPK is a master energy sensor, responding to altered cellular energy charge by activating catabolism and inhibiting anabolism. Active AMPK phosphorylates ULK1 at S555 to induce autophagy-mediated mitochondrial and energetic homeostasis (Egan et al., 2011). We found that AK4 knockdown induced ULK1 S555 phosphorylation, consistent with AMPK activation status (Figure 6C). AMPK was similarly induced by AK4 knockdown in U20S and U251 cells (Figure 6D), suggesting that normal AK4 function impacts AMPK activation across multiple cell types. Together, these data implicate AK4 as an important regulator of cellular homeostasis through regulation of cellular energy charge and AMPK activation.
Figure 6. AK4 Regulates Cellular Energy Charge and AMPK Signaling.

(A), ATP levels, ADP/ATP ratio, and phospho-AMPK were measured after 72 hours in the indicated medias (n=6 for each condition). Error bars represent SD values.
(B) ADP/ATP ratio was measured in cells transfected with control or AK4 siRNAs (n=3 for each condition). Error bars represent SD values.
(C,D) Western blot analyses of AMPK signaling pathway from cells transfected with control or AK4 siRNAs. Blots are representative of three independent experiments.
See also Figure S3.
Discussion
The results described here provide the most comprehensive assessment of mitochondrial proteins involved in cellular bioenergetics to date. While some previous reports investigate the effects of metabolic inhibitors on bioenergetics with respect to cell viability and oxygen consumption and glycolytic rates (Giordano et al., 2012; Gohil et al., 2010), our study maps the contributions of individual mitochondrial proteins to ATP levels in response to four distinct fuel sources. This analysis was performed in both glycolytic and OXPHOS conditions, providing the relative contribution of each protein in response to multiple fuel sources. While the work described here does not comprehensively define all mitochondrial proteins contributions to bioenergetics in all metabolic environments, the approach and results generated by this study provide a workable platform to expand our knowledge of cellular bioenergetics on a large scale. Future studies using similar approaches in a larger number of cell types and bioenergetic substrates can add to our knowledge of the similarities and differences in bioenergetic determinants between disparate cell types and fuel sources. The etiology of many diseases includes altered energetics and mitochondrial dysfunction, yet the genetic basis for many of these diseases remains unknown. Importantly, this study will provide a valuable resource for identifying the genetic determinants of altered mitochondrial-dependent energetics. It provides additional rationale for identifying potential drug candidates for diseases which preferentially rely on specific metabolic programs (e.g., the relative contributions of glycolytic and OXPHOS metabolic programs may vary between cell and cancer type (Fan et al., 2013; Zu and Guppy, 2004)).
Many of the specific genes and functions associated with mitochondrial-dependent ATP levels in this study fit logically within the current understanding of bioenergetics and mitochondrial biology. Knockdown of genes and functions that consume ATP generally increased ATP levels (e.g., ATPases and vesicular trafficking proteins), while knockdown of the mitochondrial machinery required for protein synthesis would be expected to disrupt normal mitochondrial function and adversely affect ATP levels in mitochondrial-dependent nutrient conditions. In agreement with this, silencing mitochondrial ribosomal proteins generally decreased ATP levels in pyruvate and glutamine conditions. However, in the glycolytic condition, which is able to maintain normal ATP levels without OXPHOS (Figure 1B, Figure 4), knockdown of ribosomal proteins and tRNA synthetases increased ATP levels. This likely reflects the fact that protein synthesis requires ATP consumption, and decreasing mitochondrial-specific ATP consumption in a context which does not require mitochondrial-dependent ATP production will tip the bioenergetic balance towards an overall increase in cellular ATP levels. An overall comparison of commonly-altered functions between the nutrient conditions reveals a similarity between glucose and galactose and between pyruvate and glutamine (Figure S2A). As mentioned above, this may reflect the common routes by which these carbon sources are incorporated in to metabolic pathways. Alternatively, this may be similar to the situation described above wherein inhibiting energetically expensive mitochondrial functions in fuels that do not rely exclusively on OXPHOS leaves a positive balance of ATP, while inhibiting these same functions in fuels that exclusively rely on mitochondrial metabolism is detrimental to overall ATP balances. Future studies following carbon flux in each of these conditions may shed additional light on the mechanisms driving these observations.
