

Abstract
We have devised a class of isothermal reactions for amplifying DNA. These homogeneous reactions rapidly synthesize short oligonucleotides (8–16 bases) specified by the sequence of an amplification template. Versions of the reactions can proceed in either a linear or an exponential amplification mode. Both of these reactions require simple, constant conditions, and the rate of amplification depends entirely on the molecular parameters governing the interactions of the molecules in the reaction. The exponential version of the reaction is a molecular chain reaction that uses the oligonucleotide products of each linear reaction to create producers of more of the same oligonucleotide. It is a highly sensitive chain reaction that can be specifically triggered by given DNA sequences and can achieve amplifications of >106-fold. Several similar reactions in this class are described here. The robustness, speed, and sensitivity of the exponential reaction suggest it will be useful in rapidly detecting the presence of small amounts of a specific DNA sequence in a sample, and a range of other applications, including many currently making use of the PCR.
The invention of the PCR changed the practice of molecular biology. It has become a mainstay of biological research and diagnostics in providing a method for the rapid detection, isolation, and measurement of DNA sequences through their specific amplification. There are currently two widely used methods for amplifying specific DNA sequences: PCR (1, 2) and the rolling-circle amplification method (3–5). The PCR method is the simpler and more flexible of these and has the added advantage of being geometric rather than linear in character, so that amplification levels of 106 or more can be achieved. It is by far the most widely used amplification method in biology. It has the disadvantage relative to the isothermal rolling-circle amplification method, however, of needing a temperature cycling protocol to achieve amplification. This imposes instrumentation constraints on the PCR method that make it more complex and limit the rate of the amplification to the temperature cycling schedule. Another limitation of the rate of PCR derives from the nature of the reaction itself in that a maximum 2-fold amplification can be achieved in each cycle. Advances in speed, accuracy, and sensitivity, in addition to simplicity, would be most welcome for applications in biology and medicine.
We report here a class of isothermal reactions for amplifying DNA that overcomes all the above disadvantages of PCR. This class includes a linear amplification method, which is fundamental to the others, and several versions of an exponential amplification scheme. These reactions are simple, flexible, and require no special cycling of conditions. They depend entirely for their rate of amplification on the molecular parameters governing the interactions of the molecules in the reaction. Because of the balance between the thermal properties of the DNA oligonucleotides and the enzymes used, the optimum temperature of the reaction with these enzymes is 60°C (see Materials and Methods). The exponential version of the method, designated the exponential amplification reaction (EXPAR), is an isothermal molecular chain reaction in that the products of one reaction catalyze further reactions that create the same products.
Materials and Methods
Oligonucleotides and Enzymes.
Sequences used in experiments described in the text:
Oligonucleotides used in the linear reaction as described in Figs. 1 and 2: ITAtop, 5′-CCGATCTAGTGAGTCGCTC-3′; NBbt12, 5′-ACGACTGGAACTGAGCGACTCACTAGATCGG-3′; NBbt16, 5′-ACCTACGACTGGAACTGAGCGACTCACTAGATCGG-3′; NBbt20, 5′-TGAAACCTACGACTGGAACTGAGCGACTCACTAGATCGG-3′.
Fig 1.

(a) The cycle of the synthesis and release of the amplified oligonucleotide is shown schematically. On the upper strand is indicated the recognition site for the enzyme N BstNB (5′-GAGTC-3′) and the specific nicking site four bases downstream on this strand. The oligonucleotide produced is indicated in blue, the primer in green, and the template in red. The lengths of the template and amplified oligo are shown (Upper Left). (b) The results of a linear amplification reaction where the primer template produces a 12 mer as the full-length product. The primer template was present at 1 μM in a 50-μl reaction (see Materials and Methods), and the yield of the reaction products is shown. The duplex used a top strand (ITAtop) of 16 nucleotides and a bottom strand (NBbt12) of 28 nucleotides that produced a 12 mer.
Fig 2.

