

Abstract
High-density lipoproteins (HDLs) and their major protein, apoA-I, remove excess cellular cholesterol and protect against atherosclerosis. However, in acquired amyloidosis, non-variant full-length apoA-I deposits as fibrils in atherosclerotic plaques; in familial amyloidosis, N-terminal fragments of variant apoA-I deposit in vital organs damaging them. Recently, we used the crystal structure of Δ(185-243)apoA-I to propose that amyloidogenic mutations destabilize apoA-I and increase solvent exposure of the extended strand 44-55 that initiates β-aggregation. Here we test this hypothesis by exploring naturally occurring human amyloidogenic mutations, W50R and G26R, within or close to this strand. The mutations caused small changes in the protein’s α-helical content, stability, proteolytic pattern, and protein-lipid interactions. These changes alone were unlikely to account for amyloidosis, suggesting the importance of other factors. Sequence analysis predicted several amyloid-prone segments that can initiate apoA-I misfolding. Aggregation studies using N-terminal fragments experimentally verified this prediction. Three predicted N-terminal amyloid-prone segments, mapped on the crystal structure, formed an α-helical cluster. Structural analysis indicates that amyloidogenic mutations or Met86 oxidation perturb native packing in this cluster. Together, the results suggest that structural perturbations in the amyloid-prone segments trigger α-helix-to-β-sheet conversion in the N-terminal ~75 residues forming the amyloid core. Polypeptide outside this core can be proteolysed to form 9-11 kDa N-terminal fragments found in familial amyloidosis. Our results imply that apoA-I misfolding in familial and acquired amyloidosis follows a similar mechanism that does not require significant structural destabilization or proteolysis. This novel mechanism suggests potential therapeutic interventions for apoA-I amyloidosis.
Keywords: High-density lipoprotein, α-helix to β-sheet conversion, amyloid self-recognition elements or “hot spots”, apoA-I oxidation and proteolysis, triglyceride reduction therapies
INTRODUCTION
High-density lipoproteins (HDLs) are protein-lipid nano-assemblies that transport lipids in plasma and protect against atherosclerosis [1,2]. Amino acid sequences of the major HDL protein, apoA-I (243 a. a.), and other apolipoproteins contain 11/22-mer tandem repeats with high propensity to form amphipathic α-helices whose apolar faces are optimized for lipid surface binding [5]. Plasma levels of HDL and apoA-I correlate inversely with the incidence of atherosclerosis [1-3], although this relationship is complex; reduced levels of apoA-I and HDL are not always associated with atherosclerosis, while elevated levels do not necessarily confer additional cardioprotection [6,7]. The cardioprotective effect of apoA-I and HDL is due mainly to their central role in reverse cholesterol transport whereby HDLs remove excess cholesterol from peripheral tissues to the liver [1-3,8,9]. At the critical early step of this complex process apoA-I promotes lipid efflux from cells to generate nascent HDL. Nascent “discoidal” HDL particle is envisioned as a cholesterol-containing phospholipid bilayer with protein α-helices wrapped around the edge (supplemental Fig. S1) [2-4]. Following maturation, HDL acquires a spheroidal shape with an apolar core of cholesterol esters and triglycerides (Fig. S1).
Most plasma apoA-I circulates on HDL, providing a flexible structural scaffold and an important functional ligand [3,4,10,11]. This HDL-bound protein is in dynamic equilibrium with monomeric lipid-poor or lipid-free protein (further referred to as “free”) that comprises ~5% of circulating apoA-I [2,12-15]. Free apoA-I is either generated de novo or dissociates from HDL during metabolic remodeling [12]. This labile transient species is thought to be particularly cardioprotective, as it provides the primary acceptor of cell cholesterol at the critical step of reverse cholesterol transport [13-16]. Nevertheless, shifting the population distribution from HDL-bound to free apoA-I is not necessarily beneficial [13,16-18]. In fact, unlike HDL whose structural stability is conferred by high kinetic barriers [19], free apoA-I has marginal thermodynamic stability [18,20] that not only facilitates its high metabolic activity [20,21] but also accelerates its proteolysis and clearance [2,13]. Moreover, free apoA-I can misfold and deposit as fibrils in amyloidosis [22,23].
The most common form of apoA-I amyloidosis is acquired [23]. Extracellular fibrillar deposits of non-variant apoA-I containing full-length protein or its N-terminal fragments have been found in tissues and in atherosclerotic plaques of most patients undergoing atherectomy [24-26]. Although the physiological role of these deposits is not entirely clear, they are proposed to be pathogenic [17,23-30]. In vitro studies report that apoA-I fibrils are cytotoxic [28] and can activate arterial macrophages [22,23], while in vivo studies report that apoA-I deposition correlates with increased vulnerability of atherosclerotic plaques [17,29,30]. Importantly, mild oxidation, which is proposed to contribute to atherogenesis, can induce amyloid formation by non-variant full-length human apoA-I in vitro and, probably, in vivo [31]. Together these findings indicate that apoA-I deposition in acquired amyloidosis is pro-atherogenic [23,27].
Familial apoA-I amyloidosis (AApoAI) is an autosomal dominant disorder in which mutant apoA-I deposits as fibrils in vital organs (kidney, liver, heart, etc.) leading to organ damage and ultimate failure [22,32-34]. There is no cure for AApoAI and the only available treatment is end-stage organ transplant. In contrast to many other amyloid diseases that involve protein overproduction, AApoAI patients have lower than normal plasma levels of apoA-I and HDL [6,32] resulting from reduced secretion and/or enhanced degradation of apoA-I [35-37]. Paradoxically, AApoAI is not associated with cardiovascular disease [34-36], suggesting that low levels of HDLs may be compensated by their improved function. Wide variation in the clinical presentation of the disease associated with the same mutation, from relatively mild to severe symptoms [34], suggests that AApoAI is influenced by additional factors such as lipids [38-40]. To identify these factors and to establish the much-needed therapeutic targets for this life-threatening disease, it is necessary to elucidate the molecular mechanism of apoA-I misfolding, from highly α-helical (up to 80% helix on HDL and nearly 60% helix in solution [3,10,20]) to the predominantly cross-β-sheet conformation in amyloid.
The prevailing concept in the field is that apoA-I proteolysis triggers release of the N-terminal fragments that misfold to form amyloid in AApoAI. This concept is based upon proteomic analysis of the patient-derived fibrillar deposits that contained residue fragments 1-83 to 1-100 of variant apoA-I in the amyloid core [22,32]. Of the ~20 known AApoAI mutations, most are located within the N-terminal 100 residues (“inside” mutations); in addition, several “outside” AApoAI mutations are located in residues 170-178 [32]. Heterogeneity of the patient-derived N-terminal fragments suggests multiple cleavage sites between residues 83 and 100. The origin of this cleavage is unclear, and was proposed to involve broad-specificity proteases such as matrix metalloproteinases (MMPs) [22,32]. In vitro studies of the recombinant 1-93 fragments characteristic of cardiac AApoAI showed that the mutation-induced differences in the aggregation kinetics do not correlate with the phenotype of the disease and cannot explain its mechanism [41]. These negative results prompted Raimondi and colleagues to propose that the main effect of AApoAI mutations is to promote the release of the N-terminal proteolytic fragments [41]. However, no experimental data supporting this effect were reported.
The notion that proteolysis of apoA-I precedes its misfolding in AApoAI [32,37,41-43] has several drawbacks. First, it is unclear why apoA-I is cleaved between residues 83 and 100. These residues form well-ordered helical structure in lipid-bound and in free apoA-I [10,44,45], which is expected and observed to be protected from proteolysis. In fact, broad-specificity proteases readily cleave free apoA-I at multiple sites, preferentially in its flexible C-terminal and central regions after residue 120, rather than between residues 83 and 100 [22,46,47]. Second, efforts to detect apoA-I N-terminal fragments in circulation have been unsuccessful [32]. Third, apoA-I proteolysis is not a prerequisite for fibril formation, as demonstrated by the ability of the full-length protein to form amyloid fibrils in vivo [48] and in vitro upon Met oxidation [31]. Consequently, it is possible that amyloid formation can precede apoA-I proteolysis rather than follow it [31, 49]. Our work addresses the molecular mechanism of such misfolding.
To elucidate the misfolding mechanism, we used the 2.2 Å resolution x-ray crystal structure of the C-terminally truncated human lipid-free apoA-I, Δ(185-243)apoA-I [10], which to-date provides the only atomic-level insight into the structure and function of apoA-I in solution and on HDL.* Importantly, Δ(185-243)apoA-I contains all sites of known AApoAI mutations (Fig. 1A), providing an excellent model for understanding the structural implications of these mutations [38]. Our previous structure-based studies suggested that AApoAI mutations may have multiple effects. They can potentially decrease the protein’s affinity for lipid and thereby promote dissociation of free apoA-I from HDL; they can potentially destabilize the helix bundle structure in free apoA-I and thereby increase its susceptibility to proteolysis; in addition, they can increase solvent accessibility of the extended residue segment 44-55 that, we proposed, may initiate α-helix to β-sheet conversion [38].
Figure 1.
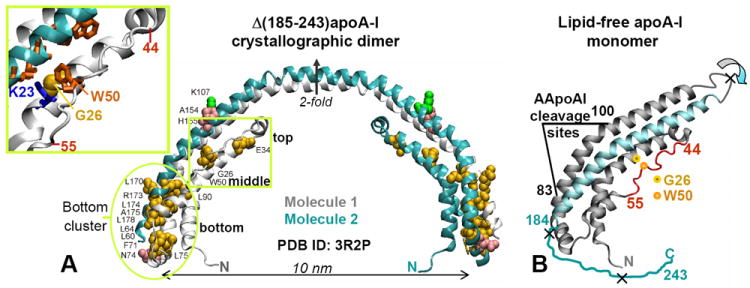
Locations of the AApoAI mutations in the structure of free apoA-I. (A) X-ray crystal structure of human lipid-free Δ(185-243)apoA-I (PDB ID 3R2P) solved by Mei and Atkinson [10] with the sites of all known AApoAI mutations mapped [38]. The structure is a crystallographic dimer stabilized by two symmetry-related four-helix bundles. Dimer-forming molecule 1 (gray) and 2 (teal) and the crystallographic two-fold axis relating them are shown. Residues that are mutated in AApoAI are shown by spheres: point mutations (yellow), deletions (green), and frame shifts (pink). Oval encircles the “bottom” half of the helix bundle where most of the AApoAI point mutations are located. Boxed region contains the sites of all known AApoAI point mutations located in the middle (G26R and W50R) or near the top (E34K) of the helix bundle. Insert shows zoomed-in structure in this boxed region; side chains of G26, W50 and K23 are shown. (B) Proposed structure of lipid-free apoA-I monomer obtained from the dimer structure via the domain swapping (indicated by circular arrows) of residues 121-184 (in teal) from molecule 2 to molecule 1 around the 2-fold axis [10]. Thin irregular line represents C-terminal residues 185-243 that are largely unfolded in solution and have been deleted from Δ(185-243)apoA-I to facilitate crystallization [10]. Extended segment in residues 44-55 is in red. The cleavage sites between residues 83 and 100, which are characteristic of the protein fragments found in patient-derived AApoAI deposits, are indicated; X indicate major cleavage sites upon processing of free apoA-I by the broad-specificity proteases. Locations of the two mutations studied in this work, G26R and W50R, are shown.
