

Abstract
Most modern speech recognition uses probabilistic models to interpret a sequence of sounds. Hidden Markov models, in particular, are used to recognize words. The same techniques have been adapted to find domains in protein sequences of amino acids. To increase word accuracy in speech recognition, language models are used to capture the information that certain word combinations are more likely than others, thus improving detection based on context. However, to date, these context techniques have not been applied to protein domain discovery. Here we show that the application of statistical language modeling methods can significantly enhance domain recognition in protein sequences. As an example, we discover an unannotated Tf_Otx Pfam domain on the cone rod homeobox protein, which suggests a possible mechanism for how the V242M mutation on this protein causes cone-rod dystrophy.
Protein domains are the structural, functional, and evolutionary units of proteins. A protein can be regarded as a sequence of its domains. Given a new protein sequence, for instance from a genome project, the domains can be recognized on the basis of the similarity of sections of the amino acid sequence to known domain members. Similarly, speech recognition aims to identify the sequence of words in a continuous speech signal. However, in both cases, detection of individual constituent domains or words is impeded by noise. Fortunately, extra information is available from “context”: the presence of other words before and after the word in question. It has previously been observed that protein domains form a limited set of pairwise combinations (1). The presence of such combinations has been used for a number of purposes, for example, to predict protein cellular localization (2).
Speech recognition has been greatly facilitated by the application of statistical models including hidden Markov models (3, 4). Once the acoustic signal has been parsed into discrete units, the statistical approach is to build two types of model: for each word there is a phonetic model for the emission of phonemes, based on observed pronunciation patterns; and above the phonetic model there is a language model for the emission of a sequence of words, based on word use patterns. To recognize a given sentence, the method seeks the sequence of words D that maximizes the probability of the sentence given the acoustic evidence A and the language model M. This probability can be split (using Bayes' rule) into a word term based on the phonetic model (first term), and a context term, based on the language model (second term):
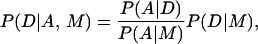 |
assuming that A is independent given D of the language model M.
We adapt this approach to domain recognition by using Eq. 1 to search for the domain sentence D, which maximizes the probability of the domain sentence given the amino acid sequence A and the context model M. We split the terms in this equation and introduce the terms P(Di) to represent the prior probability of the ith domain:
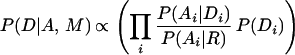 |
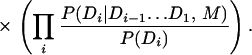 |
Note that we have replaced P(A|M), which is a constant given the signal, independent of our interpretation of the sequence, by another constant, P(A|R): the probability of the sequence being generated independently residue by residue according to a baseline composition model R. This also explains any sequence not allocated to domains. We have also assumed conditional independence given Di of each subsequence Ai on every other subsequence Aj and domain Dj. Finally, note that it is equivalent to maximize
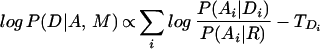 |
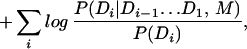 |
with domain score threshold TDi =log (1/P(Di)).
Hidden Markov models have been successfully applied to identifying protein domains within amino acid sequences (5–7). In the hmmer package (8) written by Sean Eddy and currently used by the protein domain family database Pfam (9), a domain is recognized as real if the domain log-odds ratio is greater than a manually curated threshold,
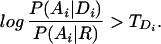 |
Comparison of Eqs. 3 and 4 reveals that the standard approach is essentially equivalent to ignoring the context term on the right side of Eq. 1. Our approach is to maximize Eq. 3, by using a Markov model as the language model.
Methods
Language Model.
The domain model, M, is a Markov model. Begin and end states are included in the modeling, to capture associations of domains with the beginning and end of proteins. A Markov model of order k asserts that the conditional probability of the ith domain given all preceding domains only depends on the k preceding domains:
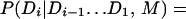 |
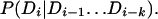 |
We first restrict M to be a first-order Markov model. We estimate the transition probabilities in Eq. 5 by using the observed counts in the Pfam database (denoted by N), by using the background frequency P(Di) to smooth our probability estimates:
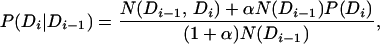 |
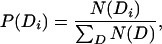 |
where αN(Di−1) can be regarded as the size of a pseudocount population, and we set α = 0.1. Note that the sum in Eq. 7 is over all domain occurrences in the Pfam database.
