

Abstract
Objective
To investigate the role of Nrf2 in the pathogenesis of hepatic ischemia-reperfusion (I/R) injury.
Summary Background Data
Hepatic I/R injury is a serious complication that leads to liver failure after liver surgery. NF-E2-related factor 2 (Nrf2) is a transcription factor that plays a critical role in protecting cells against oxidative stress. Therefore, it is suggested that Nrf2 activation protects the liver from I/R injury.
Methods
Wild-type (WT) and Nrf2-deficient mice were treated with 15-deoxy-Δ12, 14-prostaglandin J2 (15d-PGJ2), or a vehicle. Subsequently, these mice were subjected to 60 min hepatic 70% ischemia followed by reperfusion. Liver and blood samples were collected to evaluate liver injury and mRNA expressions.
Results
After hepatic I/R, Nrf2-deficient livers exhibited enhanced tissue damage, impaired GSTm1, NQO1, and GCLc inductions, disturbed redox state, and aggravated TNF-α mRNA expression in comparison to WT livers. 15d-PGJ2 treatment protected the livers of WT mice from I/R injury via increased expressions of GSTm1, NQO1 and GCLc, maintained redox status, and decreased TNF-α induction. These effects induced by 15d-PGJ2 were not seen in the livers of Nrf2−/− mice and were not annulled by PPARγ antagonist in Nrf2+/+ mice, suggesting that the protective effect of 15d-PGJ2 is mediated by Nrf2-dependent antioxidant response.
Conclusions
Nrf2 plays a critical role in the mechanism of hepatic I/R injury and would be a new therapeutic target for preventing hepatic I/R injury during liver surgery.
Introduction
Interruption of hepatic blood inflow to decrease blood loss during liver surgery such as hepatic resection and transplantation causes hepatic ischemia and subsequent reperfusion that result in massive hepatocyte injuries. Ischemia-reperfusion (I/R) liver injury is a severe, unfavorable postsurgical complication associated with high morbidity and mortality. A number of studies have demonstrated that generation of reactive oxygen species (ROS) is associated with hepatic I/R injury.1–4 During the early phase of I/R, ROS causes hepatocyte damage through lipid peroxidation, protein oxidation, mitochondrial dysfunction, and DNA damage.2, 5 Subsequently, Kupffer cells and accumulated neutrophils are activated in response to hepatocyte death and cause liver inflammation.3 Thus, regulation of ROS is suggested as a new therapeutic strategy for hepatic I/R injury.
Nrf2 (NF-E2-related factor 2) is a transcription factor associated with various intracellular signaling that protects organs against oxidative stress.6–11 In physiological conditions, Nrf2 is retained in cytoplasm by binding to its inhibitor, Keap1. Various endogenous or exogenous stimuli dissociate Nrf2 from Keap1 that leads to the nuclear translocation of Nrf2, resulting in transcriptional activation of antioxidant responsive element (ARE)-regulated genes, such as glutathione-S-transferases (GSTs), NADPH, quinine oxidoreductase 1 (NQO1), and glutamate cysteine ligase (GCL).12 A number of studies have shown that depletion of Nrf2 increases susceptibility to toxin-induced liver injury,13–16 all of which provide strong evidence for Nrf2 as a hepatoprotective factor for liver injury. However, the involvement of Nrf2 in hepatic I/R injury has not been investigated to date.
Here, we demonstrate that Nrf2 plays a crucial role in the protection of hepatic I/R injury. We also found that treatment with 15-deoxy-Δ12,14–prostaglandin J2 (15d-PGJ2) —a derivative of omega-6 polyunsaturated fatty acids that is produced from the non-enzymatic dehydration of PGD217—protected livers from I/R injury via activation of Nrf2. Our results provide insight into the amplification of Nrf2 activation as a powerful interventional strategy to protect livers from I/R insults during and after surgical procedures.
Materials and Methods
Model of Hepatic Ischemia/ Reperfusion Injury
Male 9 to 11-week-old wild-type (WT) male mice (C57BL/6 mice; Japan SLC, Tokyo, Japan) and Nrf2 knockout male mice on C57BL/6 background were used in this study. Nrf2 knockout mice/C57BL6J (RBRC01390) were provided by RIKEN BRC, which is participating in the national Bio-Resource Project of the MEXT, Japan. The protocol for animal experiments in this study has been approved by the Animal Research Committee in Akita University (approval number: a-1-2213). All subsequent animal experiments adhered to the “Regulation for Animal Experimentation,” of the Akita University. Mice were anesthetized with pentobarbital sodium. After midline laparotomy (2cm), partial hepatic ischemia was induced by clamping the vessels to the left and median lobes of the liver using an atraumatic clip to hinder blood supply to the liver. After a 60-minute ischemia, the clip was removed to accomplish reperfusion. The abdomen was closed in layers, and the animals were allowed to recover in their cages. Some mice were injected intravenously with vehicle (10% DMSO) or 0.3mg/kg 15d-PGJ2 (Enzo Chemical Co., St. Louis, MO) 3 hours prior to ischemia. To block PPARγ activity, a separate group of mice was intraperitoneally injected with 1.0mg/kg of GW9662 (Wako, Osaka, Japan) 30 minutes prior to 15d-PGJ2 administration, as described elsewhere.18 At several indicated time points, mice were sacrificed to collect the liver and blood samples.
Serum enzyme analyses
Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were measured by FUJI DRI-CHEM SLIDE system (Fujifilm, Tokyo, Japan).