These proposed effects are consistent with the recent hydrogen-deuterium exchange (HX) mass spectrometry (MS) studies of full-length apoA-I monomer in solution by Phillips and colleagues who showed that the most common AApoAI mutation, apoA-I Iowa (G26R), greatly reduced protection of the N-terminal ~90 residues, suggesting loss of their α-helical structure [28]. The authors suggested that this helical unfolding augments proteolysis at residue 83 [28]. Contrary to this suggestion, proteolytic studies by Ramella and colleagues reported that recombinant wild type (WT) apoA-I and its selected AApoAI mutants such as G26R are degraded to a similar extent by MMP-12 [22]. This result contrasts with a previous report that G26R mutation promotes processing of the His-tagged apoA-I by non-specific proteases, chymotrypsin and V8, to produce the N-terminal fragment [42]. In sum, the effects of the AApoAI mutations on the protein cleavage are not entirely clear. It is also unclear why all known AApoAI mutants are cleaved between residues 83 and 100, why some destabilizing mutations in apoA-I are amyloidogenic while others are not [6,22,32,50-52]), or why Met oxidation promotes amyloid fibril formation by the full-length non-variant apoA-I [31].
To address these questions, here we report the structural stability, proteolytic susceptibility and lipid interactions of full-length recombinant human apoA-I containing two common AApoAI point mutations in the middle of (W50R) or adjacent to (G26R) the extended strand 44-55 (Fig. 1). Previous biophysical studies of full-length AApoAI mutants have been limited to lipid-free G26R and L178H [22,42,43,53,54]. We report the effects of the mutations on the stability of the full-length protein in solution and on reconstituted HDL (rHDL), and map the residue segments predicted to promote or prevent amyloid formation on the crystal structure of Δ(185-243)apoA-I. The results explain, for the first time, why the N-terminal residues form the amyloid core in AApoAI and why oxidation of specific Met promotes misfolding of the full-length apoA-I. Moreover, we propose a common molecular mechanism for apoA-I misfolding in familial and acquired amyloidosis, which has potentially important therapeutic implications.
Results and Discussion
Effects of G26R and W50R Mutations on ApoA-I Stability and Proteolysis
The structure and stability of full-length lipid-free apoA-I was analyzed by circular dichroism (CD). Far-UV CD spectra showed that G26R mutation caused only ~3% reduction in the α-helical content, while W50R mutation did not (Fig. 2A). Both mutations caused a decrease in the melting temperature by about 3 °C, suggesting a small reduction in stability (Fig. 2B). This is consistent with the previous studies reporting reduced helical content and reduced stability of free G26R apoA-I [22,42,43]. In addition, our melting data showed that G26R mutation reduced the unfolding cooperativity, which can augment protein misfolding [55], while W50R did not (Fig. 2B). W50R showed comparable or smaller effects than G26R on the secondary structure, stability and unfolding cooperativity of free apoA-I (Fig. 2), suggesting that the W50-K23 π-cation interaction observed in the crystal structure [10] contributes little to the overall conformation and stability of free protein in solution. Notably, similar studies of several naturally occurring non-amyloidogenic mutants [6,50-52], including apoA-I Milano (R173C) and Nichinan (ΔE235), showed comparable or larger decrease in the helical content, structural stability and unfolding cooperativity of free apoA-I [50-52]. Therefore, such a decrease alone does not necessarily lead to amyloidosis.
Figure 2.
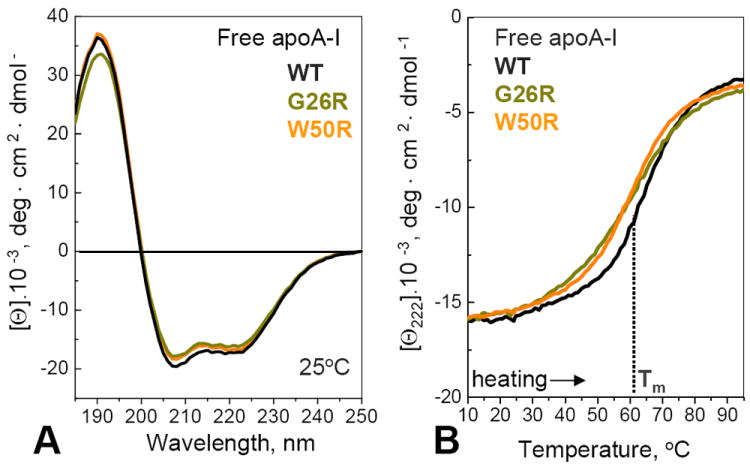
Secondary structure and stability of lipid-free apoA-I analyzed by far-UV CD. (A) Far-UV CD spectra of recombinant human WT, G26R and W50R apoA-I, recorded at 25 °C. The α-helix content estimated from these spectra is 52% for WT, 51% for W50R, and 49% for G26R, with approximately 3% accuracy. (B) Heating data of free proteins recorded at a rate of 80 °C/h show a small but significant decrease in the melting temperature, Tm, upon mutations. In addition, G26R shows a more gradual heat-induced change in CD signal indicating reduced cooperativity. Heating and cooling data (not shown to avoid overlap) superimpose.
To test whether AApoAI mutations promote the release of the N-terminal 9-11 kDa proteolytic fragments, we performed limited tryptic digestion of free protein whose products have been well-characterized for WT apoA-I [56] but not for its AApoAI mutants. The time course of proteolysis was monitored by using SDS PAGE (Fig. 3). Trypsin and other broad-specificity proteases have multiple cleavage sites in apoA-I, and initially process WT at two flexible sites, one near G185, G186 and another in the central part (so-called helix 5), producing ~22 kDa and 14 kDa fragments, respectively, followed by generation of shorter fragments resulting mainly from proteolysis of the flexible C-terminal part [42,46,47,56,57]. Figure 3 shows formation of the major fragments at comparable rates in WT and mutant apoA-I. There were also subtle differences; perhaps most relevant to AApoAI was the observation that W50R apoA-I showed a well-defined ~9 kDa band (Fig. 3C, arrow) that was not detected in WT (Fig. 3A) and was less well-defined in G26R apoA-I (Fig. 3B). This was the only band matching in size the N-terminal fragments in AApoAI deposits. In sum, G26R and W50R mutations had little effect on the overall rate of proteolysis, and G26R did not greatly enhance the release of the 9-11 kDa fragments (Fig. 3A, B), consistent with the previous proteolytic studies of this mutant using MMP-12 [22].
Figure 3.
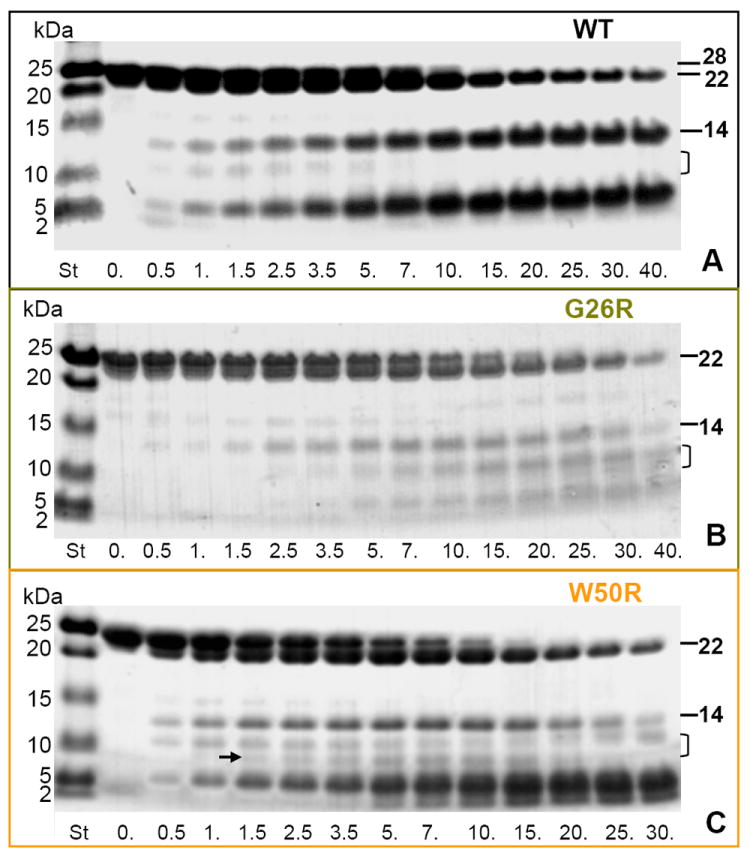
Time course of limited tryptic digestion of lipid-free WT and mutant apoA-I. The proteins were incubated with trypsin for 0-40 min (indicated on the lanes), the products were separated on SDS PAGE using Tris-Tricine system, and the gels were stained with Denville blue protein stain. Molecular weight standards (St) are indicated. Full-length apoA-I (28 kDa) and its two major proteolytic fragments, 22 kDa (cleaved near G186) and 14 kDa (cleaved in the middle of the molecule between residues 121 and 142) are indicated. Bracket shows the range where the acidic N-terminal 9-11 kDa fragments are expected to migrate. Arrow in panel C shows a band that may contain such a fragment.
Effects of G26R and W50R Mutations on the Lipoprotein Formation and Disintegration
To test the ability of free proteins to bind to a phospholipid bilayer and remodel it into rHDL, we monitored clearance of multilamellar vesicles of the model phospholipid, dimyristoyl phosphatidylcholine (DMPC), at 24 °C by turbidity (Fig. 4A). Lipid clearance was slightly retarded for G26R and slightly increased for W50R as compared to the WT apoA-I. Upon completion of DMPC clearance after 24 h incubation, the final turbidity leveled off at the similar value of 0.001 a. u. for the three apoA-I variants, which is consistent with our observation of similar-size particles formed at this stage (described below). Therefore, AApoAI mutations did not necessarily decrease the protein’s ability to bind to a phospholipid surface and form rHDL.
Figure 4.
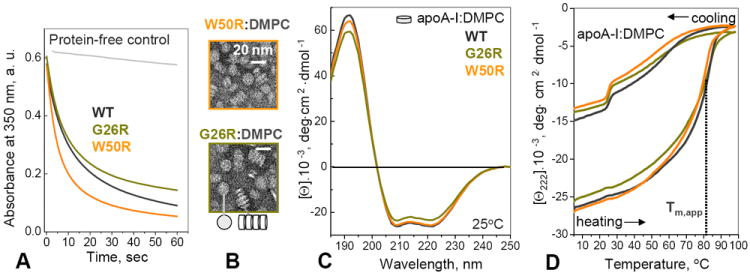
Effects of mutations on apoA-I affinity for lipid. (A) Time course of DMPC clearance by WT and mutant apoA-I. The proteins were added to aqueous suspensions of DMPC liposomes at t=0, and the remodeling of these liposomes into smaller lipoproteins was monitored by turbidity. (B) Electron micrographs on negatively stained “discoidal” complexes reconstituted from apoA-I and DMPC. The complexes are seen face-up or stacked on edge in rouleaux (bottom). (C) Far-UV CD spectra of apoA-I:DMPC complexes at 25 °C. The α-helix content estimated from these spectra is 74% for WT, 73% for W50R, and 67% for G26R, with ~3% accuracy. (D) Heating and cooling data of apoA-I:DMPC complexes recorded at a rate of 80 °C/h show a thermodynamically irreversible transition that was previously shown to involve protein dissociation and unfolding and lipoprotein fusion [19,21]. The apparent transition temperature, Tm,app, which corresponds to the first derivative maximum in the heating data, dΘ222/dT, is within 2 °C for the complexes containing WT, G26R and W50R apoA-I, suggesting similar structural stability. Kinetic temperature-jump studies strongly support this conclusion and show that G26R and W50R mutations do not significantly change structural stability of apoA-I:DMPC complexes (Fig. S2).
