

Abstract
Abasic sites are ubiquitous DNA lesions that are mutagenic and cytotoxic but are removed by the base excision repair pathway. DNA polymerase β carries out two of the four steps during base excision repair, including a lyase reaction that removes the abasic site from DNA following incision of its 5′-phosphate. DNA polymerase β is overexpressed in cancer cells and is a potential anticancer target. Recently, DNA oxidized abasic sites that are produced by potent antitumor agents were shown to inactivate DNA polymerase β. A library of small molecules whose structures were inspired by the oxidized abasic sites was synthesized and screened for their ability to irreversibly inhibit DNA polymerase β. One candidate (3a) was examined more thoroughly and modification of its phosphate backbone led to a molecule that irreversibly inactivates DNA polymerase β in solution (IC50 ~ 16 μM), and inhibits the enzyme’s lyase activity in cell lysates. A bis-acetate analogue is converted in cell lysates to 3a. The bis-acetate is more effective in cell lysates, more cytotoxic in prostate cancer cells than 3a, and potentiates the cytotoxicity of methyl methanesulfonate between 2- and 5-fold. This is the first example of an irreversible inhibitor of the lyase activity of DNA polymerase β that works synergistically with a DNA damaging agent.
Introduction
Base excision repair (BER) is a primary mechanism for maintaining genome integrity. A large variety of modified nucleotides resulting from DNA oxidation and alkylation are removed by glycosylases.1 Some BER glycosylases are bifunctional and cleave DNA at a transiently formed abasic site (AP) via a lyase process.2 In other instances AP sites are produced as metastable intermediates. AP sites are also generated via spontaneous hydrolysis of native and damaged nucleotides. DNA polymerase β (Pol β) plays an integral role in BER by excising the remnant of an AP site following 5′-incision by apurinic endonuclease I (Ape1), and subsequently filling in the single nucleotide gap (Scheme 1). Pol β’s vitality to genome integrity is manifested by the observation that cells lacking both alleles of the gene for this enzyme are embryonic lethal, and knocking down Pol β activity sensitizes cells to DNA damaging agents.3 Consequently, Pol β has attracted interest as a target for antitumor therapy. Inhibiting Pol β potentiates the cytotoxic effects of DNA damaging agents and can be cytotoxic in its own right. We wish to report on a series of Pol β inhibitors whose design was inspired by DNA lesions that irreversibly inactivate the enzyme by targeting its lyase active site.4–7
Scheme 1.
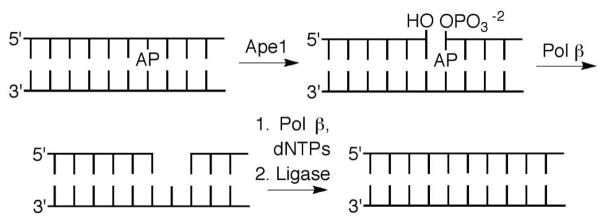
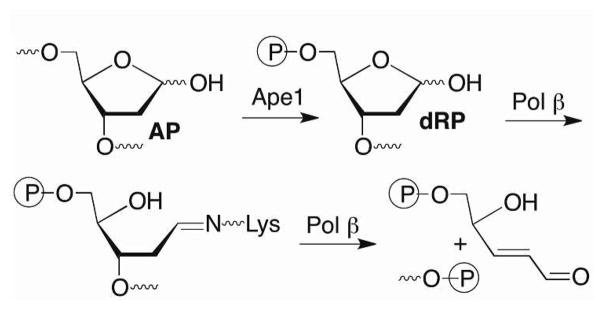
Pol β is a bifunctional enzyme that contains an 8 kDa lyase active site separate from its polymerase active site.8–10 The enzyme excises the 5′-phosphorylated 2-deoxyribose (dRP) produced upon Ape1 incision of DNA containing an AP site (Scheme 2). Lys72 is the primary amine responsible for Schiff base formation, although the enzyme retains some lyase activity when this amino acid is mutated.11–14 Lys84, which is also present in the lyase active site is postulated to substitute for Lys72 in the mutated enzyme, albeit with much lower efficiency. Following Schiff base formation, dRP elimination leaves a single nucleotide gap that contains the appropriate end groups for DNA synthesis (by Pol β) and ligation to complete repair (Scheme 1). Part of the attraction of Pol β as a potential therapeutic target is that it is over expressed in a variety of cancer cells.15–17 In addition, Pol β variants are found in a large percentage of tumors.18–20 Some of the variants exhibit reduced activity and may contribute to tumorigenesis by decreasing genomic stability.
Scheme 2.