Exponential amplification reactions. (a) Diagram of the reaction scheme for the exponential amplification of oligonucleotides. The segments in red represent the sequence complement of the oligonucleotide sequence to be amplified, the signal sequence (shown in blue). The amplification template, t, consists of two copies of the signal complement flanking the nicking enzyme recognition site, shown as a light blue box, and a spacer sequence, shown as a green segment. The signal oligonucleotide (labeled σ) is produced in the linear amplification cycle for each amplification template created. The labels on each structure in the figure correspond to the symbols used for their concentrations in the equations. (b) MS measurement results for the reaction. The oligonucleotide concentration (M) of the oligonucleotide (σ in the equations) was measured as described in Materials and Methods. The initial point is not measurable in the mass spectrometer and is the initial concentration introduced into the reaction. The template oligos and trigger oligos are shown in Materials and Methods. Solution of the differential equations in the text describing the mass-action kinetics of the reaction scheme shown in a. The kinetic parameters used for the solution (see Materials and Methods) were: r = 0.4 sec−1; a = 2 × 10−5 M−1·sec−1; c = 2 sec−1. The theoretical curves are shown as heavy lines. Parameter c was chosen to give a reasonable fit to the data, although the curve is not very sensitive to this parameter. The other parameters are determined as described. The initial (trigger) concentrations were chosen to match the curves in b. The σ curve for the higher concentration of trigger (10−11) is indicated by the blue line. The curve of σ for the lower concentration corresponds to the lower curve (green). (c) “Real-time” fluorescence monitoring of the EXPAR reaction. The reaction was carried out under the conditions of Fig. 3. The trigger oligonucleotide, σ, was present at 10−5 μM at time 0. The fluorescence of SYBR green was monitored every 30 sec in six independent identical reactions. The error bars indicate the standard deviations of these reactions at each time point.
Oligonucleotides used in the exponential reaction described in Figs. 3 and 4: template oligo, ceap, 5′-CCTACGACTGGaacaGACTCACCTACGACTGGAP-3′; trigger, seqS, 5′-ACCAGTCGTAGG-3′ (spacer bases are indicated in lowercase; P indicates phosphate group; the nicking enzyme site or its complement is indicated by an underline in all above sequences).
Fig 3.

Triggering mechanisms for the EXPAR chain reaction. Schematic representation of a mechanism for producing the initial oligonucleotides from naturally occurring nicking sites in targeted DNA. The trigger template (green) is made up of sequences matching the target DNA shown in yellow (s0, s1, the nicking site and the 4-base spacer). A tilde over a sequence symbol indicates its complement.
Fig 4.

Two alternative EXPAR schemes that can be used for different applications. “Direct EXPAR” is the scheme described in Fig. 3 in shorthand form, in which the trigger sequence (blue) is exponentially amplified using the template (red). The “copy EXPAR” scheme consists of two parts. The upper bracket represents a template with a nicking site in the reverse orientation, relative to those in Fig. 3 and Left. This template amplifies the complement of the triggering sequence (including the 5′ overhang). The lower bracket represents the exponential amplification of that complement, now containing a copy of the 5′ overhang on its 3′ end (described in the text). The bases represented by the yellow and purple circles in the copy EXPAR section indicate complementary bases. The base represented by the green circle in template b indicates another base variant.
Note that the trigger oligo above (seqS) is one base longer than that produced by the primer template. This enables us to distinguish the initial trigger from the amplified sequence and does not affect its ability to prime effectively.
Oligonucleotides were synthesized by Midland Certified Reagent Company (Midland, TX), MWG Biotech (High Point, NC), or Sigma–Genosys (The Woodlands, TX). The oligonucleotides were routinely checked by time-of-flight MS [using LCT from Micromass (Manchester, U.K.); see below].
All enzymes were purchased from New England Biolabs. The DNA polymerase used was Vent exo- (6, 7). The nicking enzyme (N.BstNBI) has a specific activity of ≈106 units/mg (H.-M. Kong, personal communication).
All HPLC components (water and acetonitrile) were purchased from Fisher Scientific. Dimethyl-butylamine was purchased from Sigma–Aldrich, and a salt was made by addition of acetic acid (Sigma–Aldrich) to pH 7.1. The 2 M stock solution was filtered by using a 0.2-μm nylon filter.
Linear Amplification Reaction.
