

Abstract
Chronic infections with hepatitis B virus (HBV) and/or hepatitis C virus (HCV) are the major causes of cirrhosis globally. It takes 10-20 years to progress from viral hepatitis to cirrhosis. Intermediately active hepatic inflammation caused by the infections contributes to the inflammation-necrosis-regeneration process, ultimately cirrhosis. CD8+ T cells and NK cells cause liver damage via targeting the infected hepatocytes directly and releasing pro-inflammatory cytokine/chemokines. Hepatic stellate cells play an active role in fibrogenesis via secreting fibrosis-related factors. Under the inflammatory microenvironment, the viruses experience mutation-selection-adaptation to evade immune clearance. However, immune selection of some HBV mutations in the evolution towards cirrhosis seems different from that towards hepatocellular carcinoma. As viral replication is an important driving force of cirrhosis pathogenesis, antiviral treatment with nucleos(t)ide analogs is generally effective in halting the progression of cirrhosis, improving liver function and reducing the morbidity of decompensated cirrhosis caused by chronic HBV infection. Interferon-α plus ribavirin and/or the direct acting antivirals such as Vaniprevir are effective for compensated cirrhosis caused by chronic HCV infection. The standard of care for the treatment of HCV-related cirrhosis with interferon-α plus ribavirin should consider the genotypes of IL-28B. Understanding the mechanism of fibrogenesis and hepatocyte regeneration will facilitate the development of novel therapies for decompensated cirrhosis.
Keywords: Liver cirrhosis, Hepatitis B virus, Hepatitis C virus, Evolution, Immune cells, Signaling pathway, Hepatic stellate cells, Antiviral therapy
Core tip: Hepatic inflammation caused by viral infections contributes to the inflammation-necrosis-regeneration process, ultimately cirrhosis. Immune selection of some hepatitis B virus mutations in the evolution towards cirrhosis seems different from that towards hepatocellular carcinoma. Hepatic stellate cells and macrophages are important for the fibrogenesis. Antiviral treatment is generally effective in reducing the morbidity of decompensated cirrhosis. The standard of care for the treatment of hepatitis C virus-related cirrhosis with pegylated interferon-α and ribavirin should consider the genotypes of IL-28B. Stem cell-based therapy can be an option for the treatment of decompensated cirrhosis patients who fail to respond to antiviral treatment.
INTRODUCTION
Liver cirrhosis represents a worldwide public health problem and its mortality rate increases stably these years. Alcoholic hepatitis, non-alcoholic steatohepatitis, and chronic infection with hepatitis C virus (HCV) are the major causes of cirrhosis in Western countries, while chronic infection with hepatitis B virus (HBV) contributes greatly to cirrhosis in HBV-endemic areas. Chronic HBV and HCV infections account for 57% of cirrhosis cases globally[1]. There are about 300 million subjects chronically infected with HBV and 130-170 million subjects chronically infected with HCV worldwide. About one million people die from diseases related to chronic HBV and/or HCV infections each year, mostly end-stage liver diseases, namely, decompensated cirrhosis, liver failure, and hepatocellular carcinoma (HCC). In East Asia where HBV genotypes B and C are endemic, HBV genotype B is more apt to cause acute infection in young people and to be cleared than genotype C; whereas HBV genotype C leads to higher persistence following an acute course and is more apt to cause cirrhosis and HCC compared to genotype B[2-5]. HBV genotype C is associated with a lower rate of spontaneous hepatits B e antigen (HBeAg) seroconversion than HBV genotype B. Cirrhosis is specifically found in HBeAg-negative subjects and more frequently found in subjects with genotype C than in those with genotype B. In HBeAg-negative subjects, high viral load is frequently associated with abnormal alanine aminotransferase (ALT) level, while ALT abnormality is more frequent in those with cirrhosis than those without[6]. In addition, older age (longer duration of infection), high levels of HBV DNA, habitual alcohol consumption, and concurrent infection with HCV, hepatitis D virus or human immunodeficiency virus are significantly associated with an increased rate of cirrhosis[7]. Most (70%-80%) HCV infections may persist and about 30% of individuals with persistent HCV infection will develop end-stage liver diseases, including cirrhosis and HCC. According to the most recent classification criteria, HCV variants are classified into 6 genotypes, of which HCV-1b is the most common one worldwide[8]. HCV genotype 1b has been associated with an increased risk of HCC in both European and Asian populations[9,10], but it remains uncertain if this genotype of HCV is specifically related to fibrosis and cirrhosis. An interesting cross-sectional study carried out in mainland China indicated that co-infection with HBV, low HCV viral load, low serum ALT, high serum aspartate aminotransferase, diabetes, and high pickled food consumption were significantly associated with the risk of cirrhosis in HCV-infected patients[11].