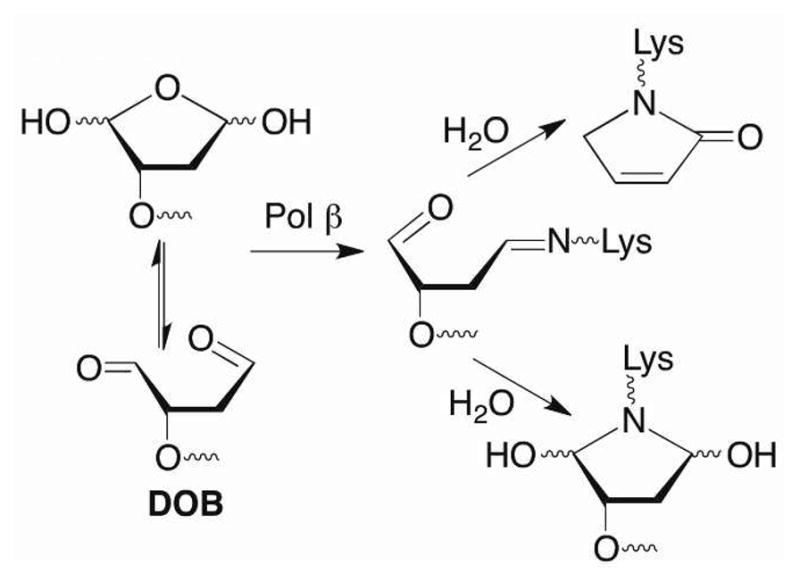
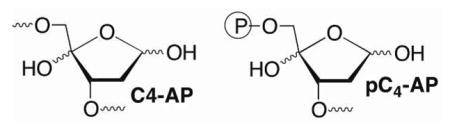
Natural and unnatural products have been tested as inhibitors of Pol β and the related enzyme, Pol λ, which is believed to act as a back up for Pol β in BER.21–26 Some of these molecules are believed to target the lyase domain. The inhibitors described below were designed to mimic the interaction between Pol β and a DNA lesion, 2-phosphato-1,4-dioxobutane (DOB), which is produced by a family of potent cytotoxic antitumor antibiotics following C5′-hydrogen atom abstraction.27,28 DOB efficiently inactivates Pol β (and Pol λ).4–6 Radiolabeling experiments, liquid chromatography, and mass spectral analyses of protease digests indicate that the 1,4-dicarbonyl inactivates Pol β in two ways (Scheme 3). DOB forms a stable lactam following condensation with Lys72 or Lys84, elimination, and dehydration. The lesion also forms a stable adduct without undergoing DNA cleavage. pC4-AP that is produced upon Ape1 incision of C4-AP also contains a 1,4-dicarbonyl and inactivates Pol β and Pol λ.6,7 We hypothesized that small, DNA-like molecules containing such a 1,4-dicarbonyl motif would inactivate Pol β upon binding.
Scheme 3.
Results and Discussion
Design and synthesis of small molecule DOB mimics as potential irreversible inhibitors of Pol β
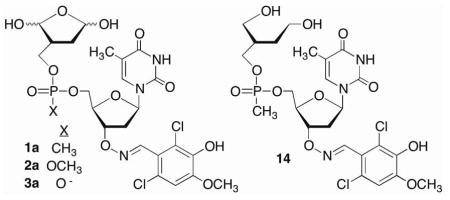
A library of nucleotide inhibitors containing the 1,4-dicarbonyl group that is present in the DOB and pC4-AP lesions that irreversibly inhibit DNA polymerase β was conceived (Scheme 4). A thymidine nucleotide was incorporated to make the molecule DNA-like. Since Pol β exhibits no selectivity for one nucleotide over another, thymidine was chosen for synthetic simplicity, as it has no exocyclic amines that require protection. Initially, the phosphate diester in DNA was replaced by a more lipophilic methyl phosphonate (1). The molecules contained a carbon between the ring containing the 1,4-dicarbonyl and the methyl phosphonate to improve their chemical stability in aqueous solution by preventing elimination. Finally, structural diversity was introduced in the form of an oxime linkage at the 3′-terminus. The oxime group is easy to incorporate and has proven to be a useful means for introducing structural diversity (represented by “R”) into chemical libraries of enzyme inhibitors.29,30
Scheme 4.
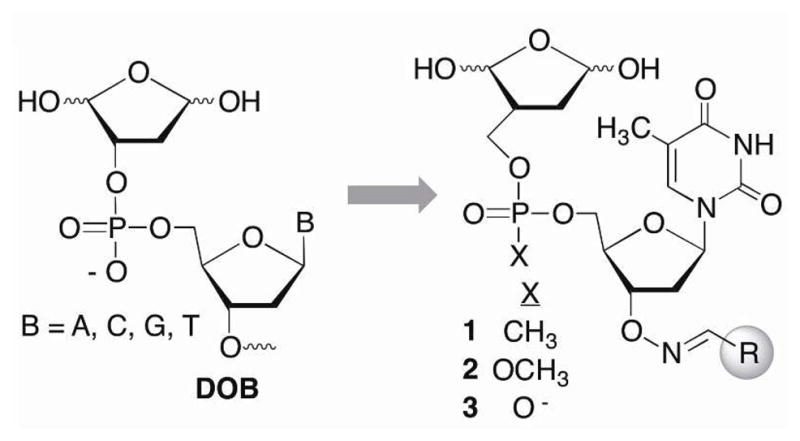
We anticipated unmasking the 1,4-dialdehyde of 1 in the final step after the library was prepared containing single compounds in individual wells of microtiter plates (Scheme 5). The choice of acetal protecting group was important, as it needed to be cleaved rapidly under mild conditions. We chose to use the pentenyl group that has been very useful in carbohydrate synthesis for glycosidic bond formation and is cleaved rapidly under mild oxidizing conditions. 31 The alkoxyamine (7) was the last common intermediate in the library synthesis and was apportioned into the wells of the microtiter plates. The methyl phosphonate coupling to produce 6 was carried out using the phosphonamidite of the thymidine component (5) and the primary alcohol of the protected 1,4-dicarbonyl (4). The coupling yields were higher using this approach due to poor solubility of the corresponding 5′-hydroxy-3′-phthalimide substituted thymidine (9). Following removal of the phthalimide protecting group using hydrazine, chemical diversity was introduced by reacting 7 separately with one of 232 aldehydes overnight in DMSO and acetic acid. DMSO was removed from the crude mixture of 8 under vacuum prior to deprotecting the bis-acetal with N-bromosuccinimide.
