

Abstract
Levomilnacipran (1S, 2R-milnacipran) is a potent and selective serotonin and norepinephrine reuptake inhibitor; an extended-release (ER) formulation allows for once-daily dosing. This phase III study (NCT01034462) evaluated the efficacy, the safety, and the tolerability of 40 to 120 mg/d of levomilnacipran ER versus placebo in the treatment of patients (18-80 y) with major depressive disorder. This multicenter, randomized, double-blind, placebo-controlled, parallel-group, flexible-dose study comprised a 1-week single-blind, placebo run-in period; an 8-week double-blind treatment; and a 2-week double-blind down-taper period. The primary efficacy parameter was total score change from baseline to week 8 on the Montgomery-Åsberg Depression Rating Scale (MADRS); the secondary efficacy was the Sheehan Disability Scale. Analysis was performed using the mixed-effects model for repeated measures on a modified intent-to-treat population. A total of 434 patients received at least 1 dose of double-blind treatment (safety population); 429 patients also had 1 or more postbaseline MADRS assessments (modified intent-to-treat population). The least squares mean differences and 95% confidence interval were statistically significant in favor of levomilnacipran ER versus placebo for the MADRS total score (−3.095 [−5.256, −0.935]; P = 0.0051) and the SDS total score (−2.632 [−4.193, −1.070]; P = 0.0010) change from baseline to week 8. Adverse events were reported in 61.8% of the placebo patients and in 81.6% of the levomilnacipran ER patients. Frequently reported adverse events (≥5% in levomilnacipran ER and twice the rate of placebo) were nausea, dizziness, constipation, tachycardia, urinary hesitation, hyperhidrosis, insomnia, vomiting, hypertension, and ejaculation disorder. In conclusion, there was a statistically significant difference in the score change from baseline to week 8 between levomilnacipran ER and placebo on several depression rating scales, reflecting symptomatic and functional improvement; treatment was generally well tolerated.
Key Words: major depressive disorder, SNRI, depression, selective serotonin and norepinephrine reuptake inhibitor, antidepressants
Major depressive disorder (MDD) is a leading cause of global disease burden; by 2030, it is projected that it will be only second to human immunodeficiency virus/acquired immunodeficiency syndrome in worldwide burden of disease.1 Some defining symptoms of MDD are thought to be related to reduced noradrenergic activity exacerbating the depressed mood state.2 These symptoms, which include fatigue, lack of motivation, decreased concentration, and lassitude, complicate the depressive state by impairing social and occupational functioning.
An inability to correlate changes in the biological indices that accompany improvement in MDD has restricted the definition of treatment response to a reduction in the number and the severity of symptoms. However, the complex nature of depression is better suited to a multifaceted concept of recovery and wellness that includes broader outcome criteria and improvement in key domains, including both symptomatic and functional improvement.3
Research suggests that changes in symptom severity and improvement in functional impairment may occur asynchronously, with functional improvement often lagging behind changes in symptoms.4,5 As such, symptom improvement may provide an early sign of treatment response, whereas functional improvement may be a better indicator of meaningful change.5 Independent assessments of symptoms and functioning in MDD may improve clinical research by providing greater insight into the relationship between symptoms and functioning.
Levomilnacipran (1S, 2R-milnacipran) is a potent and selective serotonin and norepinephrine reuptake inhibitor (SNRI); an extended-release (ER) formulation was developed to allow for once-daily dosing. Levomilnacipran has approximately 2-fold greater potency for norepinephrine than serotonin reuptake inhibition. Compared with duloxetine or venlafaxine, levomilnacipran has more than 10-fold higher selectivity for norepinephrine relative to serotonin reuptake inhibition.6 Studies suggest that antidepressants with a prominent noradrenergic component may be especially effective in improving symptoms related to functioning2,7,8; as such, levomilnacipran ER may be an antidepressant that can provide both symptomatic and functional efficacy.
Levomilnacipran is the more active enantiomer of milnacipran, an SNRI that is approved for the treatment of fibromyalgia in the United States (prescribing information: Savella [milnacipran hydrochloride], 2011; Forest Laboratories, Inc, St Louis, MO). Milnacipran is not approved for the treatment of MDD in the United States; it is approved for that use in many countries outside the United States. Milnacipran studies in MDD were conducted more than a decade ago, and no head-to-head trials have been performed; as such, no valid comparison between levomilnacipran ER and milnacipran clinical data can be made.
Milnacipran is a racemic mixture of levomilnacipran (1S, 2R-milnacipran) and F2696 (1R, 2S-milnacipran); the 2 enantiomers are not interconvertible.9 Regulatory guidelines in the United States and Europe recommend development of enantiomers over racemic drugs where appropriate,10 for a variety of reasons.11 Levomilnacipran exhibited high affinities for human recombinant transporter proteins for norepinephrine and serotonin (Ki = 92.2 and 11.2 nM, respectively) that were at least 10 times higher than the other enantiomer, F2696.6 Moreover, levomilnacipran was substantially more potent than was F2696 in an animal model of antidepressant activity and in vitro functional assays.6 Levomilnacipran also has pharmacokinetic advantages relative to F2696. When the milnacipran racemate was administered to human subjects, levomilnacipran was eliminated more slowly than was F2696, with higher maximum concentration (Cmax) and area under the curve (AUC) values. The elimination half-life was ∼9 hours for levomilnacipran and 6 hours for F2696. The AUC0-∞ of levomilnacipran was ∼85% greater than that of F2696. Plasma clearance was ∼2 times greater for F2696 than for levomilnacipran. The pharmacokinetic results are consistent with those of a milnacipran human mass balance study, in which the elimination rate for F2696 was higher than for levomilnacipran.9 Given the favorable characteristics of this enantiomer, levomilnacipran was selected for development as an antidepressant.
Levomilnacipran ER is approved for the treatment of MDD in adults. Efficacy and safety have been evaluated in 4 additional randomized, double-blind, placebo-controlled studies. In 1 flexible-dose (75-100 mg/d of levomilnacipran ER) study12 and 2 fixed-dose (40, 80, and 120 mg/d of levomilnacipran ER13 and 40 and 80 mg/d of levomilnacipran ER14) studies, differences in scores on the primary efficacy parameter were statistically significant in favor of levomilnacipran ER over placebo. In a second flexible-dose study, 40 to 120 mg/d of levomilnacipran improved depressive symptoms but did not achieve statistically significant separation from placebo.15 Levomilnacipran ER was generally well tolerated in all 4 studies. The objective of this study (NCT01034462) was to evaluate the efficacy, the safety, and the tolerability of 40 to 120 mg/d of flexibly dosed levomilnacipran ER versus placebo in the treatment of patients with MDD.
MATERIALS AND METHODS
This study was conducted at 23 US study centers between December 2009 and December 2011 in full compliance with the Food and Drug Administration guidelines for Good Clinical Practice and the ethical principles of the Declaration of Helsinki. All patients provided written informed consent before the initiation of any study procedures.