The above observations concern comparisons of the few mitochondrial functions whose perturbation commonly altered ATP levels in all conditions. Interestingly, a gene-by-gene comparison shows that galactose and pyruvate conditions are more similar than galactose and glucose conditions (Figure S2B), which is a more commonly recognized comparison (Gohil et al., 2010; Marroquin et al., 2007; Rossignol et al., 2004; Warburg et al., 1967). This may again reflect the fact that the above functions represent the common routes of metabolic flux shared between galactose and glucose, while the overall metabolic economy of galactose is more similar to pyruvate (OXPHOS) on a gene-by-gene basis. It is also worth noting that we found many uncharacterized proteins to be associated with significant changes in ATP levels. The functional significance of a large portion of proteins that localize to mitochondrial is unknown. This study provides the first step towards characterizing these proteins.
Knockdown of genes from the TCA cycle did not consistently alter ATP levels in our screen with the exception of the succinate dehydrogenase complex which also functions in the electron transport chain. This is perhaps due to the many well-known and recently described biochemical routes by which cells can circumvent perturbations of the TCA cycle (Cheng et al., 2011; DeBerardinis et al., 2007; Le et al., 2012; Marin-Valencia et al., 2012; Metallo et al., 2012; Mullen et al., 2012; Wise et al., 2011). However, an unanticipated result of this study was the general increase in glycolytic bioenergetics and flux observed upon knockdown or chemical inhibition of core OXPHOS machinery, a phenomenon essentially opposite of the Crabtree effect (Crabtree, 1929; Diaz-Ruiz et al., 2011). Potential explanations for this include an unidentified ETC-flux sensing mechanism which signals to accelerate glycolytic flux upon ETC impairment. Alternatively, in the context of ETC impairment, NADH levels may increase as a function of reduced Respiratory Complex I activity. In this case, increasing glycolytic flux and the conversion of pyruvate to lactate by lactate dehydrogenase could serve to restore the NAD+/NADH ratio. Each of these scenarios would provide cells a mechanism for rapid adaptation in heterogeneous metabolic environments, similar to the Crabtree effect. Additional studies comparing the capacities of normal and diseased cells for this response may provide valuable insight into the mechanistic basis of specific diseases.
Phosphorylation is another potential mechanism by which ETC activity may be regulated (Arachiche et al., 2008; Covian and Balaban, 2012; Lee et al., 2005; Samavati et al., 2008). Here, we compile the known phosphorylation sites on the core ETC machinery and provide the relative contributions of the mitochondrial kinome and phosphatome to ATP levels. Our studies identify the uncharacterized phosphatase, PPTC7, as a candidate for phosphoregulation of the ETC. Interestingly, while this manuscript was in preparation, the yeast homologue of PPTC7, PTC7, was found to be associated with regulating ubiquinone biosynthesis, and thus ETC activity, through dephosphorylating the ubiquinone biosynthetic enzyme, Coq7 (Martin-Montalvo et al., 2013).