Scheme 5a a.

Key a) i. tetrazole, CH3CN, 25 °C ii. t-BuOOH b) N2H4, THF, 25 °C c) RCHO, AcOH, DMSO, 37 °C d) NBS, CH3CN, −5 °C
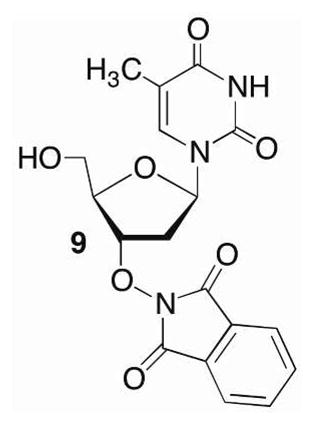
The methyl phosphonamidite (5) was prepared from previously reported 9 under standard phosphitylation conditions in 60% yield.32 The requisite hydroxymethyl compound (4) was prepared from 10 using a strategy employed previously for the synthesis of photolabile DOB precursors (Scheme 6).33,34 The hydroxymethyl group was introduced via an aldol condensation with formaldehyde that was ultimately trapped as the silyl ether of the formacetal (11) using a chiral catalyst derived from proline.35 Substituting Selectfluor® for N-bromosuccinimide, which was used in the synthesis of the DOB precursor, to induce cyclization of 11 by oxidatively cleaving the 1,3-dithiane resulted in a significant improvement in yield, albeit as an inseparable mixture of diastereomers of 12.36 Two of the 4 diastereomers were separable upon desilylation to 4. Although the stereochemistry of the acetals were unimportant with respect to the properties of the inhibitor candidates, working with less complex mixtures facilitated characterizing subsequent intermediates in the synthetic sequence. A library containing 232 members was synthesized from 4 (Scheme 5) and screened for Pol β inhibition.
Scheme 6a a.
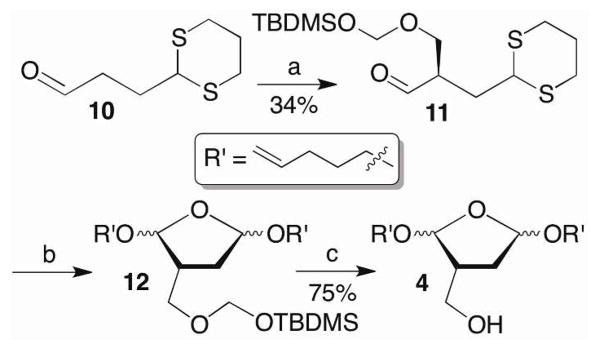
Key a) i. CH2O, prolinol catalyst, toluene, 25 °C ii. TBDMSCI b) Selectfluor, pent-4-en-1-ol, CH3CN, 25 °C c) TBAF, THF, 0 °C
Screening inhibitor candidates using a strand displacement assay
Inactivation of Pol β’s lyase activity by DOB or C4-AP also shuts down the enzyme’s ability to extend a primer via strand displacement synthesis.5,7 Consequently, we speculated that successful small molecules that inactivate the lyase activity would also shut down polymerase activity. This enabled us to use a previously reported fluorescence assay (Scheme 7) in which a ternary substrate (13, 50 nM) containing TAMRA at the 3′-terminus of the displaced strand and quencher (BHQ-2) at the 5′-terminus of the template strand was subjected to Pol β (10 nM) and dTTP (100 μM).37,38 Strand displacement synthesis results in fluorescence by TAMRA, and Pol β inhibition is reflected by decreased fluorescence relative to control lacking inhibitor.
Scheme 7.
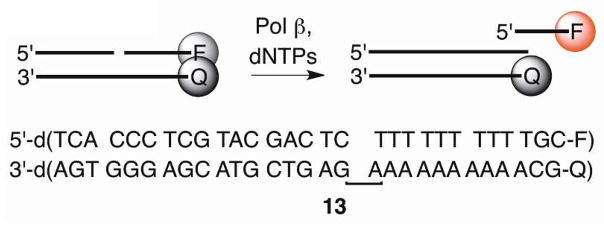
The 232 candidates38 (50 μM) were screened using this method. Although the underivatized oxime (50 μM) had no effect on polymerase activity (data not shown), several candidates exhibited significant inhibition in the strand displacement assay. Of these, the molecule derived from 2,6-dichloro-3-hydroxy-4-methoxy benzaldehyde (1a) was most effective at reducing fluorescence. Purified 1a significantly inhibited Pol β in the strand displacement assay at as low as 10 μM (Figure 1A). The reduction product (14) had no effect on enzyme activity (data not shown), indicating that the 1,4-dicarbonyl is required for inhibition. Furthermore, the extent of inhibition was dependent upon the preincubation time (Figure 1B), a property that is consistent with irreversible inactivation.
Figure 1.