Histopathology
Liver samples were fixed in 4% paraformaldehide and embedded in paraffin. Liver sections were stained with hematoxylin and eosin.
Isolation and culture of primary hepatocytes
Primary hepatocytes were isolated from WT C57BL/6 mice and cultured as described previously.19
Immunofluorescence
Primary hepatocytes were stimulated with 6µM 15d-PGJ2 or DMSO for 1 hour. For Nrf2 detection, cells were fixed with 4% paraformaldehide, and permeabilized with 0.1% Triton X-100. Subsequently, cells were blocked with 5% BSA in PBS and incubated overnight with rabbit polyclonal anti-Nrf2 antibody (1:50 dilution, Santa Cruz Biotechnology, Santa Cruz, CA) at 4°C. After washing, cells were incubated with Alexa Flour 594-conjugated chicken anti-rabbit IgG (Invitrogen, Carlsbad, CA). DAPI was used for nuclear staining. After washing, cells were mounted in fluorescent mounting medium and examined using the confocal microscope (Zeiss, Jena, Germany).
Real-Time Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA extraction and real-time RT-PCR were performed as described previously.19 The primer sequences for RT-PCR were shown in Supplemental table1.
Western blot analysis
Whole liver extracts were prepared and subjected to Western blot analysis as previously described,19 using antibodies specific for NQO1, GCL catalytic subunit (GCLc) (Abcam; Cambridge, UK), and β-actin (Sigma, St Lois, MO).
TdT-Mediated dUTP Nick-end Labeling (TUNEL) Assay
Terminal deoxynucleotidyl transferase-mediated dUTP-nick-end labeling (TUNEL) was performed using an “in situ cell detection kit” (Roche) according to the manufacture’s instructions. The percentage of TUNEL-positive hepatocytes represents the number of TUNEL-positive hepatocytes/ total number of hepatocytes in five randomly chosen 400× fields on each slide.
Glutathione analysis
Glutathione levels of the liver tissue were measured by using Glutathione Assay Kit (Cayman Chemical company, Ann Arbor, MI) according to the manufacture’s instructions.
MDA analysis
Malondialdehyde (MDA) concentrations of the liver tissue were measured by the NWLSS Malondialdehyde assay kit (Northwest Life Sciences Specialities, Vancouver, Canada) according to the manufacture’s instructions.
Statistics
Data are expressed as means ±SEM. Student’s 2-tailed t test was used to evaluate the differences between the vehicle- and 15d-PGJ2-treated mice within a single genotype as well as between genotypes. In addition, the difference between before and after I/R of the vehicle-treated mice was also evaluated. P< 0.05 was considered statistically significant.
Results
Hepatic I/R injury was exacerbated in Nrf2−/− mice and prevented by the treatment with 15d-PGJ2 in WT mice
To investigate the role of Nrf2 on hepatic I/R injury, we used Nrf2−/− mice and their Nrf2+/+ littermates as WT control. Serum AST and ALT levels were significantly elevated in Nrf2−/− mice compared to Nrf2+/+ mice after 6 hours of reperfusion (AST: 1758 ± 376 vs 857 IU/L ± 84 IU/L, p<0.05; ALT: 2893 ± 450 IU/L vs 1835 ± 158 IU/L, p<0.05). Since this result suggested a preventive role of Nrf2 in the progression of hepatic I/R injury, we hypothesized that amplification of Nrf2 would have a therapeutic effect on hepatic I/R injury. To test this hypothesis, Nrf2+/+ and Nrf2−/− mice were treated with 15d-PGJ2— an Nrf2 activator—before I/R (Fig. 1A). In Nrf2+/+ mice, 15d-PGJ2 pretreatment significantly reduced the elevation of serum AST and ALT levels 6 hours after reperfusion. In contrast, treatment with15d-PGJ2 had no effect on the elevation of serum transaminase levels in Nrf2−/− mice following hepatic I/R. We further examined whether treatment with 15d-PGJ2 activates Nrf2 in primary hepatocytes using immunofluorescence for Nrf2 (Fig. 1B). DMSO-treated hepatocytes showed diffuse and faint staining, whereas the cells treated with 15d-PGJ2 had a strong staining for Nrf2 in the nucleus, demonstrating the induction of nuclear translocation of Nrf2 by 15d-PGJ2 treatment. These results indicate that pre-administration of 15d-PGJ2 activates Nrf2 and protects livers against I/R injury in an Nrf2-dependent manner.
Figure 1. Nrf2-deficient mice are susceptible to hepatic I/R injury and 15d-PGJ2 has a protective effect in an Nrf2-dependent manner.


(A) Serum concentrations of AST/ ALT were measured 6 hours after reperfusion. Data represent means ± SEM. *, p<0.05 vs. vehicle treated Nrf2+/+ mice. n=10 per group. (B) Nrf2 nuclear translocation in primary hepatocytes isolated from Nrf2+/+ mice was determined by immunofluoroscence. Original magnification was ×100. The figures are representative of 4 independent experiments.
Oncotic necrosis was enhanced in Nrf2−/− mice
Histology was then assessed to evaluate the degree of cell death in each mouse. H&E staining of WT livers 6 hours after reperfusion exhibited gross necrosis of hepatocytes characterized by cell swelling, karyolysis, loss of basophilic staining, and vacuolization in centrilobular area (Fig. 2A). A number of nuclei and cytoplasm of hepatocytes were positive for TUNEL staining (Fig. 2B). The massive DNA fragmentation in hepatocytes suggests apoptosis and necrosis. Consistent with increased serum levels of liver enzymes in Nrf2−/− mice after I/R, the area of necrosis and the number of TUNEL-positive cells were greater in Nrf2−/− livers than Nrf2+/+ livers (Fig. 2C). In addition, treatment with 15d-PGJ2 markedly reduced the extent of necrosis and TUNEL-positive hepatocytes in Nrf2+/+ mice, but not in Nrf2−/− mice. These results indicate that Nrf2 plays a protective role and its preactivation has a therapeutic effect on hepatic I/R injury.
