

Abstract
Basal cell carcinoma (BCC) is the most common human cancer. We have demonstrated previously that topical application of the retinoid prodrug tazarotene profoundly inhibits murine BCC carcinogenesis via RARγ-mediated regulation of tumor cell transcription. Since topical retinoids can cause adverse cutaneous effects and since tumors can develop resistance to retinoids, we have investigated mechanisms downstream of tazarotene’s anti-tumor effect in this model. Specifically we have used (i) global expression profiling to identify and (ii) functional cell-based assays to validate the PI3K/AKT/mTOR pathway as a downstream target pathway of tazarotene’s action. Crucially, we have demonstrated that pharmacologic inhibition of this downstream pathway profoundly reduces murine BCC cell proliferation and tumorigenesis both in vitro and in vivo. These data identify PI3K/AKT/mTOR signaling as a highly attractive target for BCC chemoprevention and indicate more generally that this pathway may be, in some contexts, an important mediator of retinoid anti-cancer effects.
Introduction
BCCs comprise approximately one third of all cancers in the United States with an incidence that is increasing by 4–5% per year and with a cost to Medicare alone of $400 million/year (1). Inappropriate hyperactivation of hedgehog (HH) signaling is the pivotal abnormality that underlies BCC carcinogenesis in both sporadic and inherited (basal cell nevus [Gorlin] syndrome; BCNS; OMIM #109400) cases and generally is caused by mutational inactivation of the tumor suppressor gene PATCHED 1 (PTCH1) or by gain-of-function mutations in the HH pathway effector SMOOTHENED (SMO) (2). Like PTCH1+/− BCNS patients, Ptch1+/− mice exposed to clinically relevant environmental mutagens develop multiple BCCs, thus providing an accurate and practical model of human BCCs (3, 4).
Endogenous retinoids have a major role in skin homeostasis (5), and pharmacologic doses of oral retinoids can inhibit BCC carcinogenesis in humans, albeit with significant systemic toxicities (6). In the epidermis, the major retinoid receptors are retinoic acid receptors (RAR) α and γ and retinoid X receptor (RXR) α. Previously we found that topical application of tazarotene (Tazorac™), an anti-acne retinoid prodrug that is converted into tazarotenic acid and specifically activates RARβ and γ (7), robustly prevents BCCs in Ptch1+/− mice (8, 9). In addition, a pharmacologic pan-RAR antagonist that inhibits physiological RAR signaling in skin increases the BCC burden (8). These observations suggest that physiological retinoid signaling in the skin restrains BCC carcinogenesis and are consistent with the observation that long term topical tazarotene cures 30–50% of sporadic human BCCs (10). Approximately 60% of untreated visible BCCs arising in Ptch1+/− mice have partial or total loss of RARγ expression, suggesting that receptor loss may underlie the resistance of some BCCs to tazarotene. We have assessed the chemopreventive efficacy of topical tazarotene in a Phase II clinical trial (Identifier: NCT00489086, ClinicalTrials.gov) (see accompanying manuscript). Although topical tazarotene may be found to be an effective agent for long-term chemoprevention of human BCCs under some circumstances, some patients are deterred from using it optimally because topical retinoids irritate the skin, and this irritation may account for some of the disparity between the robust results in mice and the comparatively disappointing effects in humans.
Therefore, we have investigated downstream pathways mediating tazarotene’s anti-BCC chemopreventive efficacy to search for novel therapeutic strategies that (i) might be tolerated better and (ii) might overcome retinoid resistance.
Materials and Methods
Global gene changes with tazarotene
BCC cell line treatment and RNA extraction: We cultured ASZ001 cells as described previously (11). Cells at 80% confluency were incubated in serum-free media (154-CF medium containing 0.05 mM CaCl2 and 1x Penicillin/Streptomycin) for 2 h. Working concentrations of tazarotene and DMSO were prepared as follows: for tazarotene the powder was dissolved in 100% DMSO at a stock concentration of 10 mM and diluted to 10 µM working concentration in 154-CF medium containing 0.05 mM Ca2+ and 1X penicillin/streptomycin. We then incubated cells with 10 µM tazarotene or 0.1% DMSO for 10 or 24 h in a 5% CO2 incubator. Four replicates were carried out for each treatment, and the three replicates with the best RNA integrity were analyzed with Affymetrix gene expression arrays. We analyzed a further replicate in which ASZ001 cells were incubated for 48 h in 0.1% DMSO or 10 µM tazarotene using the Wst-1 cell proliferation assay (Roche Applied Science, IN) to confirm cell proliferation inhibition by 10 µM tazarotene. After the incubation, cells were washed in phosphate-buffered saline and harvested in 1 ml of Trizol for RNA extraction (Invitrogen, CA). Total RNA was re-purified using RNeasy RNA isolation columns (Qiagen, CA), and RNA integrity was confirmed with the RNA Bioanalyzer (Ambion/Applied Biosystems, CA). Two micrograms of total RNA were used for cRNA amplification using the MessageAmp™ II-Biotin Kit (Ambion/Applied Biosystems, CA). Briefly, reverse transcription of total RNA was carried out using an oligo (dT) primer bearing a T7 promoter using ArrayScript™. Second-strand synthesis using the cDNA was carried out and purified for use as a template for in vitro transcription in a reaction containing biotin-modified UTP and T7 RNA Polymerase (Ambion/Applied Biosystems, CA). Biotin-labeled, amplified RNA (aRNA) was then purified for gene expression analysis on Mouse Genome 430A 2.0 Array GeneChips (Affymetrix, CA), at the Gladstone Genomics Core Facility (San Francisco, CA). Assessment of hybridization quality was also performed (Gladstone Genomics Core Facility, San Francisco, CA) using the Bioconductor software affyPLM, which fitted a specified robust linear model to the probe level data. Preprocessing of the data was performed using an Bioconductor Robust Multiarray Analysis (RMA) algorithm to correct for background; the data were normalized using the quantile method (12) to generate lists of statistically significant, differentially expressed (DE) genes (comparing tazarotene and DMSO vehicle control groups). After correcting for the false discovery rate, Partek Genomics Suite was used for Principal Component analysis and hierarchical clustering, and DE genes that were significantly differentially expressed were further analyzed using bioinformatic software from Ingenuity® Systems (CA) and Stratagene (CA).