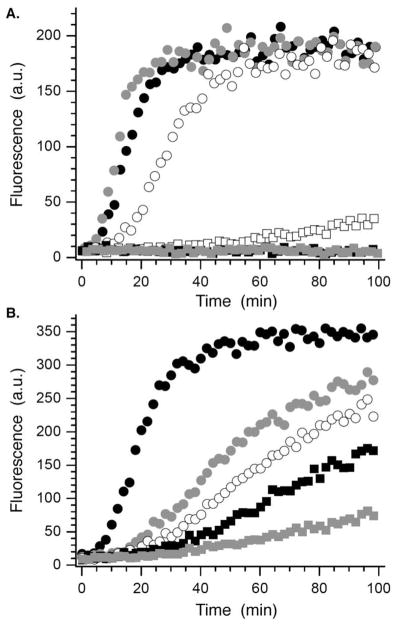
Inhibition of strand displacement synthesis in 13 by 1a. A) Dependence on [1a] (μM): 0, ●; 1,
 ; 5, ○; 10, □; 25, ■; 50,
; 5, ○; 10, □; 25, ■; 50,
 . B) Dependence on preincubation time of 1a (10 μM) with Pol β. No inhibitor, ●; Preincubation time 42: 5,
; 20, ○; 40, ■; 60,
.
. B) Dependence on preincubation time of 1a (10 μM) with Pol β. No inhibitor, ●; Preincubation time 42: 5,
; 20, ○; 40, ■; 60,
.
Direct examination of Pol β lyase inactivation by 1a
The ability of 1a to inhibit Pol β’s dRPase activity was examined using 3′-32P-15 in which the oligonucleotide containing dRP was labeled. The DNA substrate was added to the reaction following preincubation of 1a with Pol β and subsequent 100-fold dilution. The lyase reaction was monitored by gel electrophoresis. A logarithmic plot of activity in the presence of 1a (up to 30 μM) relative to when no inhibitor is present versus preincubation time decays linearly at each concentration over the range tested (Figure 2A).39 In addition, Pol β activity was not restored following dialysis of the enyzme-inhibitor solution for up to 3 days (Figure 2B).40,41 Both observations are consistent with irreversible inhibition of Pol β. However, we were unable to characterize the modified Pol β by mass spectrometry.
Figure 2.

Irreversible inhibition of Pol β by 1a. A) Relative Pol β lyase activity on 32P-15 as a function of [1a] and preincubation time of inhibitor with enzyme. B) Normalized lyase activity of Pol β on 32P-15 upon dialysis following incubation with or without 1a ([1a] (μM): 0, ■; 50,
).

Effects of phosphate backbone modification on Pol β inactivation
Using 1a as a lead we explored the effect of modifying the phosphorous backbone on inhibitor activity by synthesizing the phosphate triester (2a) and phosphate diester (3a) analogues. These candidates were prepared from 4 and 9 via a similar manner as 1a.38 Phosphate triester 2a exhibited comparable inhibition activity as 1a using the strand displacement assay (Scheme 7).38 This backbone motif was also useful for establishing the necessity of the 1,4-dicarbonyl for irreversible inactivation, as monoaldehdye 16 had no effect on Pol β lyase activity.38
In contrast, introducing the negative charge present in DNA (3a) produced a more potent inhibitor. Direct measurement of Pol β lyase activity showed that 3a was at least 2-fold more potent than 1a and exhibited an IC50 of ~21 ± 1 μM (Figure 3). The IC50 for 1a was 42 ± 5 μM.38 Furthermore, as for the methyl phosphonate, dialysis of Pol β incubated with 3a confirmed that inhibition was irreversible, and reduction to the diol (17) provided additional affirmation that the 1,4-dialdehyde was required for inactivation.38
Figure 3.
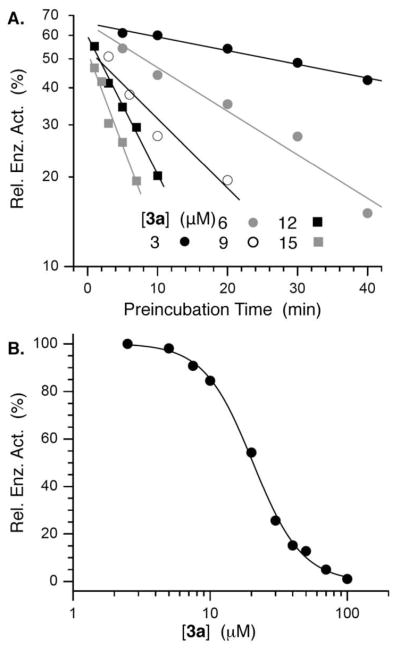
Irreversible inhibition of Pol β by 3a. A) Relative Pol β lyase activity on 32P-15 as a function of [3a] and preincubation time of inhibitor with enzyme. B) IC50 of Pol β inactivation following 30 min preincubation with 3a.
Selectivity and robustness of Pol β inactivation by small molecule DOB analogues
There are more than one dozen polymerases in a human cell. Because we do not have access to each of these, we used the Klenow fragment of DNA polymerase I from E. coli, which is often used as a model polymerase to test the inhibitor’s (3a) selectivity. While 3a (10 μM) almost completely eliminated Pol β’s ability to carry out strand displacement synthesis using 13 (Scheme 7) and dTTP, it had no effect on the Klenow fragment’s activity under the same conditions.38 However, the efficacy of the 1,4-dicarbonyl containing inhibitor (1a) was compromised by thiols, presumably due to nucleophilic addition to the ring-opened 1,4-dicarbonyl form (Scheme 3, Figure 4). For instance, preincubation (20 min) of 1a (50 μM) with glutathione (5 mM) resulted in similar Pol β activity as 25 μM inhibitor in the absence of thiol.38
Figure 4.

Effect of glutathione (GSH) on Pol β inhibition by 1a. A) No GSH B) [GSH] = 5 mM. [1a] (μM): 0, ●; 5, □; 10,
; 25,
; 50, ■.