Since both HBV and HCV are preferentially hepatotropic, not directly cytopathic, virus-caused liver damage is attributed to immune-mediated mechanisms. Under inflammatory microenvironment caused by the infections, the continuous infiltration of immune cells and the secreted inflammatory cytokines result in liver damage. The hepatic lobule reconstruction following the damage promotes hepatic fibrosis and eventually leads to cirrhosis. Cirrhosis represents a consequence of wound-healing response to chronic stimulation. It is generally believed that cirrhosis is the outcome of interaction between liver damage and tissue repair. Hepatic satellite cells (HSCs) play a critical role in the progression of cirrhosis. However, the mechanisms by which HSCs modulate fibrogenesis and cirrhosis are less clarified. Active viral replication and intermediate inflammatory reactions facilitate hepatic fibrogenesis but the evolution of fibrogenesis might depart from HCC evolution[12]. In this paper, we focus on virus-related cirrhosis and summarize recent progress on its molecular basis. Moreover, we also discuss possible therapeutic options for hepatic fibrosis, early cirrhosis, and even decompensated cirrhosis.
MOLECULAR BASIS OF VIRUS-RELATED CIRRHOSIS
Immune response and liver damage
Roles of immune cells and cytokines: The immune system is a complicated dynamic network constituted by various immune cells and cytokines. T and B lymphocytes, macrophages (macrophages residing in the liver are also called Kuffer cells), NK cells, neutrophils, HSCs, NKT cells, dendritic cells (DCs), and mast cells are all important in the maintenance of chronic inflammation. CD8+ cytotoxic T lymphocytes (CTL) and CD4+ T helper lymphocyte subpopulations [Th1, Th2, Th17, and regulatory T (Treg) cells] also play key roles in maintaining chronic inflammation. The immune effectors not only play critical roles in the HBV and HCV clearance, but also participate in liver damage. Toll-like receptors (TLR)-3 and -7 can recognize the viruses and induce the production of type I interferon (IFN) (IFN-α/β), proinflammatory cytokines and chemokines to inhibit the viruses[13-15], whereas TLR-4 activation by lipopolysaccharides (LPS) in HSCs enhances TGF-β signaling and hepatic fibrosis[16]. TGF-β, a pleiotropic cytokine produced by immune and non-immune cells, has receptors on several cell types. It induces fibrosis via increasing Th17 and HSCs and reducing NK cell activation[17]. A study using TLR-9-deficient mouse model has demonstrated that TLR-9 is also involved in liver fibrosis[18]. As important antiviral cytokines activated in the initial immune response, IFN-α/β can inhibit viral replication and lead to death of the infected hepatocytes by inducing the expression of multiple IFN-stimulated genes (ISG), including protein kinase R (PKR), Mx proteins, ISG-15, RnaseL/2’,5’-oligoadenylate synthetase (2’,5’-OAS) and RNA helicases[19,20]. Additionally, it may activate the neighboring immune cells, including macrophages, NK cells, NKT cells and DCs[21-23]. Both NK cells and CD8+ CTLs exert immunoregulatory functions via direct, non-MHC-restricted cytotoxic mechanisms and cytokine production[24,25]. IFN-γ and tumor necrosis factor-α (TNFα) produced by NK cells and CD8+ CTLs can not only inhibit the viruses, but also cause liver damage through TNF-related apoptosis-inducing ligand (TRAIL)-mediated death of hepatocytes[25,26]. Moreover, CD8+ CTLs can exert the bystander killing effect through perforin, Fas/Fas ligand and TNFα pathways[27], thus causing a wide range of hepatocellular apoptosis. IFN-γ-induced chemokines such as CXC chemokine ligands CXCL9, CXCL10, and CXCL11 can induce the migration of nonspecific mononuclear cells into the liver[28], which are unable to control the infection but result in sustained liver damage. In addition, the antibody response has also been associated with the extent of liver injury during HCV chronic infection, and anti-E2 antibodies can mediate liver damage via antibody-dependent cell-mediated cytotoxicity (ADCC)[29], but it just occurs after a prolonged HCV infection. The contributions of natural HCV antibodies to liver damage and fibrosis progression still need to be determined.