The conditions for the linear reaction were: 85 mM KCl/25 mM Tris⋅HCl (pH 8.8, 25°C)/2.0 mM MgSO4/5 mM MgCl2/10 mM (NH4)2SO4/0.1% (vol/vol) Triton X-100/0.5 mM DTT/0.4 units/μl N.BstNBI nicking enzyme/0.05 units/μl Vent exo− polymerase/400 μM dNTPs (Epicentre, Madison, WI)/10 μg/ml BSA/0.05 μM template and primer olignucleotides [NBtop and NB12 (equimolar) in ultrapure water that is nuclease-free (Ambion, Austin, TX)]. These conditions correspond to 1 part Thermopol buffer and 0.5 parts N.BstNBI buffer as supplied by New England Biolabs. Reactions were assembled at 4°C, initiated by transferring to a preheated thermocycler at 60°C, and stopped by incubation at 4°C. No further manipulations were performed before placement on the autoinjector for the HPLC-MS, which is held at 4°C.
Exponential Amplification Reaction.
The exponential reactions were also carried out at 60°C, temperature controlled to within 0.1°C. The exponential reaction conditions were as follows: the same as described above for the linear reaction except with 0.1 μM template oligonucleotide only (unless otherwise noted). Triggering oligonucleotides were added as described for each experiment. In the case of fluorescence monitoring, SYBR green (Molecular Probes) was added to 5× concentration (SYBR green is supplied by the manufacturer at 10,000×).
Chromatography and MS.
The chromatography system was an Agilent (Palo Alto, CA) 1100 Series HPLC composed of a binary pump, degasser, a column oven, a diode array detector, and thermostated microwell plate autoinjector. The column is a Waters Xterra MS C18, incorporating C18 packing with 3.5 μM particle size, with 125-Å pore size, 2.1 mm × 20 mm. The column was run at 30°C with a gradient of acetonitrile in 5 mM dimethyl-butylamine acetate (DMBAA). As a check on the complete release of the signal oligo during the chromatography and injection, we ran the column at 50°C after incubating the sample briefly at 95°C. We saw no increase in the oligo yield over our standard conditions. Buffer A is 5 mM DMBAA, and buffer B is 5 mM DMBAA and 50% (V/V) acetonitrile. The MS was a Micromass LCT time-of-flight instrument. Samples were run in electrospray negative mode, ranging from 800 to 2,000 amu, 1-sec scan time. Analysis of the HPLC-MS data made use of the software supplied by the manufacturer.
Oligonucleotides are known to exhibit different ionization efficiencies, which in our measurements would be translated into sequence-specific differences in measured oligo concentration. A survey of a range of >80 different 12 mers indicated that the variation between sequences attributable to this difference is <30%. Almost all relevant quantitative comparisons are with the same oligo sequence. It is necessary, however, to calibrate for ionization efficiencies for quantitative comparisons between different sequences.
Real-Time Fluorescence Measurement.
All fluorescence measurements reported here were made on an MJ Opticon instrument (MJ Research, Waltham, MA) by using software supplied by the manufacturer. The real-time measurements on this instrument were made by using an isothermal protocol with a 30-sec interval read beginning 10 sec after the lid and chamber reached 60°C.
The Exponential Reaction Equations.
The simplified mass action equations use the following variables: a, the annealing rate between the product oligonucleotide concentration, σ, and the amplification template concentration, τ; χ, the concentration of the transient complex between σ and τ; π, the concentration of the primer template formed by extension of the complex; c, the rate of conversion of χ to π; r, the rate of oligonucleotide production (σ) by each primer template. The equations, using the simplifying assumptions that annealing is a single-step bimolecular reaction and that the conversion of χ into π can be represented as a simple effective rate, are then,
 |
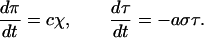 |
σ can easily be shown from these equations to exhibit exponential behavior. The exponential phase occurs before the template becomes depleted, but after π reaches a steady ratio with χ. In this regime, the equation for σ has the approximate solution, σ ≈ σoeβt, where β = (τ(0)ar/2)1/2 − aτ(0).
A more direct method is simply to solve the above equations computationally using a direct finite difference method. The results of a computational solution of the equations in Fig. 2b show clearly the regime in which the exponential solution applies.
Results
Linear Amplification.