Dynamic Programming Algorithm.
The space of all potential domain assignments for a particular protein is large. Hence we need to design algorithms that concentrate on searching through probable domain assignments. Our approach is to first run hmmer against the protein for each Pfam family. We keep those hits that have a hmmer e value <1,000. In this way, we obtain a list d = d1 … dm of potential domains, ordered by end position, with corresponding amino acid sequences a1 … am. Our search space is now all possible combinations of domains in this list. We optimize the search through this reduced space by using a dynamic programming technique.
We want to find the domain sentence D = D1 … Dn, a sublist of d with corresponding amino acid sequences A1 … An, which maximizes the protein log-odds score S(D), where
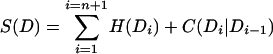 |
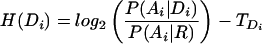 |
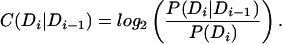 |
Note that H(Di) is just the hmmer score for the domain, and that C(Di|Di−1) is the transition score. We denote the begin and end states as D0, Dn+1, respectively, so that C(D1|D0) is the transition from begin state and C(Dn+1|Dn) is the transition to end state. We set H(Dn+1) = 0 as the end state contributes no sequence-based score. We use the curated Pfam “gathering” threshold for TDi.
We define Di to be the highest scoring domain sentence that ends in domain di without overlaps. The following recursion relation then applies:
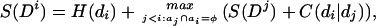 |
where the condition aj ∩ ai = φ ensures that the maximizing sentence does not contain domain overlaps, which requires tracking the protein coordinates of domains. We then set
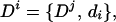 |
where Dj maximizes Eq. 11. Repeated application of Eqs. 11 and 12 for i = 1 … m + 1 gives the maximizing sentence D = Dm+1 required by Eq. 8 (again, we use the convention that dm+1 is the end state, so that Dm+1 is interpreted as the maximizing sentence ending with the end state).
We note that Pfam uses a “sequence score” threshold in addition to the domain score threshold outlined in Eq. 4. This thresholding is equivalent to a threshold on the sum of bit scores contributed by all instances of a domain on a protein. As our method applies Pfam thresholds, we must also apply a similar filter as a postprocessing step to retain consistency with Pfam. We first calculate the maximizing domain sentence, as described above. We then distribute each transition score equally between the source and target domain of the transition and add these scores to their respective hmmer scores. Finally, for each Pfam family in the maximizing sentence, we sum the modified scores for each instance of the family and compare this sum with the Pfam sequence score threshold: families that do not meet the threshold are removed from the annotated domain sentence.
Extension to Variable-Order Markov Model.
The approach of using a fixed-order Markov model has a significant drawback: the lengths of commonly occurring domain architectures are not fixed; some patterns are first order (CBS domains often occur in pairs), whereas many patterns have a higher order (the group of RNA polymerase RBP1 domains commonly occur in groups of seven). Restricting to a fixed-order Markov model will degrade the ability of the model to recognize patterns of arbitrary length. Instead, for each proposed context Dj from Eq. 11 in the dynamic programming algorithm, we choose a different order k for M, which is the maximum order for which we still have observations in the database. More precisely, labeling Dj = D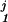 … D
… D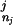 we choose the order k to be the largest order for which we have a nonzero training set count (D
we choose the order k to be the largest order for which we have a nonzero training set count (D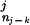 … D
… D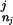 ). As this does not depend on the current domain di, Eq. 5 still defines a consistent probability distribution over domains. In practice, to cut down on memory requirements, we restrict the order of the model to k ≤ 5.
). As this does not depend on the current domain di, Eq. 5 still defines a consistent probability distribution over domains. In practice, to cut down on memory requirements, we restrict the order of the model to k ≤ 5.
This approach is an example of decision tree modeling that is commonly used in language modeling. Decision trees partition domain histories Dj into equivalence classes Φ1 … ΦM with a corresponding probability distribution P(Di|Φl). Our approach partitions on the basis of the longest domain context that has been observed in the training set. It is straightforward to develop more complicated decision rules, which remains a subject for further investigation. Our approach is also similar to the interpolated Markov chain approach used by Salzberg et al. (10) in gene prediction.
