

Abstract
Schizophrenia is characterized by alterations in cortico-limbic processes believed to involve modifications in activity within the prefrontal cortex (PFC) and the hippocampus. The nucleus accumbens (NAc) integrates information from these 2 brain regions and is involved in cognitive and psychomotor functions that are disrupted in schizophrenia, indicating an important role for this structure in the pathophysiology of this disorder. In this study, we used in vivo electrophysiological recordings from the NAc and the PFC of adult rats and the MAM developmental disruption rodent model of schizophrenia to explore the influence of the medial PFC on the hippocampal–accumbens pathway. We found that, in MAM-treated rats, tetanization of hippocampal inputs to the NAc produce opposite synaptic plasticity compared with controls, which is a consequence of alterations in the hippocampal–mPFC pathway. Moreover, we show that administration of the D2-receptor-blocking antipsychotic drug sulpiride either systemically or directly into the mPFC reverses the alterations in the MAM rat. Therefore, specific disruptions in cortical and hippocampal inputs in the MAM-treated rat abnormally alter plasticity in subcortical structures. Moreover, our results suggest that, in the presence of antipsychotic drugs, the disrupted plasticities are normalized, supporting a role for this mechanism in antipsychotic drug action in schizophrenia.
Keywords: dopamine, long-term potentiation, nucleus accumbens, prefrontal cortex, schizophrenia
Introduction
The pathophysiology of schizophrenia likely involves alterations within a number of brain circuits. The nucleus accumbens (NAc) is the central structure for integration of contextual stimuli processed in the hippocampus and executive functions from the prefrontal cortex (PFC), modulated by the dopamine system from the ventral tegmental area (VTA) to control goal-directed behavior (Mogenson et al. 1988; Grace 2000). Studies suggest that dysfunction within the circuit involving the PFC, the hippocampus, and the NAc and its dopaminergic afferents could play a central role in the pathophysiology of schizophrenia (O'Donnell and Grace 1998; Grace et al. 2007).
Schizophrenia patients show a substantial loss of parvalbumin-containing interneuron staining in different brain structures, such as the PFC, which is proposed to contribute to cognitive disruption or negative symptoms (Lewis and Moghaddam 2006). The loss of these interneurons presumably creates a disruption in the modulation of cortical inputs to the NAc, further disrupting this pathway. In addition, schizophrenia patients are reported to exhibit hyperactivity within hippocampal circuits (Heckers et al. 1998; Malaspina et al. 1999; Medoff et al. 2001; Molina et al. 2005). Hippocampal activity is directly correlated with positive symptoms in schizophrenia (Liddle et al. 1992; Molina et al. 2003), and hyperactivity in the ventral subiculum (vSub) of the hippocampus (Weiss et al. 2006) is proposed to disrupt NAc responses and lead to hyperactivity of the dopamine pathway (O'Donnell et al. 1999; Lodge and Grace 2007).
Several animal models that use developmental disruption have been advanced as models of schizophrenia, such as the neonatal ventral hippocampal lesion (Lipska et al. 1993; O'Donnell et al. 2002), the maternal immune activation (Smith et al. 2007), or the methylazoxymethanol acetate (MAM; Moore et al. 2006) models, which show similar behavioral phenotypes. This last model, in particular, has been characterized in our laboratory (Moore et al. 1999; Moore et al. 2006) and involves exposure of the rat fetus to a DNA methylating agent, MAM. Fetal MAM exposure selectively disrupts cortical development and leads to anatomical and behavioral abnormalities in the adult offspring that are highly consistent with schizophrenia in humans, such as thinning of limbic cortices with increased neuronal packing density (Moore et al. 2006), increased response to psychostimulants (Flagstad et al. 2004; Moore et al. 2006; Lodge and Grace 2007), increased sensitivity to stress (Goto and Grace 2006), decreased prepulse inhibition of startle, and deficits in latent inhibition (Flagstad et al. 2004; Lodge et al. 2009). We have previously shown hyperactivity within the hippocampus in adult offspring treated with MAM (Lodge and Grace 2007). Moreover, this hyperactivity drives the hyperdopaminergic state that may underlie psychosis in this disorder. This is consistent with imaging studies showing hyperactivity within the hippocampus that correlates with psychosis in schizophrenia patients (Silbersweig et al. 1995).
This circuit also has central roles in goal-directed behavior; thus, plasticity in ventral hippocampal inputs provide strong facilitation of reward-driven learned behavior (Goto and Grace 2005a, 2005b; Sesack and Grace 2010), whereas this is opposed by the PFC input to allow behavioral flexibility (Goto and Grace 2005a, 2005b). Previously, we found that the mPFC regulates information flow within the vSub–NAc circuit (Goto and Grace 2005a, 2005b) and provides a permissive role in facilitating vSub-driven spike firing in the NAc (Belujon and Grace 2008). Moreover, when the vSub–NAc pathway is potentiated, the mPFC is no longer necessary to enable vSub–NAc drive, presumably allowing a faster response without the intervention of higher cognitive processes (Belujon and Grace 2008). These studies provided insight into the functional regulation of NAc circuits in the normal condition. However, this regulatory mechanism is likely substantially disrupted in schizophrenia. Given the involvement of the PFC and the vSub in schizophrenia, and the central position of the NAc in the cortico-basal ganglia-thalamo-cortical loop, we examined synaptic plasticity in the vSub–NAc pathways in the MAM model of schizophrenia and its modulation by the mPFC.
Materials and Methods
Animals
All animal experiments were performed in accordance with the guidelines outlined in the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh.
Methylazoxymethanol Acetate
MAM- and saline-treated rats were prepared as described previously (Moore et al. 2006; Lodge and Grace 2007). Briefly, timed pregnant female Sprague Dawley rats (Hilltop) arrived at gestational day (GD) 15 were housed individually under a 12-h light/dark cycle with food and water ad libitum with temperature (22 °C) and humidity (47%) held constant. On GD17, dams were injected either with the mitotoxin MAM (diluted in saline, 22 mg/kg, i.p.) or with saline (1 mL/kg, i.p.). Male pups were weaned on postnatal day 21 and housed in groups of 2–3 with littermates, and all females were removed from the litter. Multiple MAM- and saline-treated litters were used for each condition of the study. It should be noted that in previous studies, cross-fostering did not impact the MAM phenotype (Moore et al. 2006).
Extracellular Recordings
MAM/saline offspring at least 3 months old were injected with chloral hydrate (400 mg/kg, i.p.) and mounted in a stereotaxic frame (Kopf). Anesthesia was maintained by subsequent i.p. injections of chloral hydrate as needed to maintain suppression of the hindlimb compression withdrawal reflex. Moreover, rats were implanted with a catheter in the lateral tail vein to allow for intravenous (i.v.) injections. The internal temperature of the rats was held constant at 37 °C using a temperature-controlled heating pad (Fine Science Tools). In vivo extracellular recordings were performed using WPI glass tubing (2 mm outer diameter; 1.12 inner diameter) that was pulled on a vertical puller (PE-2, Narashige, Japan) and broken back under microscopic control to an impedance of 12–16 MΩ and filled with a 2% Chicago Sky Blue solution in 2 M NaCl. Microelectrodes were lowered through the NAc (A/P +1.5 mm from bregma; M/L +1.1 mm from midline; D/V −5 to −7.5 mm from the dura) or the mPFC (A/P +3.2 mm from bregma; M/L +0.5 mm from midline; −3 to −5 mm from dura).