Study Design
This phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group, flexible-dose study compared 40 to 120 mg/d of levomilnacipran ER with placebo in adult outpatients with MDD. The patients were randomized by a computer-generated list of numbers and assigned to identically appearing treatment that corresponded to the sequence of randomization numbers; investigators and patients were blinded to treatment assignment.
This study consisted of a 1-week single-blind, placebo run-in period followed by an 8-week double-blind treatment period and a 2-week double-blind down-taper period. At the end of screening, patients meeting entry criteria were randomized on a 1:1 basis to placebo or levomilnacipran ER. The patients assigned to receive levomilnacipran ER were initiated at 20 mg/d for days 1 and 2 and to 40 mg/d beginning on day 3. Dosage increase from 40 to 80 mg/d was allowed at the end of week 1 or 2, and an increase from 40 to 80 mg/d or 80 to 120 mg/d was allowed at the end of week 4 on the basis of patient response (<50% improvement in the Montgomery-Åsberg Depression Rating Scale [MADRS] total score from baseline) and tolerability. No dosage increase was allowed after week 4; dosage decrease to the previous level was possible if significant tolerability issues developed. The patients who completed the 8-week, double-blind treatment period or who prematurely discontinued from the study were eligible to enter the 2-week double-blind down-taper period.
Inclusion Criteria
Male or female outpatients, 18 to 80 years of age, inclusive, who met the criteria for MDD according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision, with an ongoing major depressive episode of at least 4 weeks’ duration were eligible to participate in this study. Major depressive disorder diagnosis was confirmed by the Mini International Neuropsychiatric Interview16 and a computerized depression diagnostic interview. Additional criteria included a score of 30 or higher on the clinician-rated MADRS17 and a score 26 or higher on the self-rated MADRS. Additional inclusion criteria included normal or abnormal results that were judged as not clinically significant by the investigator on physical examinations, clinical laboratory test results, and electrocardiogram (ECG); serum pregnancy test negative for β-human chorionic gonadotropin for the female patients and urine screening negative for alcohol and prohibited substances; and body mass index (BMI) between 18 and 40, inclusive.
Key Exclusion Criteria
Patients who met the criteria for an axis I disorder other than MDD within 6 months of screening or who had a lifetime history of other major psychiatric diagnoses (eg, manic/hypomanic episode, depressive episode with psychotic features, substance abuse/dependence within the previous 6 months, borderline or antisocial personality disorders, or cognitive disorders) were excluded; comorbid generalized anxiety disorder, social anxiety disorder, and/or specific phobias were allowed. Additional exclusion criteria included a history of nonresponse to adequate treatment with at least 2 antidepressants or intolerance or hypersensitivity to milnacipran or other SNRIs, selective serotonin reuptake inhibitors, or selective noradrenergic reuptake inhibitors. Suicide risk, defined as suicide attempt within the past year, a score of 5 or higher on MADRS item 10 (suicidal thoughts), or significant risk as judged by the investigator or determined by information obtained from the Columbia–Suicide Severity Rating Scale (C-SSRS),18 was an additional reason for exclusion. Patients with medical conditions that might interfere with the conduct of the study, confound the interpretation of study results, or endanger well-being (eg, history or evidence of malignancy or any significant hematologic, endocrine, cardiovascular, respiratory, renal, hepatic, gastrointestinal, or neurologic disease) were excluded. The use of concomitant psychotropic medications (except eszopiclone zaleplon, zolpidem for insomnia) was prohibited, as was treatment with any other investigational product during the study or within 3 months of screening.
Efficacy Assessments
The MADRS was assessed at all study visits (screening [week −1]; baseline [week 0]; and weeks 1, 2, 4, 6, and 8), and the Sheehan Disability Scale (SDS)19 was assessed at weeks 0, 4, 6, and 8.
In addition, the 17-item Hamilton Rating Scale for Depression (HAMD17)20 (all study visits), the Clinical Global Impressions-Severity (weeks 0, 1, 2, 4, 6, and 8) and the Clinical Global Impressions-Improvement (CGI-I) (weeks 1, 2, 4, 6, and 8),21 and the Motivation and Energy Inventory–Short Form (MEI-SF) (weeks 0, 4, 6, and 8)22 were also evaluated. A battery of cognitive tests, including the Cognitive Drug Research System for Attention23 and the Bond-Lader Visual Analogue Scales of Mood and Alertness,24 was completed by the patient using a dedicated computer and response box (weeks −1, 0, and 8).
Safety Assessments
Adverse events (AEs), judged by intensity (mild, moderate, and severe) and relationship to study drug, and serious AEs (SAEs) were recorded at baseline and all subsequent study visits. Clinical laboratory tests, including hematology, chemistry, and urine drug screen (weeks −1, 4, and 8); vital signs (all study visits); and ECG (weeks −1, 1, 4, and 8) were performed. The C-SSRS was administered at all study visits to assess suicidal ideation and behavior.
Statistical Analyses
The safety population consisted of all patients who were randomized and received at least 1 dose of the double-blind study drug; the modified intent-to-treat (ITT) population consisted of all patients in the safety population who also had at least 1 postbaseline MADRS total score assessment. The percentage of premature discontinuations was compared between the treatment groups using the Fisher exact test.
The primary efficacy parameter was change from baseline to week 8 in MADRS total score; the primary analysis was performed using the mixed-effects model for repeated measures (MMRM) approach, with treatment group, pooled study center, visit, and treatment group-by-visit interaction as factors and baseline value and baseline-by-visit interaction as covariates. An unstructured covariance matrix was used to model the covariance of within-patient scores; the Kenward-Roger approximation,25 based on all postbaseline scores using only observed cases without imputation of missing values, was used to estimate denominator degrees of freedom.
Sensitivity analyses were performed on the primary parameter using the last observation carried forward (LOCF) approach to impute missing postbaseline values and the pattern mixture model (PMM). In the LOCF analysis, the treatment groups were compared using an analysis of covariance model, with the treatment group and pooled study center as factors and the baseline MADRS total score as the covariate. The PMM approach, based on non–future-dependent missing value restrictions,26 was performed to assess the robustness of the primary MMRM results to the possible violation of the missing-at-random missingness assumption.
The secondary efficacy parameter was change from baseline to week 8 on SDS total score; the SDS total score was calculated only for patients with valid scores on all 3 SDS subscale domains (work, family life, and social life). Subscale scores were calculated on the basis of nonmissing observations for each domain. The method used for SDS analysis was similar to that for the primary outcome analysis; LOCF sensitivity analysis was also performed.
For most additional efficacy parameters, change from baseline to week 8 was analyzed using MMRM and LOCF approaches, similar to those used for the primary efficacy parameter. Composite scores on cognitive tests (power of attention, continuity of attention, cognitive reaction time, reaction time variability, alertness, calmness, and contentment) were analyzed using the LOCF approach only.
The MADRS response (≥50% total score reduction from baseline) and remission (total score ≤10) rates at week 8 were analyzed using logistic regression, with treatment group and the MADRS baseline score as explanatory variables. The SDS response (total score ≤12 and score ≤4 on each item) and remission (total score ≤6 and score ≤2 on each item)27 rates were also evaluated and analyzed using logistic regression. Additional analyses included the number needed to treat (NNT) for the MADRS and the SDS response and remission.