To estimate the transition probabilities in Eq. 5, we extend Eq. 6, again by using the observed counts in the Pfam database (denoted by N), but now recursively interpolating lower-order transition probabilities in the form of pseudocounts,
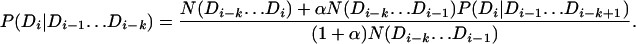 |
The terms P(Di|Di−1) and P(Di) are given by Eqs. 6 and 7. The pseudocount population αN(Di−k … Di−1) is set by fixing α = 0.1.
In the case of an arbitrary-order Markov model, we apply the same search strategy as Eqs. 11 and 12, replacing C(Di|Di−1) in Eq. 8 with
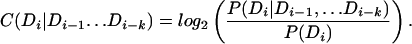 |
The recursion relation (Eq. 11) no longer holds, and hence we cannot guarantee that this method will always find the domain sentence that maximizes Eq. 8. However, the method has been found to still work well in practice.
Databases.
The protein database used is Swiss-Prot40 + TrEMBL18. The Pfam database release 7.7 was used both for training the model and searching against the protein database. Pfam is a database of multiple sequence alignments and hidden Markov models (9). Release 7.7 contains 4,832 families, with matches to 74% proteins in Swiss-Prot40 + TrEMBL18 (53% of the residues).
Results
We implement a first-order and variable-order Markov model (see Methods). The first-order Markov model found 8,591 extra occurrences of Pfam families in proteins from Swiss-Prot40 + TrEMBL18 compared with those previously annotated in the Pfam database, covering 266,321 residues. However, the variable-order Markov model found 15,263 additional domains, covering 610,010 residues, showing that by using a flexible amount of context information increases the power of the method by nearly a factor of 2. This coverage is equivalent to the last 15.6% of Pfam families (753 of 4,832 families) and corresponds to 0.64% sequence coverage. The additional occurrences are limited to 605 Pfam families, of which 212 families contribute 95% of additional hits. A complete list of additional domain occurrences is available in Table 2, which is published as supporting information on the PNAS web site, www.pnas.org.. The discussion from here on is based on the results of the variable-order model.
Fig. 1 displays the families that the method detects. Our method particularly enhances detection of short Pfam families: the additional occurrences have an average length of 40 residues, compared with the database average of 155 residues. This difference is caused by the over-representation of repeats in short Pfam families (and hence better contextual information) and a lower sequence-based signal-to-noise ratio for short families.
Fig 1.

Domain occurrences among the top 20 context families. The bars shows the absolute number of additional predictions; the line shows the percentage increase in that family.
Fig. 2 shows several examples of domains found by this method. Two TPR domains are found on the SR68_HUMAN protein, which has no TPR domains annotated in any of the protein databases. This protein is known to interact with SR72_HUMAN in the signal recognition particle (11), which itself has a pair of annotated TPR domains. As TPRs are protein–protein interaction motifs, we suggest that the interaction between SR68 and SR72 may be mediated by this region. On the previously unannotated E2BG_CAEEL protein, we find an NTP transferase domain, followed by three hexapep repeats, all raised above the noise by their mutual compatibility.
Fig 2.

Examples of additional context domains, indicated by rectangles. Standard Pfam domains are indicated by angled boxes.