Electrophysiological signals were amplified (×1000) and filtered (low pass: 400 Hz, high pass: 16 kHz) using a Fintronics amplifier (model WDR-420). Recording were displayed on an oscilloscope (B&K precision) and transferred via a Powerlab interface (AD Instruments) to a computer equipped with LabChart v.7 software. Neuronal activity with a signal-to-noise ratio >3 was recorded and used for analyses.
Single-pulse, low-frequency, and high-frequency stimulations (LFS and HFS, respectively) were applied via a concentric bipolar stimulating electrode (NEX-100X; Rhodes Medical Instruments) to the fimbria (saline rats: A/P −1.6 mm from bregma; M/L +1.3 mm from the midline; D/V −4.5 mm from the dura; MAM rats: A/P −2 mm from bregma; M/L +1.3 mm from midline; D/V −4.2 mm from dura). A chemotrode (combination of a stimulating electrode and guide cannula) was placed in the mPFC at a 15° angle (for saline rats: A/P +2.9 mm from bregma; M/L +1.9 mm from midline; D/V −3 mm from dura (Belujon and Grace 2008); MAM: A/P +3.2 mm from bregma; M/L +1.3 mm from midline; D/V −3.6 mm from dura). Coordinates used in MAM-treated rats were adjusted to compensate for the reduction in brain size (Flagstad et al. 2004).
Drugs and Drug Infusions
Tetrodotoxin (TTX) and Dulbecco's PBS (dPBS) were obtained from Sigma-Aldrich. Following a baseline period, 0.5 µL of TTX (1 µM dissolved in 0.5 µL of dPBS), sulpiride (3 µg in 0.5 µL) or dPBS was infused locally into the mPFC at a rate of 0.5 µL/min. Additionally, following a baseline period, sulpiride was injected intravenously (5 mg/kg) (Floresco et al. 2001; Belujon and Grace 2008).
Stimulation Protocol
While searching for a responsive NAc neuron, the fimbria and mPFC received alternating single-pulse stimuli delivered using a dual-output stimulator (S8800; Grass technologies) (intensity: 1 mA; pulse width: 0.25 ms) with the fimbria receiving the first stimulation and the mPFC stimulated 100 ms after, and repeated every 2s. Once a putative monosynaptically activated cell was found to be responsive, a similar stimulation procedure was used (pulse duration: 0.25 ms; every 2s), and the current administered to the fimbria was adjusted to evoke an action potential approximately 50% of the time (an action potential every 2 stimulations). Spike probabilities were measured by dividing the number of spikes observed by the number of stimuli in 5-min intervals. After recording stable baseline activity, 1 of 6 experimental paradigms was administered for NAc recordings: HFS (20 Hz; 10 s at suprathreshold) or LFS (5 Hz; 500 pulses at suprathreshold) of the fimbria pathway, infusion of TTX/vehicle into the mPFC, HFS to the fimbria followed by TTX/vehicle infusion into the mPFC, injection of sulpiride/vehicle i.v. followed by HFS to the fimbria, and sulpiride/vehicle infusion into the mPFC followed by HFS to the fimbria. For mPFC recordings, a single stimulation of the fimbria was applied and responsive mPFC neurons were recorded. After recording stable baseline activity, 1 of 2 experimental paradigms was administered: HFS to the fimbria or injection of sulpiride/vehicle i.v. followed by HFS to the fimbria. No more than 1 neuron per animal was recorded, except for the study of single-pulse stimulation in mPFC neurons where 32 neurons were recorded in 14 rats.
Histology
Following the cessation of each experiment, electrode placement was verified via electrophoretic ejection of Chicago Sky Blue dye into the recording site (−20 μA constant current: 20–30 min). To verify the stimulation electrode placement, a 10s pulse at 200 µA was administered. Rats were euthanized with a lethal dose of chloral hydrate (additional 400 mg/kg) and brains were removed following decapitation. The tissue was fixed in 8% paraformaldehyde for at least 48 h and then transferred to a 25% sucrose solution for cryoprotection. Once saturated, brains were frozen and sliced coronally at 60 µM thick using a cryostat (Leica Frigocut 2800) and mounted onto gelatin-chromalum-coated slides. Tissue was stained with combination of neutral red and cresyl violet.
Analysis
Data were analyzed using a 1-way ANOVA with repeated measures followed by the Holm–Sidak test, with time as the within-subject factor. Multiple comparisons were analyzed using a 2-way ANOVA followed by the Holm–Sidak test, with treatment as the between-subject factor and time as the within-subject factor.
Results
Response of NAc Neurons to Afferent Activation
Placements of stimulating and recording electrodes are shown in Supplemental Figure 1. The mean baseline spike probability and the latency of fimbria-evoked responses were not statistically different between MAM- and saline-treated animals (t-test; P = 0.09 and P = 0.45, respectively; Table 1). The current used to obtain a 50% spike probability baseline for fimbria-evoked responses was significantly higher in MAM-treated animals than in saline-treated animals (Table 1; P < 0.05).
Table 1.
Mean (±SEM) of the current used to obtain a 50% spike probability baseline, the latency to spike, and the baseline spike probability for saline-treated and MAM-treated animals for fimbria-evoked responses
| Saline (n = 32) | MAM (n = 77) | |
|---|---|---|
| Current to 50% spike probability (µA) | 523.3 ± 55.83 | 706.6 ± 46.65* |
| Latency to spike (ms) | 8.10 ± 0.06 | 8.39 ± 0.39 |
| Baseline spike probability | 0.55 ± 0.02 | 0.50 ± 0.02 |
*P < 0.05.
Opposite Effects of High-Frequency Stimulation, but Not Low-Frequency Stimulation, of vSub–NAc Pathway on Synaptic Plasticity in MAM Rats Versus Controls
It is now well established that HFS to vSub afferents induces a long-term potentiation (LTP) in the vSub–NAc pathway in normal animals, which is, at least in part dopamine dependent (Floresco et al. 2001; Goto and Grace 2005a, 2005b; Belujon and Grace 2008). As MAM-treated animals show altered activity of the dopamine system from the VTA (Lodge and Grace 2007), we tested for changes in dopamine-dependent synaptic plasticity in MAM-treated rats. The effect of HFS on vSub inputs was tested by stimulating the fimbria and assessing evoked responses in the NAc (Fig. 1A–D). In saline-treated animals, HFS led to a significant increase in fimbria-evoked spike probability in the NAc (Fig. 1A,C,D; baseline spike probability: 0.46 ± 0.04 and post-HFS: 0.80 ± 0.03; F = 19.373; P < 0.05; n = 5). In contrast, in MAM-treated animals, a significant decrease in fimbria-evoked spike probability was seen immediately following tetanic stimulation (Fig. 1B,C,D; baseline spike probability: 0.43 ± 0.04 and post-HFS: 0.24 ± 0.04; F = 4.594; P < 0.05; n = 11). There was a significant difference between MAM and saline animals on the percentage change of fimbria-evoked spike probability (2-way ANOVA, Holm–Sidak post hoc, F = 71.513; P < 0.05). Therefore, HFS of the fimbria had opposite effects on the fimbria-evoked responses in saline versus MAM rats; inducing LTP in the saline rats but depression in the MAM rats.