Figure 2. Oncotic necrosis caused by hepatic I/R is exacerbated in Nrf2−/− mice, but it is alleviated in 15d-PGJ2-treated Nrf2+/+ mice.



Six hours after reperfusion, liver tissues were harvested. (A) Liver sections were stained with hematoxylin/eosin. The figures are representative of 6 independent experiments. Original magnification was ×400 (inset, ×800). (B) DNA fragmentation was assessed by TUNEL staining. (C) TUNEL–positive hepatocytes were counted in 5 high-power (×400) fields. The numbers were expressed as the percentage of TUNEL-positive hepatocytes of the total hepatocytes in the fields. Data represent means ± SEM. *, p<0.05 vs. vehicle-treated Nrf2+/+ mice. N.S, not significant. n=6 per group.
Expressions of antioxidant enzymes were reduced in Nrf2−/− mice
To examine hepatic Nrf2 activation, we measured mRNA levels of Nrf2-dependent antioxidant genes including GST Mu1 (GSTm1), NQO1, and GCLc in the liver. Before ischemia, the expressions of GSTm1, NQO1, and GCLc mRNA were significantly reduced in the livers of Nrf2−/− mice compared with those of Nrf2+/+ mice (Fig. 3A, B, and C). The expressions of these mRNA were significantly increased in 15d-PGJ2-treated Nrf2+/+ livers, but were not altered in 15d-PGJ2-treated Nrf2−/− livers. These results indicate that 15d-PGJ2-mediated Nrf2 activation induces the expressions of GSTm1, NQO1, and GCLc. Three hours after reperfusion, the expressions of these mRNA were markedly elevated in Nrf2+/+ livers; however, no differences in mRNA levels were observed between the vehicle- and 15d-PGJ2-treated Nrf2+/+ mice. In Nrf2−/− livers, however, the elevations of these genes after reperfusion were not significant either in vehicle- or in 15d-PGJ2-treated mice. These results indicate that hepatic I/R stimulate Nrf2 activation and the following induction of GSTm1, NQO1, and GCLc mRNA expressions. To confirm whether the levels of these mRNAs correlate with their respective protein expressions, the protein levels of NQO1 and GCLc were investigated as representative Nrf2-target antioxidant enzymes (Fig.3E). Western blot shows consistent kinetics between these two protein expressions and mRNA expressions.
Figure 3. The expression of antioxidant genes is reduced in Nrf2−/− mice, and it is augmented in 15d-PGJ2-treated Nrf2+/+ mice.


Total hepatic RNA and protein were extracted before ischemia and after I/R. (A) GSTm1, (B) NQO1, (C) GCLc, and (D) HO-1 mRNA expressions were quantified by real-time RT-PCR. The expression was normalized as a ratio to GAPDH mRNA as a housekeeping gene. Data represent mean ± SEM. (E) The protein expressions of NQO1 and GCLc were evaluated by Western blot analysis. The figure shown is representative of 4 independent experiments. (*, p<0.05 vs. vehicle-treated Nrf2+/+ control mice. #, p<0.05 vs. vehicle-treated Nrf2+/+ I/R mice. N.S, not significant. n=5 per group).
We also measured the mRNA expression of Heme oxygenase 1 (HO-1)—a well-known protective gene for hepatic I/R injury— because HO-1 has also been reported to be one of Nrf2-target genes.11, 20, 21 However, the expression pattern of HO-1 mRNA was completely different from those of GSTm1, NQO1, and GCLc mRNA (Fig. 3D). Basal levels were similar in two genotypes, while the expression was upregulated to a greater extent in Nrf2−/− mice after hepatic I/R. In addition, 15d-PGJ2 treatment did not affect the expression of HO-1. These results indicate that HO-1 is induced by Nrf2-independent mechanisms.
Oxidative stress and TNF-α expression were aggravated in Nrf2−/− mice
To investigate the redox state of the livers, we measured GSH and GSSG contents in the liver (Supplemental Table 2). Hepatic GSH levels decreased 3 hours after reperfusion in all groups. Six hours following reperfusion, GSH levels recovered slightly in Nrf2+/+ livers, whereas in Nrf2−/− livers, the decreased GSH levels sustained. Six hours after reperfusion, the GSH levels of Nrf2+/+ livers treated with 15d-PGJ2 recovered to that of a normal level under healthy conditions. However, no such recovery in GSH levels were observed in 15d-PGJ2 treated Nrf2−/− livers 6 hours following reperfusion. While hepatic GSSG levels were increased in all groups following I/R, Nrf2+/+ mice had lower GSSG levels than Nrf2−/− mice after reperfusion, although the difference did not reach statistical significance. In addition, 15d-PGJ2 did not affect hepatic GSSG levels in both genotypes of mice. For better understanding of oxidative stress status in the livers of each mouse, the GSH/GSSG ratio and hepatic levels of MDA—a biomarker of lipid peroxidation—were evaluated (Fig. 4A and B). Under normal conditions, no significant differences in the hepatic GSH/GSSG ratio and MDA level were observed between the 2 groups. However, decrease in the GSH/GSSG ratio and increase in the MDA level after hepatic I/R were significantly greater in Nrf2−/− mice than in Nrf2+/+ mice, suggesting that Nrf2 deficiency leads to the augmentation of oxidative stress induced by I/R. Treatment with 15d-PGJ2 significantly suppressed the decrease in GSH/GSSG ratio and increase in MDA levels observed after reperfusion in Nrf2+/+ mice, but not in Nrf2−/− mice. These results suggest that activation of Nrf2 is critical to prevent oxidative stress during hepatic I/R.