Design and synthesis of a proinhibitor
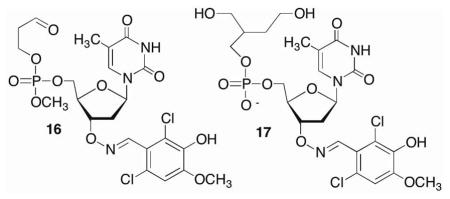
The adverse effect of glutathione on 1a led us to design bisacetate 18 as a potential proinhibitor. We postulated that 18 would be converted into 3a by cellular esterases.42,43 Proinhibitor 18 was synthesized from 3-hydroxymethylfuran (19) and 9 (Scheme 8). Following Pb194 oxidation and hydrogenation, 18 was coupled with the methyl phosphoramidite obtained from 9. Selective cleavage of the phthalimide group yielded alkoxyamine 21, which was conjugated to 2,6-dichloro-3-hydroxy-4-methoxybenzaldehyde prior to revealing the phosphate diester (18). As expected, 18 had no effect on Pol β lyase activity because the electrophilic carbonyl groups are masked (data not shown).
Scheme 8a a.
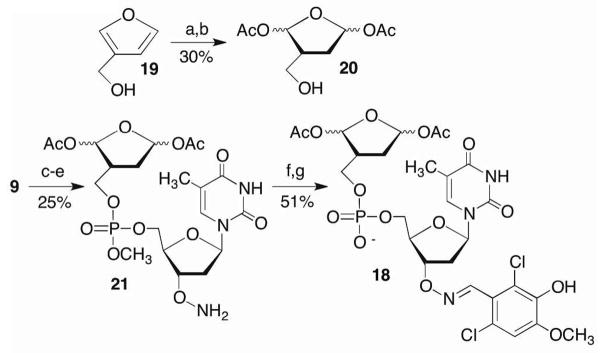
Key a) Pb(OAc)4 b) H2/Rh c) Phosphitylation d) i. S-Ethyl-1H-tetrazole, 20 ii. t-BuOOH e) N2H4 f) 2,6-Dichloro-3-hydroxy-4-methoxybenzaldehyde, AcOH g) Demethylation
The ability of 18 to provide more effective inhibition than 3a directly was examined in prostate cancer cell (DU145) lysates. TLC analysis revealed that the bisacetate was converted to 3a within 5 min in DU145 lysate. The ability of 3a and 18 to inhibit DU145 lysate lyase activity was then examined using 3′-32P-15 as substrate (Figure 5). Lyase activity inhibition was significantly greater by bisacetate 18 than 3a. Almost complete inhibition was achieved using 150 μM of 18, while more than 30% lyase activity remained following incubation with the same concentration of 3a. Varying the preincubation time of 18 with the cell lysate between 5 and 60 min showed that inactivation was complete by 15 min.
Figure 5.

Effect of 3a and proinhibitor 18 on DU145 lysate lyase activity. A) Comparison of 3a and 18 as a function of concentration. Preincubation time: 1 h, [32P-15] = 200 nM. B) Effect of preincubation time on the ability of 18 (50 μM) to inhibit lyase activity.
Selective targeting of Pol β in cell lysates
The selectivity of 3a for Pol β was examined further by incubating various cell lysates of mouse embryonic fibroblasts (MEFs) with proinhibitor 18. Pol λ−/− and Pol β−/− lysates were obtained from the MEFs of mice (in a C57BL/6 background) that were established in two different laboratoaries.44,45 Pol β−/−/Pol λ−/− lysates were obtained from the MEFs of double knockout mice that were generated by breeding Pol β+/− and pol λ+/− heterozygote mice together.21 Pol λ WT and pol β WT cells were matched to Pol λ null and Pol β null, respectively. Cell lines that differ from wild type by the presence or absence of these polymerases are of interest because there is evidence that Pol λ acts as a back-up for Pol β during BER, although its lyase activity in vitro is considerably weaker than that of Pol β.6,21,22 Consequently, it would be useful to know if 3a distinguished between the two enzymes. The lyase activity on 3′-32P-15 of various MEF cell lysates was examined in the absence and presence of 18 (50 μM). Lyase reaction rates on 3′-32P-15 were measured following preincubation of the lysate with 18 (or buffer). Preincubation with proinhibitor 18 reduced the rate of the lyase reaction almost 2-fold in lysates obtained from wild type cells. As shown in Figure 6 this effect was similar in both WT cells as expected (even though their individual rates were different).38 Also the effect of 18 on lyase activity in cells lacking Pol λ (Pol λ −; Pol λ−/−/Pol β+/+) was statistically indistinguishable (p = 0.54) from reactivity in the wild type cells, suggesting that the inhibitor does not signficantly inhibit this enzyme.
Figure 6.

Effect of proinhibitor 18 (50 μM) on lyase activity (3′-32P-15) of various cell lysates from mouse embryonic fibroblasts.
The effect of 18 in lysates lacking Pol β (Pol β −; Pol β−/−/Pol λ+/+)was clearly different (p < 0.05). The overall lyase activity in lysates lacking Pol β (in the absence of 18) was signficantly lower than in those obtained from the wild type or Pol λ deficient cells, indicating that Pol β enzyme was the major contributor to the lyase reaction with 3′-32P-15. More importantly, 18 had a much smaller effect in Pol β deficient cells. The lyase activity was reduced less than 30% in the presence of 18. The effect of 18 on lyase activity in the double knock-out (Pol β/λ−; Pol β−/−/Pol λ−/−) cells was within experimental error of that in the Pol β deficient (Pol β−; Pol β−/−/Pol λ+/+) cells, providing additional evidence that the inhibitor is selective for Pol β over Pol λ. However, the observation that 18 has even a small effect on the lyase reaction in the double knock-out or Pol β deficient cell lysates indicates that one or more other enzymes are affected by the proinhibitor (Figure 6).