All statistical tests were 2-sided hypothesis tests performed at the 5% level of significance; confidence intervals (CIs) were 2-sided 95% CIs.
Safety parameters were summarized for the safety population using descriptive statistics; no inferential statistics comparing the treatment groups were performed. Clinical laboratory values, vital signs, and ECG values were considered potentially clinically significant (PCS) if these met either low or high PCS criteria. The QT interval was corrected for heart rate using the Bazett formula [QTcB = QT/(RR)½] and the Fridericia formula [QTcF = QT/(RR)⅓].
RESULTS
Patient Disposition and Demographic Characteristics
A summary of the patient disposition and reasons for premature discontinuation is presented in Table 1. The incidence of premature discontinuation due to AEs was higher for levomilnacipran (7.8%) than placebo (3.2%), but the difference was not statistically significant (P = 0.0567); the incidence of discontinuations for all other reasons was similar in both groups.
TABLE 1.
Patient Disposition and Discontinuation
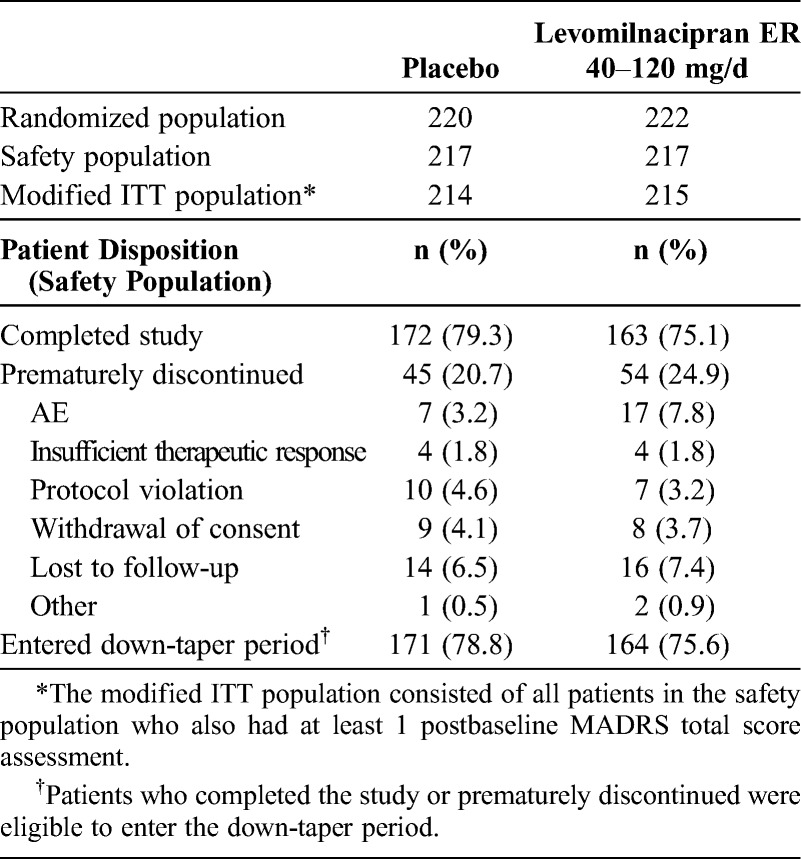
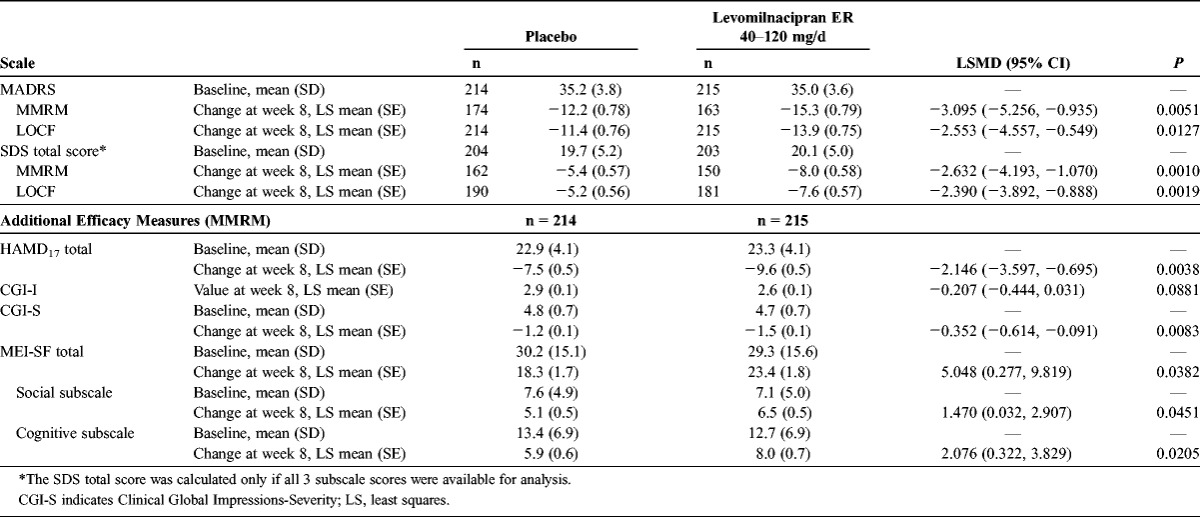
The placebo and levomilnacipran ER groups were similar with regard to baseline demographic characteristics and MDD characteristics; in each treatment group, the mean age was 45 years, and approximately 65% of the patients were women. The baseline scores for all efficacy measures were similar between the groups (Table 2). The mean MADRS baseline score was 35 in both groups, a score that is higher than the usual cutoff threshold for severe depression,28 indicating that the patients had at least moderate to severe depression.
TABLE 2.
Baseline Efficacy Values and Change From Baseline at Week 8 (Modified ITT Population)
Exposure: Duration and Dosing
The median duration of treatment was 56 days for both treatment groups. For the patients in the levomilnacipran ER group, the overall mean daily dose was approximately 73 mg; 21.2% of the patients received 40 mg/d, 34.1% received 80 mg/d, and 44.2% received 120 mg/d as the final daily dose. No dose adjustments were allowed after week 4.
Analysis of Efficacy
Primary Efficacy Parameter
A statistically significant difference in MADRS total score change from baseline to week 8 was observed in favor of levomilnacipran ER over placebo (MMRM); the difference remained statistically significant when MADRS change from baseline was analyzed using the LOCF approach (Table 2). The PMM analyses tested the missing-at-random assumption of the imputation method and supported the robustness of the primary results for levomilnacipran ER. In a by-week analysis of MADRS total score change, statistical separation from placebo occurred at week 4 for levomilnacipran ER (P = 0.0486) and persisted through week 8 (Fig. 1).
FIGURE 1.