Dysfunction of T cells: In the phase of chronic HBV and HCV infections, specific T cells present a condition of dysfunction due to the persistent exposure to high levels of viral antigens that exceed the capacity of host immunity. It may be induced by the following mechanisms. First, genetic predisposition of immune-related genes contributes to persistence of HBV and/or HCV infections. Genetic polymorphism of class II human leukocyte antigen (HLA) has been associated with HBV[30] and HCV persistence/clearance[31], possibly because this genetic predisposition affects T cell function upon HBV infection. Genetic polymorphisms of other immune-related genes, notably, interleukin-28B (IL28B), have closely been associated with HCV clearance, progression, and treatment response[32,33]. Second, antigen presentation by DCs and macrophages may be impaired, resulting in ineffective priming of T cells or deficient maintenance of antigen-experienced T cells[34]. Additionally, dysfunctional specific T cells can express the inhibitory receptor programmed death-1 (PD-1), which inhibits immune activity and induces the apoptosis of T cells[35-37]. Moreover, HCV core protein can inhibit T cell proliferation by binding to the complement receptor gC1qR[38,39]. During the prolonged infection, the expression of chemokine (C-C) motif ligand (CCL) 17 and CCL22, attractors for Tregs[40,41], can inhibit the proliferation and function of T cells and other immune cells[42]. The dysfunctional T cells can hardly eliminate HBV or HCV. On the contrary, the consistent but insufficient immune responses break up several balances between the immune cells or cytokines, such as Th1/Th2 cells or Th17/Treg cells, neutrophils/lymphocytes, neutrophils/CD8+ T cells, and Th1/Th2 cytokines[43,44], which induces a high immune pressure and results in the evolution of the viruses during chronic infection.
Role of inflammatory signaling pathways
There are multiple inflammatory signaling pathways and molecules involved in the sustained inflammation caused by chronic infection, including nuclear factor-κB (NF-κB), Wnt/β-catenin, TGF-β/Smad, RAF/MEK/ERK, JAK/STAT, PI3K-AKT/PKB, Ras-MAPK, and Vitamin A[45-48]. NF-κB as a dimeric transcription factor can be activated by proinflammatory stimuli, such as TNFα or interleukin-1β (IL-1β)[48]. The activated NF-κB signaling pathway thereafter induced the expression of a series of growth factors and cytokines to regulate the inflammatory response. IL-6 can be released by macrophages and regulate the proliferation and differentiation of liver fibroblasts[49]. The PI3K-AKT/PKB and Ras-MAPK pathways are also important because they are involved in the activation of HSCs. Platelet-derived growth factor (PDGF) can lead to the Ras-MAPK activation by binding its receptor, and the activation of protein kinase C (PKC) family through PI3K-AKT/PKB eventually induces cell proliferation and HSC activation[50]. These signaling pathways and molecules may play an active role in the pathogenesis of HBV- and HCV-related cirrhosis, thus serving as therapeutic targets and prognostic markers.