The effects of 3a and 18 in prostate cancer cells (DU145)
The superior performance of 18 compared to 3a in cell lysates was also evident in studies using DU145 cells. For instance, ~0.01% of the DU145 cells survived treatment with 40 μM 18, whereas 12% of the cells survived treatment with the same concentration of 3a (Figure 7A). We postulate that greater stability of 18 than 3a to nucleophiles in the extracellular matrix, resulting in a higher concentration of molecule delivered to the cell, is one source of its greater efficacy. The above cell lysate experiments suggesting that the small molecule DOB mimics inhibit Pol β lyase activity, combined with the encouraging intracellular activity of 18 led us to examine its ability to potentiate the cytotoxicity of a DNA damaging agent whose effects would require repair by Pol β. BER of DNA alkylated by methyl methanesulfonate (MMS) proceeds through an abasic site (AP, Scheme 1). Consequently, DU145 cell survival was measured as a function of MMS concentration, without and with 18 at a concentration (20 μM) where the proinhibitor itself results in ~45% cell death. After normalizing the fraction of surviving DU145 cells by taking into account the cytotoxicity of 18, plotting cell survival as a function of MMS concentration (Figure 7B) reveals a clear potentiation (Figure 7C) of the alkylating agent’s cytotoxicity at 0.2 mM and above. The cytotoxicity of 18 (20 μM) and MMS (200 μM) is more than 2-fold greater than one would expect if the two agents were not acting synergistically. The synergistic effect of 18 and MMS is even greater at higher MMS concentrations but is difficult to quantify above 0.3 mM MMS where one observes a 5-fold potentiation, due to the small numbers of surviving cells. Interestingly, the level of potentiation observed by 18 is comparable and even slightly greater than that seen in cells in which either the Pol β gene is removed or its expression is knocked down using siRNA.46,47
Figure 7.
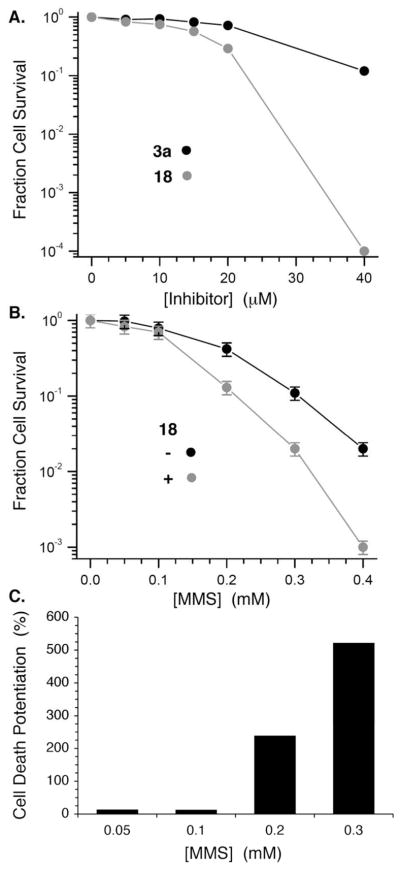
Effect of 3a and proinhibitor 18 on DU145 prostate cancer cells. A) Survival fraction as a function of inhibitor concentration. B) Effect of 18 (20 μM) on methyl methanesulfonate (MMS) cytotoxicity. C) Potentiation of MMS cytotoxicity by 18 (20 μM).
Conclusions
The kinetic experiments described above demonstate that small molecules containing a 1,4-dicarbonyl, the same functional group present in DNA lesions that is responsible for inactivating Pol β (and Pol λ), irreversibly inhibit the lyase activity of this enzyme.17 To our knowledge, these are the first suicide inhibitors that target the lyase activity of Pol β. Due to the lack of MS evidence, we cannot unequivocally state that the inhibitor modifies the lysine(s) involved in Schiff-base formation. Furthermore, the IC50 of 3a is comparable to the best of previously reported Pol β inhibitors.17 Importantly, experiments in cell lysates derived from a variety of mouse embryonic fibroblasts lacking neither, one, or both Pol β or Pol λ demonstrate that 18, which is enzymatically converted to 3a in lysates selectively inhibits the lyase activity of the former over the latter. Although other enzymes that contribute to the lyase reaction of 32P-15 are affected more weakly by 18, overall the experiments involving mouse embryonic fibroblast lysates support inhibition of the targeted Pol β in cells. Pol λ is believed to back up BER by Pol β. Hence, developing molecules that selectively inhibit one of these enzymes over the other is useful for probing the enzymes’ roles in DNA repair in cells.48,49 This suggests that 18 could be a useful tool for examining the effects of Pol β in cells. Furthermore, 18 functions as well or better than other molecules at potentiating the effects of a DNA damaging agent (MMS) in cells.3 To our knowledge, this is the first example of an irreversible inhibitor of Pol β that potentiates the cytotoxicity of a DNA damaging agent.3,17 Future generations of DNA repair inhibitors that potentiate MMS and/or other DNA damaging agents at even lower concentrations are desirable. Finally, there is a resurgence in interest in molecules that covalently modify their biological targets.50–52 The approach described here may be useful for inhibiting other DNA repair processes.53
Supplementary Material
Acknowledgments
We are grateful for generous financial support from the National Institute of General Medical Sciences (GM-063028) to MMG and the National Cancer Institute (CA-058236) to TLD. We thank Professor James Stivers (JHU) for providing access to the aldehyde library and for helpful discussions, and Dr. Sam Wilson (NIEHS) and Dr. Julie Horton (NIEHS) for providing the mouse embryonic fibroblasts.