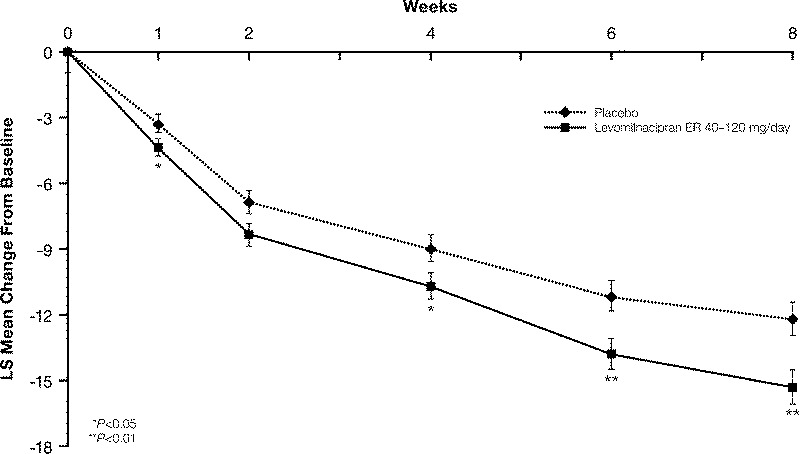
Montgomery-Åsberg Depression Rating Scale total score change from baseline (least squares [LS] mean [SE]) to week 8 (Modified ITT population, MMRM).
Secondary Efficacy Parameter
A statistically significant difference in SDS total score (MMRM) for levomilnacipran ER over placebo was observed at week 8 (Table 2); the scores at week 8 were also significantly different in favor of levomilnacipran ER on the SDS work, family life, and social life subscales (Fig. 2). The statistically significant difference from placebo in SDS total score was observed for levomilnacipran ER starting from the first SDS assessment (week 4).
FIGURE 2.
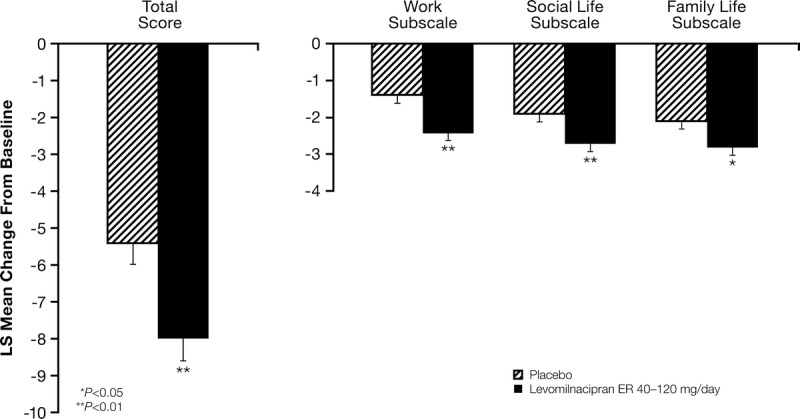
Sheehan Disability Scale change from baseline (least squares [LS] mean [SE]) to week 8 (Modified ITT population, MMRM).
Additional Efficacy Parameters
At week 8, the least squares mean difference (LSMD) was statistically significant in favor of levomilnacipran ER versus placebo on many additional efficacy measures (Table 2). The difference in mean change in MEI-SF total score was statistically significant for levomilnacipran ER versus placebo, indicating greater motivation and energy for the levomilnacipran ER patients compared with the placebo patients. Significantly greater differences in scores on the MEI-SF social and cognitive subscales were also seen for levomilnacipran ER versus placebo (Table 2). A statistically significant difference between the treatment groups was not observed in CGI-I score at week 8.
On the battery of attention-related cognitive tests, the difference in change from baseline to week 8 was statistically significant in favor of levomilnacipran ER versus placebo on the continuity of attention composite score (LSMD [95% CI], 1.817 [0.599, 3.036]; P = 0.0036); differences for levomilnacipran ER versus placebo trended toward statistical significance on the power of attention composite score (P = 0.0666) and reaction time variability composite score (P = 0.0600) but not on the cognitive reaction time composite score (P = 0.2958). Differences in the composite scores for alertness of attention, calmness, and contentment did not reach statistical significance for levomilnacipran ER versus placebo on any factors (data not shown).
Significantly more levomilnacipran ER patients (41.9%) than placebo patients (29.4%) met the MADRS response criteria (≥50% decrease from baseline; P = 0.0083); the proportion of levomilnacipran ER (17.2%) and placebo (18.2%) patients who achieved remission (MADRS total score ≤10) was similar between the groups (P = 0.7255). A significantly greater proportion of levomilnacipran ER patients (44.2%) compared with placebo patients (28.9%) achieved response on the SDS (total score ≤12 and score ≤4 on each item; P = 0.0009); the proportion of patients who achieved SDS remission (total score ≤6 and score ≤2 on each item) was not significantly different for the levomilnacipran ER versus placebo patients (21.0% vs 16.3%; P = 0.1859). The NNT for 1 additional levomilnacipran ER patient to achieve response was 8 on the MADRS and 7 on the SDS.
Safety and Tolerability
Adverse Events
During the double-blind treatment, the incidence of treatment-emergent AEs (TEAEs) was higher for the levomilnacipran ER patients than the placebo patients; the incidence of commonly reported TEAEs (≥5% and twice the rate of placebo) is shown in Table 3. Nausea, the most frequently-occurring TEAE in the levomilnacipran ER group was reported in 21.7% of the levomilnacipran ER patients compared with 3.7% of the placebo patients. The incidence of newly emergent AEs in the double-blind down-taper period was similar (approximately 10%) in both treatment groups.
TABLE 3.