We identified adenylate kinase 4 (AK4) as a key regulator of cellular ATP levels and energy-sensing signaling pathways and found it to be associated with glioma patient survival. It is unlikely that the increase in ATP observed by AK4 knockdown is solely due to a loss in mitochondrial adenlyate kinase maintenance of ATP, ADP, and AMP concentrations. Cells typically maintain ATP, ADP, and AMP concentrations at ratios of 100:10:1, respectively. Therefore, conversion of all ADP and AMP stores to ATP would only raise ATP levels by approximately 10%, while AK4 knockdown consistently increases ATP levels by more than 25%. Additionally, we find that AK4 knockdown concomitantly increases the cellular ADP/ATP ratio, indicating that ADP (and likely AMP) levels increase along with ATP. One possible explanation is that AK4 loss and associated loss of mitochondrial adenylate kinase function activates a feedback mechanism which induces adenosine biosynthesis, effectively increasing all adenosine nucleotide pools. Adenosine nucleotide biosynthesis is known to affect AMPK activation status in flies (Stenesen et al., 2013). Initial metabolic profiling experiments show a trend of increased AMP and ADP upon AK4 knockdown, although not statistically significant (Figure S3A). Our observations that modulating AK4 levels impinges upon the energetic stress-sensing AMPK pathway is also consistent with its proposed association with cellular stress responses (Edhager et al., 2014; Kong et al., 2013; Liu et al., 2009). In addition, we find AK4 to be negatively correlated with glioma patient survival, in accordance with an analysis of lung cancer expression profiles that found high AK4 expression to correlate with poor patient survival (Jan et al., 2012). This previous report suggested that AK4 regulates gene transcription to mediate its effects on cancer cell oncogenic potential, although the mechanism by which a mitochondrial adenylate kinase regulates gene transcription remains unknown. Our findings raise the possibility that AK4’s impact on bioenergetics and metabolism may contribute to cancer biology by maintaining the proper balance of adenosine nucleotides and other metabolites. Indeed, metabolic profiling revealed that siRNA-mediated depletion of AK4 alters the cellular concentrations of several intermediates of central carbon metabolism (Figure S3B). Altered utilization of central carbon metabolites and switches in bioenergetic programs by cancer cells are becoming recognized as critical components of the oncogenic potential of many cancers (Hitosugi et al., 2012; Locasale et al., 2011; Metallo and Vander Heiden, 2013; Mullen et al., 2012; Oliva et al., 2010; Possemato et al., 2011; Son et al., 2013); thus, AK4’s involvement in these processes may represent its association with cancer patient survival.
Similar to the effects that we observe upon AK4 knockdown, Snijder et al. recently reported that siRNA-mediated knockdown of AK5 in HeLa cells also increased proliferation (Snijder et al., 2012), and other groups have implicated nucleotide biogenesis in oncogenic potential (Stenesen et al., 2013; Wawrzyniak et al., 2013). Knockdown of additional nucleotide kinases in our study also increased ATP levels (e.g., NME proteins). Thus, regulation of nucleotide composition is an emerging area of interest with respect to cellular bioenergetics and disease biology.
The results presented here provide a useful resource relating individual mitochondrial proteins to cellular bioenergetics, identify a previously unknown mechanism of metabolic adaptation, and identify AK4 as a gene with a potentially important role in maintaining cellular energy levels and AMPK signaling.
EXPERIMENTAL PROCEDURES
A full explanation of all methods used can be found within the Supplemental information.
RNAi Screen
Cells were transfected in each nutrient condition in duplicate with a pool of 2 siRNAs per gene and subjected to CellTiter-Glo and CyQUANT assays 72 hours after siRNA transfection. siRNA knockdown throughout the experiments presented here reduced target mRNA levels from 80-98% (Figure S1D). Full experimental methods can be found within the Supplemental Information.
ATP/cell Measurements
Growth media was removed following treatments, and a room-temperature solution of Cell Titer-Glo, Opti-MEM, and CyQUANT was immediately added to each well. Luminescence and fluorescence readings were consecutively taken after a 10 minute room-temperature incubation.
Cytoscape
Cytoscape version 3.0.2 was used to visualize network connectivity (Cline et al., 2007; Shannon et al., 2003).
Electron Transport Chain Phosphorylation Sites
Protein post-translational modifications data presented in Table S4 were obtained from the online systems biology resource PhosphoSite on 09.10.2013.
Proliferation Index
Real-time cell proliferation was determined using the xCelligence system, which utilizes an electric current to determine cellular attachment to an electrode-containing plate. Cells were plated in glucose media in a 96-well electrode-containing plate, allowed to enter exponential growth, then transfected with the indicated siRNAs. The electrical impedance, caused by cell adhesion to the plate, is directly proportonial to cell number and is numerically reported.