HBV and HCV escape strategies in chronic infection
Spontaneous clearance of the viruses can be achieved in some infected subjects by an efficient immune response. However, following the early immune response, 70%-80% of HCV infected patients will develop chronic infection[51] and 8.5% of adults with acute hepatitis B will develop chronic infection[2]. Chronic infection of HBV occurs frequently in those who acquired the infection perinatally (90%) or during childhood (20%-30%), when the immune system is thought to be immature[12]. It implies that HBV and HCV can produce a series of strategies to evade the clearance. Under immune pressure, HBV and HCV can evade the immune clearance through persistent viral replication and mutations, making themselves preferably adapt to the inflammatory microenvironment. However, HBV and HCV have different mutation patterns conforming to their distinct viral structures. HCV replicates at a rate of 1010-1012 virions per day. The RNA-dependent RNA polymerase lacks a proofreading function, favoring the Darwinian selection of viral variants by humoral and cellular immune responses[52]. During the phase of acute infection, there is a high rate of nonsynonymous and synonymous substitutions due to the high levels of selective pressure exerted by antibodies and activated T cells, but the rate decreases at continuous infection stage[53]. The HCV E2 glycoprotein is thought to be a major target for HCV-specific antibody. The nucleotide substitutions at the hypervariable HCV E2 region 1 during anti-HCV seroconversion in acutely HCV-infected individuals are intensively correlated with the viral evasion. HBV exists in the form of covalently closed circular DNA (cccDNA) in the nuclei of hepatocytes and exhibits higher frequencies of mutations than other DNA viruses due to the spontaneous error rate of reverse transcriptase. The rate of HBV mutation in HBeAg-positive patients is 1.5 × 10-5-5 × 10-5 nucleotide substitutions/site per year[54], and is even higher in HBeAg-negative patients. Under immune pressure, hypermutation provides viruses a choice not only for growth advantage, but also for long-term survival. APOBEC3, an important member of the APOBEC family of cellular cytidine deaminases, can inhibit HBV through a series of editing-dependent and -independent mechanisms. However, it is also involved in the viral genetic diversification and evolution[12]. The expression of activation induced deaminase, an APOBEC3 paralog, has been observed in a variety of chronic inflammatory syndromes including HCV infection. Introduction of exogenous APOBEC3G into HCV-infected Huh7.5 human hepatocytes inhibits HCV replication; knockdown of endogenous APOBEC3G enhances HCV replication[55]. Some APOBEC3 genes in primary human hepatocytes are up-regulated by IFN-α and IFN-γ. Up-regulation of APOBEC3 by inflammation inhibits HCV genome synthesis via viral editing. Two to five APOBEC3 genes are significantly up-regulated in cirrhotic livers in following order: HCV ± HBV-related cirrhosis > HBV-related cirrhosis > alcoholic cirrhosis, compared to normal livers. In HBV-related cirrhosis, HBV genome is particularly edited by APOBEC3G, and APOBEC3G is the dominant deaminase in vivo with up to 35% of HBV genomes being edited[56]. Hepatic inflammation might provide a “fertile field” to facilitate “viral mutation-selection-adaptation” evolutionary process via activating a series of enzymes including APOBEC3 and promote immune escape of HBV and HCV during chronic infection.