Figure 1.

HFS, but not LFS of the fimbria has opposite effect on accumbens plasticity in saline- and MAM-treated animals. (A and B) Representative example of extracellular recordings from NAc neuron activity evoked by fimbria stimulation before and after HFS to the fimbria in saline-treated animals (A) and MAM-treated animals (B). Twenty-five overlaid consecutive traces are shown with the numbers demonstrating the number of evoked spikes for 25 stimulations. Scale: 10 mV, 2 ms. (C) Mean percent change (±SEM) in fimbria-evoked spike probability, normalized to the baseline, after HFS to the fimbria in saline-treated (black circles) and MAM-treated (gray circles) animals (*P < 0.05; arrow indicates the time of stimulation). (D) Mean percent change in fimbria-evoked responses (±SEM) following HFS in saline-treated (black bar) and MAM-treated (gray bar) animals. (E) Mean percent change (±SEM) in fimbria-evoked spike probability, normalized to the baseline, after LFS to the fimbria in saline-treated (black circles) and MAM-treated (gray circles) animals (arrow indicates time of stimulation). (F) Mean percent change in fimbria-evoked responses (±SEM) following LFS in saline-treated (black bar) and MAM-treated (gray bar) animals.
In a separate group of rats, LFS was administered to the fimbria in both saline- and MAM-treated animals (Fig. 1E,F). LFS led to a significant decrease in fimbria-evoked spike probability in the NAc in both saline- and MAM-treated rats (F = 13.201 and F = 2.444, respectively; P < 0.05). There was no difference between MAM and saline-treated rats (2-way ANOVA, Holm–Sidak post hoc; F = 0.181; P = 0.673. Therefore, with LFS of the fimbria, fimbria-evoked spiking in the NAc exhibited a similar type of depression in both MAM and saline rats.
The vSub Potently Activates the mPFC in Addition to the NAc in MAM-Treated Rats
Besides a regulatory role of synaptic plasticity in the NAc, DA is also known to regulate synaptic plasticity in the mPFC (Gurden et al. 2000; Huang et al. 2004). We therefore assessed in MAM-treated rats possible changes in synaptic plasticity in the mPFC. First, single-pulse stimulation of the fimbria was used to evoke spike discharge in mPFC neurons (representative example Fig. 2B). In saline-treated animals, the majority of neurons responded to a single fimbria stimulation (1 mA) with a single spike (n = 15 of 17 neurons in 7 rats; representative example Fig. 2B, 2 of 17 neurons showed multiple spikes per stimulation). However, in MAM-treated animals, a single-pulse stimulation induced a burst of spikes in the mPFC in the majority of neurons (n = 17 of 19 neurons in 7 rats, representative example Fig. 2B; 2 of 19 neurons showed only a single spike per stimulation). Thus, the fimbria-evoked spike probability in MAM-treated rats was significantly higher than in saline-treated animals (Fig. 2C; saline: 0.75 ± 0.17; MAM: 1.46 ± 0.20; t-test; P < 0.05; n = 17 neurons and n = 19 neurons, respectively). HFS to the fimbria had no effect on evoked spiking in saline-treated rats (Fig. 2D,E; baseline: 0.44 ± 0.07; post-HFS: 0.49 ± 0.08; F = 0.69; P = 0.69; n = 6), whereas a significant increase in evoked spike activity was observed in MAM-treated animals (Fig. 2D,E; baseline: 0.56 ± 0.06; post-HFS: 0.72 ± 0.06; F = 4.63; P < 0.05; n = 6). Moreover, the difference between the fimbria-evoked response of MAM- and saline-treated rats was significant (2-way ANOVA, Holm–Sidak post hoc; F = 5.496; P < 0.05). It has been shown previously that HFS to the mPFC lead to a depression of the vSub–NAc synaptic plasticity in normal rats (Goto and Grace 2005a, 2005b). Therefore, we tested whether, in the MAM rat, the sustained activation of the mPFC by the vSub may underlie the vSub–NAc long-term depression (LTD) observed after HFS.
Figure 2.

HFS of fimbria induces abnormal plasticity in the mPFC in MAM-treated animals. (A) Schematic showing the placements of recording electrodes in the mPFC for saline-treated (black triangles) and MAM-treated (gray squares) rats, shown as coronal sections of the rat brain, taken from Paxinos and Watson (Paxinos and Watson 1996). (B) Representative example of extracellular mPFC recordings showing the response of pyramidal neurons to single fimbria stimulation (1 mA) in saline-treated animals (left) and MAM-treated animals (right). Scale: 10 mV, 10 ms (C) Scatterplot showing the spike probability over 5min in saline- and MAM-treated rats (black triangles and gray squares, respectively) evoked by 1 mA stimulation. (D) Mean percent change (±SEM) in fimbria-evoked spike probability, normalized to baseline, following HFS to the fimbria in saline-treated (black triangles) and MAM-treated (gray squares) animals (*P < 0.05; arrow indicates time of stimulation). (E) Mean of the percent change in fimbria-evoked responses (±SEM) following HFS in saline-treated (black bar) and MAM-treated (gray bar) animals.
Inactivation of the mPFC Has Opposite Effect in Saline- and MAM-Treated Animals
We have previously reported a modulatory role of the mPFC on the hippocampal drive of the NAc, in that the mPFC facilitates single-pulse drive of the vSub–NAc circuit (Belujon and Grace 2008), but repeated mPFC stimulation will lead to LTD in the vSub–NAC pathway (Goto and Grace 2005a, 2005b). Moreover, we have demonstrated here that MAM-treated rats show hyper-responsivity of the mPFC in response to fimbria stimulation, and altered vSub–NAc synaptic plasticity. Therefore, we hypothesize that the hippocampal overdrive of the mPFC in MAM rats might be responsible, at least in part, for the altered synaptic plasticity observed in the vSub–NAc pathway. To first test this hypothesis, the effect of inactivating the mPFC on vSub–NAc afferents in both saline- and MAM-treated animals was examined (Fig. 3). Inactivation of the mPFC with TTX led to a 60% decrease in the probability of evoking spikes in NAc neurons in saline-treated animals (Fig. 3A,C,D; baseline spike probability: 0.58 ± 0.06; post-TTX spike probability: 0.31 ± 0.08; F = 6.887; P < 0.05; n = 4), which is consistent with our previous study (Belujon and Grace 2008). In contrast, in MAM-treated rats, infusion of TTX into the mPFC led to a potentiation of the vSub–NAc pathway, with an approximate 50% increase in spike probability (Fig. 3B,C,D; baseline: 0.53 ± 0.05 and post-TTX: 0.75 ± 0.04; F = 6.387; P < 0.01; n = 12). Infusion of dPBS (0.5 µL) into the mPFC had no effect on the fimbria-evoked spike probability, excluding an effect of the infusion itself (Fig. 3C; F = 1.87; P = 0.10; n = 5). Moreover, after inactivation of the mPFC, the fimbria-evoked spike probability in the NAc was significantly different between MAM and saline rats (2-way ANOVA, Holm–Sidak post hoc; F = 27.817; P < 0.05). Therefore, the inactivation of the mPFC had opposite effects on vSub–NAc spike probability in MAM versus saline rats, increasing vSub–NAc drive in MAM rats but decreasing it in saline rats.
Figure 3.