Generation of stably-transfected Myr-HA-AKT1 and HA-AKT1 cell lines
One million log phase ASZ001 cells were transfected with pLNCX-Myr-HA-AKT1 and pLNCX-HA-AKT1 (13) (Addgene# 903 and #901 respectively), using program T29 of the Amaxa Nucleoporation system (Amaxa-Lonza, MD) (11). As a negative control, plasmid # 903 was digested with restriction enzymes to remove the anti-hemagglutinin (HA) tag and most of the AKT1 open reading frame, generating pLNCX’. Cells were then mixed with 154-CF complete media and replated. Media was removed after 24 h to remove the dead cells, and living cells were allowed to recover for 1–2 days and then at 70% confluency were passaged and replated at a lower density. After 4 days, ASZ001 cells containing the pLNCX-Myr-HA-AKT1, pLNCX-HA-AKT1 or pLNCX’ were selected using G418 (Life Technologies, CA) at 1 mg/ml (a dose that killed non-transfected ASZ001 cells) to generate myr-HA-AKT1, HA-AKT1 and pLNCX’ cell lines, respectively. After at least 1 month of G418 selection, cells were expanded for cell proliferation experiments.
In vivo pharmacologic PI3K inhibitor drug efficacy testing
Mice
Ptch1+/− K14-CreERT2 p53fl/fl mice treated with tamoxifen (0.1 mg/day) for 3 days at age 1.5 months and 4 Gray (Gy) of X-rays at age 8 weeks were given drugs by oral gavage 5 days/week from age 13 weeks until age 21 weeks when a dorsal skin biopsy (1 cm × 1 cm) was obtained (9). Mice were then monitored for the first visible BCC, and visible BCC burden was compared at age 28 weeks. Mice that died or were euthanized for unrelated causes were censored in the study.
In vivo drug treatments
Small molecule PI3K inhibitors GDC-0941 (a gift from Genentech-Roche), XL147 and XL765 (gifts from Exelixis) were given by gavage at 50 mg/Kg daily, 100 mg/Kg and 30 mg/Kg/twice a day, respectively.
Tazarotene-resistant allograft assessment
A tazarotene-resistant clone was established by challenging BCC allografts with high dose of tazarotene orally (10mg/kg daily, 5 days per week) for 3 weeks followed by a low dose of tazarotene (2mg/kg daily, 5 days per week) for 8 weeks. One resistant clone was further transplanted into 3 naïve NOD/SCID mice (2 sites per mouse). When allografts became palpable (age 7 weeks), one mouse each received treatment with vehicle, tazarotene (5mg/kg daily, 5 days per week), or XL765 (30mg/kg twice daily, 5 days per week). Tumor volume was assessed for each mouse.
Results
Tazarotene induces gene expression changes in murine BCC cells in vitro
To explore tazarotene’s anti-BCC mechanism of action, we began by assessing global gene expression changes induced by tazarotene in ASZ001 cells, a line derived from a Ptch1+/− mouse BCC. We treated log phase cells in serum-free conditions for 10 h or 24 h in triplicate with 0 or 10 µM tazarotene, a concentration that inhibits their proliferation by at least 50% after 48 h and does not inhibit the proliferation of either the immortalized murine non-tumorigenic keratinocyte cell line C5N or of a murine medulloblastoma cell line (11). We selected these timepoints for analysis based on detectable increases in expression of Crabp2, a direct RA target gene (11) at 10 h of incubation and the first detectable reduction of cell proliferation at 24 h of incubation with 10 µM tazarotene (data not shown). We extracted RNA and converted it to cDNA, which we then hybridized to Mouse Genome 430A 2.0 Array GeneChips (Affymetrix, CA) containing 20,000 probes representing 14,000 transcripts. Each incubation time point and treatment group was hybridized at the same time (i.e. hybridized under similar conditions).
Preprocessing of the raw data from the arrays indicated that for each experimental group (i.e. tazarotene or DMSO treated) the technical replicates were similar to one another and of good quality (data not shown). The quantile-normalized array data for treatments with vehicle and tazarotene and for both incubation timepoints were compared by Principal Component Analysis using the Partek Genomics Suite software (Figure 1A), which indicated a high degree of concordance between the replicates and demonstrated that tazarotene treatment significantly perturbed the gene expression profiles of ASZ001 cells at both 10 h and 24 h, compared to DMSO treatment, at both time points (Figure 1B). After adjustment for the false discovery rate (FDR), comparison to the 10 h or 24 h DMSO control gene sets generated statistically significant (p < 0.05) lists of DE genes (Figure 1C): tazarotene treatment at 10 h gave 279 DE genes and expressed sequence tags (ESTs), which after disregarding replicate probes generated a list of 240 DE genes of which 193 were upregulated and 47 were downregulated (p < 0.05). Tazarotene treatment at 24 h yielded 649 DE genes, excluding gene/EST replicates, of which 146 were upregulated and 503 were downregulated (p < 0.05). The top 30 gene probes (including replicates) with the most up- and downregulated expression at 10 h are listed in Supplementary Table S1 and Supplementary Table S2, respectively, as are the top DE gene probes after 24 h tazarotene treatment (Supplementary Tables S3 and S4, respectively). The greater number of upregulated than of downregulated genes at 10 h is consistent with a direct transcriptional activator effect of RARs, which fully dissociate from corepressors/silencing mediators and bind to coactivators in the presence of a retinoid hormone agonist such as tazarotenic acid to activate retinoid-target genes (14, 15). Indeed, known RA target genes such as Tgm2, Dhrs3, and Rai3, were upregulated after 10 h tazarotene incubation (Figure 1D, and data not shown). At 24 h however, more genes were downregulated than upregulated, likely as a result of the secondary effects of tazarotene treatment. We designated genes whose expression was altered by 10 h as ‘early’ and those whose expression was altered at 24 h but not at 10 h as ‘late’. We used qPCR to confirm the altered expression of a selection of the 10 h DE genes - Fst, Tgm2, Trib3, Eif4ebp1 (4EBP1), Gadd45a, Ndrg1 and Dtx4 (Figure 1D). To investigate whether tazarotene specifically downregulates HH signaling, we searched the DE gene lists for known direct HH target genes (i.e. genes that contain the consensus Gli binding site) such as Gli1, Ptch1, Hhip1, Nmyc1, Ccnd1, Ccnd2, Grem1, Fst and Pthlh (16). Of these genes, only Fst and Pthlh were downregulated at 10 h (Figure 1D, and data not shown). Fst was also downregulated at 24 h. Other genes that are strongly associated with HH signaling are FoxM1, Ccnd1, and Gas1. We found that FoxM1 