To test the effects of AApoAI mutations on rHDL stability and apoA-I dissociation, WT and mutant proteins were reconstituted into complexes with either DMPC or palmitoyl-oleoyl phosphatidylcholine (POPC) and cholesterol, and structure and stability of these complexes were analyzed by CD. Figures 4B, C and S2 show the results for apoA-I:DMPC complexes. Negative stain EM showed that W50R and G26R readily formed rHDL with DMPC (Fig. 4B) which were comparable in size to similar WT-containing complexes. Far-UV CD spectra of these complexes showed that G26R caused a ~5% reduction in the protein α-helical content while W50R did not (Fig. 4C). Far-UV CD melting data showed comparable apparent melting temperatures for the WT and mutant rHDLs (Fig. 4D). Because lipoprotein stability is subject to kinetic control, we carried out kinetic temperature-jump experiments that are particularly sensitive to small changes in lipoprotein stability (see Methods for details) [19,21]. The results clearly showed that G26R and W50R mutations have no detectable effect on the stability of apoA-I:DMPC complexes (Fig. S2). Similar studies of apoA-I complexes with POPC and cholesterol, which better represent the composition of nascent HDL, also clearly showed that G26R and W50R mutations did not reduce α-helical content or stability of apoA-I on rHDL (data not shown). Since rHDL destabilization involves lipoprotein fusion and apoA-I dissociation [19,21], we conclude that G26R and W50R mutations do not promote apoA-I dissociation from rHDL.
Summary of the Experimental Studies of G26R and W50R apoA-I
Our results clearly show that AApoAI mutations can either slightly reduce (G26R) or slightly increase (W50R) protein’s ability to recruit lipids and form rHDL (Fig. 4A). Further, the mutations do not destabilize apoA-I on rHDL and do not promote dissociation of free protein (Fig. 4D, S2), suggesting that they do not necessarily shift the population distribution from HDL-bound to free apoA-I. Moreover, the effects of G26R and W50R mutations on the conformation and stability of free apoA-I (Fig. 2) are comparable or smaller than the destabilizing effects of certain non-amyloidogenic mutations such as Milano or Nichinan [6,50-52], indicating that such destabilization alone is insufficient to cause amyloidosis. Finally, G26R mutant shows little increase in the production of the 9-11 kDa fragments that could potentially contribute to amyloidosis; the exact nature of such fragments will be determined once the proteases involved in apoA-I cleavage in AApoAI have been clearly identified. Together, our results suggests that the amyloidogenic properties of the apoA-I mutants result, at least in part, from factors other that their reduced affinity for lipid, reduced stability of the dissociated proteins, or increased production of their N-terminal fragments. This unexpected result prompted us to search for additional triggers of protein misfolding in AApoAI.
Sequence Analysis Predicts Amyloid-Prone Segments in apoA-I
Protein misfolding is commonly initiated by short self-recognition elements that are four- to ten-residue segments with high propensity to self-aggregate into β-sheets and act as “hot spots” in amyloid formation [58-62]. To identify such segments in apoA-I, we used the consensus algorithms AmylPred and AmylPred2 [63,64] that utilize five and 11 sequence-based prediction methods, respectively (supplemental Doc. S1, Table S1 and Doc. S3; for detail see [64]). Using the consensus analysis helps optimize the balance between the specificity and sensitivity in the hot spot prediction. Here, high specificity means low probability of predicting too many amyloid-prone segments (few false positives) while high sensitivity means low probability that such segments are mistakenly omitted (few false negatives; see [64] for details). In AmyPred2, the prediction specificity is ~85% for consensus agreement by five out of 11 methods, and increases with increasing the number of hits [64]. Hence, the hot spots predicted by more than five hits (called “major” in this work) have better than 85% specificity. Consensus agreement by five or fewer hits can also reveal potentially important amyloidogenic segments [64], such as the “minor” hot spots described below. In addition, we used the PASTA server [65] to predict amyloidogenic segment pairings in a parallel or antiparallel β-sheet (Table S2, Fig. S3).
The application of AmylPred and AmylPred2 to apoA-I sequence predicted two major and two minor amyloid hot spots containing residues 14-22 (LATVYVDVL), 53-58 (VTSTFS), 69-72 (QEFW) and 227-232 (VSFLSA), in the following rank order: (14-22) > (227-232) > (53-58) > (69-72) (Fig. 5, Table S1). Each of these segments is rich in large hydrophobic residues, contains at least one aromatic residue in the middle, may contain an acidic group but hardly any basic groups, and has other characteristics of amyloidogenic sequences. PASTA predicted three strongest hot spots in the same rank order, (14-22) > (227-232) > (53-58) (Fig. S3). Similar amyloidogenic segments were predicted in other studies [63,66]. In sum, segment 14-22 was the strongest hit predicted by up to 11 out of 12 methods (Table S1), suggesting better than 90% prediction specificity. Experimental validation of this N-terminal hot spot is described below. The fourth and only hot spot located outside the N-terminal half is predicted in residues 227-232 from the C-terminal part that forms the primary lipid binding site in apoA-I.
Figure 5.
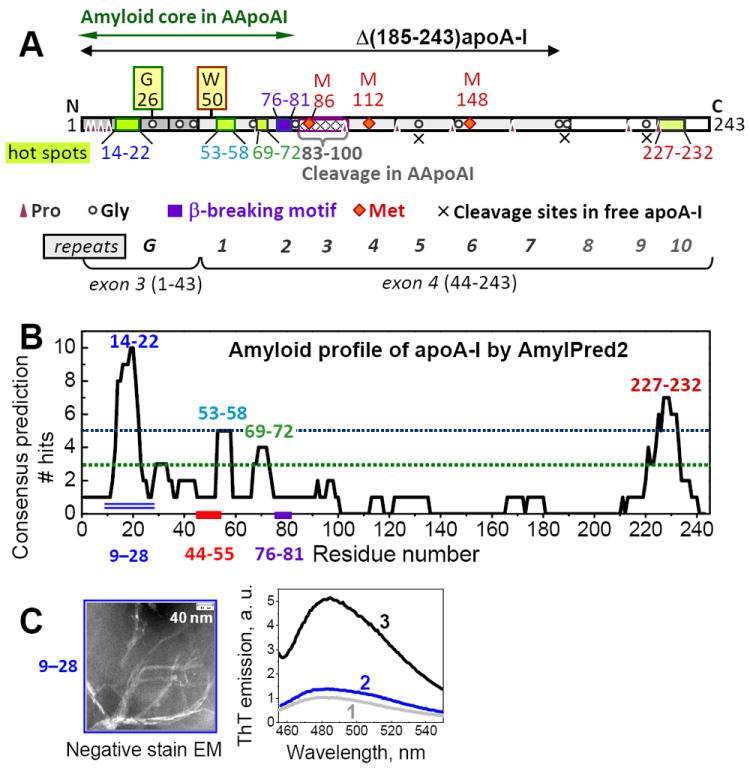
Amino acid sequence of apoA-I and its segments predicted and observed to promote amyloid formation. (A) Residues 1-43, encoded by exon 3, form N-terminal G-repeats (dark grey). Residues 44-243, encoded by exon 4, form ten 11/22-mer tandem repeats with high helical propensity, 1-10, punctuated by Pro or Gly. Repeats 1-7 are in dark grey and 8-10 are in white. Positions of Pro and Gly are indicated. Green boxes show amyloid “hot spots” formed by residues 14-22 (LATVYVDVL), 53-58 (VTSTFS), 69-72 (QEFW), and 227-232 (VSFLSA) predicted by using AmylPred [63,64] (B). Violet box shows the putative β-breaking motif (EKETEG) in residues 76-81. Green double arrow shows the amyloid core in the N-terminal fragments forming AApoAI deposits. Dashed box shows cleavage sites between residues 83 and 100 characteristic of AApoAI deposits. X show cleavage sites characteristic of free apoA-I in solution. Positions of G26R and W50R mutations are indicated. Red diamonds show methionines. Black double arrow indicates Δ(185-243)apoA-I construct whose 2.2Å resolution crystal structure [10] is shown in Figure 1. (B) Output of AmylPred2 consensus sequence analysis of human apoA-I plotted as a number of algorithms predicting β-aggregating propensity (from the total of 11) versus residue number. Segments predicted by a consensus of more than five hits (above the blue dotted line) are termed “major amyloid hot spots”; those predicted by three to-five hits (between the two dotted lines) are termed “minor hot spots”. Double line indicates 20-residue peptide fragment 9-28 predicted to have the highest amyloid-forming propensity. (C) Amyloid fibrils formed by synthetic fragment 9-28 observed by negative stain EM and ThT fluorescence. Fibrils were formed upon incubation of peptide solution in 100 mM Na phosphate buffer at pH 7.4, 22 °C for 6 days, and were analyzed as described in Methods. Fluorescence was measured from buffered solutions containing 10 μM ThT alone [1] or in the presence of 100 μM peptide that was either freshly dissolved (2) or formed fibrils (3) upon incubation at 22 °C for 17 days as described in Methods.
In addition to amyloidogenic segments, the apoA-I sequence contains several short motifs that are expected to hamper the β-sheet propagation. The longest of such motifs, EKETEG (residues 76-81), is predicted to have a β-breaking potential resulting from the Coulombic repulsion among three proximal Glu located on the same side of the β-strand, followed by the β-breaking Gly. This repulsion is expected to diminish at acidic pH upon Glu protonation and potential carboxyl pair formation. In sum, the sequence analysis suggests that the N-terminal part of apoA-I has high propensity to form amyloid that encompasses the first 75 residues but may not readily propagate beyond the β-breaking segment 76-81 at near-neutral pH.
Validation of the Predicted Amyloid Hot Spots
Several lines of experimental evidence support the predicted amyloid-forming segments in the N-terminal part of apoA-I. First, the AmylPred2 and PASTA profiles of apoA-I suggest that the amyloid-forming propensity resides largely within the first ~100 residues (Fig. 5, S3). This prediction is validated by the ability of the recombinant apoA-I fragment 1-83 and 1-93 to readily form amyloid fibrils in cell culture and/or in vitro [41,66], and is consistent with the finding of residue fragments 1-83 to 1-100 in patient-derived AApoAI deposits [22,32]. Second, synthetic apoA-I fragment 46-59, which contains a minor predicted hot spot 53-58, forms amyloid fibrils in vitro [67], validating this prediction. Third, recent studies report that fragment 1-43 that contains the major predicted hot spot, 14-22, readily forms amyloid fibrils in vitro at pH 7.4 [66]. In comparison, fragment 44-65 that contains a minor hot spot 46-59 is less prone to amyloid formation, while fragment 66-83 that contains the predicted pH-dependent β-breaking motif in residues 76-81 does not form amyloid at pH 7.4 [66]. There results are in excellent agreement with the sequence-based predictions.
To further validate the major predicted hot spot 14-22, we explored amyloid formation by a synthetic 20-residue peptide fragment containing apoA-I residues 9-28, which is centered on segment 14-22 and is predicted to have the highest amyloid propensity in the N-terminal half of apoA-I (Fig. 5B, double line). Peptide solutions of 0.1-0.5 mg/ml concentrations in 100 mM Na phosphate buffer at pH 7.4 were incubated at 22 °C for up to 17 days. Negative stain EM showed fibril formation after 6 days, followed by visible precipitation after 10 days. The fibrils were approximately 10 nm in width and hundreds of nm in length, and often formed clusters (Fig. 5C, left). Binding of a diagnostic dye, thioflavine T (ThT), followed by fluorescence measurements showed a large increase in ThT emission upon the peptide aggregation leading to fibril formation (Fig. 5C, right). In sum, electron microscopy and ThT binding / fluorescence indicate amyloid fibril formation by apoA-I fragment 9-28 that encompasses the major predicted amyloid hot spot, 14-22, further validating this prediction.