To produce an amplification reaction, we need to devise a cyclic chain of reactions that will restore the reactants to their initial state after each synthesis of the molecule to be amplified. The linear amplification reaction described here provides such a cycle whose sequence specificity derives from template-dependent synthesis of the oligonucleotide to be amplified. The reaction synthesizes short oligonucleotides whose cycle of reactions depends on the idea that, at the reaction temperature, oligonucleotides above a certain length form stable duplexes, whereas those below this critical length form unstable duplexes that dissociate readily. By arranging a specific single-strand nicking site and nicking enzyme and a compatible DNA polymerase (6, 7) as described in Fig. 1, a cycle of polymerization and subsequent oligonucleotide release is created. This cycle depends on the nicking reaction cleaving a phosphodiester bond to create an oligonucleotide that is below the threshold of stability in a duplex and is thereby released from the duplex, thus regenerating the initial primer template. The synthesized oligonucleotide is fully stable at 60°C when it is covalently joined to the rest of the upper stand, as it is immediately after its synthesis, but is only transiently stable as a 12 mer after the nicking reaction. Therefore, when the bond is cleaved at the nicking site, the oligonucleotide dissociates recreating a primer template, ready for elongation. This cycle thus generates oligonucleotides that are complementary to the template beyond the nicking site (shown in blue in Fig. 1a).
When the nicking enzyme is present with a compatible polymerase, the reaction proceeds around the cycle shown in Fig. 1a, and amplification of the product oligonucleotide occurs. In Fig. 1b, we show the results of one of these reactions. The experiment was devised to produce a 12 mer as its amplified product. The products of the reaction were analyzed on the LC-MS system after the indicated incubation times at 60°C (see Materials and Methods). Because the exact masses of all of the relevant molecules are known, the relative concentrations of all of the components, including the amplified oligonucleotide, can be directly measured. The yield of oligonucleotide is perhaps best characterized in this case as the number of molecules produced per primer template per second. For the experiment shown in Fig. 1, this initial rate is about one molecule (12 mer) per primer template every 2.5 sec, or ≈0.4 molecules per primer template ⋅ sec. Note that the reaction slows down noticeably after 10 min or so. This is consistent with the reaction rate declining exponentially, as if an essential component of the reaction is being inactivated. We expect the nicking enzyme is responsible, as the optimum temperature (≈55°) of the enzyme is lower than the 60°C of the reaction, and preliminary experiments show a clear difference in the rate decline between different starting nicking enzyme concentrations: more enzyme makes the reaction stay linear longer. Further experiments, however, are needed to verify this hypothesis. An extensive set of experiments (data not shown) show that the absolute initial rate of the reaction is proportional to the primer-template concentration, as expected, over a wide range of concentrations. The balance between the nicking enzyme and the DNA polymerase is more complex.
To investigate this relationship, we examined the dependence of the reaction yield (12-mer product) on the amounts of the two enzymes. It is clear that the reaction is completely dependent on the presence of both enzymes, the template, and the primer oligonucleotide (data not shown), but the yield is a complex function of the amounts of both enzymes. What we find is that for small amounts of NE, there is a broad range of low reaction yields. At higher NE concentrations, there is a sharp maximum as a function of polymerase concentration. In addition, it is clear from the data (published as supporting information on the PNAS web site, www.pnas.org) that we can modulate the yield of partial products by changing the ratio of the enzymes. Although we do not know precisely how the enzymes interact, cooperate, or compete with one another, it is clear that there are optimal concentration ranges of both enzymes. In amplifying an oligonucleotide, we see that the extension leads to some partial products (see supporting information on the PNAS web site) identified by their masses to be the result of incomplete elongation of the primer to the full length of the template. The reaction favors 12 mers as partial products for reasons not well understood but may have to do with the structural details of the DNA–polymerase complex for this distributive polymerase (7). Tuning the reaction conditions via the enzyme concentrations thus appears to be important for maximizing the yield of any particular product.
Exponential Amplification.