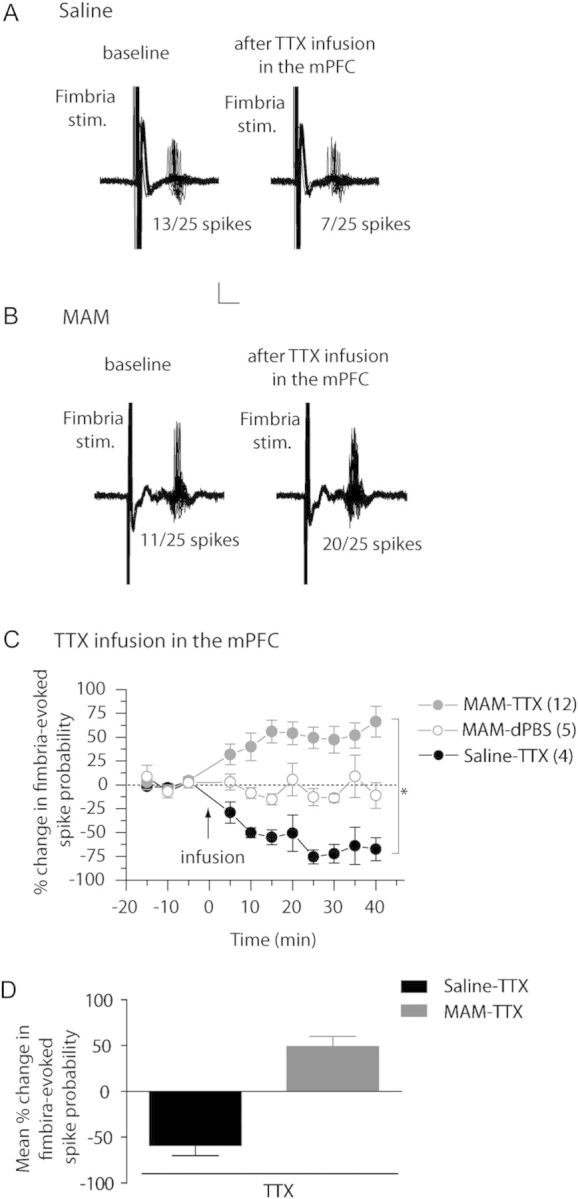
Inactivation of the mPFC has opposite effect in the NAc of saline- and MAM-treated animals. (A and B) Representative example of extracellular recordings from accumbens neurons evoked by fimbria stimulation before and after infusion of tetrodotoxin (TTX) in the mPFC in saline-treated animals (A) and MAM-treated animals (B). Twenty-five overlaid consecutive traces are shown with the numbers demonstrating the number of evoked spikes for 25 stimulations. Scale: 10 mV, 5 ms. (C) Mean percent change (±SEM) of the fimbria-evoked spike probability, normalized to the baseline, after infusion of TTX (filled circle) or dPBS (open circles) into mPFC in saline-treated (black circles) and MAM-treated (gray circles) animals (*P < 0.05; arrow indicates the time of infusion). (D) Mean of the percent change in fimbria-evoked responses (±SEM) following infusion of TTX in saline-treated (black bar) and MAM-treated (gray bar) animals.
Tetanic Stimulation-Induced LTD of vSub–NAc Neuronal Response in MAM Animals is Reversed by Inactivation of the mPFC
The previous data suggest that in the MAM model, removing the mPFC influence would disinhibit the vSub–NAc pathway. Therefore, we tested the effect of inactivating the mPFC after HFS of the fimbria in MAM rats.
In MAM-treated animals, HFS of the fimbria produced an LTD in the vSub–NAc pathway, which was reversed by inactivating the mPFC with TTX. The inactivation of the mPFC led to an increase in spike probability in fimbria-evoked responses, returning the spike probability to baseline levels (Fig. 4A,B; baseline spike probability: 0.47 ± 0.05; post-HFS: 0.24 ± 0.02; post-TTX: 0.35 ± 0.06). Thus, the fimbria-evoked spike probability after infusion of TTX was within a 95% confidence interval (CI) of the pre-HFS baseline (baseline: 0.47 ± 0.05; post-TTX: 0.35 ± 0.06; CI: 0.34–0.59; n = 8). Additionally, infusion of dPBS into the mPFC after HFS to the fimbria did not alter the fimbria-evoked spike probability, excluding the possibility of an infusion effect (Fig. 4A,B; post-HFS: 0.30 ± 0.06; post-dPBS: 0.22 ± 0.03; CI: 0.18–0.42; n = 5), and there was a significant difference between the spike probabilities after TTX and dPBS infusions in the mPFC in MAM-treated rats (2-way ANOVA, Holm–Sidak post hoc; F = 5.602; P < 0.05; Fig. 4B). In summary, in the MAM-treated rats, the HFS-induced LTD in the vSub–NAc pathway was reversed upon mPFC inactivation.
Figure 4.

Abnormal accumbens plasticity following HFS to fimbria is reversed in MAM-treated animals by inactivating the mPFC. (A) Mean percent change (±SEM) in fimbria-evoked spike probability, normalized to baseline, following tetanization of the fimbria and infusion of TTX (closed circles) or vehicle, dPBS (open circles) in the mPFC in MAM-treated animals (*P < 0.05; arrows indicate time of stimulation and infusion). (B) Mean of the percent change in fimbria-evoked responses (±SEM) following infusion of vehicle, dPBS (white bar), or TTX (gray bar) in MAM-treated animals.
It should be noted that consistent with previous results (Belujon and Grace 2008), there was no change in fimbria-evoked NAc responses after infusion of TTX into the mPFC 20min following tetanization of the fimbria in saline-treated animals (Supplemental Figure 2; baseline spike probability: 0.49 ± 0.02; post-HFS: 0.66 ± 0.06; post-TTX: 0.83 ± 0.04; n = 8). The fimbria-evoked spike probability was within a 95% CI of the spike probability post-tetanus/pre-TTX (20min pre-TTX: 0.84 ± 0.04; post-TTX: 0.83 ± 0.04; CI: 0.65–1.0; n = 8).
Injection of the D2 Antagonist Sulpiride Reverses vSub–NAc LTD and Induces LTP in MAM-Treated Rats Following HFS
We reported here that the fimbria-evoked synaptic plasticities in the mPFC and in the NAc, which are partially dopamine dependent, were altered in MAM-treated animals. Considering the major role of D2 receptors in schizophrenia, we examined the effect of manipulation of D2 receptors by systemic injection and local infusion of the D2 receptor antagonist sulpiride. First, sulpiride was injected intravenously 10min prior to fimbria tetanization (Fig. 5A,B). In the NAc of MAM-treated animals, the injection of sulpiride had no effect on fimbria-evoked spike probability until HFS was administered, after which an LTP was observed (baseline: 0.48 ± 0.05; post-sulpiride: 0.50 ± 0.08; post-HFS: 0.69 ± 0.06; F = 3.219; P < 0.05; n = 5), whereas HFS post-saline injection induced an LTD (baseline: 0.53 ± 0.00; post-sulpiride: 0.59 ± 0.07; post-HFS: 0.36 ± 0.10; F = 8.002; P < 0.05; n = 5). The effect of injection of sulpiride on the fimbria-evoked spike probability after HFS was significantly different in comparison with saline injection (2-way ANOVA, Holm–Sidak post hoc; F = 4.278, P < 0.05). Thus, systemic injection of sulpiride reversed the altered vSub–NAc synaptic plasticity in MAM-treated animals.