Our method also predicts a previously unannotated Tf_Otx domain in the cone rod homeobox protein (CRX), in Homo sapiens, Rattus norvegicus, and Mus musculus (Fig. 2). CRX is a 299-aa homeodomain transcription factor that is expressed primarily in the rod and cone receptors of the retina (12, 13). CRX is highly conserved among mammalian species. CRX is known to share homology with Otx1 and Otx2 and contains a homeodomain near the N terminus followed by a glutamine-rich region, a basic region, a WSP motif, and an Otx-tail motif. Our TF_Otx prediction extends over the unannotated region amino acids 164–250. This region encloses a valine to methionine mutation at position 242 associated with autosomal dominant cone rod dystrophy, which leads to early blindness (14, 15). Recent research demonstrates that a region coinciding with our prediction (amino acids 200–284) is essential for transcriptional activation of the photo-receptor genes and supports the hypothesis that the V242M mutation acts by impairing this transactivation process (16). An analysis of the multiple alignment of the TF_Otx domains (Fig. 3) demonstrates the existence of two subfamilies of the domain, the first of which has a methionine at position 22 and contains all Otx1 proteins, the second of which has a valine at position 22 and contains all Otx2 proteins. Furthermore, the CRX V242M mutation aligns with this position and hence transfers the CRX TF_Otx domain from the Otx2 to Otx1 subfamily. Finally, we note that it has been demonstrated that both Otx2 and CRX transactivate the interphoto receptor binding protein (IRBP) (17), although this has not been demonstrated for Otx1. We therefore suggest that the V242M mutation loss of function is caused by loss of IRBP transactivation ability, and conversely that this position in the TF_Otx motif is critical for IRBP transactivation.
Fig 3.

Part of multiple alignment of TF_Otx domain in members of the Otx1 and Otx2 subfamilies. Position 22 in this alignment corresponds to position 242 on CRX_HUMAN. This position is methionine for all members of the Otx1 subfamily, whereas it is valine for all members of the Otx2 subfamily.
The predictions of our method have been validated by a psi-blast (18) test (Table 1). For each novel predicted domain occurrence, psi-blast was used to generate a set of similar sequence fragments. These sequences were then searched for matches to Pfam families. For 30.7% of novel domain occurrences psi-blast found matches that are annotated in Pfam. In 90.0% of these the majority of annotations matched the identified family; a further 7.6% had at least one match to the correct family; 0.8% matched a related family, and for the remaining 1.5% all matches were to incorrect families. By inspection, the assignment caused by the language modeling method of this article appears to be correct for the overwhelming majority of the 7.6% and 0.8% matches and many of the 1.5% matches. Therefore we suggest that the false-positive rate is no more than a few percent. Because many of the 69.3% novel predictions for which psi-blast does not find a match have higher scores than those for which it does, this finding also indicates our approach can detect matches that psi-blast does not.
Table 1.
blast results for new positives predicted by model
| Result | No. | Percentage |
|---|---|---|
| psi-blast does not find match in Pfam family | 10,575 | 69.3 |
| Majority of matches to correct Pfam family | 4,220 | 27.6 |
| Majority of matches to incorrect family | ||
| Has one match to correct family | 358 | 2.3 |
| Has matches to related family | 38 | 0.3 |
| All matches to unrelated families | 72 | 0.5 |
The Pfam database maintains for each domain hit an e-value score as well as a domain bit score. The e-value score for a domain is the number of hits that would be expected to have a score greater than or equal to the score of the domain in a random database of the same size. It is calculated for each Pfam family by fitting an extreme value distribution (EVD) to the bit scores of hits to that family against a set of randomly generated proteins as implemented in the hmmcalibrate program of the hmmer package.
It is important to note that the e-value score does not directly affect the assignment of domains; that is, it is not used as a threshold. Rather, bit score thresholds for domain assignment in Pfam are manually curated. However, it is desirable to consider the effect of language modeling on the significance of hits to Pfam families, because these are used by end users when evaluating marginal hits.
We explore here one potential way of calculating modified e-value scores, incorporating language modeling. The question we ask is: in a database of randomly generated proteins, what is the distribution of scores to a given Pfam family, using the language model methodology outlined above? We require that our hmm passes at least once through the Pfam family in question. We then attribute transition scores from the language model to the hits and fit an EVD to these modified scores. Note that in almost all cases the language model uses a start → domain → end architecture as it finds no other domains with scores above threshold to include in the calculation. Also, in this case, all of the start-to-domain and domain-to-end transition scores are attributed to the domain.