expression was downregulated after 24 h of tazarotene treatment and not at 10 h, suggesting it is an indirect target of tazarotene signaling. Gas1 was downregulated at 10 and 24 h while Ccnd1 was not represented. However other cyclins – Ccna2, Ccnf, Ccne2, Ccnb1 and Ccnb2 – were downregulated at 24 h (data not shown), suggesting that these late DE genes are indirect targets of tazarotene-mediated signaling and that at least part of tazarotene’s anti-BCC efficacy is via blocking of cell cycle progression at the G2/M checkpoint. DE genes such as Tgm2, Dtx4, Eif4ebp1, Fst and Trib3 were represented more than once in the DE gene lists (i.e. by replicate probes on the microarrays), suggesting that these genes are likely to be ‘real’ targets of tazarotene-mediated signaling (Figure 1D, left graph). However, 10 µM tazarotene treatment of both ASZ001 and the murine medulloblastoma cell line Med1 (a cell line whose proliferation is not altered by 10 µM tazarotene treatment) upregulated Tgm2 expression in both cell lines, i.e. irrespective of whether or not tazarotene inhibited cell proliferation (data not shown). This suggested that Tgm2 upregulation was not related to tazarotene’s anti-proliferative effects, but rather to direct effects of retinoid transcriptional activation of target genes involved in other skin biofunctions. Therefore, since (i) the numbers of DE genes from tazarotene treatment at both 10 h and 24 h were relatively high, and (ii) changes in individual DE genes such as Tgm2 suggest biological processes that may be irrelevant to tazarotene’s anti-cancer effects, we used bioinformatic software to identify tazarotene-altered pathways and functions. Bioinformatic analyses of the 10 h and 24 h DE genes using Stratagene’s Pathway Architect™ software indicated that the DE genes with the most connections to other DE genes in their respective lists were associated with VEGF (Supplementary Table S5) and insulin-like growth factor-insulin receptor/phosphatidylinositol 3-kinase/AKT (IGF-IR/PI3K/AKT) signaling (Supplementary Tables S5, S6). Of note, the IGF-IR/PI3K/AKT pathway was represented in the analyses of both 10 h and 24 h DE genes lists. Gene network analyses using the Ingenuity® Systems software suggested three top networks at 10 h, one of which, again, was the IGF-IR/PI3K/AKT pathway, indicating that the latter may be a central downstream functional “node” involved in BCC inhibition by tazarotene (Figure 2) The other two ‘top’ networks identified by Ingenuity® Systems software were cholesterol metabolism and the mitogen activated protein kinase (MAPK) pathway, the latter of which has been found to interact with HH signaling (17, 18) (data not shown). Bioinformatic functional clustering analyses of the late DE genes using Ingenuity® Systems software suggested that many of these were linked to biological processes such as cancer, cell cycling, and DNA recombination and repair (p < 0.0001) (Supplementary Table S7). Also, a ‘top canonical pathway’ cluster analysis using the same software suggested that the 24 h DE genes affected by tazarotene treatment were associated with metabolism and cell cycle processes (p < 0.0001) (Supplementary Table S8), which are known to be regulated by IGF-IR/PI3K/AKT signaling (19). Moreover, the observation that DE genes such as Trib3, Eif4ebp1, and Igfbp3, all of which are negative regulators of the IGF-IR/PI3K/AKT pathway, were upregulated by tazarotene suggests that at least part of tazarotene’s anti-BCC effect is via inhibition of the IGF-IR/PI3K/AKT pathway with consequent decrease in cell growth and proliferation.
Figure 1. Global gene expression analysis of tazarotene-treated ASZ001 cells indicated that the expression of a relatively large number of genes was altered significantly.

(A) Principal component analysis of the Affymetrix array data showed that treatment with 10 µM tazarotene for 10 h or 24 h significantly altered the gene expression profile of ASZ001 in a manner that was distinct from treatment with 0.1% DMSO for 10 h and 24 h (* p < 0.05). The red triangles and squares represent genes from replicate samples treated with 0.1% DMSO for 10 and 24 h, respectively, while the blue triangles and squares represent genes from replicates treated with 10 µM tazarotene for 10 and 24 h, respectively. (B) Heat-map analyses of the standardized array data (using Partek Genomics Suite) showed that the 10 and 24 h tazarotene-treated samples had similar gene expression profiles, which were distinct from the 10 and 24 h DMSO-treated samples: expression levels of 4292 genes were significantly altered with 10 µM tazarotene treatment compared to treatment with 0.1% DMS0 (* p < 0.05, ANOVA test followed by FDR correction). (C) Using Bioconductor software, MA-plot analyses of all gene probes showed that a relatively large number of genes were differentially expressed between tazarotene- and DMSO-treated samples. The genes whose expression was statistically significant altered (* p < 0.05) sat as outliers (red dots) from the majority of the gene probes (grey dots) that were either not, or only modestly, differentially expressed. For lists of the most differentially expressed genes, see Supplementary Tables S1–S4. (D) Graphical presentation of a number of statistically significant gene probes from the 10 h Bioconductor gene set (left graph), and taqman real-time qPCR validation of the altered expression with tazarotene treatment (right graph) (* p < 0.05). The different colored bars for a gene in the microarray expression data graph indicated that replicates gene probes that were significantly differentially expressed (left graph; P denotes an Affymetrix gene probe). For Tgm2, in tazarotene-treated ASZ001 cells four DE gene-probes were represented in the 10 h Bioconductor list in a statistically significant manner (* p < 0.05). Taqman PCR confirmed the differential expression of the selected 10 h DE genes, whose expression is shown as fold change compared to the DMSO-treated ASZ001 samples.
Figure 2. Bioinformatic analyses suggest that PI3K/AKT signaling may be a key downstream pathway of tazarotene signaling.

Bioinformatic network analysis of the tazarotene-treated 10 h DE genes suggested that a significant number of the DE genes connected to the PI3K/AKT pathway as a central downstream node. The shaded shapes indicated the DE genes (* p < 0.05). The different shapes assigned to each molecule that was represented in the 10 h DE list, indicate the type/function of the protein as classified by the IPA analysis software (Ingenuity®, CA).