Figure 5.

Administration of the D2 antagonist, sulpiride, reverses vSub–NAc LTD, and vSub–mPFC LTP in MAM-treated rats. (A) Mean percent change (±SEM) in fimbria-evoked spike probability in NAc neurons, normalized to baseline, following i.v. injection of sulpiride (closed circles) or saline (open circles) and tetanization of the fimbria in MAM-treated animals (*P < 0.05; arrows indicate time of injection and stimulation). (B) Mean of the percent change in fimbria-evoked responses (±SEM) after HFS to fimbria following an injection of sulpiride (gray bar) or saline (white bar) in MAM-treated animals. (C) Mean percent change (±SEM) in fimbria-evoked spike probability in NAc neurons, normalized to baseline, following local infusion of sulpiride (closed circles) or dPBS (open circles) in the mPFC and tetanization of the fimbria in MAM-treated animals (*P < 0.05; arrows indicate time of infusion and stimulation). (D) Mean of the percent change in fimbria-evoked responses (±SEM) after HFS to fimbria following local infusion of sulpiride (gray bar) or dPBS (white bar) in mPFC in MAM-treated animals. (E) Mean percent change (±SEM) in fimbria-evoked spike probability in mPFC neurons, normalized to baseline, following i.v. injection of sulpiride (closed squares) or saline (open squares) and HFS to the fimbria in MAM-treated animals (*P < 0.05; arrows indicate time of injection and stimulation). (F) Mean of the percent change in fimbria-evoked responses (±SEM) following HFS of the fimbria after injection of sulpiride (gray bar) or saline (white bar) in MAM-treated animals. (G) Mean percent change (±SEM) in fimbria-evoked spike probability in mPFC neurons, normalized to baseline, following i.v. injection of sulpiride and HFS to the fimbria in Saline-treated animals SAL; arrows indicate time of injection and stimulation). (H) Mean of the percent change in fimbria-evoked responses in saline animals (±SEM) following HFS of the fimbria alone (white bar) or after injection of sulpiride (black bar).
It should be noted that, in saline animals, systemic injection of sulpiride had no clear effect on the fimbria-evoked spike probability after HFS (baseline: 0.48 ± 0.03; post-sulpiride: 0.56 ± 0.06; post-HFS: 0.50 ± 0.05; 1-way ANOVA, F = 0.476; P = 0.882; n = 7, Fig. 5G,H).
Local Infusion of Sulpiride in the mPFC Reverses vSub–NAc LTD in MAM-Treated Rats Following HFS
We have shown in the MAM model (Goto and Grace 2006) and other animal models of schizophrenia (Tseng et al. 2006) that the effects of DA in the mPFC are altered, in that activation of VTA afferents produces excitation in the schizophrenia model but inhibition in the control case. Given the data showing increased vSub drive of the mPFC in MAM animals, and due to the effect seen when intravenously injecting sulpiride in these animals, we examined whether D2 receptors in the mPFC are responsible for the mPFC-mediated attenuation of vSub–NAc transmission in the MAM rat. Consistent with this prediction, we found that infusion of sulpiride in the mPFC prevented the HFS-induced LTD previously observed in the vSub–NAc pathway (Fig. 5C,D). Thus, in MAM-treated rats, HFS had no effect on the fimbria-evoked spike probability in comparison with baseline when preceded by sulpiride infusion. The spike probability after HFS was within a 95% CI of the pre-HFS baseline (baseline: 0.43 ± 0.06; post-sulpiride: 0.40 ± 0.03; post-tetanus: 0.43 ± 0.11; CI: 0.49–0.737; n = 6). Furthermore, infusion of dPBS into the mPFC did not alter the HFS-induced LTD in MAM-treated rats (baseline: 0.58 ± 0.04; post-dPBS: 0.57 ± 0.06; post-HFS: 0.28 ± 0.04; F = 4.864; P < 0.05; Fig. 5C,D). Moreover, there was a significant difference between spike probability after HFS with preinfusion of sulpiride and with preinfusion of dPBS in MAM-treated animals (F = 4.447; P < 0.05). These data suggest that the pathological effect of DA in the mPFC of MAM-treated rats is responsible for the abnormal attenuation of vSub–NAc drive.
Injection of Sulpiride Occludes the HFS-Induced LTP in vSub–mPFC in MAM-Treated Animals
Based on the previous finding that the abnormal plasticity in the NAc is, at least partially, dopamine dependent and acting through D2 receptors in the mPFC, we investigated whether the sustained activation of the mPFC by the vSub can also be manipulated with sulpiride. Therefore, we again injected sulpiride intravenously 10min before HFS to the fimbria and recorded evoked responses in the mPFC. There was no effect on fimbria-evoked spike probability in the mPFC following the injection of sulpiride (Fig. 5E,F; baseline spike probability: 0.72 ± 0.08; post-sulpiride: 0.81 ± 0.12; CI: 0.40–1.00; n = 5). However, the HFS-induced LTP previously observed in the MAM-treated rat was prevented by the injection of sulpiride, yielding a response to HFS in MAM-treated animals that was similar to that seen in saline-treated animals (as seen in Fig. 2D,E). Thus, the spike probability after HFS was within a 95% CI of the pre-HFS baseline (Fig. 5E,F; baseline: 0.72 ± 0.08; post-HFS: 0.66 ± 0.13; CI = 0.40–1.00; n = 5). In addition, vehicle injection did not alter the HFS-induced LTP in MAM-treated rats (baseline: 0.33 ± 0.02; post-vehicle: 0.34 ± 0.01; post-HFS: 0.50 ± 0.06; F = 3.158; P < 0.05; Fig. 5E,F). The effect of injection of sulpiride on the fimbria-evoked spike probability after HFS was significantly different in comparison with saline injection (F = 3.144; P < 0.05). Therefore, these data suggest that the sustained activation of the mPFC observed in MAM rats to fimbria HFS is also dependent on D2 receptors, as is the sustained depression to fimbria HFS observed in the NAc.
Discussion
The vSub–NAc system is regulated in complex ways by several inter-related cortical and limbic circuits, and these relationships are altered in the MAM model of schizophrenia. Thus, in previous studies, we have shown that the mPFC and the vSub can exert both cooperative and antagonistic interactions within the NAc in normal animals. Specifically, hippocampal HFS causes LTD in the PFC–accumbens pathway and HFS in the mPFC induces LTD in the hippocampus–accumbens pathway (Goto and Grace 2005a, 2005b). In contrast, the vSub is dependent on the mPFC to phasically drive NAc neurons (Belujon and Grace 2008). Such an interaction exerted potent impacts on goal-directed behavior, in that induction of LTP in the vSub–NAc circuit potently facilitated the learning of reward-driven behavior, but activation of the PFC reversed this to allow behavioral flexibility.
However, this relationship is substantially altered in the MAM model of schizophrenia (Fig. 6). In schizophrenia patients, a hypofunctionality of the PFC has been described (Molina et al. 2005), and this disruption in activation may be due to an increased basal activity state of the PFC. As a result, continued activation of the vSub, instead of facilitating learning, would lead to a disruption of experience-dependent learned behavior, secondary to altered mPFC modulation of vSub–NAc drive. Our data suggest that this was due to abnormally increased activity in the mPFC suppressing part of the vSub–NAc drive via competition with the vSub.