Notably, the two major hot spots, 14-22 and 227-232, are predicted strongly to form parallel intermolecular β-sheet in amyloid (Doc. S2, Table S2). In agreement with this prediction, FTIR spectra of fibrils formed by fragment 1-43 lack the diagnostic peak near 1690 cm-1 (66) and hence, are indicative of a parallel β-sheet, validating the parallel β-sheet predicted in this region of apoA-I (Table S2, Fig. S3). Spectroscopic data show that parallel in-register β-sheet is the major structural motif in protein fibrils containing β-strands longer than four residues [68,69], such as those predicted for apoA-I. Notably, parallel β-sheet was observed by EPR and by solid-state NMR in the fibrils of two related proteins, apoC-II [70] and α-synuclein [71,72], that contain lipid surface-binding 11-mer sequence repeats homologous to those in apoA-I [5]. These observations support the prediction of the parallel β-sheet as the preferred structural motif in all these apolipoprotein amyloids.
Mapping Amyloid Hot Spots on the Crystal Structure of Δ(185-243)apoA-I
The three N-terminal amyloid hot spots predicted by AmylPred, together with the β-breaking segment, were mapped on the proposed structure of apoA-I monomer (Fig. 6), which was derived from the crystal structure of Δ(185-243)apoA-I via the domain swapping around the dimer two-fold axis [10]. In the crystal structure all these segments are α-helical, except for the second hot spot, 53-58, that partially overlaps with the extended strand, 44-55, and the following α-helix, 56-64. The first and strongest hot spot, 14-22, forms an α-helix kinked at Y18, and is packed against the second hot spot. The third hot spot, 69-75, and the following the β-breaker, 76-81, form a short helix at the bottom of the bundle. In the crystal structure, this well-ordered region forms extensive interactions in the “bottom” hydrophobic cluster [10] (Fig. 6, oval). All three predicted N-terminal hot spots are located in or near this cluster. We propose that such tertiary packing, together with the predominantly α-helical conformation of these amyloid-prone segments, helps protect free apoA-I from misfolding. Similarly, the amyloid hot spots in other proteins often adopt native α-helical structure protecting them from misfolding [60].
Figure 6.

Residue segments predicted to promote or prevent amyloid formation mapped on the structure of lipid-free apoA-I monomer. Three N-terminal amyloid hot spots predicted in residues 14-22, 53-58, and 69-72 are mapped on the crystal structure of free Δ(185-243)apoA-I [10]. The fourth predicted “hot spot”, 227-232, is in the flexible C-terminal region (irregular line) that forms the primary lipid binding site in apoA-I driving its adsorption to phospholipid surface [18]. Residues 76-81 that are expected to form a β-breaking motif at pH 7 are in purple. Extended segment 44-55 is in red.
The C-terminal region containing the fourth hot spot, 227-232, was truncated from Δ(185-243)apoA-I to facilitate crystallization. This region forms the primary lipid binding site in apoA-I ([10,45] and references therein) and is largely α-helical on the lipid. In the low-resolution crystal structure of the N-terminally truncated apoA-I, which is thought to represent the lipid-bound conformation, residues 228-232 form the beginning of the C-terminal α-helix [44]. We posit that this highly α-helical conformation, together with the rapid protein adsorption via the C-terminal end to the phospholipid surface [18], help protect the C-terminal hot spot from initiating apoA-I misfolding.
The extended strand 44-55 observed in the crystal structure of Δ(185-243)apoA-I [38] partially overlaps with the minor hot spot, 53-58, suggesting that this strand has modest amyloid-forming propensity, consistent with the relatively polar character of residues 44-55, LKLLDNWDSVTS. X-ray crystallography and HX/MS studies indicate high solvent accessibility of residues 44-55 in the free protein [10,28,45] (Fig. S4). This contrasts with the amyloid-prone segments in globular proteins, which are usually protected by extensive native packing [60]. We propose that, despite its native strand-like geometry [38], segment 44-55 has limited amyloidogenic propensity which, together with the W50-K23 π-cation interaction that may restrict conformational mobility of this segment in free apoA-I [10], helps protect it from β-aggregation.
Previous studies reported that AApoAI mutations reduce the α-helical propensity of apoA-I [32] and perturb the radial distribution of charged and apolar residues characteristic of apolipoprotein α-helices [38]. Sequence-based prediction algorithms including AmylPred2 and PASTA showed no significant increase in the amyloid-forming propensity upon W50R, G26R or other AApoAI mutations. This suggests that the primary effect of such mutations is on the native conformation of apoA-I.
Revised Mechanism of Misfolding in AApoAI Mutants
The results reported here, together with the previous HX/MS analyses of WT and G26R apoA-I [45] and in vitro amyloid formation by apoA-I fragments [66, 67], suggest that protein misfolding in AApoAI is triggered by perturbations of the native structure in the N-terminal amyloid hot spots, increasing their solvent exposure and/or mobility. In the crystal structure of free protein, G26 is juxtaposed to W50 in the middle of the extended segment 44-55 (Fig. 1A). This segment is loosely packed and forms few specific interactions with the rest of the protein molecule. These interactions probably contribute little to the overall protein stability, as suggested by minimal effects of W50R mutation on the structure and stability of free protein (Fig. 2). Notably, W50R mutation replaces the π-cation attraction, W50-K23 (Fig. 1A), with the Coulombic repulsion, R50-K23, which is expected to perturb the local conformation and dynamics of the strand 44-55. This perturbation can propagate from the mutation site at residue 50 to the nearby hot spot, 53-58, perturbing its interactions with the adjacent major hot spot, 14-22. We propose that this perturbed hot spot packing triggers amyloid formation by W50R apoA-I.
A similar mechanism probably contributes to amyloid formation by G26R apoA-I. In G26R mutant, Coulombic repulsion between K23 and R26, which are adjacent in the α-helix (Fig. 1A), would also disrupt the π-cation interaction, W50-K23, and once again perturb the local conformation and dynamics of the extended segment 44-55 and the nearby hot spots. Importantly, HX/MS studies of free apoA-I by Phillips and colleagues showed that G26R mutation greatly increases solvent accessibility and dynamics in the first ~90 residues [45]. We note that residues 1-90 encompass all predicted N-terminal amyloid hot spots (Fig. 5, S4). Hence, we propose that the destabilization of the helical structural in the N-terminal amyloid hot spots triggers the misfolding of G26R apoA-I. In addition, G26R slightly reduces the overall structural stability [22,28] and cooperativity of free apoA-I (Fig. 2B) and eliminates Gly26 that may be β-blocking [42]. Finally, both G26R and W50R reduce the net negative charge on apoA-I, particularly on its acidic N-terminal part, which generally favors β-aggregation [60,73]. A combination of these factors probably contributes to amyloid formation by G26R and W50R apoA-I.
We speculate that other AApoAI mutations promote amyloid formation via a combination of similar factors. In fact, 11 out of 14 known sites of AApoAI point mutations are located in the bottom half of the helix bundle in direct contact with one or more N-terminal hot spots (Fig. 1A, oval; Fig. 6). Close inspection of the sites of these mutations suggests that they perturb native packing in these hot spots. Of the remaining three AApoAI point mutations, two (G26R and W50R) are located near the middle of the helix bundle (Fig. 1A, boxed region); structural perturbations caused by these mutations are described above. The remaining AApoAI point mutation, E34K, is unusual as it is located near the top of the helix bundle distant from the predicted hot spots (Fig. 1A). E34K decreases the net negative charge by 2, the largest decrease among AApoAI mutations, which is expected to favor β-aggregation [60,73]. Other AApoAI mutations involve deletions and frame shifts [32,34,38] that potentially alter the helix registry in the bundle or truncate it, which can be detrimental to protein stability [74].
Of the nearly 20 known AApoAI mutations, only one most conservative substitution, F71Y, is located in the middle of the minor predicted hot spot, 69-72. This raises a possibility that the disruptive mutations inside the hot spots promote apoA-I misfolding and sequestration in inclusion bodies [63,75,76] and thereby interfere with the secretion of the mutant protein [36]).
In sum, of the 14 known AApoAI point mutations, at least 13 are expected to perturb local packing in the N-terminal hot spots in free apoA-I. Such a perturbation was observed by HX/MS in G26R, the most common AApoAI mutant [45] (Fig. S4). This observation strongly supports our hypothesis that local perturbations of the native structure and dynamics in the N-terminal hot spots in free apoA-I trigger its misfolding in AApoAI. These perturbations can be synergistic with other factors, such as reduced overall structural stability and cooperativity (Fig. 2B), but do not require them. Hence, our proposed mechanism helps uncouple apoA-I destabilization from misfolding and explain why many mutations, such as Milano or Nichinan, reduce structural stability of free apoA-I but do not cause amyloidosis [6,22,32,50-52].
Role of Methionine Oxidation in Amyloid Formation by ApoA-I
Our mechanism helps explain why Met oxidation causes misfolding of full-length non-variant apoA-I [27,31]. Apolipoprotein methionines are thought to serve as natural antioxidants for lipids, since these residues are highly susceptible to oxidation [77,78]. Met oxidation can alter protein function, stability, and propensity to form amyloid [31,78]. ApoA-I contains three methionines, M86, M112 and M148, that are located in the apolar lipid-binding faces of the α-helices, and can be converted into Met sulfoxides (MetO) in vivo. Replacement of the apolar Met with polar MetO in these locations is expected and observed to diminish protein affinity for lipid and decrease protein stability in solution [79-81], probably because of the perturbed hydrophobic core packing in the helix bundle. The effect of Met oxidation on the structural stability and lipid binding of apoA-I is expected to resemble certain AApoAI mutations, such as L60R, L64R or L178H, that also replace apolar with polar or basic groups in the hydrophobic core of the helix bundle [38] Hence, similar to these mutations, Met oxidation is expected to augment amyloid formation by apoA-I. In fact, full-length non-variant human plasma apoA-I forms amyloid upon Met oxidation in vitro, suggesting that mild oxidation in vivo can promote apoA-I deposition in atherosclerotic plaques [31].
The role of individual Met oxidation in apoA-I misfolding is unclear. To clarify this role, we examined Met packing in the crystal structure of Δ(185-243)apoA-I (Fig. 7). M86 is located in the “bottom” hydrophobic cluster, with the Met side chain forming extensive interactions with the amyloid hot spots. First, well-ordered Sδ and Cγ atoms of M86 are only 4–5 Å away from Cβ of Y18 in the middle of the major hot spot, 14-22 (Table S3). Second, Sδ in M86 is packed in the predominantly hydrophobic region close to carbonyl carbons from residues 173, 174 and 177 located in the helical segment 170-178 (Table S2) that contains “outside” point mutations in AApoAI (Fig. 7). This segment interacts directly with all N-terminal hot spots. Polar MetO in this well-ordered hydrophobic region is expected to perturb these interactions and thereby promote apoA-I misfolding.
Figure 7.