Figure 4. Oxidative stress and induction of TNF-α are aggravated in Nrf2−/− mice, but these are alleviated by 15d-PGJ2 only in Nrf2+/+ mice.

Before ischemia and 3 to 6 hours after reperfusion, (A) GSH/GSSG ratio, (B), MDA contents, and (C) TNF-α mRNA in the liver tissues were measured. Data represent mean ± SEM. (*, p<0.05 vs. vehicle-treated Nrf2+/+ control mice. #, p<0.05 vs. vehicle-treated Nrf2+/+ I/R mice. N.S, not significant. n=4 to 5 per group).
Because oxidative stress activates a redox-sensitive transcription factor NF-κB and the activation of NF-κB induces the expression of proinflammatory mediators, we assessed the changes of TNF-α mRNA expression during hepatic I/R. Basal expression levels of TNF-α were similar between the 2 groups. Six hours after reperfusion, TNF-α expression was significantly increased in Nrf2−/− mice, but was suppressed in 15d-PGJ2-pretreated Nrf2+/+ mice (Fig. 4C). These results suggest that Nrf2 plays an important role in prevention of excessive inflammatory response after reperfusion.
The expression of PPARγ was reduced in Nrf2−/− mice
Peroxisome proliferator-activated receptors (PPARs) are the members of the nuclear hormone receptor superfamily and classified into three isoforms, α, β, and γ.17 Of these three isoforms, PPARγ, is known to have an anti-inflammatory effect. Because 15d-PGJ2 is a natural ligand for PPARγ, we measured the hepatic expression of PPARγ mRNA in each group. The levels of PPARγ mRNA were significantly lower in Nrf2−/− mice than in Nrf2+/+ mice (Fig. 5A). In addition, 15d-PGJ2 treatment significantly enhanced the mRNA expression of PPARγ in Nrf2+/+ mice, but not in Nrf2−/− mice, suggesting that the expression of PPARγ is, at least in part, Nrf2-dependent. To evaluate whether protective effect of 15d-PGJ2 against hepatic I/R was mediated by PPARγ enhancement, 15d-PGJ2-injected Nrf2+/+ mice were pretreated with GW9662—a selective PPARγ antagonist—to block PPARγ activity. GW9662 suppressed PPARγ activation: pretreatment with GW9662 inhibited upregulation of PPARγ target genes—CD36, ATP-binding cassette transporter A1 (ABCA1), ABCG1, and liver X receptor α (LXRα)—uniformly in the liver of 15d-PGJ2-treated Nrf2+/+ mice (Fig. 5B). In spite of effective blocking of PPARγ, serum AST and ALT levels after hepatic I/R were not different from those without GW9662 pretreatment (AST: 878 ± 259 IU/L with GW9662 vs 906 ±139IU/L without GW9662, p>0.05; ALT: 1240 ±254 IU/L with GW9662 vs 1360 ±314IU/L without GW9662, p>0.05, n=5 per group). These results indicate that 15d-PGJ2 indeed enhance the expression and activation of PPARγ in an Nrf2-dependent manner, however the protective function of 15d-PGJ2 against hepatic I/R is not PPARγ-dependent.
Figure 5. The mRNA expression of PPARγ is Nrf2-dependent, and GW9662 suppresses the upregulation of PPARγ target genes induced by 15d-PGJ2.
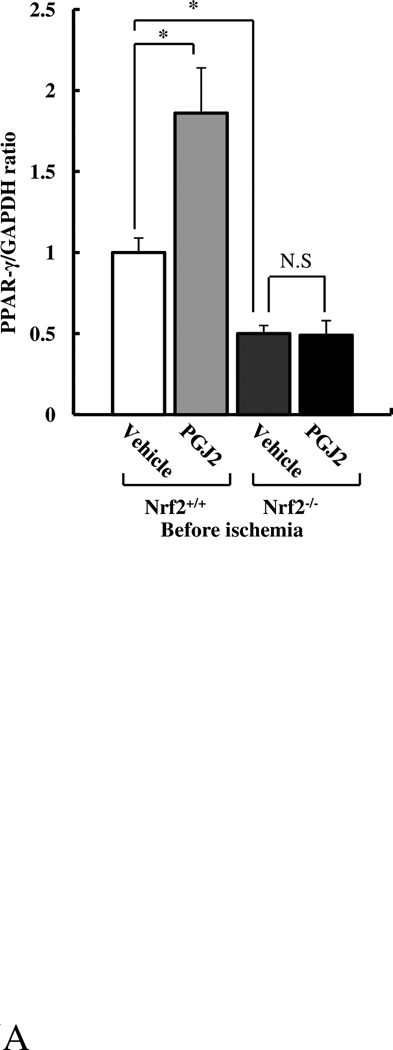

Total hepatic RNA was extracted before ischemia. (A) PPARγ and (B) CD36, AGCA1, ABCG1, and LXRα mRNA expressions were quantified by real-time RT-PCR. Data represent mean ± SEM. (*, p<0.05 vs. vehicle-treated Nrf2+/+ control mice. #, p<0.05 vs. 15d-PGJ2-treated Nrf2+/+ mice. N.S, not significant. n=6 per group).