Figure 6.
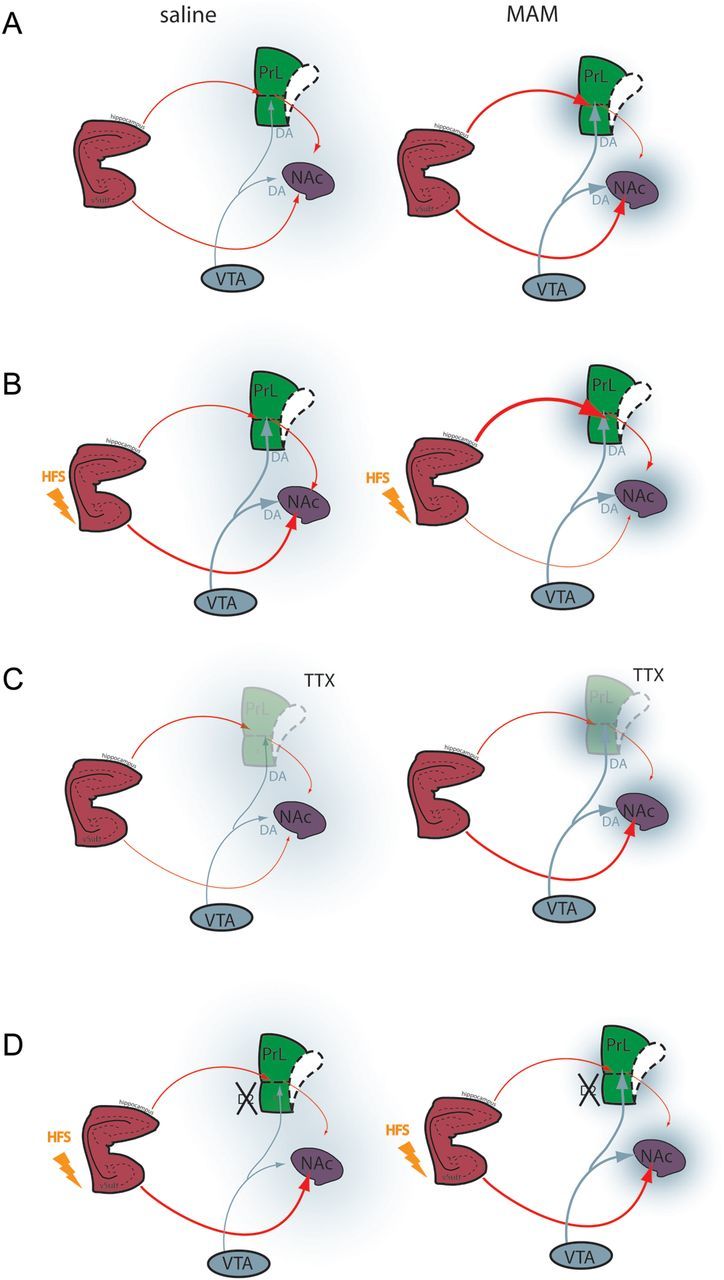
Schematic illustrating vSub–mPFC–NAc interactions in saline-and MAM-treated animals. (A) In saline rats, there is a balance between vSub and mPFC inputs to the NAC modulated by dopamine afferents from the VTA. In the MAM rats, there is a disruption of this balance due to hyperactivity in the vSub, leading to a hyperdopaminergic state and hypofunctional mPFC. (B) Tetanization of the fimbria increases the vSub–NAc pathway in saline animals (left) but induced a decrease in vSub–NAc drive and a hyperactive vSub–mPFC pathway in MAM rats (right). (C) Inactivation of the mPFC reduces the vSub drive of NAc neurons in saline rats (left) whereas in MAM rats, it increases the drive, demonstrating pathological phasic antagonistic action of the vSub and the mPFC on NAc neurons in our model. (D) Infusion of antipsychotic drug has no effect on the vSub–NAc HFS-induced synaptic plasticity in saline rats (Belujon and Grace 2008) (left) but normalizes the vSub–NAc and vSub–mPFC pathological synaptic plasticity in MAM rats (right). vSub, ventral subiculum of the hippocampus; PL, prelimbic area of the mPFC; IL, infralimbic area of the mPFC; NAc, nucleus accumbens; VTA, ventral tegmental area; DA, dopamine; TTX, tetrodotoxin; HFS, high-frequency stimulation.
LTP and long-term depression constitute changes in the strength of synaptic connections and constitute a major role in information storage within neuronal networks.
Alteration of the synaptic plasticity in the vSub–NAc–PFC network may contribute to the pathological condition that underlies some aspects of schizophrenia. Indeed, we found that, in addition to altered phasic activation, induction of synaptic plasticity is substantially altered in the MAM rat. In the present study, HFS of the fimbria in saline-treated animals induced LTP (>30 min) in the vSub–NAc pathway. This result is consistent with previous data from our laboratory (Goto and Grace 2005a, 2005b; Belujon and Grace 2008) and from others (Floresco et al. 2001). Conversely, in MAM-treated animals, the same stimulation induced the opposite effect on synaptic plasticity in the vSub–NAc pathway (LTD). We have previously shown that the hippocampus is hyperactive in MAM animals (Lodge and Grace 2007). Thus, in our MAM model, it is possible that hyperactivity of the hippocampus induces a ceiling effect that prevents proper encoding of new information. As a consequence, the pathological decrease observed in MAM animals after HFS could represent a depotentiation of an already heightened pathway. Interestingly, LFS induced changes in the vSub–NAc pathways that were comparable between MAM- and saline-treated rats, suggesting alteration of synaptic plasticity in MAM animals is limited to the HFS-induced plasticity. Thus, LFS appears to be ineffective in restoring the baseline state of the circuit in the MAM rats.
The altered synaptic plasticity observed in the MAM rat appears to be driven by altered responsiveness in the vSub–PFC circuit. Thus, in normal rats, phasic mPFC activation potentiates the vSub–NAc pathway, whereas sustained activation of the mPFC results in attenuation of this pathway (Goto and Grace 2005a, 2005b). In this study, we found that fimbria tetanization produces a sustained and potent activation of the mPFC in MAM animals, but has no such effect in saline rats. These data are consistent with previous work showing a more robust DA-facilitated tetanization-induced LTP in the vSub–mPFC pathway in the MAM rat (Goto and Grace 2006). Moreover, the present study shows that inactivating the mPFC in MAM animals produced the opposite effect of that seen in controls; that is, mPFC inactivation increased the ability of the vSub to drive spike firing in the NAc in MAM animals, whereas in normal animals, an engagement of the mPFC–NAc pathway was necessary for the activation of NAc by the vSub. Thus, removal of the PFC influence would disinhibit the vSub–NAc pathway in MAM rats.