We have devised a simple way to use the above-described linear amplification to create an exponential amplification reaction. It has several variants that can be adapted for different uses. The key idea is to arrange it so that the oligonucleotide product of the linear reaction serves to create a new primer that in turn anneals to a target template and creates a new primer template, which in turn produces more of the same oligonucleotide product, creating a chain reaction. Our simplest scheme for doing this is depicted in Fig. 2a. The scheme depends on our observation that even though the product oligonucleotide is unstable as a duplex, it will form a transient duplex molecule with its complement, and this transient duplex can act as a primer for extension by the DNA polymerase. Once extension of the oligonucleotide has occurred, the duplex is stabilized by the additional complementary duplex section and will not readily dissociate. Extending the primer thus creates a stable primer template that will produce oligonucleotide products in a linear fashion (Fig. 1). To create these new duplexes, we need only provide a ready supply of complementary oligonucleotides we call amplification templates. The key feature of these single-stranded oligonucleotides is that they contain two copies in tandem of the complement of the oligonucleotide product to be amplified, separated by the complement of the nicking enzyme recognition site (3′-CTCAG-5′) and a four-base spacer (on the 5′ side). When the transient duplex (formed with the first of these copies, by hybridization with the complementary oligonucleotide) is extended, a stable new primer template is created. This primed template will then continue to produce oligonucleotide product via the linear amplification cycle as described above (nicking after the four-base spacer, dissociating the oligonucleotide, and reelongating the primer) as long as the enzymes remain active and dNTPs are available. One might ask what happens when a transient duplex is formed with the second copy of the complementary sequence (Fig. 1 Right). The thermodynamics of the situation are essentially the same as after the extended product has been nicked. The key difference is that there can be no extension to stabilize the duplex by elongating it, because it provides no primer template structure for the polymerase, and it rapidly dissociates. If the amplification templates are present at a high concentration (experiments reported here use 0.01–0.1 μM amplification template oligos), we can rapidly create primer-template structures that will produce product oligonucleotide at an accelerating rate. In our reactions, we take the important precaution of blocking the 3′ ends of the template oligonucleotides (with 3′ PO4 groups, for all of the experiments reported here, or tethered amines) to prevent spurious self-priming by pairs of template molecules. We have seen no such spontaneous priming in any of our experiments to date (data not shown). Finally, it is clear that when all of the template has been converted into primer template, the exponential reaction kinetics must shift to a linear amplification mode.
To examine the kinetics of amplification, we carried out the full exponential reaction in the presence of differing initial amounts of amplifying oligonucleotide (σ in Fig. 2a) and measured the amounts of σ at a number of time points with the mass spectrometer. We find that the oligonucleotide amplifies approximately exponentially for the first 2 min or so, as shown by the data points in Fig. 2b. Note that this amplification proceeds approximately exponentially until the concentration of σ approaches the concentration of the template pool. After this point, it proceeds in an approximately linear fashion, as expected. The total amplification of ≈106 to 107 in shown in Fig. 2. In end-point measurements from the same reaction, for a range of starting concentrations, we find that the amplification levels are all in the range of 106.
Although it may be intuitive that there will be an approximately exponential increase in the product oligonucleotide in a chain reaction that proceeds as described, it is informative to look carefully at the mass action reaction equations. If we write out these equations making the simplest assumptions, we can show that indeed the kinetics of product generation are predicted to be exponential in character, while the template lasts (see Materials and Methods). Solutions of the mass action equations using parameters estimated from our experimental results are shown as heavy lines in Fig. 2b.
Because the EXPAR reaction is rapid and simple, it is potentially appealing as a “real-time” reaction in which the amplification is monitored in the reaction volume during the reaction itself. To test this possibility, we carried out the reaction as described above with the addition of a “double-strand-specific” dye, SYBR-green, to the reaction (see Materials and Methods), and the reaction was carried out on a temperature-controlled fluorescence reader with which measurements of fluorescence were made at regular time intervals. The fluorescence in this case is generated during the amplification reaction, not by the presence of the amplified oligonucleotide itself, but rather from the double-stranded primer templates produced during the reaction (π in Fig. 2a). The results of this experiment are shown in Fig. 2c. MS measurements show that the amplification in this case was ≈106- to 107-fold.
Triggering Mechanisms.
To initiate the exponential reaction, we need to produce from the sample the first few oligonucleotide molecules to form the first primer templates that then will start generating the amplified signal oligonucleotide. The initial oligonucleotide products must be accurate representations of the sequence to be amplified. The mechanism by which these first few oligonucleotides are produced is called the triggering mechanism for EXPAR. There are several ways to do this, each of which requires that we provide a 3′OH group-terminated strand of DNA that can anneal with a complementary template to form a primer template with the proper configuration and sequence. One of these triggering schemes, and probably the simplest, relies on the natural occurrence of nicking enzyme recognition sites (5′-GAGTC-3′) in the DNA of interest. For example, as is shown in Fig. 3, a linear amplifier of a genomic sequence can be created by providing an oligonucleotide (trigger template, shown as the green line) that is complementary to the genomic DNA flanking a specific nicking site. When the trigger template is annealed to the genomic DNA, this creates a structure that the nicking enzyme can convert to a primer-template structure, similar to the linear amplification structure shown in Fig. 1. This structure will then produce oligonucleotide corresponding to the sequence to the right of the nicking site. This oligonucleotide is then used as the trigger for a subsequent exponential amplification reaction. Because the nicking enzyme recognition sequence occurs naturally in both bacterial and human DNA at the expected frequency for a five-base sequence (about 1 in 1,000), potential trigger sites abound in DNA and provide a wide range of target sites for triggering reactions. This simple scheme is the one demonstrated here. Triggering does not depend on occurrence of these sites, but for simplicity they are used in this demonstration of the triggering reaction.