Discussion
Numerous studies have revealed strong evidence for critical involvement of ROS in hepatic I/R injury.1, 3, 4, 23 Therefore, inhibition of ROS production and scavenging ROS have been expected as therapeutic approaches. Nrf2— a transcription factor mediating the expression of many endogenous antioxidants—is known to play an important role in the cytoprotection against oxidative stress. Recent studies have shown that Nrf2 also exerts anti-inflammatory effect through regulation of NADPH oxidase and pro-inflammatory signaling.7, 24, 25 An accumulating number of reports have revealed that Nrf2-null mice are highly susceptible to hepatic injuries induced by chemicals, methionine-choline-deficient diet, sepsis, and cytokines, indicating that Nrf2 protects the liver through multiple cytoprotective pathways.7, 13, 14 From this perspective, it is reasonable to establish therapeutic strategies targeting Nrf2. In this study, we demonstrate that Nrf2 confers the livers with protection against I/R. During reperfusion phase, GSTm1, NQO1, and GCLs —representative Nrf2-target genes which are reported to play protective role as an antioxidant and/or anti-inflammatory molecule in various diseases12, 26—were markedly increased in the livers of Nrf2+/+ mice. In contrast, in the livers of Nrf2−/− mice, the induction of these mRNAs and protein were inhibited; instead, accentuation of liver damage, the deterioration of oxidative stress, and excess expression of TNF-α mRNA were seen. Collectively, these results strongly suggest that Nrf2 activation during hepatic I/R functions to eliminate ROS and suppress inflammatory response through induction of these antioxidant genes.
We also demonstrated that 15d-PGJ2 administration reduce oxidative stress and TNF-α mRNA expression that consequent in less hepatic damage upon hepatic I/R in Nrf2+/+ mice. This beneficial effect of 15d-PGJ2 was not seen in Nrf2−/− mice, suggesting that the protective effect of 15d-PGJ2 is Nrf2 dependent. Given the ability of Nrf2 to regulate wide array of antioxidant genes, it is conceivable that induction of multiple Nrf2-responsive genes before ischemic insult is sufficient to protect liver damage caused by I/R. In our study, GSTm1, NQO1, and GCLc mRNA expression were significantly upregulated in 15d-PGJ2-treated Nrf2+/+ mice prior to ischemia, although the expression of these genes reached to similar extent after I/R. The expressions of NQO1 and GCLc proteins were also increased by 15d-PGJ2 in Nrf2+/+ livers before ischemia and were maintained high level after reperfusion. We observed remarkable differences in hepatocyte protection after I/R between the mice treated with and without 15d-PGJ2. Therefore, production of antioxidant proteins driven by upregulation of antioxidant genes before ischemia through Nrf2 preactivation might contribute to the protective mechanism.
It is well known that increased ROS triggers the onset of mitochondrial permeability transition, resulting in the induction of both apoptotic and necrotic cell death.27 Previous studies revealed that oncotic necrosis is predominant mode of cell death after I/R of the liver because ATP depletion blocks apoptotic signaling, which is required for caspase activation.27, 28 In agreement with previous reports, damaged hepatocytes were diffusely stained by TUNEL assay, and these hepatocytes did not meet the morphological criteria of apoptosis (cell shrinkage, chromatic condensation and margination, and apoptotic bodies) in our study. In addition, TUNEL positive hepatocytes were located in the confluent area around pericentral regions where oxygen supply is low. These findings are characteristic in the histology for oncotic necrosis of the liver tissue. It has been reported that this type of cell death is also observed in acetaminophen overdose-mediated liver injury.29 The formation of N-acetyl-p-benzoquinone imine—a cytotoxic reactive metabolite, which leads to GSH consumption, mitochondrial damage, ATP depletion, and ROS formation—has been postulated to be one of the mechanisms of oncotic necrosis. Because the activation of Nrf2 is also known to protect against acetaminophen-induced hepatotoxicity,13, 16, 30 it is suggested that Nrf2 may have an ability to confer the liver with protection in various pathological conditions that cause oncotic necrosis through oxidative stress.
GSH is the most abundant antioxidant in hepatocytes and plays a central role in maintaining redox homeostasis.31, 32 Because GSH can detoxify ROS such as hydrogen peroxide, hypochlorous acid, and peroxynitirite, GSH is expected to protect the organs from oxidative stress.33, 34 N-acetylcysteine (NAC)—a thiol-containing synthetic compound that increases intracellular GSH level—is mainly used for treating acetaminophen poisoning.35, 36 NAC has also been reported to replenish hepatic GSH store, thereby reducing hepatic I/R injury in many experimental studies. However, there is no clinical evidence stating that the use of NAC is effective as a protective agent to attenuate liver injury in patients undergoing liver surgery. One explanation of this discrepancy is that glutathione has a very short half-life in human livers.37 Other antioxidative strategies against hepatic I/R, the use of exogenous antioxidant enzymes such as catalase or superoxide dismutase has also been investigated.4 Since these enzymes cannot maintain their bioavailability when administered intravenously, the protective effect has been proved using gene delivery system, which is difficult to apply clinically with present technologies. In this study, we showed that the expression levels of GCLc—GSH-synthesizing enzyme—was higher in Nrf2+/+ mice than in Nrf2−/− mice. GCLc level was further increased by the pretreatment with 15d-PGJ2 in Nrf2+/+ livers. Consistent with the level of GCLc expression after reperfusion, GSH content was sustained low in Nrf2−/− mice and recovered well in 15d-PGJ2-pretreated Nrf2+/+ mice. This fact suggests that Nrf2-dependent induction of GCLc confers effective regulation of GSH.