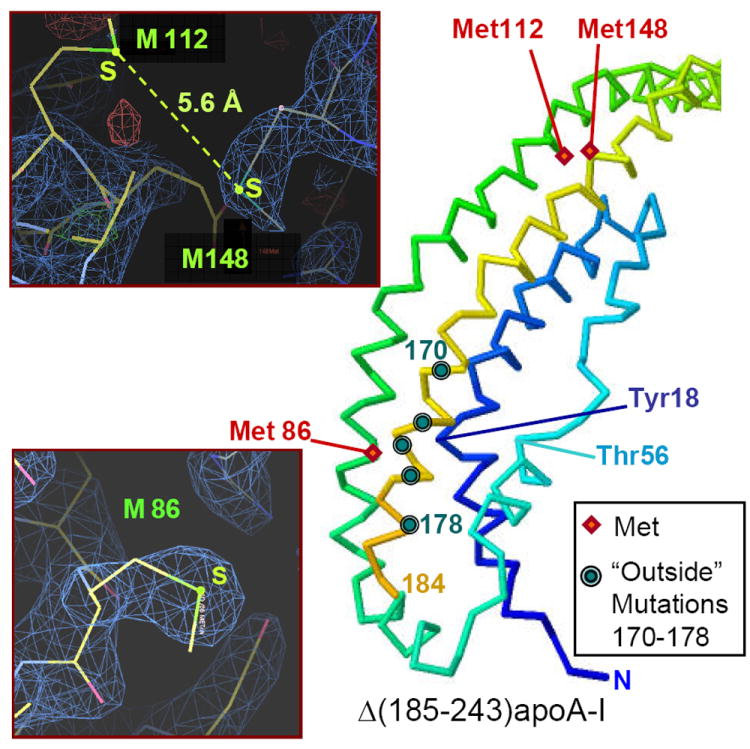
Locations of the thee methioines in free apoA-I. Positions of M86, M112, and M148 (red diamonds) are mapped on the crystal structure of Δ(185-243)apoA-I. Polypeptide chain is in rainbow colors from N- to C-terminus (blue to red). Circles indicate sites of all known “outside” point mutations in AApoAI, including L170, R173, L174, A175 and L178 (top to bottom). Main chain of Tyr18 located in the middle of the major spot, 14-22 (blue), and its selected nearest neighbors that include Met86 side chain are indicated. Inserts: Electron density map showing M86 (bottom) and M112 and M148 (top). The 2Fo-Fc map was generated by using the x-ray diffraction data (observed amplitudes, Fo) and the refined 2.2Å crystal structure of Δ(185-243)apoA-I [10] (calculated amplitudes, Fc). Stick model shows atomic structure. Sulfur positions are in yellow-green, electron density is in blue.
In contrast, M112 and M148 are located at the top of the helix bundle, remote from the amyloidogenic point mutations or hot spots (Fig. 7). M112 and M148 sulfurs are less than 6 Å apart. Unlike M86, M112 and M148 side chains are not well ordered in the crystal structure, as evident from the electron density map (Fig. 7 left) and the temperature factors of Sδ (B=60 Å2 in M86 as compared to ~95 Å2 in M112 and M114). These details help explain several reports that M112 and M148 are more susceptible to oxidation in vivo and in vitro than M86 ([80,82] and references therein). They also explain why in apoA-I isolated from plasma of hyperglycemic patients, M112 and M148 correlate in their oxidation degree [82]. We propose that spatial proximity of the M112 and M148 sulfurs in the free protein explains correlation in their oxidation degrees, while sequestration of the M86 side chain in the well-ordered hydrophobic cluster explains its reduced susceptibility to oxidation.
In sum, our structural analysis shows that in free apoA-I, M86 is located near the bottom of the helix bundle where it forms extensive hydrophobic interactions with Y18 and other groups from the major amyloid hot spot, 14-22 (Table S3). M86 oxidation is expected to perturb these interactions. In contrast, oxidation of M112 and M148, which are located at the top of the helix bundle, probably has little direct effect on the packing of the hot spots near its bottom. Therefore, we propose that oxidation of M86 is the likely trigger of apoA-I misfolding in atherosclerotic plaques.
Common Mechanism of apoA-I Misfolding in Familial and Acquired Amyloidosis
Taken together, the results reported here suggest that apoA-I misfolding in familial and in acquired amyloidosis is triggered by perturbed native structure in the amyloid hot spots, increasing their mobility and solvent accessibility. On HDL, all hot spots are protected from misfolding by the protein-lipid interactions, with additional protection conferred by the helical packing in the antiparallel “double-belt” conformation of apoA-I dimer on HDL [4,10,11,44]. This antiparallel helical packing (Fig. 1) must be disrupted prior to formation of the parallel intermolecular β-sheet predicted for apoA-I amyloid (Fig. 8, Table S2). This underscores the importance of free monomeric apoA-I as a direct progenitor of amyloid.
Figure 8.
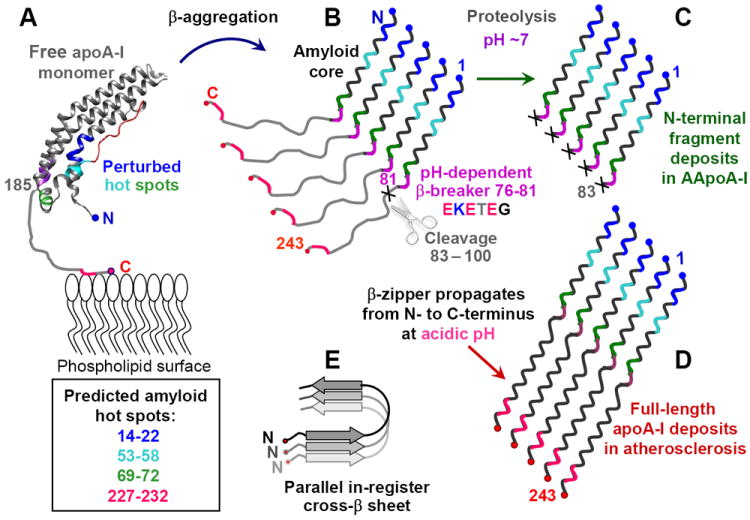
Hypothetical pathway of apoA-I misfolding in familial and in acquired amyloidosis. (A) Free apoA-I monomer, generated de-novo or dissociated from HDL, forms a four-helix bundle containing residues 1-184 (helix ribbon) followed by the partially unstructured C-terminal residues 185-243 (thin line) containing the primary lipid binding site. Four predicted amyloid hot spots are color-coded. AApoAI mutation in familial amyloidosis or M86 oxidation in acquired amyloidosis may perturb native structure in these hot spots and initiate β-aggregation. This leads to formation of the protease-resistant amyloid core in the first 75 residues containing N-terminal amyloid hot spots (in blue, teal and green) (B). In the parallel in-register β-sheet predicted for apoA-I, each hot spot (color-coded) stacks in register against its counterparts from the adjacent molecules. At pH~7, the β-breaking EKETEG residues 76-81 probably impede the amyloid core propagation towards the C-terminus and hamper the in-register stacking of the similarly charged groups. As a result, amyloid core stops at residues 76-81, leading to protein cleavage shortly thereafter. This explains why amyloid core in the patient-derived AApoAI deposits contains residue segments 1-83 to 1-100 (C). (D) At acidic pH, partial protonation of the adjacent Glu is expected to diminish the β-breaking potential of the EKETEG motif. This facilitates the β-sheet propagation towards the C-terminus and formation of fibrils containing full-length apoA-I, such as those found in atherosclerotic plaques. In the resulting parallel in-register β-sheet structure, different hot spots within the molecule act in synergy (D), while similar secondary structural elements from different molecules (in different shades of gray, E) are stacked in register.
In free apoA-I, the N-terminal hot spots are normally protected from misfolding by the interactions within the helix bundle [10,45] (Fig. 6). The C-terminal hot spot located in the primary lipid binding site is probably protected by its helical native conformation and by the bound lipids [18]. We propose that AApoAI mutations or Met86 oxidation perturb native packing in one or more of the N-terminal hot spots in free apoA-I, and thereby lessen their protection from misfolding. This local perturbation can be synergistic with other amyloidogenic factors such as reduced protein affinity for lipid, reduced stability and cooperativity of free protein structure, increased susceptibility to oxidation and proteolysis, reduced net charge and solubility, removal of the β-breaking groups such as Pro or Gly, etc. [73].
Our hypothetical mechanism is supported by multiple lines of experimental evidence and helps resolve several important issues. First, it uncouples structural destabilization of free apoA-I from local perturbations in its amyloid-prone segments, and explains why some but not all destabilizing apoA-I mutations are amyloidogenic [6,32]. A similar concept may apply to other globular proteins whose amyloidogenic propensity correlates with the structural stability but is not tightly coupled to it [73,76].
Second, the proposed mechanism explains why patient-derived AApoAI deposits contain N-terminal proteolytic fragments cleaved between residues 83 and 100 [22,32,41], even though in native apoA-I these residues form ordered α-helical structure that is protected from proteolysis [10,44,45]. We propose that the intermolecular parallel β-sheet readily propagates through the N-terminal 75 residues that are predicted and observed to be amyloidogenic. At near-neutral pH, the gate-keeping motif EKETEG in residues 76-81 is expected to impede the propagation of the β-zipper. Hence, the N-terminal ~75 residues form the protease-resistant amyloid core; polypeptide outside this core can be readily proteolyzed. In sum, we propose that apoA-I cleavage can occur after the amyloid core formation in AApoAI, although the current proteolytic studies cannot exclude the accepted pathway in which apoA-I is partially cleaved prior to misfolding. The former mechanism explains why the attempts to find the 9-11 kDa N-terminal fragments of apoA-I in circulation have been unsuccessful [32], and suggests that the proteolytic environment at the site of the amyloid deposition determines the cleavage site in AApoAI.
Third, the proposed mechanism suggests why arterial deposits in acquired amyloidosis commonly contain full-length apoA-I. At acidic pH reaching pH 5.5 in deep atherosclerotic plaques [83], partial protonation of the adjacent Glu in the EKETEG residues 76-81 is expected to reduce their β-breaking propensity and facilitate the β-sheet propagation from the N- to the C-terminus, to encompass all four amyloid hot spots in the full-length apoA-I (Fig. 8D).
Potential clinical implications
The concept that amyloid core formation is the primary event that does not necessarily involve proteolysis suggests, for the first time, that apoA-I misfolding in familial and in acquired amyloidosis proceeds via similar molecular mechanisms and, hence, is potentially influenced by similar factors. We hypothesize that HDL proteins, lipids and other factors that affect apoA-I dissociation from HDL can contribute to apoA-I misfolding in amyloid. Such contributing factors may possibly include serum amyloid A protein that can displace apoA-I from HDL during acute inflammation [84,85]; free fatty acids whose increase in HDL promotes apoA-I dissociation and lipoprotein fusion in vitro and, probably, in vivo [12,86]; increased HDL triglycerides whose lipolysis promotes apoA-I dissociation from HDL in vitro and in vivo [84,87], etc. The latter idea is supported by the observation that apoA-I fibril deposition in atherosclerotic plaques of patients undergoing atherectomy correlates positively only with patient’s age and plasma triglycerides [25]. This observation prompts us to speculate that therapies targeting HDL triglycerides and other factors that promote apoA-I dissociation from HDL in vivo may help hamper apoA-I amyloidosis.
Materials and Methods
Proteins and lipids
Recombinant human WT apoA-I and its two common naturally occurring AApoAI mutants, G26R and W50R, were produced by using His6-MBP-TEV expression system in E-coli and purified by FPLC as previously described [10]. The proteins contained one additional N-terminal Gly from the TEV cleavage site [10]. The lyophilized proteins were refolded in standard buffer that was used throughout this work (10 mM Na phosphate, pH 7.4) containing 0.25 mM Na EDTA. Protein stock solutions were stored in the dark at 4 °C and used in four weeks. DMPC, POPC and unesterified cholesterol were 95+% purity from Avanti Polar Lipids (Alabaster, AL, USA). Trypsin was from Sigma (St. Louis, MO, USA). All chemicals were of highest purity analytical grade.
Lipoprotein reconstitution and lipid clearance
DMPC complexes with apoA-I were obtained by incubating protein solution with the lipid suspension (1 mg/ml protein, 4 mg/ml lipid in standard buffer) at 24 °C overnight [18]. Complexes of apoA-I with POPC and unesterified cholesterol were obtained by thin film evaporation using apoA-I : POPC : cholesterol molar ratio of 1:80:4 as previously described [18]. Lipoprotein formation was confirmed by negative stain EM using a CM2 transmission electron microscope (Philips Electron Optics, Eindhoven, the Netherlands) as previously described ([18,19] and references therein).