This process was carried out on two Pfam families: WD40 and pkinase as shown in Fig. 4. We see two different types of behavior. In one case, pkinase commonly occurs in a singleton pattern and hence hits to random proteins typically have their scores enhanced slightly by the language model, so that the EVD shifts to the right. However, real hits also have their scores enhanced, furthermore, in the case of a single domain protein, the increase will be the same as the shift in the EVD, so that the significance of the hit remains unchanged. In contrast, hits to the pkinase domain in atypical contexts will not have their scores enhanced, so their significance will decrease. On the other hand, WD40 commonly occurs in repeats of five to eight units, so that singleton random hits are penalized under the language model (by about four bits) and so the EVD shifts to the left. The language model enhances the score of real hits (as they do occur in the appropriate repeating pattern), thus providing the compound effect of increasing the score of real hits and increasing the significance of hits at a given score. To summarize, the effect of language modeling on significance scores appears to be either neutral, in the case in which the scores of random and real hits are shifted by the same amount, or more discriminatory, in the case of decreasing random scores and increasing real scores.
Fig 4.

EVD curves calculated for pkinase and WD40 Pfam domains. The solid lines are the standard EVD curves calculated by using HMMER. The dashed lines use our language modeling method and hence take contextual information into account. For almost all sequences, this process results in the addition to the forced match to the domain of interest of begin-to-domain and domain-to-end transitions. WD40 is commonly found in groups of five to eight tandem repeats, so that single random WD40 hits are penalized by the language model. The WD40 EVD shifts 4.0 bits to the left. On the other hand, pkinase often occurs by itself on a protein, and hence random single pkinase repeats gain slightly under the language model. The pkinase EVD shifts 1.1 bits to the right.
We note here a weakness of calculating modified significance scores in this way. Namely, that it may be inappropriate to consider random proteins for calculating the language model component of the score, particularly as this leads to random hits effectively having no nonstart/end context. Rather, we are interested in nonmatching amino acid sequences occurring within real proteins, so we should sample real protein contexts. However, doing this correctly remains a topic for further investigation.
Discussion
We have demonstrated that significant improvement in protein domain detection is possible through the use of language modeling. We have shown several examples in which the increased predictive power has discovered domains that further understanding of both human disease and biology, and we expect there will be many others. From a theoretical point of view, this method is important as it provides a fully integrated prediction of domain annotation for a given protein, evaluating in a strictly probabilistic fashion the appropriate tradeoff between amino acid signal strength and contextual information. Lastly, from a pragmatic perspective, the method significantly increases sequence coverage.
Further improvements to the language models are possible, motivated by similar techniques in speech recognition. Modifications to the decision trees used to classify domain contexts are possible; for example, we could classify domain contexts on the basis of the longest potentially noncontiguous preceding subsequence that is also observed in the training database. Alternatively, standard classification techniques to learn optimal decision trees can be used. Other annotated regions on the protein could be used in our search, for example, regions of low complexity. Explicitly modeling the length distribution between domains may also increase sensitivity. Lastly, alternative classes of generative grammars may be used, although it remains unclear which level is appropriate for domain modeling. The language modeling could also be adapted to take into account nested domains, although this problem is not shared with speech recognition.
Extra information regarding the protein may also prove to be a useful guide in domain annotation. It has been shown that different species have markedly different domain repertoires and that the diversity of domain combinations increases with organism complexity (19). Techniques from speech recognition can be used to formally integrate information regarding protein species and localization.
This method may also be applicable to the discovery of cis-regulatory motifs (CRMs) and transcription factor (TF) binding sites. Identification of TF binding sites using weight matrices is difficult, as they can lie kilobases away from the transcription start site, and the motifs occur often at random throughout the genome. Several authors have built organizational models that take motif positioning and orientation into account (20, 21), whereas others have attempted to identify CRMs on the basis of high local density of potential binding sites (22). Our approach is related to some of these methods and may provide an alternative strategy.
Supplementary Material
Acknowledgments
The Wellcome Trust Sanger Institute is supported by the Wellcome Trust. L.C. holds a Leslie Wilson Scholarship from Magdelene College Cambridge and a Cambridge Australia Trust studentship.