Overexpression of AKT1 in ASZ001 cells reduces the in vitro anti-BCC effect of tazarotene
To investigate whether BCCs that arise in our Ptch1+/− K14-CreER2 p53floxed/floxed mice have activated PI3K/AKT signaling, we assessed expression of phosphorylated (Ser473) (activated) AKT (p-AKT) immunohistochemically. We detected activated AKT at low to moderate levels in 50% (4/8) of visible IR-induced BCCs in these mice (Figure 3A), a finding similar to that of the moderate levels of p-AKT activity detected in human BCCs (20). Similarly, we also detected activated AKT signaling in our ASZ001 BCC cell line (Figure 3A).
Figure 3. Akt activity was detected in murine BCCs and BCC cell line, ASZ001, and over-expression of AKT1 in ASZ001 cells reduced the in vitro anti-BCC effect of tazarotene.
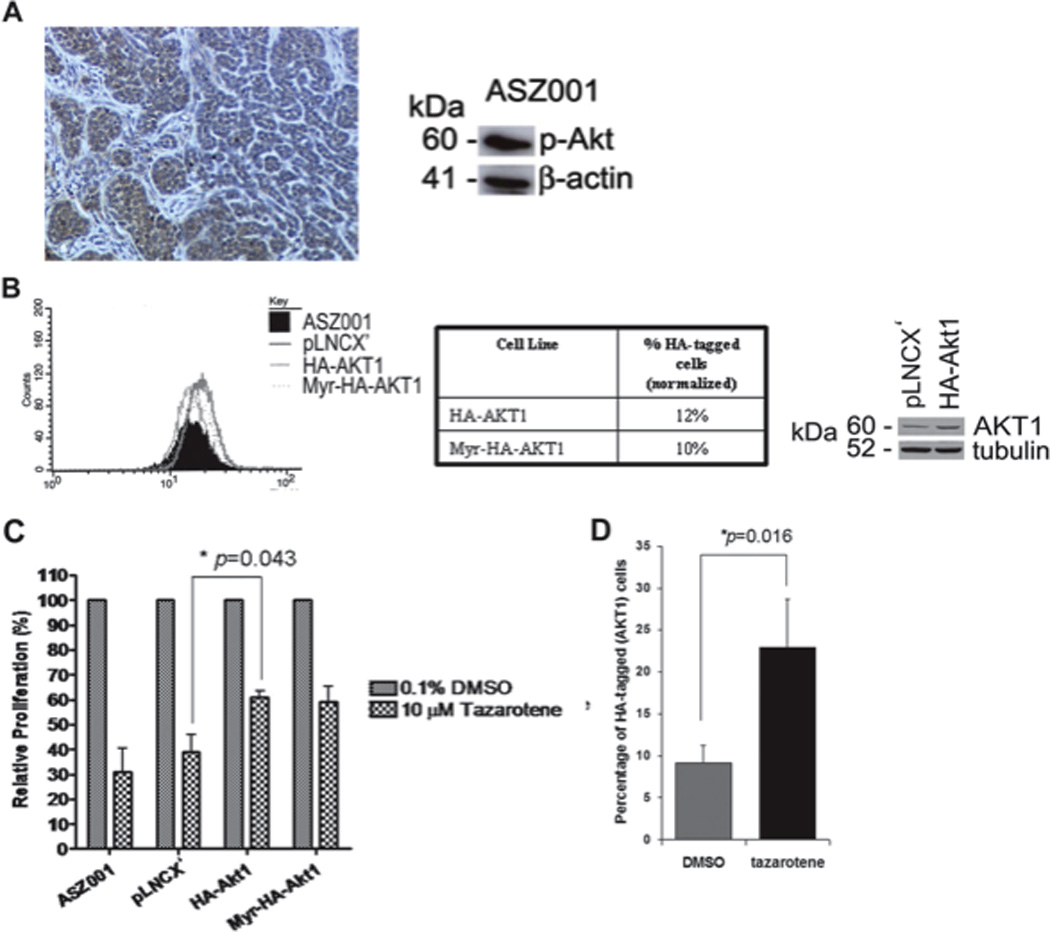
(A) Expression of phosphorylated Akt (p-Akt) in a murine BCC (left panel) and ASZ001 cell line (right panel). Immunohistochemistry with antibodies against p-Akt on a representative visible untreated murine BCC showed p-Akt immunoreactivity in the tumor nests of BCC (left panel, scale bar = 100 µM). The murine BCC cell line ASZ001 also had showed detectable p-Akt activity (right panel). (B) ASZ001 cells transfected with HA-tagged AKT1 or myristolyated AKT1 were selected with G418 to generate stable HA-AKT1 expressing cell lines (left panel). FACS analyses demonstrated that both stable cell lines generated expressed heterogeneous levels of AKT1 as measured by the HA-tag levels and indicated that only 10 and 12% of cells that were stably transfected with the AKT1 constructs expressed high levels of HA-myr-AKT1 and HA-AKT1, respectively (middle table). Slightly higher levels of AKT1 in the HA-AKT1 cell line compared to the negative control cell line (pLNCX’) were confirmed by Western blotting with antibodies against AKT1. Western blotting for tubulin (on the same blot) indicated that similar amounts of protein were loaded (right panel). (C) 10 µM tazarotene treatment of these cell lines for 48 h showed that the cell lines overexpressing AKT1 were partially resistant to tazarotene compared to the negative control cell line, pLNCX’ (* p < 0.05, Student t test). (D) FACS analysis of the HA-AKT1 cell line after 48 h incubation in 10 µM tazarotene or 0.1% DMSO vehicle showed that tazarotene enriched the population of HA-AKT1 cells as measured by HA-tag levels, suggesting that AKT1 overexpressing cells were more resistant to tazarotene treatment (* p < 0.05, Student t test).