[3-13C] Lactate Measurement
Lactate production was measured with gas chromatography-mass spectrometry on media from cells cultured for 6 hr in glucose free medium containing dialyzed FBS supplemented with 10 mM D-[1,6-13C]-glucose (Cambridge Isotope Labs). Full experimental methods can be found within the Supplemental Information.
ADP/ATP Ratio Measurement
ApoSENSOR ADP/ATP Ratio Bioluminescent Assay from BioVision (Milpitas, CA) was used according to manufacturer’s specifications.
Antibodies and Immunoblotting
Cells grown in the indicated culture conditions were washed with cold PBS and harvested on ice in cold lysis buffer. After normalization of concentration, samples were then separated on Tris-glycine polyacrylamide gels, transferred to nitrocellulose membranes, blocked, and probed with primary antibodies. Membranes were incubated in enhanced chemiluminescent reagent, exposed to film, and developed for signal.
Metabolomics
Sample preparation
1.5ml of extraction solvent (mixture of methanol, chloroform and water) with internal standards is added to cell culture plates and cells are scraped off from plate and transferred to eppendorf tubes. Sample tubes were vertex briefly, sitting on ice for 5min, then centrifuge at 15000 xg for 5min. Supernatant containing metabolites are transferred to autosampler vials for LC-MS analysis. A series of calibration standards were prepared along with samples to quantify metabolites.
LC-MS analysis
Agilent 1200 chromatographer, Luna NH2 HILIC (hydrophilic interaction chromatography) column was used for chromatographic separation. Mass spectrometer: Agilent 6520 series time-of-flight mass spectrometer.
Data were processed by MassHunter workstation software, version B.06.
Statistical Analysis
Data represent biological replicates and are depicted as means ± s.d. Student’s t-test was used to determine significance.
Supplementary Material
Highlights.
Defined essential genes and mitochondrial functions associated with bioenergetics.
Uncovered a novel mechanism of metabolic plasticity in glycolytic conditions.
Identified AK4 as important for cellular energy homeostasis and AMPK signaling.
ACKNOWLEDGEMENTS
We thank members of the MacKeigan laboratory for their insights and critical feedback. This work was supported by the National Institutes of Health National Cancer Institute grants R01CA138651 (J.P.M.), R01CA157996 (R.J.D.), F32CA159709 (N.J.L.). This work utilized Core Services supported by grant DK097153 of NIH to the University of Michigan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Acin-Perez R, Gatti DL, Bai Y, Manfredi G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab. 2011;13:712–719. doi: 10.1016/j.cmet.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arachiche A, Augereau O, Decossas M, Pertuiset C, Gontier E, Letellier T, Dachary-Prigent J. Localization of PTP-1B, SHP-2, and Src exclusively in rat brain mitochondria and functional consequences. J Biol Chem. 2008;283:24406–24411. doi: 10.1074/jbc.M709217200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breuer ME, Koopman WJ, Koene S, Nooteboom M, Rodenburg RJ, Willems PH, Smeitink JA. The role of mitochondrial OXPHOS dysfunction in the development of neurologic diseases. Neurobiol Dis. 2013;51:27–34. doi: 10.1016/j.nbd.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, DeBerardinis RJ. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A. 2011;108:8674–8679. doi: 10.1073/pnas.1016627108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S, Moncada S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proc Natl Acad Sci U S A. 2011;108:21069–21074. doi: 10.1073/pnas.1117500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covian R, Balaban RS. Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. Am J Physiol Heart Circ Physiol. 2012;303:H940–966. doi: 10.1152/ajpheart.00077.