Footnotes
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
ASSOCIATED CONTENT
Supporting Information. Experimental procedures for all experiments, cell lysate kinetics, fluorescence assays of inhibition, chart of aldehydes used to generate library, NMR spectra of new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T. DNA Repair and Mutagenesis. 2. ASM Press; Washington, D.C: 2006. [Google Scholar]
- 2.Stivers JT, Jiang YL. Chem Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 3.Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. Cell Res. 2008;18:48–63. doi: 10.1038/cr.2008.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guan L, Greenberg MM. J Am Chem Soc. 2010;132:5004–5005. doi: 10.1021/ja101372c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Guan L, Bebenek K, Kunkel TA, Greenberg MM. Biochemistry. 2010;49:9904–9910. doi: 10.1021/bi101533a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens AJ, Guan L, Bebenek K, Kunkel TA, Greenberg MM. Biochemistry. 2013;52:975–983. doi: 10.1021/bi301592x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jacobs AC, Kreller CR, Greenberg MM. Biochemistry. 2011;50:136–143. doi: 10.1021/bi1017667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsumoto Y, Kim K. Science. 1995;269:699–702. doi: 10.1126/science.7624801. [DOI] [PubMed] [Google Scholar]
- 9.Matsumoto Y, Kim K, Katz DS, Feng JA. Biochemistry. 1998;37:6456–6464. doi: 10.1021/bi9727545. [DOI] [PubMed] [Google Scholar]
- 10.Prasad R, Beard WA, Strauss PS, Wilson SH. J Biol Chem. 1998;273:15263–15270. doi: 10.1074/jbc.273.24.15263. [DOI] [PubMed] [Google Scholar]
- 11.Deterding LJ, Prasad R, Mullen GP, Wilson SH, Tomer KB. J Biol Chem. 2000;275:10463–10471. doi: 10.1074/jbc.275.14.10463. [DOI] [PubMed] [Google Scholar]
- 12.Prasad R, Batra VK, Yang XP, Krahn JM, Pedersen LC, Beard WA, Wilson SH. DNA Repair. 2005;4:1347–1357. doi: 10.1016/j.dnarep.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 13.Prasad R, Beard WA, Chyan JY, Maciejewski MW, Mullen GP, Wilson SH. J Biol Chem. 1998;273:11121–11126. doi: 10.1074/jbc.273.18.11121. [DOI] [PubMed] [Google Scholar]
- 14.Feng JA, Crasto CJ, Matsumoto Y. Biochemistry. 1998;37:9605–9611. doi: 10.1021/bi9808619. [DOI] [PubMed] [Google Scholar]
- 15.Husain I, Arteaga CL, Srivastava DK, Wilson SH. Carcinogenesis. 1999;20:1049–1054. doi: 10.1093/carcin/20.6.1049. [DOI] [PubMed] [Google Scholar]
- 16.Starcevic D, Dalal S, Sweasy JB. Cell Cycle. 2004;3:998–1001. [PubMed] [Google Scholar]
- 17.Barakat K, Gajewski M, Tuszynski JA. Curr Top Med Chem. 2012;12:1376–1390. doi: 10.2174/156802612801319070. [DOI] [PubMed] [Google Scholar]
- 18.Donigan KA, Sun K-w, Nemec AA, Murphy DL, Cong X, Northrup V, Zelterman D, Sweasy JB. J Biol Chem. 2012;287:23830–23839. doi: 10.1074/jbc.M111.324947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nemec AA, Donigan KA, Murphy DL, Jaeger J, Sweasy JB. J Biol Chem. 2012;287:23840–23849. doi: 10.1074/jbc.M112.362111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Donigan KA, Hile SE, Eckert KA, Sweasy JB. DNA Repair. 2012;11:381–390. doi: 10.1016/j.dnarep.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braithwaite EK, Kedar PS, Stumpo DJ, Bertocci B, Freedman JH, Samson LD, Wilson SH. PLoS One. 2010;5:e12229. doi: 10.1371/journal.pone.0012229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Braithwaite EK, Prasad R, Shock DD, Hou EW, Beard WA, Wilson SH. J Biol Chem. 2005;280:18469–18475. doi: 10.1074/jbc.M411864200. [DOI] [PubMed] [Google Scholar]
- 23.Gao Z, Maloney DJ, Dedkova LM, Hecht SM. Bioorg & Med Chem. 2008;16:4331–4340. doi: 10.1016/j.bmc.2008.02.071. [DOI] [PubMed] [Google Scholar]
- 24.Nakamura R, Takeuchi R, Kuramochi K, Mizushina Y, Ishimaru C, Takakusagi Y, Takemura M, Kobayashi S, Yoshida H, Sugawara F, Sakaguchi K. Org & Biomol Chem. 2007;5:3912–3921. doi: 10.1039/b710944j. [DOI] [PubMed] [Google Scholar]
- 25.Strittmatter T, Bareth B, Immel TA, Huhn T, Mayer TU, Marx A. ACS Chem Biol. 2011;6:314–319. doi: 10.1021/cb100382m. [DOI] [PubMed] [Google Scholar]