Association of HBV mutations with cirrhosis
HBV DNA consists of four overlapping open reading frames that encoding the envelope (pre-S/S), core (precore/core), polymerase (P), and X proteins, respectively. HBV preS region consists of preS1 and preS2 domains and they are essential for the immune responses because they contain several epitopes for T and/or B cells. In our previous studies, we defined wild-type nucleotides and mutations of HBV genotypes B and C, respectively. A nucleotide with the highest frequency in the sequences of HBV from asymptomatic HBsAg carriers (ASCs) seropositive for HBeAg was termed a wild-type nucleotide because HBeAg-positive HBV has been traditionally treated as a wild-type strain[6,57,58]. Under this definition, we found some of the cirrhosis-related HBV mutations (compared to ASCs and CHB patients) and HCC-related HBV mutations (compared to HBV-infected subjects without HCC) in the preS and enhancer II (EnhII)/basal core promoter (BCP)/precore regions. In the preS region, 81.0% of preS1 wild-type nucleotide hotspots of HBV genotype C are significantly associated with an increased risk of cirrhosis, as compared with CHB; 90.5% of preS1 mutations of HBV genotype C are significantly associated with an increased risk of HCC, compared to cirrhosis. Furthermore, wild-type nucleotides A2964C and T3116C are independently associated with an increased risk of cirrhosis, whereas their mutation counterparts C2964A and C3116T are independently associated with an increased risk of HCC[57]. In the EnhII/BCP/precore region of HBV genotype C, C1673T, A1726C, A1727T, C1730G, C1766T, T1768A, C1773T, and C1799G are significantly associated with an increased risk of cirrhosis, compared to CHB; whereas these mutations are inversely associated with HCC risk, compared to cirrhosis. T1768A, A1762T/G1764A, and A1846T are independently associated with an increased risk of cirrhosis[57]. The frequencies of G1652A, T1673C, and G1730C increase successively from the ASC state to CHB to cirrhosis, but significantly decrease in HCC[58]. A study conducted in Taiwan has demonstrated that multiple HBV mutations including preS deletion, T1762/A1764, T1766 and/or A1768 are more often found than a single mutation patterns in the process of progression to cirrhosis[59]. The evidence indicates that immune selection of some viral mutations in HBV evolution towards cirrhosis seems different from that in the evolution towards HCC. Even though, several important HCC-related HBV mutations, namely, preS deletion, C1653T, T1753V, and A1762T/G1764A successively increase in their frequencies during HBV evolution from ASC state, CHB, and cirrhosis to HCC[58,60]. This reflects the robustness of HBV preS deletion, C1653T, T1753V, and A1762T/G1764A in predicting the entire evolutionary process from ASC state to HCC. Although cirrhosis is an important risk factor of HCC, most of the virus-related cirrhosis patients do not develop HCC. The difference in immune selection of viral mutants between evolutionary processes towards cirrhosis and HCC should be further elucidated.
Our previous study has demonstrated that genetic predisposition to HLA-DP function play an important role in immune selection of cirrhosis-related HBV mutations and a significant effect of HBV mutations on cirrhosis is selectively evident in those with HLA-DP genotypes that promote HBV persistence[58]. HLA class II loci are mainly associated with spontaneous clearance of HBV and HCV. Interestingly, genetic polymorphisms within HLA II loci have been frequently associated with chronic HBV or HCV hepatitis, hepatic fibrosis, and the development of HCC[61]. Thus, HLA-DP affects HBV persistence, regulates immune selection of viral mutations, and influences cirrhosis risk contributed by HBV mutations.
Cirrhosis-related HBV mutations biologically affect the progression from chronic hepatitis to cirrhosis. Approximately 30.9% of S gene mutations occur in the major hydrophilic region of HBs antigen, which can change the epitopes of the three-dimensional structure, and lead to immune escape of HBV. C1766T/T1768A, an independent risk factor of cirrhosis in HBeAg-negative patients, can increase the pre-genome mRNA encapsidation and then promote viral assembly[62]. In addition, the X gene mutations such as G1386M, C1485T, and C1653T, can regulate the NF-κB signaling pathway which plays a crucial role in the progression to cirrhosis or HCC[63].
Tissue repair and liver fibrosis
Tissue repair following liver damage as a consequence of the inflammation through the accumulation of extracellular matrix (ECM) proteins mainly secreted by HSCs will eventually result in the occurrence of fibrosis. PDGF is the most potent mitogen for HSCs via the Ras-MAPK and PI3K-AKT/PKB signaling pathways[50]. Moreover, TGFα and epidermal growth factor (EGF) can also stimulate the proliferation of HSCs. The “inactive” HSCs should undergo a process of unidirectional activation, contributing to differentiate into myofibroblast-like phenotypes during fibrosis[64]. TGF-β1, the most important profibrogenic cytokine known for activated HSCs, is mediated by intracellular signaling via Smad proteins. Smad2 and Smad3 proteins are associated with the activated receptor of TGF-β1, whereas Smad7 is an effective inhibitor for TGF-β1 signaling. In addition, TGF-β1 also can increase α1 collagen mRNA stability via p38 MAPK. HSCs also can trigger an activated process via phagocytizing the apoptotic bodies induced by virus infection[65]. The c-Jun N-terminal kinase-1 (Jnk1) as a profibrotic kinase in HSCs, but not in hepatocytes, significantly contributes to liver fibrosis development[66]. JNK is involved in HSC activation and fibrogenesis and represents a potential target for antifibrotic treatments[67,68]. These fibrosis-promoting proteins will increase the secretion of fibrillar collagens, resulting in the deposition of excess fibrotic matrix. The activated HSCs also can inhibit the expression of matrix metalloproteinases (MMPs)-2, -9 and -14 which play a role because of proteolytic activity towards ECM, via promoting the expression of tissue inhibitors of metalloproteinases (TIMPs), thus inhibit the matrix degradation[69]. The characteristics of HSC-mediated fibrogenesis result in disruption of the original architecture and liver dysfunction (Figure 1).