Double-Blind Treatment: Common (≥5% and Twice the Rate of Placebo) AEs and Suicidality Based on the C-SSRS (Safety Population)
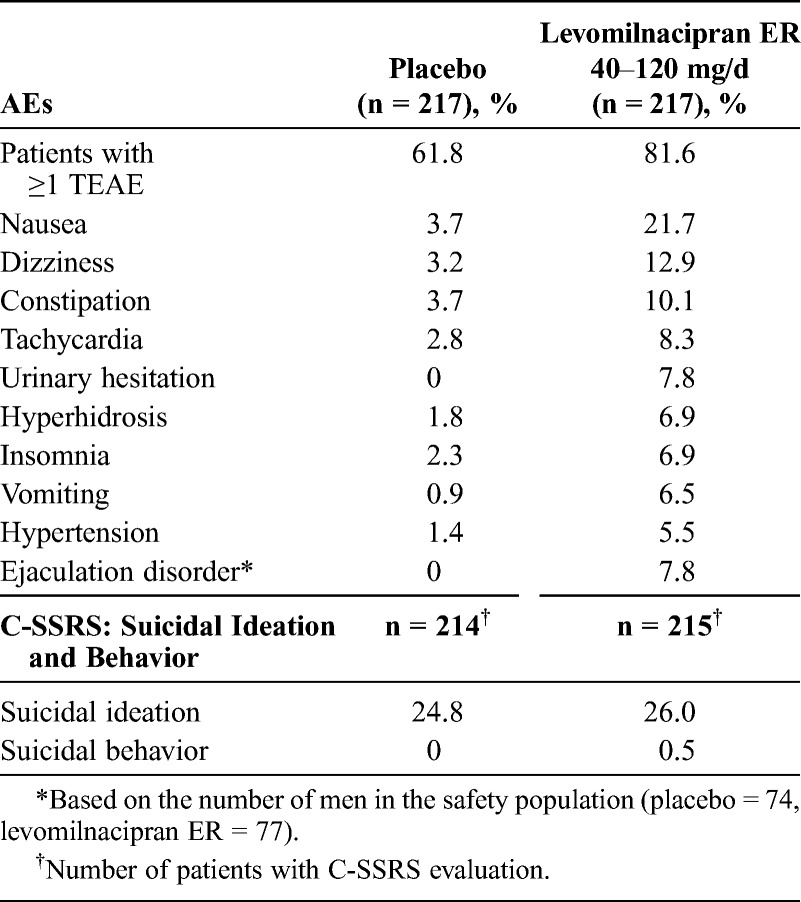
Serious AEs were reported by 8 patients during the study: 1 patient after randomization but before the start of treatment, 5 patients during the double-blind treatment, and 2 patients during the down-taper period. During the double-blind treatment, SAEs occurred in 3 placebo patients (noncardiac chest pain in 1 patient, head injury/traumatic liver injury/road traffic accident in 1 patient, and asthma/chronic obstructive pulmonary disease in 1 patient) and 2 levomilnacipran ER patients (intentional overdose/suicide attempt in 1 patient and road traffic accident/back pain/scratches in 1 patient). The SAE of intentional overdose/suicide attempt in the levomilnacipran ER patient occurred on day 9 of treatment and led to discontinuation from the study; it was considered by the investigator to be severe and unrelated to the study drug. The SAEs in 2 patients during the down-taper period were hypertension/noncardiac chest pain and suicidal ideation; both were in the levomilnacipran ER group. Adverse events that led to the discontinuation of 2 or more patients in the levomilnacipran ER group were tachycardia, nausea, insomnia, agitation, constipation, and hypertension.
Suicidal ideation and behavior based on the C-SSRS assessment during the double-blind treatment are presented in Table 3. Similar numbers of levomilnacipran ER and placebo patients had suicidal ideation during the double-blind treatment; most of the ideation was in the least severe category (“wish to be dead” and not associated with a plan and/or intent). One levomilnacipran ER patient had suicidal behavior during the double-blind treatment; this incident was also reported as an SAE of intentional overdose/suicide attempt. During the down-taper period, more patients in the placebo group (13.0%) than in the levomilnacipran ER group (9.8%) had C-SSRS–determined suicidal ideation; 1 incident of suicidal ideation in the levomilnacipran ER group was also reported as an SAE. No patients reported suicidal behavior during the down-taper period.
Laboratory Values
Mean changes for chemistry and hematology parameters were minor, and no treatment-related trends were apparent. The number of patients with PCS postbaseline laboratory values was low and generally similar between the treatment groups.
Vital Signs, Physical Findings, and ECG
The mean changes from baseline at the end of the double-blind treatment period for vital signs and physical findings are presented in Table 4. Levomilnacipran ER was associated with a mean increase of 8.5 beats per minute (bpm) in pulse rate and mean increases of 3.5 mm Hg and 3.4 mm Hg in systolic and diastolic blood pressure, respectively. The overall incidence of orthostatic hypotension was slightly higher in the levomilnacipran ER group compared with the placebo group. Most cases of orthostatic hypotension were transient, asymptomatic episodes that resolved with continued treatment and were not associated with other clinical symptoms; no patients discontinued from the study because of orthostatic hypotension. The incidence of PCS changes in vital signs during the double-blind treatment period was low in both treatment groups (Table 4).
TABLE 4.
Baseline to Endpoint and PCS Vital Sign Changes (Safety Population)
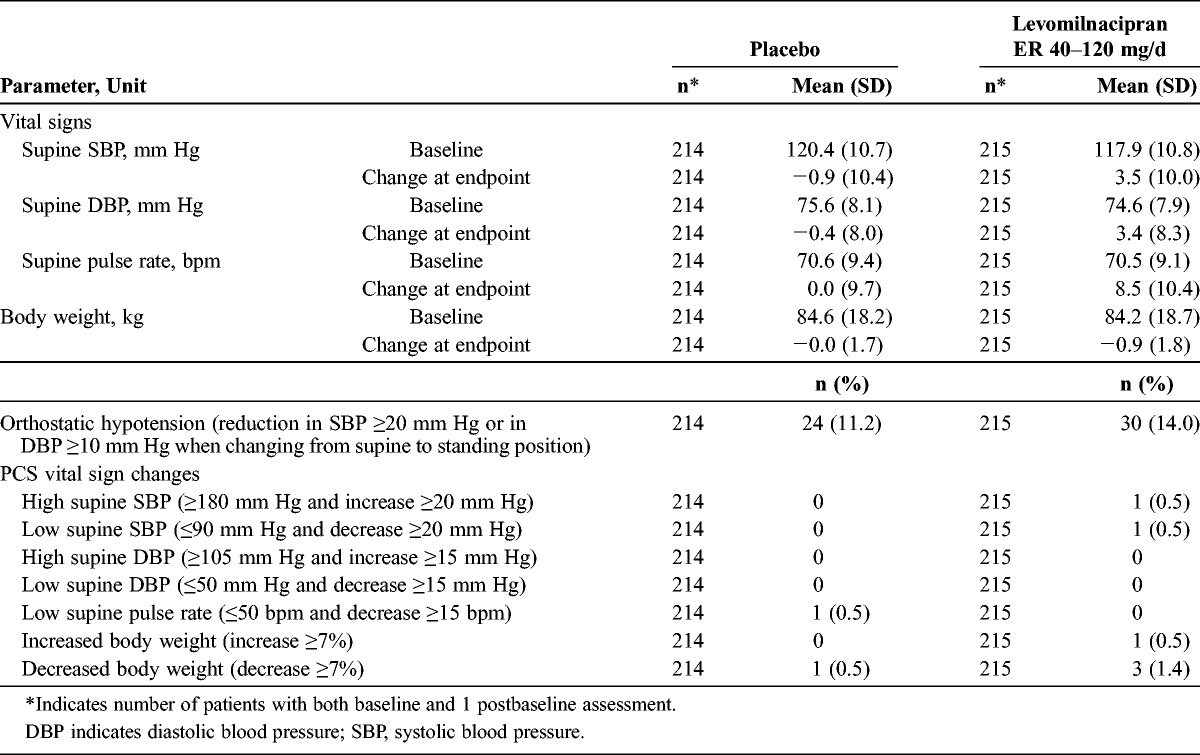
At the end of the double-blind treatment, a greater increase in ECG ventricular heart rate (13.8 bpm) was seen in the levomilnacipran ER treatment group compared with the placebo group (2.2 bpm). Consistent with the heart rate increase, a QTcB increase of 13 milliseconds was seen in the levomilnacipran ER group compared with a 1-millisecond increase in the placebo group. The mean QTcF changes were small in both groups (placebo, −1.1 milliseconds; levomilnacipran ER, −0.3 milliseconds). A QTcB interval increase of 60 milliseconds or greater was observed in 6 patients (2.8%) in the levomilnacipran ER group; no patients had a QTcF interval increase of 60 milliseconds or greater. No patients met the PCS criteria for PR (≥250 milliseconds), QTcB (>500 milliseconds), or QTcF (>500 milliseconds) interval.