To test whether tazarotene inhibits proliferation at least in part by blocking IGF-IR/PI3K/AKT signaling, constructs containing HA-tagged wildtype AKT1 (HA-AKT1) or constitutively activated (myristoylated) AKT1 (myr-HA-AKT1) were transfected into ASZ001 cells. Stable transfection resulted in a heterogeneous population with ~10–12% of the cells expressing relatively high levels of HA-AKT1 cells as measured by FACS (Figure 3B). To determine whether altering AKT activity could interfere with the anti-proliferative effects of tazarotene, we treated this mixed population of log-phase ASZ001 cells for 48 hours and measured the amount of cell proliferation and fold-enrichment of the HA-AKT1 overexpressing cells (Figures 3C, D). Transfection with myr-HA-AKT1 or HA-AKT1 significantly blunted tazarotene’s inhibtion of ASZ001 cells proliferation (Figure 3C, p < 0.05). Furthermore, tazarotene treatment significantly increased the percentage of cells expressing HA-AKT1 - by 2.5 fold: from 9% to 23% of the population (Figure 4, p < 0.05). These data strongly suggest that elevating AKT activity significantly blunts the in vitro anti-proliferative effects of tazarotene and hence that at least part of tazarotene’s anti-BCC effect is mediated via its inhibition of the PI3K/Akt pathway.
Figure 4. In vitro testing of therapeutically viable small molecule inhibitors of PI3K/AKT pathway for treating BCC.

Two therapeutically viable small molecule PI3K/AKT pathway inhibitors, XL147 and XL765, were tested in vitro, for their anti-BCC effects in the BCC cell line, ASZ001. LY294002, another PI3K/AKT pathway inhibitor, which has poor pharmacological efficacy due to its non-specific effects on other kinases, was tested for comparison. XL147 and XL765 treatment at 5 and 10 µM were the most effective against BCC cell proliferation (*** p < 0.0001, 2-way ANOVA test), while LY294002 only modestly inhibited ASZ001 cell proliferation at the highest concentration used (10 µM).
Pharmacologic PI3K inhibitors inhibit BCC proliferation in vitro
Since these data indicate that the PI3K/AKT pathway is a mechanistically important downstream target of tazarotene’s inhibition of BCC proliferation, we investigated whether pharmacologic PI3K inhibitors themselves can inhibit BCC proliferation in vitro. Specifically we assayed inhibition by treating log phase ASZ001 cells with inhibitors of PI3K - LY294002 (Eli-Lilly), XL147 (Exelixis), and XL765 (Exelixis). LY294002 is a first generation pan class I PI3K inhibitor that inhibits PI3K activity via competitive inhibition of an ATP binding site on the p85 subunit of PI3K. LY294002 not only binds to class I PI3Ks and other PI3K-related kinases, inhibiting PI3K-dependent production of the second messenger PIP3, but also to novel targets unrelated to the PI3K family (21). XL147 is a potent, orally bioavailable, specific inhibitor of class I PI3K kinases α, β, δ, and γ, which also binds the ATP-binding pocket of the catalytic domain of Class I PI3Ks and at nanomolar concentrations inhibits PI3K phosphorylation and consequent PI3K-dependent production of the second messenger PIP3 (22). Similarly, XL765 is also a bioavailable, highly selective, potent inhibitor of all four Class I PI3K isoforms with IC50 values at nanomolar concentrations in biochemical assays. However, in contrast to XL147, XL765 also inhibits mTOR and DNA-PK with IC50 values in the nanomolar range: in cellular assays, XL765 can inhibit nutrient stimulated mTOR-dependent signaling by inhibiting mTOR-dependent phosphorylation of key PI3K pathway components including AKT, the AKT substrates PRAS40 and GSK3β, p70S6K, and the p70S6K substrate S6, and 4EBP1 (23). XL765 treatment causes tumor regression, with associated decreased cellular proliferation and angiogenesis and increased apoptosis in various cancer models (24). We found that all three pharmacologic inhibitors caused dose-dependent reduction of ASZ001 cell proliferation (Figure 4). XL147 and XL765 appeared to be more effective than LY294002 in their inhibition of ASZ001, significantly reducing proliferation with an IC50 dose at approximately 5 µM and with a more pronounced inhibition at 10 µM (p < 0.0001). The IGF-IR inhibitor AG1024 (Sigma Aldrich, CO) also inhibited ASZ001 cell proliferation at 10 µM (data not shown). These data are similar to those we observed for tazarotene treatment of ASZ001 cells (11) and are consistent with the idea that much of tazarotene’s anti-BCC efficacy is mediated via inhibition of this pathway.
Pharmacologic PI3K inhibitors inhibit BCC proliferation in vivo
Having showed that inhibition of PI3K/AKT signaling can reduce BCC cell proliferation in vitro, we investigated the chemopreventive efficacy of XL147 and XL765 as well as of the additional PI3K inhibitor GDC-0941 (Genentech), a highly selective pan class I PI3K inhibitor that also has favorable pharmacokinetic and toxicological properties (25), in vivo in Ptch1+/− K14-CreER2 p53floxed/floxed mice. We administered the oral dose of each drug recommended by the manufacturers (Genentech and Exelixis). Initially, we confirmed that these doses caused no significant changes in weight and appearance in normal mice when given from ages 6 to 10–12 weeks (data not shown). We then administered the drugs for approximately 8 weeks starting at age 9 weeks (after one dose of 4 Gy X-ray irradiation at age 8 weeks), after which a dorsal skin biopsy was taken and processed for microscopic BCC analyses. As a positive control for this study, we treated three Ptch1+/− K14-CreER2 p53floxed/floxed mice topically with tazarotene and confirmed its efficacy in reducing BCC number and size (data not shown). Of the three PI3K inhibitors used in this study, we found that only XL765 inhibited both microscopic BCC number (p < 0.001) and size (p < 0.05) (Figures 5A and B) by a statistically significant amount.
Figure 5. In vivo testing of therapeutically viable small molecule inhibitors of PI3K/AKT pathway for treating BCC.

(A, B) In vivo testing of therapeutically viable PI3K/AKT pathway inhibitors against microscopic and visible BCC assessment at age 21 and 28 weeks respectively, after 8 weeks of PI3K inhibitor treatment. For microscopic BCC assessment, dorsal skin biopsies of mice given oral formulations of PI3K inhibitors XL147, XL765 or GDC-0941 were analyzed to determine the microscopic BCC number (A) and size (B). Only XL765 demonstrated significantly chemopreventive efficacy against BCC number (*** p < 0.001, Student t test) and size (* p < 0.05, Student t test) compared to the vehicle control group. Mean and SEMs are shown.
(C) A Kaplan Meier graph showed the time to the appearance of the first visible BCCs. Only XL765-treated mice had a delay in the first visible BCC appearance, although it was not statistically significant. XL147 and GDC-0941 did not appear to have any effect on the rate of BCC tumorigenesis.