Clearance kinetics of DMPC multilamellar vesicles by WT or mutant apoA-I was monitored at 24 °C by turbidity at 350 nm using a Varian Cary-300 UV/Vis spectrophotometer as described [18]. The final sample concentration was 200 μg/ml DMPC and 50 μg/ml apoA-I in standard buffer. Lipid-free apoA-I is monomeric under these conditions.
Circular Dichroism Spectroscopy
Far-UV CD and 90° light scattering data were recorded using an AVIV 400 spectropolarimeter as described ([18,88] and references therein). Briefly, melting and kinetic temperature-jump data were recorded from protein or lipoprotein solutions of 50 μg/ml protein concentration in standard buffer, placed in 2 mm path length cell. In the melting experiments, the samples were heated at a rate of 80 °C/h and α-helical unfolding was monitored at 222 nm. Thermal unfolding of free apolipoproteins in this and other studies was thermodynamically reversible, as evident from full superimposition of the heating data recorded at various scan rates and the overlap between the heating and cooling data [20,21,81,88]. To avoid overlap, only the heating data are shown for free proteins (Fig. 2B). In contrast, thermal unfolding of reconstituted HDL in this and the previous studies showed hysteresis (Fig. 4D) and a scan rate dependence indicative of a kinetically controlled transition with high activation energy [19,21,81,88]. Previously, we showed that the kinetic barriers in this transition are involved in the heat-induced apolipoprotein unfolding and partial dissociation from the lipid accompanied by lipoprotein fusion ([21,88] and references therein). In the kinetic experiments using model HDL, the unfolding was triggered by a rapid increase in temperature from 25 °C to a constant value ranging from 75 to 95 °C, and was monitored at 222 nm. The CD data were normalized to protein concentration and are reported as molar residue ellipticity, [Θ], in units of deg·cm2·dmol-1. Apolipoprotein helical content was estimated from the measured ellipticity at 222 nm, [Θ222] [20].
Proteolysis time course
Limited proteolysis was performed by incubating lipid-free WT or mutant apoA-I with trypsin (substrate:enzyme weight ratio 500:1) in standard buffer at 25 °C in a shaking water bath. Aliquots were collected at various time points during the first hour of incubation. To stop the reaction, SDS gel loading buffer was added to each aliquot, followed by immediate heating and incubation at 100 °C for 2 min. The proteolytic products were separated on SDS PAGE using 10-20% Tris / Tricine system. The gels were stained with Denville blue protein stain.
Fibril formation by a peptide fragment of apoA-I
The peptide fragment corresponding to apoA-I residues 9-28 (DRVKDLATVYVDVLKDSGRD), which encompassed the major predicted amyloid hot spot residues 14-22, was used in fibril growth experiments. The peptide was obtained commercially by solid state synthesis and purified by HPLC to 95%+ purity as previously described [88], followed by buffer exchange to remove organic solvent. The N- and C-termini of the peptide were not blocked. The amino acid sequence was confirmed by mass spectrometry. Freshly prepared stock solution (5.0 mg/ml peptide in 100 mM Na phosphate buffer, pH 7.4) was diluted to prepare samples varying in peptide concentration (0.1- 0.5 mg/ml), pH (6.8-7.4) and salt (0-150 NaCl). The samples were incubated without stirring for up to 17 days and were monitored for amyloid formation by EM and ThT binding / fluorescence. Fibril formation was consistently observed after several days of incubation at pH 7.4.
For negative stain EM, a 4 μl aliquot of peptide solution was incubated for 10 sec on a glow-discharged carbon formvar-coated copper grid, excess liquid was removed by blotting, the samples were stained with 1% w/v sodium phosphotungstate at pH 7.4, blotted, and air-dried. Images were recorded by using a CM2 transmission electron microscope.
For ThT binding, the peptide aliquots were diluted and ThT solution was added to the final concentration of 100 μM peptide, 10 μM ThT in standard buffer. After 10 min incubation, the emission spectra were recorded at 25 °C using Fluoromax-5 spectrofluorimeter. The probe was excited at 444 nm and the spectra were recorded from 450 to 550 nm with 5 nm excitation and emission slit widths.
All experiments in this study were repeated at least 3 times to ensure reproducibility.
Supplementary Material
Scheme.

Acknowledgments
We thank Cheryl England for help with protein and lipid analysis. We are indebted to Donald L. Gantz for expert help with electron microscopy, and to Dr. Haya Herscovitz for many useful discussions and help. We are grateful to Dr. Donald Puppione for his suggestion to use Rosetta-based amyloid prediction method. This work was supported by the National Institutes of Health grant GM067260 and by the institutional funds.
Abbreviations
- apoA-I
apolipoprotein A-I
- AApoAI
familial apoA-I amyloidosis
- HDL
high-density lipoproteins
- rHDL
recombinant high-density lipoprotein
- WT
wild type
- DMPC
dimyristoyl phosphatidylcholine
- POPC
palmitoyl-oleoyl phosphatidylcholine
- MetO
methionine sulfoxide
- CD
circular dichroism
- EM
electron microscopy
- HX
hydrogen deuterium exchange
- MS
mass spectrometry
- ThT
thioflavine T
- MMP
matrix metalloproteinase
Footnotes
X-ray crystal structures of full-length human apoA-I and other proteins previously published by H. M. Murphy have been discredited and should be withdrawn from the journals and the Protein Data Bank; for details see [89], http://main.uab.edu/Sites/reporter/articles/71570/ and http://classic.the-scientist.com/blog/display/56226/.
Author Contribution Statement: MD performed experiments and prepared graphics; XM produced the proteins and contributed graphics; SJ planned experiments and contributed graphics; DA provided expertise in structural analysis of apoA-I; OG planned the study, performed bioinformatics analysis and wrote the paper. All authors contributed to writing or editing the paper.
Supporting information available on-line: Cartoon representation of discoidal and spherical HDL (Fig. S1); Comparison of kinetic stability of DMPC complexes with WT and mutant human apoA-I (Fig. S2); Brief description of the algorithms used in AmylPred2 (Doc. S1, Doc. S3) and PASTA (Doc. S2) and the prediction results for apoA-I by each of these algorithms (Table S1); Sequence analysis of human apoA-I by using amyloid prediction software PASTA to assess the major “hot spots” and their preferred orientation in amyloid fibrils (Table S2); Amyloidogenic profile of apoA-I predicted by PASTA (Fig. S3); HX/MS profiles of free WT and G26R apoA-I showing the effect of the mutation on the protection of the N-terminal amyloid hot spots (Fig. S4); Nearest neighbors of Met86 sulfur in the crystal structure of Δ(185-243)apoA-I (Table S3).
References
- 1.Navab M, Reddy ST, Van Lenten BJ, Fogelman AM. HDL and cardiovascular disease: atherogenic and atheroprotective mechanisms. Nat Rev Cardiol. 2011;8:222–232. doi: 10.1038/nrcardio.2010.222. [DOI] [PubMed] [Google Scholar]
- 2.Curtiss LK, Valenta DT, Hime NJ, Rye KA. What is so special about apolipoprotein AI in reverse cholesterol transport? Arterioscler Thromb Vasc Biol. 2006;26(1):12–19. doi: 10.1161/01.ATV.0000194291.94269.5a. [DOI] [PubMed] [Google Scholar]
- 3.Phillips MC. New insights into the determination of HDL structure by apolipoproteins. J Lipid Res. 2013;54(8):2034–2048. doi: 10.1194/jlr.R034025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang R, Silva RA, Jerome WG, Kontush A, Chapman MJ, Curtiss LK, Hodges TJ, Davidson WS. Apolipoprotein A-I structural organization in high-density lipoproteins isolated from human plasma. Nat Struct Mol Biol. 2011;18(4):416–422. doi: 10.1038/nsmb.2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Segrest JP, Jones MK, De Loof H, Brouillette CG, Venkatachalapathi YV, Anantharamaiah GM. The amphipathic helix in the exchangeable apolipoproteins: A review of secondary structure and function. J Lipid Res. 1992;33:141–166. [PubMed] [Google Scholar]
- 6.Sorci-Thomas M, Thomas MJ. The effects of altered apolipoprotein A-I structure on plasma HDL concentration. Trends Cardiovasc Med. 2002;12:121–128. doi: 10.1016/s1050-1738(01)00163-3. [DOI] [PubMed] [Google Scholar]
- 7.Toth PP, Barter PJ, Rosenson RS, Boden WE, Chapman MJ, Cuchel M, D’Agostino RB, Sr, Davidson MH, Davidson WS, Heinecke JW, Karas RH, Kontush A, Krauss RM, Miller M, Rader DJ. High-Density Lipoproteins: A Consensus statement from the National Lipid Association. J Clin Lipidol. 2013;7(5):484–525. doi: 10.1016/j.jacl.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 8.Fielding CJ, Fielding PE. Molecular physiology of reverse cholesterol transport. J Lipid Res. 1995;36:211–228. [PubMed] [Google Scholar]
- 9.Rosenson RS, Brewer HB, Jr, Davidson WS, Fayad ZA, Fuster V, Goldstein J, Hellerstein M, Jiang XC, Phillips MC, Rader DJ, Remaley AT, Rothblat GH, Tall AR, Yvan-Charvet L. Cholesterol efflux and atheroprotection: advancing the concept of reverse cholesterol transport. Circulation. 2012;125(15):1905–1919. doi: 10.1161/CIRCULATIONAHA.111.066589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mei X, Atkinson D. Crystal structure of C-terminal truncated apolipoprotein A-I reveals the assembly of high density lipoprotein (HDL) by dimerization. J Biol Chem. 2011;286(44):38570–38582. doi: 10.1074/jbc.M111.260422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gursky O. Crystal structure of Δ(185-243)apoA-I suggests a mechanistic framework for the protein adaptation to the changing lipid load in Good Cholesterol: From Flatland to Sphereland via Double Belt, Belt Buckle, Double Hairpin and Trefoil/Tetrafoil. J Mol Biol. 2013;425(1):1–16. doi: 10.1016/j.jmb.2012.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rye KA, Barter PJ. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler Thromb Vasc Biol. 2004;24(3):421–428. doi: 10.1161/01.ATV.0000104029.74961.f5. [DOI] [PubMed] [Google Scholar]
- 13.Lee JY, Lanningham-Foster L, Boudyguina EY, Smith TL, Young ER, Colvin PL, Thomas MJ, Parks JS. Prebeta high density lipoprotein has two metabolic fates in human apolipoprotein A-I transgenic mice. J Lipid Res. 2004;45:716–728. doi: 10.1194/jlr.M300422-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Oram JF, Heinecke JW. ATP-binding cassette transporter A1: a cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev. 2005;85:1343–1372. doi: 10.1152/physrev.00005.2005. [DOI] [PubMed] [Google Scholar]