As mentioned above, we showed that 15d-PGJ2 suppressed hepatic I/R injury in an Nrf2-dependent manner. The function of 15d-PGJ2 was initially identified as a natural endogenous ligand for PPARγ.17 Among various functions of PPARγ, the protective properties of PPARγ against hepatic I/R was reported by Kuboki et al.38 They demonstrated that administration of rosiglitazone (synthetic PPARγ agonist) or C-peptide (PPARγ activator) maintained PPARγ activity during I/R and reduced liver damage, suggesting the protective role of PPARγ activation against I/R injury. They further showed that in hepatic I/R injury, the hepatic content of endogenous 15d-PGJ2 was reduced in association with the reduced activation of PPARγ. Our data show that the expression of PPARγ mRNA was significantly higher in the liver of Nrf2+/+ mice than Nrf2−/− mice and was increased by15d-PGJ2 treatment only in the liver of Nrf2+/+ mice. A comparable results were also reported previously by Cho et al.39 in the lung, corroborating our data showing Nrf2 dependency of PPARγ expression. In addition, we demonstrated that 15d-PGJ2 increased hepatic mRNA expressions of CD36, ABCA1, ABCG1, and LXRα—representative PPARγ target genes—, indicating that 15d-PGJ2 increased transcriptional activity of PPARγ in the liver of Nrf2+/+ mice before ischemia. Therefore, it was speculated that the hepatoprotective function of exogenous 15d-PGJ2 observed in the current study was also mediated by PPARγ via Nrf2 activation. While pretreatment with a PPARγ antagonist, GW9662, reduced the 15d-PGJ2-dependent upregulation of PPARγ target genes, it did not annul the protective effect of exogenous 15d-PGJ2 against hepatic I/R in Nrf2+/+ mice. This finding implies that PPARγ is dispensable for 15d-PGJ2-mediated liver protection in our model, but protective effect of 15d-PGJ2 on I/R injury is undoubtedly Nrf2-dependent. As a reason for this, the following mechanism is conceivable. Because protective function of PPARγ is considered to be mediated by transrepression of the inflammatory transcription factors,39,40 continuous activation of PPARγ during I/R might be necessary to suppress inflammatory response. However, 15d-PGJ2 has a very short half-life and hepatic 15d-PGJ2 level is rapidly reduced during I/R.38, 41 Therefore, there is a strong likelihood that exogenous 15d-PGJ2 enhancing PPARγ activity is rapidly decreased during hepatic I/R, failing to upkeep PPARγ activation and accordingly, its subsequent transrepression of inflammatory response. Overall, our findings indicate that the mechanism of the liver protection with exogenous 15d-PGJ2 is not mediated by PPARγ activation but other pathway under Nrf2 activation, in which we demonstrated the importance of antioxidant preinduction.
HO-1 does not seem important in Nrf2 preactivation protocol for liver protection. It has been suggested that HO-1 is an Nrf2-dependent gene in certain cells,20, 21 but some studies showed a significant induction of HO-1 in the liver of Nrf2−/− mice.42–44 In the present study, the expression of HO-1 mRNA did not respond to 15d-PGJ2 treatment even in Nrf2+/+ mice before ischemia. After hepatic I/R, HO-1 mRNA upregulation was more marked in Nrf2−/− mice than in Nrf2+/+ mice, while it did not contribute to hepatocyte protection. This finding suggests that the upregulation of HO-1 mRNA in this study are merely a sequel to I/R damage. For HO-1, in addition to ARE, the regulatory region of HO-1 contains various binding sites for transcription factors such as NF-κB and AP-1, which are activated during hepatic I/R.4, 45, 46 The complex regulatory mechanism of HO-1expression might influence the behavior of HO-1, which is distinct from that of GSTm1, NQO1, or GCLs in our model.
Our current study demonstrates that Nrf2 is an important transcription factor for the prevention of hepatocyte death during I/R. Nrf2 deficiency aggravated oxidative stress, inflammatory response, and liver damage. Preactivation of Nrf2 before ischemia significantly suppressed oxidative stress, TNFα induction, as well as the extent of liver damage caused by I/R. Development of specific and effective Nrf2 activator would provide a distinct insight into the strategy for liver protection against I/R injury.
Supplementary Material
Acknowledgments
Financial support for this study was provided by Grants-in-Aid for Scientific Research (No. 18591497 and No. 23591979) and NIH/NIEHS P42 ES010337 (Project 5).
Footnotes
The authors declare no conflict of interest.