(D, E) Mice were assessed from age 28 weeks to determine the visible BCC number and size, respectively. Again, although different control vehicles were used, there were not significantly different from each other in terms of BCC numbers and size (data not shown), therefore the vehicle groups were combined. Only XL765 treatment reduced visible BCC number and volume by statistically significant amounts at the doses tested (* p < 0.05, Student t test). Mean and SEMs are shown.
Next, we assessed whether the PI3K inhibitors had a sustained effect on tamoxifen/ionizing radiation-induced BCC growth in Ptch1+/− K14-CreER2 p53floxed/floxed mice not further treated (26). As expected, the vehicle control-treated mice developed visible BCCs from age 20 weeks, and by age 28 weeks almost all mice had a significant burden of macroscopic BCCs (Figures 5C and D). XL147 or GDC-0941 did not delay the appearance of the first visible BCC. By contrast, XL765 treatment did delay the appearance of the first visible BCC, although the delay was not statistically significant (Figure 5C). Treatment with XL765, albeit not with XL147, GDC-0941 or vehicle, significantly reduced the visible BCC number (p < 0.05) (Figure 5D). All three PI3K inhibitors reduced visible BCC size (Figure 5E). XL765 treatment produced the largest reduction (p < 0.05). GDC-0941- and XL147-treated mice also had tumors at least 50% smaller in size but this reduction did not attain statistical significance in this study.
PI3K inhibitor slows growth of Tazarotene-resistant BCC allografts
To assess whether PI3K inhibitors might reduce growth of tazarotene-resistant BCCs, we utilized our allograft model in NOD/SCID mice. We treated orally a mouse bearing an allograft made from a BCC from a Ptch1+/− K14-CreER2 p53 fl/fl mouse (that had been treated with ionizing radiation for mutagenesis and with tamoxifen to activate Cre) initially with oral tazarotene, initially at a high dose (10mg/kg 5 days/week) for three weeks and then with a lower dose (2mg/kg 5 days/week) for 8 weeks. We then transplanted cells from a tazarotene-resistant tumor into three mice and compared the growth rates when the individual recipient mice were treated with vehicle, tazarotene (5mg/kg 5 days/week), or XL-765 (30mg/kg twice daily 5 days/week) starting when the secondary transplants first became palpable (week 7). During the ensuing 10 weeks the XL-765 tumors grew and then stabilized whereas the tumors on the other two mice grew large enough to require euthanasia of the mice (Figure 6). The tazarotene-resistant tumor allografts retained imunohistochemical expression of RARγ and RXRα, and the cause of their resistance to the anti-proliferative effect of tazarotene is unknown.
Figure 6. PI3K inhibitor restrains the growth of tazarotene-resistant BCC allografts.

NOD/SCID mice bearing palpable tazarotene-resistant BCC allografts were treated orally with vehicle, tazarotene (5mg/kg daily, 5 days per week), or XL765 (30mg/kg twice daily, 5 days per week). Mice treated with XL765 had a reduction in tumor volume.
Overall, these data suggest that pharmacologic inhibition of PI3K/AKT/mTOR signaling can significantly inhibit the development of both microscopic and visible murine BCCs, that at the single dose of each tested, XL765 was considerably more effective than the other inhibitors tested, and that tumors resistant to tazarotene may retain susceptibility to PI3K inhibitors.
Discussion
We demonstrated previously the remarkable efficacy of tazarotene against murine BCC carcinogenesis, effects that likely are mediated via RARγ-mediated transcriptional activation of RA-target genes (8, 9). In this study we have identified downstream mechanisms of tazarotene’s anti-BCC effects by analyzing in vitro changes in global gene expression in a murine BCC cell line. Specifically, we found (i) gene expression changes in vitro suggesting that inhibition of the PI3K-AKT signaling pathway is a central node downstream of retinoid treatment, (ii) as in human BCCs, pathway activation in visible BCCs in our murine BCC model and in the ASZ001 murine BCC cell line, (iii) reduction of tazarotene’s anti-proliferative effects by hyperactivation of the PI3K-AKT pathway in ASZ001 cells, and (iv) reduction of both ASZ001 cell proliferation in vitro and, crucially, BCC carcinogenesis in vivo by small molecule PI3K-AKT inhibitors. Thus it is reasonable to expect that PI3K inhibitors could overcome resistance to retinoid therapy in BCCs whose resistance is due to loss of RAR expression.
Interactions between HH and IGF-IR/PI3K/AKT signaling in cancers have been identified, One possible mechanistic explanation is that the HH and IGF/PI3K/AKT pathways converge to co-regulate downstream targets. Thus, for example, HH signaling enhances N-myc expression, and PI3K/AKT signaling reduces N-myc phosphorylation and its proteolytic destruction by inhibiting GSK3 activity (27). N-myc is over-expressed not only in HH driven medulloblastomas but also in human BCCs (28). By contrast, some data indicate direct interactions between the two pathways. Thus HH signaling can enhance Igf2 transcription in some, albeit not all, contexts (29, 30) and PI3K/AKT/mTOR signaling, can enhance Gli transcriptional activity by reducing GSK3-mediated phosphorylation and degradation and by phosphorylation-mediated release from binding to the suppressor SuFU (17, 31–33) and can reduce the development of resistance to the anti-tumor effects of hedgehog inhibitors (34, 35). Contrary to our finding that the retinoid tazarotene inhibits IGF-IR/PI3K/AKT/mTOR signaling, in some contexts ATRA (which, like tazarotene, inhibits murine BCC carcinogenesis) activates PI3K/AKT signaling to induce a differentiation program (36–38). Therefore, the relationship between retinoid and PI3K/AKT signaling pathways appears to be complex and cell-type specific, but at least in murine BCCs inhibition of PI3K/AKT signaling inhibits tumor development. Thus our study not only points to enhanced PI3K/AKT signaling as a target for anti-BCC strategies but also suggests that results of topical tazarotene vs. human BCCs may already have been a “proof of principle” of this idea. Of note, mTOR inhibition appears to be effective against cutaneous carcinogenesis in at least some contexts (39, 40). Of the PI3K inhibitors we assessed, the one with the greatest in vivo anti-BCC efficacy was XL765, the only one tested that inhibits not only PI3K but also mTOR and DNA-PK. Although we do not know whether the drugs used inhibited PI3K equivalently at the single doses tested in vivo and whether the effects of XL765 were mediated in part by mechanisms other than direct inhibition of PI3K, the finding that inhibition of multiple targets in the PI3K/AKT/mTOR pathway may have better anti-tumor efficacy than does inhibition of a single target is not a new one (41, 42).