Figure 1.
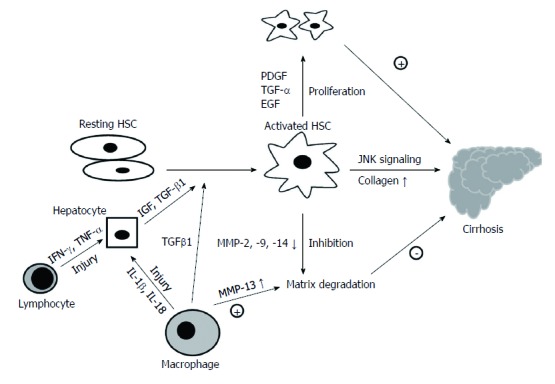
Process of hepatic satellite cells induced fibrosis. HSC: Hepatic satellite cell; IGF: Insulin-like growth factor; TGF: Transforming growth factor; PDGF: Platelet-derived growth factor; EGF: Epidermal growth factor; MMP: Matrix metalloproteinase; TNF: Tumor necrosis factor.
Macrophages also are critical for both liver damage and fibrosis. The proinflammatory cytokines interleukin-1β (IL-1β) and IL-18, which induced by macrophages in HCV-infected patients, can promote the inflammation via the NF-κB signaling pathway, resulting in liver damage[70]. Activated macrophages release growth factors, cytokines and chemokines which induce the recruitment of monocytes, thus affecting the function of HSCs and fibroblasts[71]. In particular, TGF-β1 and insulin-like growth factor can activate the fibroblasts and promote a switch in fibroblast gene expression to initiate matrix remodeling[72]. Macrophages display a key role in the different stages of fibrosis and these characteristics may be induced by either different cytokines in the microenvironment or the populations of macrophages[73]. On the other hand, macrophages can exert antifibrotic activity and promote the resolution of fibrosis by producing interstitial collagenases like MMP13[74]. In addition, the phagocytosis of apoptotic hepatocytes has been shown to inhibit the development of fibrosis. Understanding the mechanism of fibrogenesis may pave the way for the treatment of cirrhosis.
THERAPERUTIC OPTIONS FOR VIRUS-RELATED LIVER CIRRHOSIS
Without appropriate therapy for cirrhosis caused by HBV and/or HCV infections, liver injury may persist, thus facilitating the development of decompensated cirrhosis and HCC, especially in those with active viral replication. As viral replication is an important driving force of cirrhosis development, antiviral treatment should be carried out before progression into decompensated cirrhosis[75,76]. In addition, as some signaling systems, such as JNK, have been recently associated with the formation of cirrhosis[66-68], targeting these signaling systems might be an important option for the treatment of liver fibrosis and cirrhosis.
Antiviral treatment of HBV-related compensated cirrhosis
Around 30%-70% of the patients with compensated liver cirrhosis still have active viral replication, and this active viral replication promotes the progression of liver injury. Successful antiviral treatment prior to the development of cirrhosis is able to greatly reduce the morbidity and mortality of HBV-related end-stage liver diseases. Currently approved nucleos(t)ide analogs (NAs) for the treatment of HBV-related diseases include lamivudine (LAM), adefovir dipivoxil (ADV), entecavir (ETV), telbivudine (TBV), and tenofovir disoproxil fumarate (TDF)[77,78]. Antiviral therapy using newer NAs with lower resistance rates can suppress replication and re-activation of HBV, improve liver function, and restore many patients to a state of well compensated cirrhosis[79,80]. Long-term antiviral therapy can prevent the development of liver decompensation in patients with compensated cirrhosis[77]. The antiviral treatment to halt the progression of compensated cirrhosis should be carried out as early as the diagnosis has been confirmed.