To demonstrate triggering from a naturally occurring nick site, we specifically amplified certain oligonucleotides contained in cDNA. These results show that the reaction is triggered only when the trigger oligonucleotide cognate to the specific cDNA is present (see supporting information on the PNAS web site). When no trigger oligonucleotide is present, no reaction occurs, demonstrating the power of the chain reaction to detect the presence of small amounts of the cDNAs, and the strict dependence on the triggering reaction.
Discussion
The amplification scheme described here appears to have several major advantages for many research and diagnostic applications. These include the isothermal conditions required, the relative speed of the reaction, and the flexibility with which it can be triggered and elaborated into multiple coupled reactions. We have shown clearly that the linear amplification reaction can be turned into a rather simple exponential amplification scheme (EXPAR). The linear reaction itself is quite distinct from the strand displacement amplification scheme (8). It depends fundamentally on the transformation of duplex thermal stability into instability by the cleavage of a phosphodiester bond in the nicking reaction (Fig. 1). We have also demonstrated this essential distinction by experiments physically separating the polymerization step from the nicking and release steps by using the enzymes separately (data not shown).
It is difficult to be sure which step in the reaction as described limits the speed of the overall cycle of amplification and therefore the overall rate of amplification. These issues are being investigated further to optimize the reaction in speed, sensitivity, and accuracy, and adapting the reaction for a wider range of applications. It seems likely that the diffusion and annealing of the product oligonucleotides to the amplification template is a slow step, judging from the diffusion constants of oligonucleotides in this molecular weight range. At low trigger concentrations, this is expected to have a significant effect on the reaction rate, because we expect that the reaction in this case is heterogeneous.
In addition to the triggering reaction demonstrated here, we can also easily construct variant forms of the above triggering reaction using any technique that creates a discrete 3′ end in the target DNA extendable by the polymerase, by using a restriction site, for example. This fragment is then annealed to a trigger template, just as shown in Fig. 5a, except that it contains the nicking site in the single-stranded region of the oligonucleotide. Polymerization of this primed template can then create a duplex nicking site and complete the amplifying structure. The key to creating a trigger to the exponential reaction is simply to make a structure strictly dependent on the target DNA that will linearly amplify a target oligonucleotide, which can be done in many different ways.
Because the reaction is a true molecular chain reaction, once the reaction is triggered, it will proceed without change of the conditions or further stimulus. There is a concern, therefore, that the reaction may spontaneously or spuriously trigger. We take the precaution of blocking the 3′ ends of all templates present to prevent them acting as primers through mispairing with each other and do not see any spurious priming at the concentrations of template used in these reactions (up to 0.1 μM). The full range of possible amplification levels of triggering DNA sequences is not yet fully known. We routinely get 106- to 107-fold amplification within a few minutes but have observed amplification levels as high as 108-fold. The experiments here use MS and real-time fluorescence measurements to analyze and monitor the reactions and their products, but it is clear that they are amenable in practice to simple end-point fluorescence measurements as well.