As shown in this MAM model (Lavin et al. 2005) and in the neonatal ventral hippocampal lesion model (Tseng et al. 2007) of schizophrenia, there is a powerful alteration in the actions of the DA system in the mPFC. In control rats, VTA stimulation predominantly inhibits spiking in the mPFC. However, in MAM rats, the same type of stimulation facilitates spike activity (Goto and Grace 2006). This is consistent with our results showing that blockade of DA in the mPFC reverses the vSub–NAc HFS-induced LTD. However, blocking DA receptors did not restore LTP, as observed in saline rats and with systemic injection of sulpiride in MAM rats. Thus, the aberrant HFS-induced LTD in the vSub–NAc pathway in MAM rats is partially dependent on activation of D2 receptors in the PFC. Moreover, DA blockade is expected to attenuate the vSub–mPFC hyperactivation. Indeed, the injection of sulpiride i.v. prevents the vSub–PFC HFS-induced LTP in this study, confirming that sustained activation of the mPFC is D2 dependent. Importantly, this may be occurring via alterations in DA actions on D2 receptors in the mPFC as shown in models of schizophrenia (Lavin and Grace 1998; Goto and Grace 2005a, 2005b; Tseng et al. 2007), given evidence of decreased DA innervation (Akil et al. 1999) and release (Howes et al. 2012) in the dorsolateral PFC of schizophrenia patients.
Functional Implications
In control animals, synaptic plasticity observed in the vSub–NAc pathways is thought to be involved in goal-directed behavior, with a balance between limbic and cortical inputs to the NAc-modulating behavioral choice. Our hypothesis is that a disruption of this balance may contribute to the cognitive dysfunction seen in schizophrenia. Indeed, in MAM-treated animals, the vSub–NAc pathway is depressed after tetanizing the fimbria, which may affect vSub–NAc information processing and thus have an impact on cognition and behavior. Given that LTP in the vSub–NAc pathway plays a significant role in performance of goal-directed behavior (Kelley 2004; Goto and Grace 2005a, 2005b), a condition in which repeated activation would lead to LTD instead of LTP would be expected to lead to significant disruption in behavioral facilitation of a learned task. When coupled with a disruption of PFC-mediated task flexibility, such a condition would significantly interfere with adaptive behavioral processes.
There is evidence that schizophrenia patients show alteration in the function and structure of limbic and cortical regions. Postmortem studies show a reduced hippocampal volume (Nelson et al. 1998), abnormality of the hippocampal architecture (Heckers and Konradi 2002), and loss of parvalbumin GABA interneuron staining (Benes and Berretta 2001; Lewis et al. 2005). Moreover, functional imaging studies have demonstrated increased hippocampal activity during resting state and abnormal information processing during memory retrieval tasks (Medoff et al. 2001; Lahti et al. 2006; Weiss et al. 2006). In addition, our studies show that the hippocampus can potentiate phasic dopamine neuron responsivity (Lodge and Grace 2006). Together, these data suggest that hippocampal hyperactivity in schizophrenia can lead to overdrive of the NAc and might underlie the positive symptoms of the disease. Furthermore, we show that hippocampal–prefrontal relationships are also potently altered in the MAM model, which could be related to the negative symptoms and cognitive dysfunction observed in schizophrenia (O'Donnell and Grace 1998). In the MAM model, we found a D2-dependent alteration in the balance between mPFC and vSub inputs that may disrupt the interaction between goal- and context-dependent regulations of information flow within the striatum. Indeed, antipsychotic drugs may have multiple clinically significant actions in the schizophrenia patient, that is, attenuating an increased VTA–NAc DA drive leading to aberrant salience (Kapur 2003; Grace 2012) as well as aiding in restoring the balance of the cortico-limbic circuit by normalizing the PFC-mediated modulation of synaptic plasticity within the hippocampal–striatal pathway.
Supplementary Material
Supplementary material can be found at: http://www.cercor.oxfordjournals.org/.
Funding
This work was supported by United States Public Health Service Grant MH57440 (A.A.G.).
Supplementary Material
Notes
We thank Niki MacMurdo and Brandon Bizup for technical assistance and Katy Gill and Witek Lipski for critical reading and helpful discussions. Conflict of Interest: Johnson & Johnson, Lund-beck, Pfizer, GlaxoSmithKline, Puretech Ventures, Merck, Takeda, Dainippon Sumitomo, Otsuka, Lilly, Roche (A.A.G.).
References
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am J Psychiatry. 1999;156:1580–1589. doi: 10.1176/ajp.156.10.1580. [DOI] [PubMed] [Google Scholar]
- Belujon P, Grace AA. Critical role of the prefrontal cortex in the regulation of hippocampus-accumbens information flow. J Neurosci. 2008;28:9797–9805. doi: 10.1523/JNEUROSCI.2200-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Berretta S. GABAergic interneurons: implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology. 2001;25:1–27. doi: 10.1016/S0893-133X(01)00225-1. [DOI] [PubMed] [Google Scholar]
- Flagstad P, Mork A, Glenthoj BY, van Beek J, Michael-Titus AT, Didriksen M. Disruption of neurogenesis on gestational day 17 in the rat causes behavioral changes relevant to positive and negative schizophrenia symptoms and alters amphetamine-induced dopamine release in nucleus accumbens. Neuropsychopharmacology. 2004;29:2052–2064. doi: 10.1038/sj.npp.1300516. [DOI] [PubMed] [Google Scholar]
- Floresco SB, Blaha CD, Yang CR, Phillips AG. Modulation of hippocampal and amygdalar-evoked activity of nucleus accumbens neurons by dopamine: cellular mechanisms of input selection. J Neurosci. 2001;21:2851–2860. doi: 10.1523/JNEUROSCI.21-08-02851.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Dopamine-dependent interactions between limbic and prefrontal cortical plasticity in the nucleus accumbens: disruption by cocaine sensitization. Neuron. 2005a;47:255–266. doi: 10.1016/j.neuron.2005.06.017. [DOI] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Dopaminergic modulation of limbic and cortical drive of nucleus accumbens in goal-directed behavior. Nat Neurosci. 2005b;8:805–812. doi: 10.1038/nn1471. [DOI] [PubMed] [Google Scholar]
- Goto Y, Grace AA. Alterations in medial prefrontal cortical activity and plasticity in rats with disruption of cortical development. Biol Psychiatry. 2006;60:1259–1267. doi: 10.1016/j.biopsych.2006.05.046. [DOI] [PubMed] [Google Scholar]
- Grace AA. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 2000;31:330–341. doi: 10.1016/s0165-0173(99)00049-1. [DOI] [PubMed] [Google Scholar]
- Grace AA. Dopamine system dysregulation by the hippocampus: implications for the pathophysiology and treatment of schizophrenia. Neuropharmacology. 2012;62:1342–1348. doi: 10.1016/j.neuropharm.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA, Floresco SB, Goto Y, Lodge DJ. Regulation of firing of dopaminergic neurons and control of goal-directed behaviors. Trends Neurosci. 2007;30:220–227. doi: 10.1016/j.tins.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Gurden H, Takita M, Jay TM. Essential role of D1 but not D2 receptors in the NMDA receptor-dependent long-term potentiation at hippocampal-prefrontal cortex synapses in vivo. J Neurosci. 2000;20:RC106. doi: 10.1523/JNEUROSCI.20-22-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Konradi C. Hippocampal neurons in schizophrenia. J Neural Transm. 2002;109:891–905. doi: 10.1007/s007020200073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckers S, Rauch SL, Goff D, Savage CR, Schacter DL, Fischman AJ, Alpert NM. Impaired recruitment of the hippocampus during conscious recollection in schizophrenia. Nat Neurosci. 1998;1:318–323. doi: 10.1038/1137. [DOI] [PubMed] [Google Scholar]
- Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, Kapur S. The nature of dopamine dysfunction in schizophrenia and what this means for treatment: meta-analysis of imaging studies. Arch Gen Psychiatry. 2012;69:776–786. doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YY, Simpson E, Kellendonk C, Kandel ER. Genetic evidence for the bidirectional modulation of synaptic plasticity in the prefrontal cortex by D1 receptors. Proc Natl Acad Sci USA. 2004;101:3236–3241. doi: 10.1073/pnas.0308280101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160:13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Ventral striatal control of appetitive motivation: role in ingestive behavior and reward-related learning. Neurosci Biobehav Rev. 2004;27:765–776. doi: 10.1016/j.neubiorev.2003.11.015. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Holcomb HH, Tamminga CA, Carpenter WT, McMahon R. Correlations between rCBF and symptoms in two independent cohorts of drug-free patients with schizophrenia. Neuropsychopharmacology. 2006;31:221–230. doi: 10.1038/sj.npp.1300837. [DOI] [PubMed] [Google Scholar]
- Lavin A, Grace AA. Response of the ventral pallidal/mediodorsal thalamic system to antipsychotic drug administration: involvement of the prefrontal cortex. Neuropsychopharmacology. 1998;18:352–363. doi: 10.1016/S0893-133X(97)00165-6. [DOI] [PubMed] [Google Scholar]
- Lavin A, Moore HM, Grace AA. Prenatal disruption of neocortical development alters prefrontal cortical neuron responses to dopamine in adult rats. Neuropsychopharmacology. 2005;30:1426–1435. doi: 10.1038/sj.npp.1300696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol. 2006;63:1372–1376. doi: 10.1001/archneur.63.10.1372. [DOI] [PubMed] [Google Scholar]
- Liddle PF, Friston KJ, Frith CD, Hirsch SR, Jones T, Frackowiak RS. Patterns of cerebral blood flow in schizophrenia. Br J Psychiatry. 1992;160:179–186. doi: 10.1192/bjp.160.2.179. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Jaskiw GE, Weinberger DR. Postpubertal emergence of hyperresponsiveness to stress and to amphetamine after neonatal excitotoxic hippocampal damage: a potential animal model of schizophrenia. Neuropsychopharmacology. 1993;9:67–75. doi: 10.1038/npp.1993.44. [DOI] [PubMed] [Google Scholar]
- Lodge DJ, Behrens MM, Grace AA. A loss of parvalbumin-containing interneurons is associated with diminished oscillatory activity in an animal model of schizophrenia. J Neurosci. 2009;29:2344–2354. doi: 10.1523/JNEUROSCI.5419-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci. 2007;27:11424–11430. doi: 10.1523/JNEUROSCI.2847-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. The hippocampus modulates dopamine neuron responsivity by regulating the intensity of phasic neuron activation. Neuropsychopharmacology. 2006;31:1356–1361. doi: 10.1038/sj.npp.1300963. [DOI] [PubMed] [Google Scholar]
- Malaspina D, Storer S, Furman V, Esser P, Printz D, Berman A, Lignelli A, Gorman J, Van Heertum R. SPECT study of visual fixation in schizophrenia and comparison subjects. Biol Psychiatry. 1999;46:89–93. doi: 10.1016/s0006-3223(98)00306-0. [DOI] [PubMed] [Google Scholar]
- Medoff DR, Holcomb HH, Lahti AC, Tamminga CA. Probing the human hippocampus using rCBF: contrasts in schizophrenia. Hippocampus. 2001;11:543–550. doi: 10.1002/hipo.1070. [DOI] [PubMed] [Google Scholar]
- Mogenson GJ, Yang CR, Yim CY. Influence of dopamine on limbic inputs to the nucleus accumbens. Ann N Y Acad Sci. 1988;537:86–100. doi: 10.1111/j.1749-6632.1988.tb42098.x. [DOI] [PubMed] [Google Scholar]
- Molina V, Reig S, Pascau J, Sanz J, Sarramea F, Gispert JD, Luque R, Benito C, Palomo T, Desco M. Anatomical and functional cerebral variables associated with basal symptoms but not risperidone response in minimally treated schizophrenia. Psychiatry Res. 2003;124:163–175. doi: 10.1016/s0925-4927(03)00107-0. [DOI] [PubMed] [Google Scholar]
- Molina V, Sanz J, Sarramea F, Benito C, Palomo T. Prefrontal atrophy in first episodes of schizophrenia associated with limbic metabolic hyperactivity. J Psychiatr Res. 2005;39:117–127. doi: 10.1016/j.jpsychires.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry. 2006;60:253–264. doi: 10.1016/j.biopsych.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore H, West AR, Grace AA. The regulation of forebrain dopamine transmission: relevance to the pathophysiology and psychopathology of schizophrenia. Biol Psychiatry. 1999;46:40–55. doi: 10.1016/s0006-3223(99)00078-5. [DOI] [PubMed] [Google Scholar]
- Nelson MD, Saykin AJ, Flashman LA, Riordan HJ. Hippocampal volume reduction in schizophrenia as assessed by magnetic resonance imaging: a meta-analytic study. Arch Gen Psychiatry. 1998;55:433–440. doi: 10.1001/archpsyc.55.5.433. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Dysfunctions in multiple interrelated systems as the neurobiological bases of schizophrenic symptom clusters. Schizophr Bull. 1998;24:267–283. doi: 10.1093/oxfordjournals.schbul.a033325. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Greene J, Pabello N, Lewis BL, Grace AA. Modulation of cell firing in the nucleus accumbens. Ann N Y Acad Sci. 1999;877:157–175. doi: 10.1111/j.1749-6632.1999.tb09267.x. [DOI] [PubMed] [Google Scholar]
- O'Donnell P, Lewis BL, Weinberger DR, Lipska BK. Neonatal hippocampal damage alters electrophysiological properties of prefrontal cortical neurons in adult rats. Cereb Cortex. 2002;12:975–982. doi: 10.1093/cercor/12.9.975. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic Press; 1996. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 2010;35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbersweig DA, Stern E, Frith C, Cahill C, Holmes A, Grootoonk S, Seaward J, McKenna P, Chua SE, Schnorr L, et al. A functional neuroanatomy of hallucinations in schizophrenia. Nature. 1995;378:176–179. doi: 10.1038/378176a0. [DOI] [PubMed] [Google Scholar]
- Smith SE, Li J, Garbett K, Mirnics K, Patterson PH. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Amin F, Lewis BL, O'Donnell P. Altered prefrontal cortical metabolic response to mesocortical activation in adult animals with a neonatal ventral hippocampal lesion. Biol Psychiatry. 2006;60:585–590. doi: 10.1016/j.biopsych.2006.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Lewis BL, Lipska BK, O'Donnell P. Post-pubertal disruption of medial prefrontal cortical dopamine-glutamate interactions in a developmental animal model of schizophrenia. Biol Psychiatry. 2007;62:730–738. doi: 10.1016/j.biopsych.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss AP, Goff D, Schacter DL, Ditman T, Freudenreich O, Henderson D, Heckers S. Fronto-hippocampal function during temporal context monitoring in schizophrenia. Biol Psychiatry. 2006;60:1268–1277. doi: 10.1016/j.biopsych.2006.06.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.