In summary, our data suggest (i) that the PI3K/AKT/mTOR pathway is a positive effector of BCC carcinogenesis that is inhibited by tazarotene and (ii) that targeting this pathway directly may inhibit BCC carcinogenesis in tumors that are resistant to tazarotene. Also, our data suggest that short-term inhibition of this pathway can continue to inhibit BCC carcinogenesis even after treatment is completed. Therefore it might be possible to treat and/or prevent BCCs using PI3K inhibitors for a relatively short duration, thereby avoiding potential toxic side effects (e.g. interference with glucose metabolism and the immune response) that occur with the chronic use of these agents.
Supplementary Material
Acknowledgements
We thank Drs. Chris Barker, Agnes Pagnet and Yuan Yuan Xiao of the Gladstone Genomic Core facility for the microarray hybridization and primary data analyses; members of the Epstein Lab – Yefim Khaimskiy, Ben Yarin, Serena Sam, Kevin Sibucao, and Kris Chang - for their help with animal husbandry and general lab assistance; Jean Y. Tang for sharing and discussing unpublished data; Allergan for gifts of tazarotene, Douglas Laird and Exelixis for the gifts of XL147 and XL765; Lori Friedman and Genentech for the gift of GDC-0941; Alan Balmain for the gift of the C5N cell line; and Pierre Chambon and Rene Bernards for the K14CreERT2 and p53 floxed mice. We also thank Steve Martin for the use of his lab and reagents and the Sellers lab for providing the AKT1 constructs via Addgene.
Grant Support
This work was supported by NIH grant R01CA109584 (E. Epstein) and by donations from the Michael J Rainen Family Foundation. V. Calinisan Chiueh was supported by the UC Berkeley Cancer Research Laboratory.
Footnotes
Conflict of Interest: The authors have declared that no conflict of interest exists.
References
- 1.Rigel DS. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J Am Acad Dermatol. 2008;58:S129–S132. doi: 10.1016/j.jaad.2007.04.034. [DOI] [PubMed] [Google Scholar]
- 2.Epstein EH. Basal cell carcinomas: attack of the hedgehog. Nat Rev Cancer. 2008;8:743–754. doi: 10.1038/nrc2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aszterbaum M, Epstein J, Oro A, Douglas V, LeBoit PE, Scott MP, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999;5:1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- 4.Mancuso M, Pazzaglia S, Tanori M, Hahn H, Merola P, Rebessi S, et al. Basal cell carcinoma and its development: insights from radiation-induced tumors in Ptch1-deficient mice. Cancer Res. 2004;64:934–941. doi: 10.1158/0008-5472.can-03-2460. [DOI] [PubMed] [Google Scholar]
- 5.Schmuth M, Watson RE, Deplewski D, Dubrac S, Zouboulis CC, Griffiths CE. Nuclear hormone receptors in human skin. Horm Metab Res. 2007;39:96–105. doi: 10.1055/s-2007-961808. [DOI] [PubMed] [Google Scholar]
- 6.Wright TI, Spencer JM, Flowers FP. Chemoprevention of nonmelanoma skin cancer. J Am Acad Dermatol. 2006;54:933–946. doi: 10.1016/j.jaad.2005.08.062. quiz 47–50. [DOI] [PubMed] [Google Scholar]
- 7.Nagpal S, Chandraratna RA. Recent developments in receptor-selective retinoids. Curr Pharm Des. 2000;6:919–931. doi: 10.2174/1381612003400146. [DOI] [PubMed] [Google Scholar]
- 8.So PL, Fujimoto MA, Epstein EH., Jr Pharmacologic retinoid signaling and physiologic retinoic acid receptor signaling inhibit basal cell carcinoma tumorigenesis. Mol Cancer Ther. 2008;7:1275–1284. doi: 10.1158/1535-7163.MCT-07-2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.So PL, Lee K, Hebert J, Walker P, Lu Y, Hwang J, et al. Topical tazarotene chemoprevention reduces Basal cell carcinoma number and size in Ptch1+/− mice exposed to ultraviolet or ionizing radiation. Cancer Res. 2004;64:4385–4389. doi: 10.1158/0008-5472.CAN-03-1927. [DOI] [PubMed] [Google Scholar]
- 10.Bianchi L, Orlandi A, Campione E, Angeloni C, Costanzo A, Spagnoli LG, et al. Topical treatment of basal cell carcinoma with tazarotene: a clinicopathological study on a large series of cases. Br J Dermatol. 2004;151:148–156. doi: 10.1111/j.1365-2133.2004.06044.x. [DOI] [PubMed] [Google Scholar]
- 11.So PL, Langston AW, Daniallinia N, Hebert JL, Fujimoto MA, Khaimskiy Y, et al. Long-term establishment, characterization and manipulation of cell lines from mouse basal cell carcinoma tumors. Exp Dermatol. 2006;15:742–750. doi: 10.1111/j.1600-0625.2006.00465.x. [DOI] [PubMed] [Google Scholar]
- 12.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 13.Ramaswamy S, Nakamura N, Vazquez F, Batt DB, Perera S, Roberts TM, et al. Regulation of G1 progression by the PTEN tumor suppressor protein is linked to inhibition of the phosphatidylinositol 3-kinase/Akt pathway. Proc Natl Acad Sci U S A. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rochette-Egly C, Germain P. Dynamic and combinatorial control of gene expression by nuclear retinoic acid receptors (RARs) Nucl Recept Signal. 2009;7:e005. doi: 10.1621/nrs.07005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xu L, Glass CK, Rosenfeld MG. Coactivator and corepressor complexes in nuclear receptor function. Curr Opin Genet Dev. 1999;9:140–147. doi: 10.1016/S0959-437X(99)80021-5. [DOI] [PubMed] [Google Scholar]
- 16.Katoh Y, Katoh M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med. 2009;9:873–886. doi: 10.2174/156652409789105570. [DOI] [PubMed] [Google Scholar]