Reference
- 1.Jaeschke H. Reactive oxygen and ischemia/reperfusion injury of the liver. Chem Biol Interact. 1991;79(2):115–136. doi: 10.1016/0009-2797(91)90077-k. [DOI] [PubMed] [Google Scholar]
- 2.Jaeschke H, Woolbright BL. Current strategies to minimize hepatic ischemia-reperfusion injury by targeting reactive oxygen species. Transplant Rev (Orlando) 2012;26(2):103–114. doi: 10.1016/j.trre.2011.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abu-Amara M, Yang SY, Tapuria N, et al. Liver ischemia/reperfusion injury: processes in inflammatory networks--a review. Liver Transpl. 2010;16(9):1016–1032. doi: 10.1002/lt.22117. [DOI] [PubMed] [Google Scholar]
- 4.Zwacka RM, Zhou W, Zhang Y, et al. Redox gene therapy for ischemia/reperfusion injury of the liver reduces AP1 and NF-kappaB activation. Nat Med. 1998;4(6):698–704. doi: 10.1038/nm0698-698. [DOI] [PubMed] [Google Scholar]
- 5.Lemasters JJ, Qian T, He L, et al. Role of mitochondrial inner membrane permeabilization in necrotic cell death, apoptosis, and autophagy. Antioxid Redox Signal. 2002;4(5):769–781. doi: 10.1089/152308602760598918. [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi M, Yamamoto M. Nrf2-Keap1 regulation of cellular defense mechanisms against electrophiles and reactive oxygen species. Adv Enzyme Regul. 2006;46:113–140. doi: 10.1016/j.advenzreg.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 7.Thimmulappa RK, Lee H, Rangasamy T, et al. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest. 2006;116(4):984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei Y, Gong J, Yoshida T, et al. Nrf2 has a protective role against neuronal and capillary degeneration in retinal ischemia-reperfusion injury. Free Radic Biol Med. 2011;51(1):216–224. doi: 10.1016/j.freeradbiomed.2011.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanaka N, Ikeda Y, Ohta Y, et al. Expression of Keap1-Nrf2 system and antioxidative proteins in mouse brain after transient middle cerebral artery occlusion. Brain Res. 2011;1370:246–253. doi: 10.1016/j.brainres.2010.11.010. [DOI] [PubMed] [Google Scholar]
- 10.Nagai N, Thimmulappa RK, Cano M, et al. Nrf2 is a critical modulator of the innate immune response in a model of uveitis. Free Radic Biol Med. 2009;47(3):300–306. doi: 10.1016/j.freeradbiomed.2009.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao HD, Zhang F, Shen G, et al. Sulforaphane protects liver injury induced by intestinal ischemia reperfusion through Nrf2-ARE pathway. World J Gastroenterol. 2010;16(24):3002–3010. doi: 10.3748/wjg.v16.i24.3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klaassen CD, Reisman SA. Nrf2 the rescue: effects of the antioxidative/electrophilic response on the liver. Toxicol Appl Pharmacol. 2010;244(1):57–65. doi: 10.1016/j.taap.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Enomoto A, Itoh K, Nagayoshi E, et al. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 2001;59(1):169–177. doi: 10.1093/toxsci/59.1.169. [DOI] [PubMed] [Google Scholar]
- 14.Zhang YK, Yeager RL, Tanaka Y, et al. Enhanced expression of Nrf2 in mice attenuates the fatty liver produced by a methionine- and choline-deficient diet. Toxicol Appl Pharmacol. 2010;245(3):326–334. doi: 10.1016/j.taap.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morito N, Yoh K, Itoh K, et al. Nrf2 regulates the sensitivity of death receptor signals by affecting intracellular glutathione levels. Oncogene. 2003;22(58):9275–9281. doi: 10.1038/sj.onc.1207024. [DOI] [PubMed] [Google Scholar]
- 16.Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci U S A. 2001;98(8):4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scher JU, Pillinger MH. 15d-PGJ2: the anti-inflammatory prostaglandin? Clin Immunol. 2005;114(2):100–109. doi: 10.1016/j.clim.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Genovese T, Esposito E, Mazzon E, et al. Effect of cyclopentanone prostaglandin 15-deoxy-delta12,14PGJ2 on early functional recovery from experimental spinal cord injury. Shock. 2008;30(2):142–152. doi: 10.1097/SHK.0b013e31815dd381. [DOI] [PubMed] [Google Scholar]
- 19.Abe Y, Uchinami H, Kudoh K, et al. Liver epithelial cells proliferate under hypoxia and protect the liver from ischemic injury via expression of HIF-1 alpha target genes. Surgery. 2012;152(5):869–878. doi: 10.1016/j.surg.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liby K, Hock T, Yore MM, et al. The synthetic triterpenoids, CDDO and CDDO-imidazolide, are potent inducers of heme oxygenase-1 and Nrf2/ARE signaling. Cancer Res. 2005;65(11):4789–4798. doi: 10.1158/0008-5472.CAN-04-4539. [DOI] [PubMed] [Google Scholar]
- 21.Ishii T, Itoh K, Takahashi S, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275(21):16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 22.Tontonoz P, Nagy L, Alvarez JG, et al. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 23.Urakami H, Abe Y, Grisham MB. Role of reactive metabolites of oxygen and nitrogen in partial liver transplantation: lessons learned from reduced-size liver ischaemia and reperfusion injury. Clin Exp Pharmacol Physiol. 2007;34(9):912–919. doi: 10.1111/j.1440-1681.2007.04640.x. [DOI] [PubMed] [Google Scholar]