There are several variant forms of the exponential amplification scheme shown in Fig. 3. One in particular has been devised to provide an accurate copy of a polymorphic site that can subsequently be amplified. This latter scheme, illustrated in Fig. 4 in a shorthand form, is contrasted with the “direct” EXPAR scheme described above and is called “copy EXPAR.” The “copy EXPAR” scheme (Fig. 4 Right) is slightly more complex than “direct EXPAR” (Fig. 4 Left) in that there is a second template whose amplification reaction is driven by the products from the first template. The first reaction (upper bracket in Fig. 4 Right) is essentially a linear amplification of the oligonucleotide trigger with the polymorphic base on its 5′ end. Because the nicking sequence is reversed relative to the orientation shown in Fig. 3, and the template does not include this 5′ base, the replication component of the reaction (see Fig. 1) creates a primer template with a 3′ terminal base in the template that matches the polymorphic base. The amplification reaction then produces the complement of the initiating trigger with an accurate copy of the polymorphism at its 3′ end. The second bracket indicates the exponential amplification of the product of the first reaction, shown in the same shorthand as for Fig. 4 Left. The effect of the two reactions as shown is to amplify the complement of the triggering oligonucleotide (shown in blue). The scheme enables the creation of a template that carries an extra base, which can be interrogated for polymorphic variation by the mass of the resulting amplified oligonucleotide. This is shown as the yellow (or purple) disk in Fig. 4. There are two (or more) second templates available, each cognate to a different extra base. Fig. 4 shows the amplification of a sequence with the purple variant (triggered by its complement). Thus, the scheme can be used to detect and measure polymorphisms in the target DNA.
Variant forms of the amplification reaction attest to the flexibility of the method. One of the variants, just described, can be used to amplify and characterize polymorphic sites in genomic DNA. The “copy EXPAR” reaction in Fig. 4 depends for its specificity (that is, giving only the appropriate product) on a phenomenon that appears to be specific to the transient annealing and priming process that creates the primer templates. That process, in which an oligonucleotide transiently anneals to the template and is extended by the polymerase, is sharply inhibited by mispairing near the 3′ end, much more so than inhibition by mispairing of a more stable priming duplex (data not shown). It is sufficiently inhibited that we see no detectable amplification when we attempt to prime with such a mispaired oligonucleotide (data not shown).
Many potential variations of the coupled reactions are described here, including the use of one amplifying oligonucleotide to trigger another amplification reaction. There are a number of ways in which this coupling can be used. We are currently investigating several of these variations. In addition, we are currently exploring the tethering of the amplification templates to solid supports, including slides and microbeads, so that in situ amplification can be triggered. This flexibility might open up a large number of new possibilities.
Supplementary Material
Acknowledgments
This work was supported in part by the W. M. Keck Foundation, the Norris Foundation, and Defense Advanced Research Projects Agency (contract no. MDA972-02-C-0047). We are grateful to Bill Jack of New England Biolabs for useful discussions about DNA polymerases and for comments on the manuscript, and to Huimin Kong for unpublished information on the nicking enzyme N. BstNBI. We are grateful for insightful comments on the manuscript by Eric Davidson and an anonymous referee. The technology described in this paper has been licensed to Ionian Technologies Incorporated, a company in which the authors have a financial interest.
Abbreviations
EXPAR, exponential amplification reaction
References
- 1.Mullis K., Faloona, F., Scharf, S., Saiki, R., Horn, G. & Erlich, H. (1986) Cold Spring Harb. Symp. Quant. Biol. 51 Pt 1, 263-273. [DOI] [PubMed] [Google Scholar]
- 2.Saiki R. K., Scharf, S. J., Faloona, F., Mullis, K. B., Horn, G. T., Erlich, H. A. & Arnheim, N. (1985) Science 230, 1250-1354. [Google Scholar]
- 3.Fire A. & Xu, S. Q. (1995) Proc. Natl. Acad. Sci. USA 92, 4641-4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D., Daubendiek, S. L., Zillman, M. A., Ryan, K. & Kool, E. T. (1996) J. Am. Chem. Soc. 118, 1587-1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lizardi P. M., Huang, X., Zhu, Z., Bray-Ward, P., Thomas, D. C. & Ward, D. C. (1998) Nat. Genet. 19, 225-232. [DOI] [PubMed] [Google Scholar]
- 6.Morgan R. D., Calvet, C., Demeter, M., Agra, R. & Kong, H. (2000) Biol. Chem. 381, 1123-1125. [DOI] [PubMed] [Google Scholar]
- 7.Kong H., Kucera, R. B. & Jack, W. E. (1993) J. Biol. Chem. 268, 1965-1967. [PubMed] [Google Scholar]
- 8.Walker G. T., Little, M. C., Nadeau, J. G. & Shank, D. D. (1992) Proc. Natl. Acad. Sci. USA 89, 392-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.