- 17.Stecca B, Mas C, Clement V, Zbinden M, Correa R, Piguet V, et al. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc Natl Acad Sci U S A. 2007;104:5895–5900. doi: 10.1073/pnas.0700776104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie J, Aszterbaum M, Zhang X, Bonifas JM, Zachary C, Epstein E, et al. A role of PDGFRalpha in basal cell carcinoma proliferation. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:9255–9259. doi: 10.1073/pnas.151173398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 20.Lin N, Moroi Y, Uchi H, Fukiwake N, Dainichi T, Takeuchi S, et al. Significance of the expression of phosphorylated-STAT3, -Akt, and -ERK1/2 in several tumors of the epidermis. J Dermatol Sci. 2007;48:71–73. doi: 10.1016/j.jdermsci.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 21.Gharbi SI, Zvelebil MJ, Shuttleworth SJ, Hancox T, Saghir N, Timms JF, et al. Exploring the specificity of the PI3K family inhibitor LY294002. Biochem J. 2007;404:15–21. doi: 10.1042/BJ20061489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foster PG. Exelixis. Potentiating the Antitumor Effects of Chemotherapy With the Selective PI3K Inhibitor XL147. AACR-NCI-EORTIC International Conference, Molecular Targets and Cancer Therapeutics Discovery, Biology, and Clinical Applications; October 22–26, 2007.2007. [Google Scholar]
- 23.Martelli AM, Tazzari PL, Tabellini G, Bortul R, Billi AM, Manzoli L, et al. A new selective AKT pharmacological inhibitor reduces resistance to chemotherapeutic drugs, TRAIL, all-trans-retinoic acid, and ionizing radiation of human leukemia cells. Leukemia. 2003;17:1794–1805. doi: 10.1038/sj.leu.2403044. [DOI] [PubMed] [Google Scholar]
- 24.Patnaik A, LoRusso P, Tabernero J, Laird D, Aggarwal K, Papadopoupos K. Biomarker development for XL-765, a potent and selective oral dual inhibitor of PI3K and mTOR currently being administered to patients in a Phase I clinical trial. Presented at: 19th EORTC-NCI-AACR symposium on molecular targets and cancer therapeutics; San Francisco, CA, USA. 2007. [Google Scholar]
- 25.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin -4-yl-thieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. J Med Chem. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 26.Wang GY, Wang J, Mancianti ML, Epstein EH., Jr Basal cell carcinomas arise from hair follicle stem cells in Ptch1(+/−) mice. Cancer Cell. 2011;19:114–124. doi: 10.1016/j.ccr.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kenney AM, Widlund HR, Rowitch DH. Hedgehog and PI-3 kinase signaling converge on Nmyc1 to promote cell cycle progression in cerebellar neuronal precursors. Development. 2004;131:217–228. doi: 10.1242/dev.00891. [DOI] [PubMed] [Google Scholar]
- 28.Freier K, Flechtenmacher C, Devens F, Hartschuh W, Hofele C, Lichter P, et al. Recurrent NMYC copy number gain and high protein expression in basal cell carcinoma. Oncol Rep. 2006;15:1141–1145. [PubMed] [Google Scholar]
- 29.Ingram WJ, Wicking CA, Grimmond SM, Forrest AR, Wainwright BJ. Novel genes regulated by Sonic Hedgehog in pluripotent mesenchymal cells. Oncogene. 2002;21:8196–8205. doi: 10.1038/sj.onc.1205975. [DOI] [PubMed] [Google Scholar]
- 30.Villani RM, Adolphe C, Palmer J, Waters MJ, Wainwright BJ. Patched1 inhibits epidermal progenitor cell expansion and basal cell carcinoma formation by limiting Igfbp2 activity. Cancer Prev Res (Phila) 2010;3:1222–1234. doi: 10.1158/1940-6207.CAPR-10-0082. [DOI] [PubMed] [Google Scholar]
- 31.Mizuarai S, Kawagishi A, Kotani H. Inhibition of p70S6K2 down-regulates Hedgehog/GLI pathway in non-small cell lung cancer cell lines. Mol Cancer. 2009;8:44. doi: 10.1186/1476-4598-8-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Riobo NA, Lu K, Ai X, Haines GM, Emerson CP., Jr Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc Natl Acad Sci U S A. 2006;103:4505–4510. doi: 10.1073/pnas.0504337103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh RR, Cho-Vega JH, Davuluri Y, Ma S, Kasbidi F, Milito C, et al. Sonic hedgehog signaling pathway is activated in ALK-positive anaplastic large cell lymphoma. Cancer Res. 2009;69:2550–2558. doi: 10.1158/0008-5472.CAN-08-1808. [DOI] [PubMed] [Google Scholar]
- 34.Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Ding Q, Yen CJ, Xia W, Izzo JG, Lang JY, et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell. 2012;21:374–387. doi: 10.1016/j.ccr.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bastien J, Plassat JL, Payrastre B, Rochette-Egly C. The phosphoinositide 3-kinase/Akt pathway is essential for the retinoic acid-induced differentiation of F9 cells. Oncogene. 2006;25:2040–2047. doi: 10.1038/sj.onc.1209241. [DOI] [PubMed] [Google Scholar]
- 37.Doi T, Sugimoto K, Ruttenstock E, Dingemann J, Puri P. Prenatal retinoic acid upregulates pulmonary gene expression of PI3K and AKT in nitrofen-induced pulmonary hypoplasia. Pediatr Surg Int. 2010;26:1011–1015. doi: 10.1007/s00383-010-2654-x. [DOI] [PubMed] [Google Scholar]
- 38.Lopez-Carballo G, Moreno L, Masia S, Perez P, Barettino D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem. 2002;277:25297–25304. doi: 10.1074/jbc.M201869200. [DOI] [PubMed] [Google Scholar]
- 39.Athar M, Kopelovich L. Rapamycin and mTORC1 inhibition in the mouse: skin cancer prevention. Cancer prevention research. 2011;4:957–961. doi: 10.1158/1940-6207.CAPR-11-0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Euvrard S, Morelon E, Rostaing L, Goffin E, Brocard A, Tromme I, et al. Sirolimus and secondary skin-cancer prevention in kidney transplantation. The New England journal of medicine. 2012;367:329–339. doi: 10.1056/NEJMoa1204166. [DOI] [PubMed] [Google Scholar]
- 41.Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007;67:7960–7965. doi: 10.1158/0008-5472.CAN-07-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fan QW, Weiss WA. Targeting the RTK-PI3K-mTOR axis in malignant glioma: overcoming resistance. Curr Top Microbiol Immunol. 2010;347:279–296. doi: 10.1007/82_2010_67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.