Abbreviations
EVD, extreme value distribution
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Apic G., Gough, J. & Teichmann, S. A. (2001) Bioinformatics 17, S83-S89. [DOI] [PubMed] [Google Scholar]
- 2.Mott R., Schultz, J., Bork, P. & Ponting, C. (2002) Genome Res. 8, 1168-1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jelinek F., (1997) Statistical Methods for Speech Recognition (MIT Press, Cambridge, MA).
- 4.Charniak E., (1993) Statistical Language Learning (MIT Press, Cambridge, MA).
- 5.Krogh A., Brown, M., Mian, I. S., Sjölander, K. & Haussler, D. (1994) J. Mol. Biol. 235, 1501-1531. [DOI] [PubMed] [Google Scholar]
- 6.Durbin R., Eddy, S., Krogh, A. & Mitchison, G., (1998) Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids (Cambridge Univ. Press, Cambridge, U.K.).
- 7.Grundy, W. N., Bailey, T. L., Elkan, C. P. & Baker, M. E. (1997) Comput. Appl. Biosci. 397–406. [DOI] [PubMed]
- 8.Eddy S. R. (1998) Bioinformatics 14, 755-763. [DOI] [PubMed] [Google Scholar]
- 9.Bateman A., Birney, E., Durbin, R., Eddy, S. R., Howe, K. L., Sonnhammer, E. L. L., Marshal, M., Griffiths-Jones, S., Etwiller, L. & Cerruti, L. (2002) Nucleic Acids Res. 30, 276-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salzberg S. L., Pertea, M., Delcher, A. L., Gardner, M. J. & Tettelin, H. (1999) Genomics 59, 24-31. [DOI] [PubMed] [Google Scholar]
- 11.Lutcke H., Prehn, S., Ashford, A. J., Remus, M., Frank, R. & Dobberstein, B. (1993) J. Cell Biol. 121, 977-985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen S., Wang, Q. L., Nie, Z., Sun, H., Lennon, G., Copeland, N. G., Gilbert, D. J., Jenkins, N. A. & Zack, D. J. (1997) Neuron 19, 1017-1030. [DOI] [PubMed] [Google Scholar]
- 13.Furukawa T., Morrow, E. M. & Cepko, C. L. (1997) Cell 91, 531-541. [DOI] [PubMed] [Google Scholar]
- 14.Swain P. K., Chen, S., Wang, Q. L., Affatigato, L. M., Coats, C. L., Brady, K. D., Fishman, G. A., Jacobson, S. G., Swaroop, A., Stone, E., et al. (1997) Neuron 19, 1329-1336. [DOI] [PubMed] [Google Scholar]
- 15.Rivolta C., Berson, E. L. & Dryja, T. P. (2001) Hum. Mutat. 18, 488-498. [DOI] [PubMed] [Google Scholar]
- 16.Chen S., Wang, Q. L., Xu, S., Liu, I., Li, L. Y., Wang, Y. & Zack, D. J. (2002) Hum. Mol. Genet. 11, 873-884. [DOI] [PubMed] [Google Scholar]
- 17.Bobola N., Briata, P., Ilengo, C., Rosatto, N., Craft, C., Corte, G. & Ravazzolo, R. (1999) Mech. Dev. 82, 165-169. [DOI] [PubMed] [Google Scholar]
- 18.Altschul S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W. & Lipman, D. J. (1997) Nucleic Acids Res. 25, 3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rubin G. M., Yandell, M. D., Wortman, J. R., Gabor Miklos, G. L., Nelson, C. R., Hariharan, I. K., Fortini, M. E., Li, P. W., Apweiler, R., Fleischmann, W., et al. (2000) Science 287, 2204-2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galius-Durner V., Scherf, M. & Werner, T. (2001) Mamm. Genome 12, 67-72. [DOI] [PubMed] [Google Scholar]
- 21.Pavlidis P., Furey, T. S., Liberto, M., Haussler, D. & Grundy, W. N. (2001) Pac. Symp. Biocomput. 6, 151-164. [DOI] [PubMed] [Google Scholar]
- 22.Berman B. P., Nibu, Y., Pfeiffer, B. D., Tomancak, P., Celniker, S. E., Levine, M., Rubin, G. & Eisen, M. (2002) Proc. Natl. Acad. Sci. USA 99, 757-762. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.