- 24.Kong X, Thimmulappa R, Kombairaju P, et al. NADPH oxidase-dependent reactive oxygen species mediate amplified TLR4 signaling and sepsis-induced mortality in Nrf2-deficient mice. J Immunol. 2010;185(1):569–577. doi: 10.4049/jimmunol.0902315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osburn WO, Yates MS, Dolan PD, et al. Genetic or pharmacologic amplification of nrf2 signaling inhibits acute inflammatory liver injury in mice. Toxicol Sci. 2008;104(1):218–227. doi: 10.1093/toxsci/kfn079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siegel D, Bolton EM, Burr JA, et al. The reduction of alpha-tocopherolquinone by human NAD(P)H: quinone oxidoreductase: the role of alpha-tocopherolhydroquinone as a cellular antioxidant. Mol Pharmacol. 1997;52(2):300–305. doi: 10.1124/mol.52.2.300. [DOI] [PubMed] [Google Scholar]
- 27.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125(4):1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 28.Gujral JS, Bucci TJ, Farhood A, et al. Mechanism of cell death during warm hepatic ischemia-reperfusion in rats: apoptosis or necrosis? Hepatology. 2001;33(2):397–405. doi: 10.1053/jhep.2001.22002. [DOI] [PubMed] [Google Scholar]
- 29.Gujral JS, Knight TR, Farhood A, et al. Mode of cell death after acetaminophen overdose in mice: apoptosis or oncotic necrosis? Toxicol Sci. 2002;67(2):322–328. doi: 10.1093/toxsci/67.2.322. [DOI] [PubMed] [Google Scholar]
- 30.Copple IM, Goldring CE, Jenkins RE, et al. The hepatotoxic metabolite of acetaminophen directly activates the Keap1-Nrf2 cell defense system. Hepatology. 2008;48(4):1292–1301. doi: 10.1002/hep.22472. [DOI] [PubMed] [Google Scholar]
- 31.Yuan L, Kaplowitz N. Glutathione in liver diseases and hepatotoxicity. Mol Aspects Med. 2009;30(1–2):29–41. doi: 10.1016/j.mam.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 32.Davis W, Ronai Z, Tew KD. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J Pharmacol Exp Ther. 2001;296(1):1–6. [PubMed] [Google Scholar]
- 33.Liu P, Fisher MA, Farhood A, et al. Beneficial effects of extracellular glutathione against endotoxin-induced liver injury during ischemia and reperfusion. Circ Shock. 1994;43(2):64–70. [PubMed] [Google Scholar]
- 34.Knight TR, Ho YS, Farhood A, et al. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 2002;303(2):468–475. doi: 10.1124/jpet.102.038968. [DOI] [PubMed] [Google Scholar]
- 35.Prescott LF, Park J, Ballantyne A, et al. Treatment of paracetamol (acetaminophen) poisoning with N-acetylcysteine. Lancet. 1977;2(8035):432–434. doi: 10.1016/s0140-6736(77)90612-2. [DOI] [PubMed] [Google Scholar]
- 36.Jegatheeswaran S, Siriwardena AK. Experimental and clinical evidence for modification of hepatic ischaemia-reperfusion injury by N-acetylcysteine during major liver surgery. HPB (Oxford) 2011;13(2):71–78. doi: 10.1111/j.1477-2574.2010.00263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Caro L, Ghizzi A, Costa R, et al. Pharmacokinetics and bioavailability of oral acetylcysteine in healthy volunteers. Arzneimittelforschung. 1989;39(3):382–386. [PubMed] [Google Scholar]
- 38.Kuboki S, Shin T, Huber N, et al. Peroxisome proliferator-activated receptor-gamma protects against hepatic ischemia/reperfusion injury in mice. Hepatology. 2008;47(1):215–224. doi: 10.1002/hep.21963. [DOI] [PubMed] [Google Scholar]
- 39.Cho HY, Gladwell W, Wang X, et al. Nrf2-regulated PPAR{gamma} expression is critical to protection against acute lung injury in mice. Am J Respir Crit Care Med. 2010;182(2):170–182. doi: 10.1164/rccm.200907-1047OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abdelrahman M, Sivarajah A, Thiemermann C. Beneficial effects of PPAR-gamma ligands in ischemia-reperfusion injury, inflammation and shock. Cardiovas Res. 2005;65(4):772–781. doi: 10.1016/j.cardiores.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 41.Straus DS, Pascual G, Li M, et al. 15-deoxy-delta 12, 14-prostaglandin J2 inhibits multiple steps in the NF-kappa B signaling pathway. Proc Natl Acad Sci USA. 2000;97(9):4844–4849. doi: 10.1073/pnas.97.9.4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reisman SA, Yeager RL, Yamamoto M, et al. Increased Nrf2 activation in livers from Keap1-knockdown mice increases expression of cytoprotective genes that detoxify electrophiles more than those that detoxify reactive oxygen species. Toxicol Sci. 2009;108(1):35–47. doi: 10.1093/toxsci/kfn267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Okada K, Shoda J, Taguchi K, et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295(4):G735–G747. doi: 10.1152/ajpgi.90321.2008. [DOI] [PubMed] [Google Scholar]
- 44.Okawa H, Motohashi H, Kobayashi A, et al. Hepatocyte-specific deletion of the keap1 gene activates Nrf2 and confers potent resistance against acute drug toxicity. Biochem Biophys Res Commun. 2006;339(1):79–88. doi: 10.1016/j.bbrc.2005.10.185. [DOI] [PubMed] [Google Scholar]
- 45.Kurata S, Matsumoto M, Tsuji Y, et al. Lipopolysaccharide activates transcription of the heme oxygenase gene in mouse M1 cells through oxidative activation of nuclear factor kappa B. Eur J Biochem. 1996;239(3):566–571. doi: 10.1111/j.1432-1033.1996.0566u.x. [DOI] [PubMed] [Google Scholar]
- 46.Uchinami H, Yamamoto Y, Kume M, et al. Effect of heat shock preconditioning on NF-kappaB/I-kappaB pathway during I/R injury of the rat liver. Am J Physiol Gastrointest Liver Physiol. 2002;282(6):G962–G971. doi: 10.1152/ajpgi.00466.2001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.