- 15.Mulya A, Lee JY, Gebre AK, Boudyguina EY, Chung SK, Smith TL, Colvin PL, Jiang XC, Parks JS. Initial interaction of apoA-I with ABCA1 impacts in vivo metabolic fate of nascent HDL. J Lipid Res. 2008;49:2390–2401. doi: 10.1194/jlr.M800241-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavigiolio G, Geier EG, Shao B, Heinecke JW, Oda MN. Exchange of apolipoprotein A-I between lipid-associated and lipid-free states: A potential target for oxidative generation of dysfunctional high density lipoproteins. J Biol Chem. 2010;285:18847–18857. doi: 10.1074/jbc.M109.098434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki M, Wada H, Maeda S, Saito K, Minatoguchi S, Saito K, Seishima M. Increased plasma lipid-poor apolipoprotein A-I in patients with coronary artery disease. Clin Chem. 2005;51(1):132–137. doi: 10.1373/clinchem.2004.041012. [DOI] [PubMed] [Google Scholar]
- 18.Jayaraman S, Cavigiolio G, Gursky O. Folded functional lipid-poor apolipoprotein A-I obtained by heating of high-density lipoproteins: Relevance to high-density lipoprotein biogenesis. Biochem J. 2012;442:703–712. doi: 10.1042/BJ20111831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mehta R, Gantz DL, Gursky O. Human plasma high-density lipoproteins are stabilized by kinetic factors. J Mol Biol. 2003;328(1):183–192. doi: 10.1016/s0022-2836(03)00155-4. [DOI] [PubMed] [Google Scholar]
- 20.Gursky O, Atkinson D. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc Natl Acad Sci USA. 1996;93:2991–2995. doi: 10.1073/pnas.93.7.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guha M, Gao X, Jayaraman S, Gursky O. Correlation of structural stability with functional remodeling of high-density lipoproteins: The importance of being disordered. Biochemistry. 2008;47(44):11393–11397. doi: 10.1021/bi8014746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ramella NA, Schinella GR, Ferreira ST, Prieto ED, Vela ME, Ríos JL, Tricerri MA, Rimoldi OJ. Human apolipoprotein A-I natural variants: Molecular mechanisms underlying amyloidogenic propensity. PLoS One. 2012;7(8):e43755. doi: 10.1371/journal.pone.0043755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ramella NA, Rimoldi OJ, Prieto ED, Schinella GR, Sanchez SA, Jaureguiberry MS, Vela ME, Ferreira ST, Tricerri MA. Human apolipoprotein A-I-derived amyloid: Its association with atherosclerosis. PLoS One. 2011;6(7):e22532. doi: 10.1371/journal.pone.0022532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mucchiano GI, Häggqvist B, Sletten K, Westermark P. Apolipoprotein A-1-derived amyloid in atherosclerotic plaques of the human aorta. J Pathol. 2001;193(2):270–275. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH753>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 25.Röcken C, Tautenhahn J, Bühling F, Sachwitz D, Vöckler S, Goette A, Bürger T. Prevalence and pathology of amyloid in atherosclerotic arteries. Arterioscler Thromb Vasc Biol. 2006;26(3):676–677. doi: 10.1161/01.ATV.0000201930.10103.be. [DOI] [PubMed] [Google Scholar]
- 26.Howlett GJ, Moore KJ. Untangling the role of amyloid in atherosclerosis. Curr Opin Lipidol. 2006;17(5):541–547. doi: 10.1097/01.mol.0000245260.63505.4f. [DOI] [PubMed] [Google Scholar]
- 27.Teoh CL, Griffin MD, Howlett GJ. Apolipoproteins and amyloid fibril formation in atherosclerosis. Protein Cell. 2011;2(2):116–127. doi: 10.1007/s13238-011-1013-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adachi E, Nakajima H, Mizuguchi C, Dhanasekaran P, Kawashima H, Nagao K, Akaji K, Lund-Katz S, Phillips MC, Saito H. Dual role of an N-terminal amyloidogenic mutation in apolipoprotein A-I: Destabilization of helix bundle and enhancement of fibril formation. J Biol Chem. 2013;288(4):2848–2856. doi: 10.1074/jbc.M112.428052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lepedda AJ, Cigliano A, Cherchi GM, Spirito R, Maggioni M, Carta F, Turrini F, Edelstein C, Scanu AM, Formato M. A proteomic approach to differentiate histologically classified stable and unstable plaques from human carotid arteries. Atherosclerosis. 2009;203(1):112–118. doi: 10.1016/j.atherosclerosis.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patel S, Chung SH, White G, Bao S, Celermajer DS. The “atheroprotective” mediators apolipoprotein A-I and Foxp3 are over-abundant in unstable carotid plaques. Int J Cardiol. 2010;145(2):183–187. doi: 10.1016/j.ijcard.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 31.Wong YQ, Binger KJ, Howlett GJ, Griffin MD. Methionine oxidation induces amyloid fibril formation by full-length apolipoprotein A-I. Proc Natl Acad Sci USA. 2010;107(5):1977–1982. doi: 10.1073/pnas.0910136107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obici L, Franceschini G, Calabresi L, Giorgetti S, Stoppini M, Merlini G, Bellotti V. Structure, function and amyloidogenic propensity of apolipoprotein A-I. Amyloid. 2006;13(4):191–205. doi: 10.1080/13506120600960288. [DOI] [PubMed] [Google Scholar]
- 33.Gomaraschi M, Obici L, Simonelli S, Gregorini G, Negrinelli A, Merlini G, Franceschini G, Calabresi L. Effect of the amyloidogenic L75P apolipoprotein A-I variant on HDL subpopulations. Clin Chim Acta. 2011;412(13–14):1262–1265. doi: 10.1016/j.cca.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rowczenio D, Dogan A, Theis JD, Vrana JA, Lachmann HJ, Wechalekar AD, Gilbertson JA, Hunt T, Gibbs SD, Sattianayagam PT, Pinney JH, Hawkins PN, Gillmore JD. Amyloidogenicity and clinical phenotype associated with five novel mutations in apolipoprotein A-I. Am J Pathol. 2011;179(4):1978–1987. doi: 10.1016/j.ajpath.2011.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rader DJ, Gregg RE, Meng MS, Schaefer JR, Zech LA, Benson MD, Brewer HB., Jr In vivo metabolism of a mutant apolipoprotein, apoA-I Iowa, associated with hypoalphalipoproteinemia and hereditary systemic amyloidosis. J Lipid Res. 1992;33(5):755–763. [PubMed] [Google Scholar]
- 36.Marchesi M, Parolini C, Valetti C, Mangione P, Obici L, Giorgetti S, Raimondi S, Donadei S, Gregorini G, Merlini G, Stoppini M, Chiesa G, Bellotti V. The intracellular quality control system down-regulates the secretion of amyloidogenic apolipoprotein A-I variants: A possible impact on the natural history of the disease. Biochim Biophys Acta. 2011;1812(1):87–93. doi: 10.1016/j.bbadis.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 37.Arciello A, De Marco N, Del Giudice R, Guglielmi F, Pucci P, Relini A, Monti DM, Piccoli R. Insights into the fate of the N-terminal amyloidogenic polypeptide of ApoA-I in cultured target cells. J Cell Mol Med. 2011;15(12):2652–2663. doi: 10.1111/j.1582-4934.2011.01271.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gursky O, Mei X, Atkinson D. Crystal structure of the C-terminal truncated apolipoprotein A-I sheds new light on the amyloid formation by the N-terminal segment. Biochemistry. 2012;51(1):10–18. doi: 10.1021/bi2017014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gorbenko GP, Kinnunen PK. The role of lipid protein interactions in amyloid-type protein fibril formation. Chem Phys Lipids. 2006;141(1–2):72–82. doi: 10.1016/j.chemphyslip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 40.Monti DM, Guglielmi F, Monti M, Cozzolino F, Torrassa S, Relini A, Pucci P, Arciello A, Piccoli R. Effects of a lipid environment on the fibrillogenic pathway of the N-terminal polypeptide of human apolipoprotein A-I, responsible for in vivo amyloid fibril formation. Eur Biophys J. 2010;39(9):1289–1299. doi: 10.1007/s00249-010-0582-2. [DOI] [PubMed] [Google Scholar]
- 41.Raimondi S, Guglielmi F, Giorgetti S, Di Gaetano S, Arciello A, Monti DM, Relini A, Nichino D, Doglia SM, Natalello A, Pucci P, Mangione P, Obici L, Merlini G, Stoppini M, Robustelli P, Tartaglia GG, Vendruscolo M, Dobson CM, Piccoli R, Bellotti V. Effects of the known pathogenic mutations on the aggregation pathway of the amyloidogenic peptide of apolipoprotein A-I. J Mol Biol. 2011;407(3):465–476. doi: 10.1016/j.jmb.2011.01.044. [DOI] [PubMed] [Google Scholar]
- 42.Lagerstedt JO, Cavigiolio G, Roberts LM, Hong HS, Jin LW, Fitzgerald PG, Oda MN, Voss JC. Mapping the structural transition in an amyloidogenic apolipoprotein A-I. Biochemistry. 2007;46(34):9693–9699. doi: 10.1021/bi7005493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adachi E, Nakajima H, Mizuguchi C, Dhanasekaran P, Kawashima H, Nagao K, Akaji K, Lund-Katz S, Phillips MC, Saito H. Dual role of an N-terminal amyloidogenic mutation in apolipoprotein A-I: destabilization of helix bundle and enhancement of fibril formation. J Biol Chem. 2013;288(4):2848–2856. doi: 10.1074/jbc.M112.428052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Borhani DW, Rogers DP, Engler JA, Brouillette CG. Crystal structure of truncated human apolipoprotein A-I suggests a lipid-bound conformation. Proc Natl Acad Sci USA. 1997;94:12291–12296. doi: 10.1073/pnas.94.23.12291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chetty PS, Mayne L, Lund-Katz S, Stranz D, Englander SW, Phillips MC. Helical structure and stability in human apolipoprotein A-I by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. 2009;106:19005–19010. doi: 10.1073/pnas.0909708106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lindstedt L, Saarinen J, Kalkkinen N, Welgus H, Kovanen PT. Matrix metalloproteinases-3, -7, and -12, but not -9, reduce high density lipoprotein induced cholesterol efflux from human macrophage foam cells by truncation of the carboxyl terminus of apolipoprotein A-I. Parallel losses of pre-beta particles and the high affinity component of efflux. J Biol Chem. 1999;274:22627–22634. doi: 10.1074/jbc.274.32.22627. [DOI] [PubMed] [Google Scholar]
- 47.Eberini I, Calabresi L, Wait R, Tedeschi G, Pirillo A, Puglisi L, Sirtori CR, Gianazza E. Macrophage metalloproteinases degrade high-density-lipoprotein-associated apolipoprotein A-I at both the N- and C-termini. Biochem J. 2002;362(3):627–634. doi: 10.1042/0264-6021:3620627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soutar AK, Hawkins PN, Vigushin DM, Tennent GA, Booth SE, Hutton T, Nguyen O, Totty NF, Feest TG, Hsuan JJ, Pepys MB. Apolipoprotein AI mutation Arg-60 causes autosomal dominant amyloidosis. Proc Natl Acad Sci USA. 1992;89(16):7389–7393. doi: 10.1073/pnas.89.16.7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Booth DR, Tan SY, Booth SE, Hsuan JJ, Totty NF, Nguyen O, Hutton T, Vigushin DM, Tennent GA, Hutchinson WL, Thompson N, Soutar AK, Hawkins PN, Pepys MB. A new apolipoprotein Al variant, Trp50Arg, causes hereditary amyloidosis. QJM. 1995;88(10):695–702. [PubMed] [Google Scholar]