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree HG. Observations on the carbohydrate metabolism of tumours. Biochem J. 1929;23:536–545. doi: 10.1042/bj0230536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Ruiz R, Rigoulet M, Devin A. The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repression. Biochim Biophys Acta. 2011;1807:568–576. doi: 10.1016/j.bbabio.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Dzeja P, Terzic A. Adenylate kinase and AMP signaling networks: metabolic monitoring, signal communication and body energy sensing. Int J Mol Sci. 2009;10:1729–1772. doi: 10.3390/ijms10041729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edhager AV, Stenbroen V, Nielsen NS, Bross P, Olsen RK, Gregersen N, Palmfeldt J. Proteomic investigation of cultivated fibroblasts from patients with mitochondrial short-chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2014 doi: 10.1016/j.ymgme.2014.01.007. [DOI] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kamphorst JJ, Mathew R, Chung MK, White E, Shlomi T, Rabinowitz JD. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322:254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- Formentini L, Martinez-Reyes I, Cuezva JM. The mitochondrial bioenergetic capacity of carcinomas. IUBMB Life. 2010;62:554–560. doi: 10.1002/iub.352. [DOI] [PubMed] [Google Scholar]
- Genzel Y, Ritter JB, Konig S, Alt R, Reichl U. Substitution of glutamine by pyruvate to reduce ammonia formation and growth inhibition of mammalian cells. Biotechnol Prog. 2005;21:58–69. doi: 10.1021/bp049827d. [DOI] [PubMed] [Google Scholar]
- Giordano S, Lee J, Darley-Usmar VM, Zhang J. Distinct effects of rotenone, 1- methyl-4-phenylpyridinium and 6-hydroxydopamine on cellular bioenergetics and cell death. PLoS One. 2012;7:e44610. doi: 10.1371/journal.pone.0044610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohil VM, Sheth SA, Nilsson R, Wojtovich AP, Lee JH, Perocchi F, Chen W, Clish CB, Ayata C, Brookes PS, et al. Nutrient-sensitized screening for drugs that shift energy metabolism from mitochondrial respiration to glycolysis. Nat Biotechnol. 2010;28:249–255. doi: 10.1038/nbt.1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg AJ, Hackett SR, Harshman LG, Clark AG. Environmental and genetic perturbations reveal different networks of metabolic regulation. Mol Syst Biol. 2011;7:563. doi: 10.1038/msb.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guppy M, Leedman P, Zu X, Russell V. Contribution by different fuels and metabolic pathways to the total ATP turnover of proliferating MCF-7 breast cancer cells. Biochem J. 2002;364:309–315. doi: 10.1042/bj3640309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao W, Chang CP, Tsao CC, Xu J. Oligomycin-induced bioenergetic adaptation in cancer cells with heterogeneous bioenergetic organization. J Biol Chem. 2010;285:12647–12654. doi: 10.1074/jbc.M109.084194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25:1895–1908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG, Hawley SA. AMP-activated protein kinase: the energy charge hypothesis revisited. Bioessays. 2001;23:1112–1119. doi: 10.1002/bies.10009. [DOI] [PubMed] [Google Scholar]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell. 2012;22:585–600. doi: 10.1016/j.ccr.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, Witzmann FA, Harris RA, Balaban RS. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45:2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornbeck PV, Chabra I, Kornhauser JM, Skrzypek E, Zhang B. PhosphoSite: A bioinformatics resource dedicated to physiological protein phosphorylation. Proteomics. 2004;4:1551–1561. doi: 10.1002/pmic.200300772. [DOI] [PubMed] [Google Scholar]
- Jan YH, Tsai HY, Yang CJ, Huang MS, Yang YF, Lai TC, Lee CH, Jeng YM, Huang CY, Su JL, et al. Adenylate kinase-4 is a marker of poor clinical outcomes that promotes metastasis of lung cancer by downregulating the transcription factor ATF3. Cancer Res. 2012;72:5119–5129. doi: 10.1158/0008-5472.CAN-12-1842. [DOI] [PubMed] [Google Scholar]