- 26.Wilson SH, Beard WA, Shock DD, Batra VK, Cavanaugh NA, Prasad R, Hou EW, Liu YA, Asagoshi K, Horton JK, Stefanick DF, Kedar PS, Carrozza MJ, Masaoka A, Heacock ML. Cell Mol Life Sci. 2010;67:3633–3647. doi: 10.1007/s00018-010-0489-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pitié M, Pratviel G. Chem Rev. 2010;110:1018–1059. doi: 10.1021/cr900247m. [DOI] [PubMed] [Google Scholar]
- 28.Goldberg IH. Acc Chem Res. 1991;24:191–198. [Google Scholar]
- 29.Jiang YL, Krosky DJ, Seiple L, Stivers JT. J Am Chem Soc. 2005;127:17412–17420. doi: 10.1021/ja055846n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung S, Parker JB, Bianchet M, Amzel LM, Stivers JT. Nat Chem Biol. 2009;5:407–413. doi: 10.1038/nchembio.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mootoo DR, Date V, Fraser-Reid B. J Am Chem Soc. 1988;110:2662–2663. [Google Scholar]
- 32.Chen F, Gaucher EA, Leal NA, Hutter D, Havemann SA, Govindarajan S, Ortlund EA, Benner SA. Proc Natl Acad Sci USA. 2010;107:1948–1953. doi: 10.1073/pnas.0908463107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kodama T, Greenberg MM. J Org Chem. 2005;70:9916–9924. doi: 10.1021/jo051666k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bournaud C, Marchal E, Quintard A, Sulzer-Mossé S, Alexakis A. Tet Asym. 2010;21:1666–1673. [Google Scholar]
- 35.Boeckman RK, Miller JR. Org Lett. 2009;11:4544–4547. doi: 10.1021/ol9017479. [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Wong CH. Tetrahedron Lett. 2002;43:4037–4039. [Google Scholar]
- 37.Dorjsuren D, Wilson DM, Beard WA, McDonald JP, Austin CP, Woodgate R, Wilson SH, Simeonov A. Nucleic Acids Res. 2009;37:e128–e128. doi: 10.1093/nar/gkp641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.See Supporting Information.
- 39.Silverman RB. The Organic Chemistry of Enzyme-Catalyzed Reactions. Academic Press; San Diego: 2000. [Google Scholar]
- 40.Brandt GS, Nemeria N, Chakraborty S, McLeish MJ, Yep A, Kenyon GL, Petsko GA, Jordan F, Ringe D. Biochemistry. 2008;47:7734–7743. doi: 10.1021/bi8004413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Serafimova IM, Pufall MA, Krishnan S, Duda K, Cohen MS, Maglathlin RL, McFarland JM, Miller RM, Frödin M, Taunton J. Nat Chem Biol. 2012;8:471–476. doi: 10.1038/nchembio.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ora M, Taherpour S, Linna R, Leisvuori A, Hietamäki E, Poijärvi-Virta P, Beigelman L, Lönnberg H. J Org Chem. 2009;74:4992–5001. doi: 10.1021/jo9005987. [DOI] [PubMed] [Google Scholar]
- 43.Jessen HJ, Schulz T, Balzarini J, Meier C. Angew Chem Int Ed. 2008;47:8719–8722. doi: 10.1002/anie.200803100. [DOI] [PubMed] [Google Scholar]
- 44.Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH. Nature. 1996;379:183–186. doi: 10.1038/379183a0. [DOI] [PubMed] [Google Scholar]
- 45.Bertocci B, De Smet A, Flatter E, Dahan A, Bories JC, Landreau C, Weill JC, Reynaud CAS. J Immunol. 2002;168:3702–3706. doi: 10.4049/jimmunol.168.8.3702. [DOI] [PubMed] [Google Scholar]
- 46.Horton JK, Joyce-Gray DF, Pachkowski BF, Swenberg JA, Wilson SH. DNA Repair. 2003;2:27–48. doi: 10.1016/s1568-7864(02)00184-2. [DOI] [PubMed] [Google Scholar]
- 47.Polosina YY, Rosenquist TA, Grollman AP, Miller H. DNA Repair. 2004;3:1469–1474. doi: 10.1016/j.dnarep.2004.05.011. [DOI] [PubMed] [Google Scholar]
- 48.Strittmatter T, Brockmann A, Pott M, Hantusch A, Brunner T, Marx A. ACS Chem Biol. 2013 doi: 10.1021/cb4007562. [DOI] [PubMed] [Google Scholar]
- 49.Kuriyama I, Miyazaki A, Tsuda Y, Yoshida H, Mizushina Y. Bioorg & Med Chem. 2013;21:403–411. doi: 10.1016/j.bmc.2012.11.024. [DOI] [PubMed] [Google Scholar]
- 50.Singh J, Petter RC, Baillie TA, Whitty A. Nat Rev Drug Discov. 2011;10:307–317. doi: 10.1038/nrd3410. [DOI] [PubMed] [Google Scholar]
- 51.Kwarcinski FE, Fox CC, Steffey ME, Soellner MB. ACS Chem Biol. 2012;7:1910–1917. doi: 10.1021/cb300337u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liu Q, Sabnis Y, Zhao Z, Zhang T, Buhrlage SJ, Jones LH, Gray NS. Chem Biol. 2013;20:146–159. doi: 10.1016/j.chembiol.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.