- 50.Alexander ET, Tanaka M, Kono M, Saito H, Rader DJ, Phillips MC. Structural and functional consequences of the Milano mutation (R173C) in human apolipoprotein A-I. J Lipid Res. 2009;50(7):1409–1419. doi: 10.1194/jlr.M800578-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kono M, Tanaka T, Tanaka M, Vedhachalam C, Chetty PS, Nguyen D, Dhanasekaran P, Lund-Katz S, Phillips MC, Saito H. Disruption of the C-terminal helix by single amino acid deletion is directly responsible for impaired cholesterol efflux ability of apolipoprotein A-I Nichinan. J Lipid Res. 2010;51(4):809–818. doi: 10.1194/jlr.M002113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weers PM, Patel AB, Wan LC, Guigard E, Kay CM, Hafiane A, McPherson R, Marcel YL, Kiss RS. Novel N-terminal mutation of human apolipoprotein A-I reduces self-association and impairs LCAT activation. J Lipid Res. 2011;52(1):35–44. doi: 10.1194/jlr.M007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chetty PS, Ohshiro M, Saito H, Dhanasekaran P, Lund-Katz S, Mayne L, Englander W, Phillips MC. Effects of the Iowa and Milano mutations on apolipoprotein A-I structure and dynamics determined by hydrogen exchange and mass spectrometry. Biochemistry. 2012;51(44):8993–9001. doi: 10.1021/bi300926j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Petrlova J, Duong T, Cochran MC, Axelsson A, Mörgelin M, Roberts LM, Lagerstedt JO. The fibrillogenic L178H variant of apolipoprotein A-I forms helical fibrils. J Lipid Res. 2012;53(3):390–398. doi: 10.1194/jlr.M020883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dumoulin M, Canet D, Last AM, Pardon E, Archer DB, Muyldermans S, Wyns L, Matagne A, Robinson CV, Redfield C, Dobson CM. Reduced global cooperativity is a common feature underlying the amyloidogenicity of pathogenic lysozyme mutations. J Mol Biol. 2005;346(3):773–788. doi: 10.1016/j.jmb.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 56.Ji Y, Jonas A. Properties of an N-terminal proteolytic fragment of apolipoprotein AI in solution and in reconstituted high density lipoproteins. J Biol Chem. 1995;270(19):11290–11297. doi: 10.1074/jbc.270.19.11290. [DOI] [PubMed] [Google Scholar]
- 57.Roberts LM, Ray MJ, Shih TW, Hayden E, Reader MM, Brouillette CG. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and –oxidized lipid-free and lipid-bound human apoA-I. Biochemistry. 1997;36(24):7615–724. doi: 10.1021/bi962952g. [DOI] [PubMed] [Google Scholar]
- 58.Ventura S, Zurdo J, Narayanan S, Parreño M, Mangues R, Reif B, Chiti F, Giannoni E, Dobson CM, Aviles FX, Serrano L. Short amino acid stretches can mediate amyloid formation in globular proteins: the Src homology 3 (SH3) case. Proc Natl Acad Sci USA. 2004;101(19):7258–7263. doi: 10.1073/pnas.0308249101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Thompson MJ, Sievers SA, Karanicolas J, Ivanova MI, Baker D, Eisenberg D. The 3D profile method for identifying fibril-forming segments of proteins. Proc Natl Acad Sci USA. 2006;103(11):4074–4078. doi: 10.1073/pnas.0511295103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tzotzos S, Doig AJ. Amyloidogenic sequences in native protein structures. Prot Sci. 2010;19(2):327–348. doi: 10.1002/pro.314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maurer-Stroh S, Debulpaep M, Kuemmerer N, Lopez de la Paz M, Martins IC, Reumers J, Morris KL, Copland A, Serpell L, Serrano L, Schymkowitz JW, Rousseau F. Exploring the sequence determinants of amyloid structure using position-specific scoring matrices. Nat Methods. 2010;7(3):237–242. doi: 10.1038/nmeth.1432. [DOI] [PubMed] [Google Scholar]
- 62.Belli M, Ramazzotti M, Chiti F. Prediction of amyloid aggregation in vivo. EMBO Rep. 2011;12(7):657–663. doi: 10.1038/embor.2011.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hamodrakas SJ. Protein aggregation and amyloid fibril formation prediction software from primary sequence: Towards controlling the formation of bacterial inclusion bodies. FEBS J. 2011;278:2428–2435. doi: 10.1111/j.1742-4658.2011.08164.x. [DOI] [PubMed] [Google Scholar]
- 64.Tsolis AC, Papandreou NC, Iconomidou VA, Hamodrakas SJ. A consensus method for the prediction of ‘aggregation-prone’ peptides in globular proteins. PLoS One. 2013;8(1):e54175. doi: 10.1371/journal.pone.0054175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Trovato A, Seno F, Tosatto SC. The PASTA server for protein aggregation prediction. Protein Eng Des Sel. 2007;20(10):521–523. doi: 10.1093/protein/gzm042. [DOI] [PubMed] [Google Scholar]
- 66.Adachi E, Kosaka A, Tsuji K, Mizuguchi C, Kawashima H, Shigenaga A, Nagao K, Akaji K, Otaka A, Saito H. The extreme N-terminal region of human apolipoprotein A-I has a strong propensity to form amyloid fibrils. FEBS Lett. 2014;588(3):389–394. doi: 10.1016/j.febslet.2013.11.031. [DOI] [PubMed] [Google Scholar]
- 67.Wong YQ, Binger KJ, Howlett GJ, Griffin MD. Identification of an amyloid fibril forming peptide comprising residues 46-59 of apolipoprotein A-I. FEBS Lett. 2012;586(13):1754–1758. doi: 10.1016/j.febslet.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 68.Tycko R. Molecular structure of amyloid fibrils: insights from solid-state NMR. Q Rev Biophys. 2006;39(1):1–55. doi: 10.1017/S0033583506004173. [DOI] [PubMed] [Google Scholar]
- 69.Margittai M, Langen R. Fibrils with parallel in-register structure constitute a major class of amyloid fibrils: molecular insights from electron paramagnetic resonance spectroscopy. Rev Biophys. 2008;41(3-4):265–297. doi: 10.1017/S0033583508004733. [DOI] [PubMed] [Google Scholar]
- 70.Teoh CL, Pham CL, Todorova N, Hung A, Lincoln CN, Lees E, Lam YH, Binger KJ, Thomson NH, Radford SE, Smith TA, Müller SA, Engel A, Griffin MD, Yarovsky I, Gooley PR, Howlett GJ. A structural model for apolipoprotein C-II amyloid fibrils: experimental characterization and molecular dynamics simulations. J Mol Biol. 2011;405(5):1246–1266. doi: 10.1016/j.jmb.2010.12.006. [DOI] [PubMed] [Google Scholar]
- 71.Der-Sarkissian A, Jao CC, Chen J, Langen R. Structural organization of α-synuclein fibrils studied by site-directed spin labeling. J Biol Chem. 2003;278:37530–37535. doi: 10.1074/jbc.M305266200. [DOI] [PubMed] [Google Scholar]
- 72.Lv G, Kumar A, Giller K, Orcellet ML, Riedel D, Fernández CO, Becker S, Lange A. Structural comparison of mouse and human α synuclein amyloid fibrils by solid-state NMR. J Mol Biol. 2012;420(1-2):99–111. doi: 10.1016/j.jmb.2012.04.009. [DOI] [PubMed] [Google Scholar]
- 73.Winkelmann J, Calloni G, Campioni S, Mannini B, Taddei N, Chiti F. Low-level expression of a folding-incompetent protein in Escherichia coli: search for the molecular determinants of protein aggregation in vivo. J Mol Biol. 2010;398(4):600–613. doi: 10.1016/j.jmb.2010.03.030. [DOI] [PubMed] [Google Scholar]
- 74.Gorshkova IN, Atkinson D. Enhanced binding of apolipoprotein A-I variants associated with hypertriglyceridemia to triglyceride-rich particles. Biochemistry. 2011;50(12):2040–2047. doi: 10.1021/bi200158b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.García-Fruitós E, Sabate R, de Groot NS, Villaverde A, Ventura S. Biological role of bacterial inclusion bodies: a model for amyloid aggregation. FEBS J. 2011;278(14):2419–2427. doi: 10.1111/j.1742-4658.2011.08165.x. [DOI] [PubMed] [Google Scholar]
- 76.Chiti F, Dobson CM. Amyloid formation by globular proteins under native conditions. Nat Chem Biol. 2009;5(1):15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- 77.Garner B, Waldeck AR, Witting PK, Rye KA, Stocker R. Oxidation of high density lipoproteins II. Evidence for direct reduction of lipid hydroperoxides by methionine residues of apolipoproteins AI and AII. J Biol Chem. 1998;273(11):6088–6095. doi: 10.1074/jbc.273.11.6088. [DOI] [PubMed] [Google Scholar]
- 78.Shao B, Cavigiolio G, Brot N, Oda MN, Heinecke JW. Methionine oxidation impairs reverse cholesterol transport by apolipoprotein A-I. Proc Natl Acad Sci USA. 2008;105(34):12224–12229. doi: 10.1073/pnas.0802025105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Anantharamaiah GM, Hughes TA, Iqbal M, Gawish A, Neame PJ, Medley MF, Segrest JP. Effect of oxidation on the properties of apolipoproteins A-I and A-II. J Lipid Res. 1988;29(3):309–318. [PubMed] [Google Scholar]
- 80.Sigalov AB, Stern LJ. Oxidation of methionine residues affects the structure and stability of apolipoprotein A-I in reconstituted high density lipoprotein particles. Chem Phys Lipids. 2001;113(1-2):133–146. doi: 10.1016/s0009-3084(01)00186-4. [DOI] [PubMed] [Google Scholar]
- 81.Jayaraman S, Gantz DL, Gursky O. Effects of protein oxidation on the structure and stability of model discoidal high-density lipoproteins. Biochemistry. 2008;47(12):3875–3882. doi: 10.1021/bi7023783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brock JW, Jenkins AJ, Lyons TJ, Klein RL, Yim E, Lopes-Virella M, Carter RE, Thorpe SR, Baynes JW DCCT/EDIC Research Group. Increased methionine sulfoxide content of apoA-I in type 1 diabetes. J Lipid Res. 2008;49(4):847–855. doi: 10.1194/jlr.M800015-JLR200. [DOI] [PubMed] [Google Scholar]
- 83.Nguyen SD, Öörni K, Lee-Rueckert M, Pihlajamaa T, Metso J, Jauhiainen M, Kovanen PT. Spontaneous remodeling of HDL particles at acidic pH enhances their capacity to induce cholesterol efflux from human macrophage foam cells. J Lipid Res. 2012;53(10):2115–2125. doi: 10.1194/jlr.M028118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cabana VG, Lukens JR, Rice KS, Hawkins TJ, Getz GS. HDL content and composition in acute phase response in three species: triglyceride enrichment of HDL a factor in its decrease. J Lipid Res. 1996;37(12):2662–2674. [PubMed] [Google Scholar]
- 85.van der Westhuyzen DR, de Beer FC, Webb NR. HDL cholesterol transport during inflammation. Curr Opin Lipidol. 2007;18(2):147–151. doi: 10.1097/MOL.0b013e328051b4fe. [DOI] [PubMed] [Google Scholar]
- 86.Jayaraman S, Gantz DL, Gursky O. Effects of phospholipase A2 and its products on structural stability of human low-density lipoprotein: Relevance to formation of LDL-derived lipid droplets. J Lipid Res. 2011;52(3):549–557. doi: 10.1194/jlr.M012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Lamarche B, Rashid S, Lewis GF. HDL metabolism in hypertriglyceridemic states: An overview. Clin Chim Acta. 1999;286(1-2):145–161. doi: 10.1016/s0009-8981(99)00098-4. [DOI] [PubMed] [Google Scholar]
- 88.Gursky O, Ranjana, Gantz DL. Complex of human apolipoprotein C-1 with phospholipid: thermodynamic or kinetic stability? Biochemistry. 2002;41(23):7373–7384. doi: 10.1021/bi025588w. [DOI] [PubMed] [Google Scholar]
- 89.Borrell B. Fraud rocks protein community. Nature. 2009;462:970. doi: 10.1038/462970a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.