- Kong F, Binas B, Moon JH, Kang SS, Kim HJ. Differential expression of adenylate kinase 4 in the context of disparate stress response strategies of HEK293 and HepG2 cells. Arch Biochem Biophys. 2013;533:11–17. doi: 10.1016/j.abb.2013.02.014. [DOI] [PubMed] [Google Scholar]
- Kurth-Kraczek EJ, Hirshman MF, Goodyear LJ, Winder WW. 5′ AMP- activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes. 1999;48:1667–1671. doi: 10.2337/diabetes.48.8.1667. [DOI] [PubMed] [Google Scholar]
- Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, Huttemann M. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- Liu R, Strom AL, Zhai J, Gal J, Bao S, Gong W, Zhu H. Enzymatically inactive adenylate kinase 4 interacts with mitochondrial ADP/ATP translocase. Int J Biochem Cell Biol. 2009;41:1371–1380. doi: 10.1016/j.biocel.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marin-Valencia I, Yang C, Mashimo T, Cho S, Baek H, Yang XL, Rajagopalan KN, Maddie M, Vemireddy V, Zhao Z, et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012;15:827–837. doi: 10.1016/j.cmet.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing media glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicol Sci. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- Martin-Montalvo A, Gonzalez-Mariscal I, Pomares-Viciana T, Padilla-Lopez S, Ballesteros M, Vazquez-Fonseca L, Gandolfo P, Brautigan DL, Navas P, Santos-Ocana C. The phosphatase Ptc7 induces coenzyme Q biosynthesis by activating the hydroxylase Coq7 in yeast. J Biol Chem. 2013 doi: 10.1074/jbc.M113.474494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metallo CM, Vander Heiden MG. Understanding metabolic regulation and its influence on cell physiology. Mol Cell. 2013;49:388–398. doi: 10.1016/j.molcel.2013.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noma T. Dynamics of nucleotide metabolism as a supporter of life phenomena. J Med Invest. 2005;52:127–136. doi: 10.2152/jmi.52.127. [DOI] [PubMed] [Google Scholar]
- Noma T, Fujisawa K, Yamashiro Y, Shinohara M, Nakazawa A, Gondo T, Ishihara T, Yoshinobu K. Structure and expression of human mitochondrial adenylate kinase targeted to the mitochondrial matrix. Biochem J. 2001;358:225–232. doi: 10.1042/0264-6021:3580225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noor E, Eden E, Milo R, Alon U. Central carbon metabolism as a minimal biochemical walk between precursors for biomass and energy. Mol Cell. 2010;39:809–820. doi: 10.1016/j.molcel.2010.08.031. [DOI] [PubMed] [Google Scholar]
- Oakhill JS, Scott JW, Kemp BE. AMPK functions as an adenylate charge- regulated protein kinase. Trends Endocrinol Metab. 2012;23:125–132. doi: 10.1016/j.tem.2011.12.006. [DOI] [PubMed] [Google Scholar]
- Oliva CR, Nozell SE, Diers A, McClugage SG, 3rd, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie GY, Landar A, et al. Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem. 2010;285:39759–39767. doi: 10.1074/jbc.M110.147504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AP, Ludwig C, Picotti P, Kogadeeva M, Aebersold R, Sauer U. Regulation of yeast central metabolism by enzyme phosphorylation. Mol Syst Biol. 2012;8:623. doi: 10.1038/msb.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, Walford GA, Sugiana C, Boneh A, Chen WK, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliarini DJ, Dixon JE. Mitochondrial modulation: reversible phosphorylation takes center stage? Trends Biochem Sci. 2006;31:26–34. doi: 10.1016/j.tibs.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Rutter J. Hallmarks of a new era in mitochondrial biochemistry. Genes Dev. 2013;27:2615–2627. doi: 10.1101/gad.229724.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo N. Mitochondrial pathology: stress signals from the energy factory. Trends Mol Med. 2014 doi: 10.1016/j.molmed.2014.01.005. [DOI] [PubMed] [Google Scholar]