DISCUSSION
Major depressive disorder is among the leading causes of global disease burden1; related factors such as functional impairment, decreased capacity to work, decreased productivity, and increased health care use greatly contribute to the overall societal and economic liability of the disorder. The cost of depression in the United States in 2000 was estimated to be $83.1 billion, with 31% of the total attributed to direct medical costs, 7% attributed to suicide-related mortality, and 62% attributed to workplace costs.29 The substantial social and economic consequences of MDD indicate that symptomatic improvement and normalizing function should be considered integral components of recovery and return to well-being.3
In this phase III study, the total score change from baseline to week 8 on the MADRS, the primary efficacy parameter, was statistically different in favor of levomilnacipran ER (−15.3) versus placebo (−12.2; P = 0.0051; MMRM); statistically significant separation from placebo started from week 4 and persisted through the remainder of the double-blind treatment. At week 8, the score differential for levomilnacipran ER and placebo exceeded the 2-point threshold that is frequently used to establish the clinical relevance of an antidepressant,30 demonstrating the meaningful effect of levomilnacipran ER treatment in this trial. Statistically significant differences in SDS total score, the secondary efficacy parameter, for levomilnacipran ER versus placebo occurred as early as week 4, the time of the first SDS assessment; a statistically significant difference versus placebo was also observed on each SDS subscale score at week 8.
Patients with depression commonly experience reduced motivation and energy as a consequence of the disorder or as a side effect of pharmacotherapy; identifying interventions that can restore vigor is an important component of effective treatment.22 Improvement in patient motivation and energy was demonstrated by a statistically significant difference in MEI-SF total score change from baseline to week 8 in favor of levomilnacipran ER compared with placebo. In addition, statistically significant differences in the MEI-SF social and cognitive subscale scores for levomilnacipran ER versus placebo demonstrated improvement in domains that are relevant to improved functional impairment.
Significant differences in score change from baseline to week 8 were observed on many additional efficacy measures. Cognitive impairment and fatigue, MDD symptoms that commonly impair daily functioning,31 were independently assessed in this trial to more fully elucidate patient progress. Because norepinephrine may be related to specific depression symptoms including attention,8 the strong noradrenergic component of levomilnacipran ER may be of particular importance in relation to improving attention and associated functional impairment. A statistically significant difference between levomilnacipran ER and placebo was observed on the continuity of attention score, and numerical improvements were noted in several of the other attention-related measures. It is possible that the short-term duration of the study might not be long enough to detect drug treatment effects on cognitive measures, and future research in this area is warranted.
The proportion of patients achieving MADRS response (≥50% reduction from baseline) was significantly greater for levomilnacipran ER compared with placebo, and the NNT for response showed that 8 patients would need to be treated with levomilnacipran ER to show 1 additional MADRS response outcome. For antidepressant agents, an NNT of 10 or lower is considered clinically relevant.32 Remission rates were not statistically different between the treatment groups.
The SDS response (total score ≤12 and score ≤4 on each item) and remission (total score ≤6 and score ≤2 on each item) criteria used in this study were based on recommendations originally made by Sheehan and Sheehan27 and supported by a database analysis of symptomatic and functional remission.33 When using mean change from baseline as an SDS outcome parameter, the SDS has been shown to be sensitive to treatment effects and can reliably discriminate between responders and nonresponders.27 In this study, significantly more levomilnacipran ER patients compared with placebo patients achieved SDS response (44.2% vs 28.9%), with an NNT of 7. Remission rates were not statistically different between the treatment groups.
Functional improvement in MDD has been shown to lag behind symptomatic improvement,3,4,33 with clinically meaningful improvement typically perceived as occurring over time. Some investigations have indicated that improvement in functioning may not peak until 4 to 8 months after treatment initiation.34–37 Findings from a large pooled dataset analysis of adult patients with MDD who had been randomized to duloxetine or placebo in 3 short-term, double-blind clinical studies33 supported earlier reports that functional improvement in MDD lags in comparison with symptomatic improvement. More patients receiving drug or placebo achieved symptomatic remission (HAMD17 ≤7 or MADRS ≤10) than functional remission (SDS ≤6 and score ≤2 on each item). In contrast, no apparent lag in functional improvement compared with symptomatic improvement was observed in the current study. Rates of MADRS and SDS response for the patients receiving drug or placebo were similar in this case, as were rates of MADRS and SDS remission. However, in light of the potential for asynchronous symptomatic and functional improvement, separate evaluations of the 2 domains may more accurately determine the overall state of wellness in patients with MDD.38
Levomilnacipran ER was generally well tolerated by the patients in this study. The overall rate of premature discontinuation was slightly higher for the levomilnacipran ER group (24.9%) than the placebo group (20.7%); 3.2% of the placebo patients and 7.8% of the levomilnacipran ER patients discontinued because of AEs. Treatment-emergent AEs were reported more frequently in the levomilnacipran ER group (81.6%) compared with the placebo group (61.8%), although most were considered mild in intensity. Nausea, the most common levomilnacipran ER–associated TEAE, was not dose dependent, had onset early in the course of treatment (week 1–2), and was transient. No SAEs that occurred during the double-blind period, including a suicide attempt reported in 1 levomilnacipran ER patient, were considered by the investigators to be related to the investigational product. Of the 2 SAEs that occurred in the down-taper period, the investigator considered 1 SAE (hypertension/noncardiac chest pain) to be related and 1 SAE (suicidal ideation) to be unrelated to levomilnacipran ER treatment.
Pulse rate and blood pressure increases were greater for the levomilnacipran ER patients compared with the placebo patients, which is consistent with the AE profile of other SNRIs.39 potentially clinically significant changes in pulse rate or blood pressure during the double-blind treatment were reported in 2 levomilnacipran ER patients and 1 placebo patient. The incidence of vital sign–related TEAEs was higher in the levomilnacipran ER group. QTcB increase was greater for the levomilnacipran ER patients compared with the placebo patients, which is consistent with the heart rate increase also observed in the levomilnacipran ER treatment group; QTcB interval increase of 60 milliseconds or greater was observed in 6 patients (2.8%) in the levomilnacipran ER group. The mean change in QTcF was similar in both groups. No patients met the PCS criteria for PR, QTcB, or QTcF interval.
The limitations of this study include the lack of an active comparator arm; the short 8-week, double-blind treatment period; and the strict inclusion and exclusion criteria that may limit the ability to generalize the findings. The level of baseline depression in the study population may be considered a confounding factor in regard to MADRS and SDS remission rates because high baseline depression can affect the interpretation of clinical study results.28,33 In this study, low and similar rates of remission for levomilnacipran ER and placebo on the MADRS or the SDS may reflect the difficulty of detecting remission differences in short-term individual studies and the extended period needed to achieve remission in a majority of patients with MDD, especially when baseline MADRS and SDS scores are high and stringent remission criteria are used.30 The use of the SDS as the prespecified secondary outcome to measure functional impairment is a strength of this study, and significant differences on SDS total and subscale scores in favor of levomilnacipran ER compared with placebo should be of great interest to the field.
Symptomatic improvement and functional improvement are both critical components of recovery from MDD. In this phase III clinical trial, score change from baseline to end of treatment on the MADRS was statistically different in favor of levomilnacipran ER compared with placebo, indicating improvement in symptomatic severity for the levomilnacipran ER patients. In addition, statistically significant differences in score change on the SDS and its subscales suggest that treatment with levomilnacipran ER compared with placebo decreases functional impairment across the domains of work, social life, and family life. The statistically significant difference in MEI-SF total and subscale scores indicated greater motivation and energy in the patients treated with levomilnacipran ER compared with placebo. Levomilnacipran ER exhibited a generally good safety and tolerability profile. This study suggests that levomilnacipran ER is a safe and effective treatment of the symptoms and functional impairments associated with MDD.
ACKNOWLEDGMENTS
The authors thank Carol Dyer, MS, and Adam Ruth, PhD, of Prescott Medical Communications Group, Chicago, IL, and a contractor for Forest Research Institute for writing assistance and editorial support for the preparation of this manuscript.
AUTHOR DISCLOSURE INFORMATION
Angelo Sambunaris has received consultant fees from Forest Research Institute, Inc. Anjana Bose, Carl P. Gommoll, Changzheng Chen, and William M. Greenberg are employees of Forest Research Institute. Anjana Bose was an employee of Forest Research Institute at time of study and is currently employed with Otsuka Pharmaceutical Development & Commercialization.
David V. Sheehan, MD, MBA, has received grant support, has been affiliated with, or has received honoraria and travel expenses related to lectures/presentations or consultant activities from the following organizations: Abbott Laboratories, Ad Hoc Committee, Treatment Drug & Assessment Research Review; Alexa; Alza Pharmaceuticals, Palo Alto, CA; American Medical Association; American Psychiatric Association Task Force on Benzodiazepine Dependency; American Psychiatric Association Task Force on Treatments of Psychiatric Disorders; American Psychiatric Association Working Group to Revise DSM III Anxiety Disorders Section; Anclote Foundation; Anxiety Disorders Resource Center; Anxiety Drug Efficacy Case; US Food and Drug Administration; Applied Health Outcomes/XCENDA; AstraZeneca; Avera Pharmaceuticals; Boehringer Ingelheim; Boots Pharmaceuticals; Bristol-Myers Squibb; Burroughs Wellcome; Cephalon; Charter Hospitals; Ciba Geigy; Committee (RRC) of NIMH on Anxiety and Phobic Disorder Projects; Connecticut & Ohio Academies of Family Physicians; Cortex Pharmaceutical; Council on Anxiety Disorders; CPC Coliseum Medical Center; Cypress Bioscience; Dista Products Company; Division of Drugs & Technology; American Medical Association; EISAI; Eli Lilly; Excerpta Medica Asia; Faxmed, Inc; Forest Laboratories; Glaxo Pharmaceuticals; GlaxoSmithKline; Glaxo-Wellcome; Hikma Pharmaceuticals; Hospital Corporation of America; Humana; ICI; INC Research; International Clinical Research (ICR); International Society for CNS Drug Development (ISCDD); Janssen Pharmaceutica; Jazz Pharmaceuticals; Kali-Duphar; Labopharm-Angellini; Layton Bioscience; Lilly Research Laboratories; Lundbeck, Denmark; Marion Merrill Dow; McNeil Pharmaceuticals; Mead Johnson; Macmillan; MAPI; Medical Outcome Systems; MediciNova; Merck Sharp & Dohme; National Anxiety Awareness Program; National Anxiety Foundation; National Depressive & Manic Depressive Association; National Institute of Drug Abuse; National Institutes of Health (NIH); Neuronetics; NovaDel; Novartis Pharmaceuticals Corp; Novo Nordisk; Organon; Orion Pharma; Parexel International Corporation; Parke-Davis; Pfizer; Pharmacia; Pharmacia & Upjohn; PharmaNeuroBoost; Philadelphia College of Pharmacy & Science; Pierre Fabre, France; Quintiles; ProPhase; Rhone Laboratories; Rhone-Poulenc Rorer Pharmaceuticals; Roche; Roerig; Sagene; Sandoz Pharmaceuticals; Sanofi-Aventis; Sanofi-Synthelabo Recherche/Sanofi Aventis; Schering Corporation; Sepracor; Shire Laboratories, Inc; Simon and Schuster; SmithKlineBeecham; Solvay Pharmaceuticals; Takeda Pharmaceuticals; Tampa General Hospital University of South Florida Psychiatry Center; University of South Florida College of Medicine; TAP Pharmaceuticals; Targacept; TGH-University Psychiatry Center; Tikvah Therapeutics; Titan Pharmaceuticals; United Bioscience; The Upjohn Company; US Congress–House of Representatives Committee; USF Friends of Research in Psychiatry, Board of Trustees, Warner Chilcott; World Health Organization; Worldwide Clinical Trials; Wyeth-Ayerst; ZARS; Zeneca Pharmaceuticals; and Neuronetics.