- Reitzer LJ, Wice BM, Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J Biol Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- Rensvold JW, Ong SE, Jeevananthan A, Carr SA, Mootha VK, Pagliarini DJ. Complementary RNA and protein profiling identifies iron as a key regulator of mitochondrial biogenesis. Cell Rep. 2013;3:237–245. doi: 10.1016/j.celrep.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee HW, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BH, Petrova-Benedict R, Buncic JR, Wallace DC. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem Med Metab Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- Ros S, Santos CR, Moco S, Baenke F, Kelly G, Howell M, Zamboni N, Schulze A. Functional metabolic screen identifies 6-phosphofructo-2-kinase/fructose-2,6- biphosphatase 4 as an important regulator of prostate cancer cell survival. Cancer Discov. 2012;2:328–343. doi: 10.1158/2159-8290.CD-11-0234. [DOI] [PubMed] [Google Scholar]
- Rossignol R, Gilkerson R, Aggeler R, Yamagata K, Remington SJ, Capaldi RA. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M. Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 2008;283:21134–21144. doi: 10.1074/jbc.M801954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shlomi T, Benyamini T, Gottlieb E, Sharan R, Ruppin E. Genome-scale metabolic modeling elucidates the role of proliferative adaptation in causing the Warburg effect. PLoS Comput Biol. 2011;7:e1002018. doi: 10.1371/journal.pcbi.1002018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijder B, Sacher R, Ramo P, Liberali P, Mench K, Wolfrum N, Burleigh L, Scott CC, Verheije MH, Mercer J, et al. Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol. 2012;8:579. doi: 10.1038/msb.2012.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son J, Lyssiotis CA, Ying H, Wang X, Hua S, Ligorio M, Perera RM, Ferrone CR, Mullarky E, Shyh-Chang N, et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature. 2013;496:101–105. doi: 10.1038/nature12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley IA, Ribeiro SM, Gimenez-Cassina A, Norberg E, Danial NN. Changing appetites: the adaptive advantages of fuel choice. Trends Cell Biol. 2014;24:118–127. doi: 10.1016/j.tcb.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenesen D, Suh JM, Seo J, Yu K, Lee KS, Kim JS, Min KJ, Graff JM. Adenosine nucleotide biosynthesis and AMPK regulate adult life span and mediate the longevity benefit of caloric restriction in flies. Cell Metab. 2013;17:101–112. doi: 10.1016/j.cmet.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Lunt SY, Dayton TL, Fiske BP, Israelsen WJ, Mattaini KR, Vokes NI, Stephanopoulos G, Cantley LC, Metallo CM, et al. Metabolic Pathway Alterations that Support Cell Proliferation. Cold Spring Harb Symp Quant Biol. 2012 doi: 10.1101/sqb.2012.76.010900. [DOI] [PubMed] [Google Scholar]
- Wallace DC. Bioenergetic Origins of Complexity and Disease. Cold Spring Harb Symp Quant Biol. 2011 doi: 10.1101/sqb.2011.76.010462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- Warburg O, Geissler AW, Lorenz S. On growth of cancer cells in media in which glucose is replaced by galactose. Hoppe Seylers Z Physiol Chem. 1967;348:1686–1687. [PubMed] [Google Scholar]
- Wawrzyniak JA, Bianchi-Smiraglia A, Bshara W, Mannava S, Ackroyd J, Bagati A, Omilian AR, Im M, Fedtsova N, Miecznikowski JC, et al. A purine nucleotide biosynthesis enzyme guanosine monophosphate reductase is a suppressor of melanoma invasion. Cell Rep. 2013;5:493–507. doi: 10.1016/j.celrep.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, Ward PS, Shay JE, Cross JR, Gruber JJ, Sachdeva UM, Platt JM, DeMatteo RG, Simon MC, Thompson CB. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of alpha-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci U S A. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007;292:C125–136. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Xun Z, Rivera-Sanchez S, Ayala-Pena S, Lim J, Budworth H, Skoda EM, Robbins PD, Niedernhofer LJ, Wipf P, McMurray CT. Targeting of XJB-5-131 to mitochondria suppresses oxidative DNA damage and motor decline in a mouse model of Huntington’s disease. Cell Rep. 2012;2:1137–1142. doi: 10.1016/j.celrep.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.