REFERENCES
- 1. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006; 3 (11): e442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kasper S, Meshkat D, Kutzelnigg A. Improvement of the noradrenergic symptom cluster following treatment with milnacipran. Neuropsychiatr Dis Treat. 2011; 7 (suppl 1): 21– 27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Israel JA. Remission in depression: definition and initial treatment approaches. J Psychopharmacol. 2006; 20 (3 suppl): 5– 10 [DOI] [PubMed] [Google Scholar]
- 4. Hirschfeld RM, Dunner DL, Keitner G, et al. Does psychosocial functioning improve independent of depressive symptoms? A comparison of nefazodone, psychotherapy, and their combination. Biol Psychiatry. 2002; 51 (2): 123– 133 [DOI] [PubMed] [Google Scholar]
- 5. McKnight PE, Kashdan TB. The importance of functional impairment to mental health outcomes: a case for reassessing our goals in depression treatment research. Clin Psychol Rev. 2009; 29 (3): 243– 259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Auclair AL, Martel JC, Assie MB, et al. Levomilnacipran (F2695), a norepinephrine-preferring SNRI: profile in vitro and in models of depression and anxiety. Neuropharmacology. 2013; 70: 338– 347 [DOI] [PubMed] [Google Scholar]
- 7. Keller M. Role of serotonin and noradrenaline in social dysfunction: a review of data on reboxetine and the Social Adaptation Self-evaluation Scale (SASS). Gen Hosp Psychiatry. 2001; 23 (1): 15– 19 [DOI] [PubMed] [Google Scholar]
- 8. Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Psychiatry. 2008; 69 (suppl E1): 4– 7 [PubMed] [Google Scholar]
- 9. Li F, Chin C, Wangsa J, et al. Excretion and metabolism of milnacipran in humans after oral administration of milnacipran hydrochloride. Drug Metab Dispos. 2012; 40 (9): 1723– 1735 [DOI] [PubMed] [Google Scholar]
- 10. FDA’s policy statement for the development of new stereoisomeric drugs. Chirality. 1992; 4 (5): 338– 340 [DOI] [PubMed] [Google Scholar]
- 11. Hutt AJ. The development of single-isomer molecules: why and how. CNS Spectr. 2002; 7 (4 suppl 1): 14– 22 [DOI] [PubMed] [Google Scholar]
- 12. Montgomery S, Mansuy L, Ruth A, et al. The efficacy and safety of levomilnacipran sustained release in moderate to severe major depressive disorder: a randomized, double-blind, placebo-controlled, proof-of-concept study. J Clin Psychiatry. 2013; 74 (4): 363– 369 [DOI] [PubMed] [Google Scholar]
- 13. Asnis G, Bose A, Gommoll C, et al. The efficacy and safety of levomilnacipran SR 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase III, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013; 74 (3): 242– 248 [DOI] [PubMed] [Google Scholar]
- 14. Bakish D, Bose A, Gommoll C, et al. Levomilnacipran ER 40 mg and 80 mg in major depressive disorder: a phase III, randomized, double-blind, fixed-dose, placebo-controlled study. J Psychiatry Neurosci. 2013. doi: 10.11503/jpn.130040. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gommoll C, Bose A, Li H, et al A randomized double-blind, placebo-controlled, flexible-dose study of levomilnacipran in patients with major depressive disorder. Poster presented at: the 24th Annual US Psychiatric and Mental Health Congress; November 7–10, 2011; Las Vegas, NV. [Google Scholar]
- 16. Sheehan DV, Lecrubier Y, Sheehan KH, et al. The Mini-International Neuropsychiatric Interview (M.I.N.I.): the development and validation of a structured diagnostic psychiatric interview for DSM-IV and ICD-10. J Clin Psychiatry. 1998; 59 (suppl 20): 22– 33 [PubMed] [Google Scholar]
- 17. Montgomery SA, Asberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979; 134: 382– 389 [DOI] [PubMed] [Google Scholar]
- 18. Posner K, Brown GK, Stanley B, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011; 168 (12): 1266– 1277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sheehan DV, Harnett-Sheehan K, Raj BA. The measurement of disability. Int Clin Psychopharmacol. 1996; 11 (suppl 3): 89– 95 [DOI] [PubMed] [Google Scholar]
- 20. Hamilton M. A rating scale for depression. J Neurol Neurosurg Psychiatry. 1960; 23: 56– 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guy W. ECDEU Assessment Manual for Psychopharmacology: Revised. Rockville, MD: US Department of Health Education & Welfare; 1976: 218– 222 [Google Scholar]
- 22. Fehnel SE, Bann CM, Hogue SL, et al. The development and psychometric evaluation of the Motivation and Energy Inventory (MEI). Qual Life Res. 2004; 13 (7): 1321– 1336 [DOI] [PubMed] [Google Scholar]
- 23. Wesnes KA, Ward T, McGinty A, et al. The memory enhancing effects of a Ginkgo biloba/Panax ginseng combination in healthy middle-aged volunteers. Psychopharmacology (Berl). 2000; 152 (4): 353– 361 [DOI] [PubMed] [Google Scholar]
- 24. Bond A, Lader M. The use of analogue scales in rating subjective feelings Br J Med Psychol. 1974; 47: 211– 218 [Google Scholar]
- 25. Kenward MG, Roger JH. Small sample inference for fixed effects from restricted maximum likelihood. Biometrics. 1997; 53 (3): 983– 997 [PubMed] [Google Scholar]
- 26. Kenward MG, Molenberghs G, Thijs H. Pattern-mixture models with proper time dependence. Biometrika. 2003; 90 (1): 53– 71 [Google Scholar]
- 27. Sheehan KH, Sheehan DV. Assessing treatment effects in clinical trials with the discan metric of the Sheehan Disability Scale. Int Clin Psychopharmacol. 2008; 23 (2): 70– 83 [DOI] [PubMed] [Google Scholar]
- 28. Nemeroff CB. The burden of severe depression: a review of diagnostic challenges and treatment alternatives. J Psychiatr Res. 2007; 41 (3–4): 189– 206 [DOI] [PubMed] [Google Scholar]
- 29. Greenberg PE, Kessler RC, Birnbaum HG, et al. The economic burden of depression in the United States: how did it change between 1990 and 2000? J Clin Psychiatry. 2003; 64 (12): 1465– 1475 [DOI] [PubMed] [Google Scholar]
- 30. Montgomery SA, Moller HJ. Is the significant superiority of escitalopram compared with other antidepressants clinically relevant? Int Clin Psychopharmacol. 2009; 24 (3): 111– 118 [DOI] [PubMed] [Google Scholar]
- 31. Kurian BT, Greer TL, Trivedi MH. Strategies to enhance the therapeutic efficacy of antidepressants: targeting residual symptoms. Expert Rev Neurother. 2009; 9 (7): 975– 984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cipriani A, Barbui C, Brambilla P, et al. Are all antidepressants really the same? The case of fluoxetine: a systematic review. J Clin Psychiatry. 2006; 67 (6): 850– 864 [DOI] [PubMed] [Google Scholar]
- 33. Sheehan DV, Harnett-Sheehan K, Spann ME, et al. Assessing remission in major depressive disorder and generalized anxiety disorder clinical trials with the discan metric of the Sheehan disability scale. Int Clin Psychopharmacol. 2011; 26 (2): 75– 83 [DOI] [PubMed] [Google Scholar]
- 34. Giller E, Jr, Bialos D, Riddle MA, et al. MAOI treatment response: multiaxial assessment. J Affect Disord. 1988; 14 (2): 171– 175 [DOI] [PubMed] [Google Scholar]
- 35. Mintz J, Mintz LI, Arruda MJ, et al. Treatments of depression and the functional capacity to work. Arch Gen Psychiatry. 1992; 49 (10): 761– 768 [DOI] [PubMed] [Google Scholar]
- 36. Paykel ES, Weissman MM. Social adjustment and depression. A longitudinal study. Arch Gen Psychiatry. 1973; 28 (5): 659– 663 [DOI] [PubMed] [Google Scholar]
- 37. Weissman MM, Klerman GL, Paykel ES, et al. Treatment effects on the social adjustment of depressed patients. Arch Gen Psychiatry. 1974; 30 (6): 771– 778 [DOI] [PubMed] [Google Scholar]
- 38. Keller MB. Past, present, and future directions for defining optimal treatment outcome in depression: remission and beyond. JAMA. 2003; 289 (23): 3152– 3160 [DOI] [PubMed] [Google Scholar]
- 39. Stahl SM, Grady MM, Moret C, et al. SNRIs: their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectr. 2005; 10 (9): 732– 747 [DOI] [PubMed] [Google Scholar]