Antiviral treatment of HBV-related decompensated cirrhosis
About 2%-5% of patients with HBV-related compensated cirrhosis developed decompensation every year[81]. The prognosis of patients with decompensated cirrhosis is usually poor, with a 5-year survival rate of 14%-35%[82]. All oral antivirals can prevent viral replication efficaciously and improve biochemical and clinical parameters in patients with viral-related decompensated cirrhosis. The selection of antivirals with high efficiency and a low rate of resistance is necessary to attain fast and enduring viral suppression. The use of LAM or TBV is restricted by drug resistance. ADV is restricted by its slower initiation of effect and potential risk of renal injury, which is fatal in decompensated patients. Furthermore, with the more use of TBV, serum creatinine phosphokinase levels frequently increase. Therefore, TBV should be used as a second-line drug for patients with decompensated cirrhosis because of the safety is not guaranteed. A meta-analysis of clinical trials on NA-naive patients with HBeAg-positive CHB has demonstrated that TDF is associated with the highest probability of achieving undetectable HBV DNA at 1 year of all NAs considered[83]. A randomized open-label study has shown that ETV has a virological efficacy precede that of ADV in HBV-related hepatic decompensation[84]. TDF and ETV are well tolerated in these patients, with an improvement in virological, biochemical and clinical parameters[85]. Recently, a meta-analysis showed that LAM and TBV significantly decrease the mortality rate and disease severity in patients with decompensated cirrhosis. Also, both of them promote HBeAg seroconversion in these patients[86]. NAs with low rates of inducing drug-resistant mutations and powerful and rapid HBV suppressive function, such as ETV or TDF, could be regarded as the first-line drugs for NAs-naive patients with decompensated cirrhosis for long-term therapy[84-91]. Even with low doses, the application of IFN-α in patients with HBV-related decompensated cirrhosis can facilitate clinical decompensation and increase the risk of bacterial infection[92]. IFN is contraindicated in patients with HBV-related decompensated cirrhosis in the era of NAs. The studies using antivirals for the treatment of cirrhosis are listed in Table 1.
Table 1.
Current regimes of antiviral treatment for virus-related cirrhosis
| Medicine used | Number of patients | Virological responses | Survival | Ref. |
| HBV-related cirrhosis | ||||
| ETV (1.0 mg/d) or ADV (10 mg/d) | 100 subjects treated with ETV, 91 subjects treated with ADV | 57% and 20% of subjects achieve HBV DNA undetectable after 48 wk (ETV and ADV, respectively) | Overall 1-yr patient survival rates were 84% and 83% (ETV and ADV, respectively) | [83] |
| TBV (600 mg/d) or LAM (100 mg/d) | 114 in the TBV group, another 114 in LAM group | 49.1% and 39.5% of subjects achieve HBV DNA undetectable after 104 wk (TBV and LAM, respectively) | Overall 1-yr patient survival rates were 94% and 88% (TBV and LAM, respectively) | [85] |
| ETV (0.5 mg/d) | 144 compensated and 55 decompensated cirrhosis patients treated with ETV | 78.5% and 89.1% of subjects achieve HBV DNA undetectable after 12 mo (compensated and decompensated, respectively) | Not analyzed | [86] |
| TDF (300 mg/d) or TDF (300 mg/d) + FTC (200 mg/d) or ETV (0.5 mg/d) | 45 in TDF group, 45 in TDF plus FTC group and 22 in ETV group | 70.5%, 87.8% and 72.7% achieve HBV DNA undetectable after 48 wk (TDF, TDF plus FTC, ETV, respectively) | Not analyzed | [84] |
| ADV (10 mg/d) | 226 wait-listed and 241 post-LT patients | 59% and 40% achieve HBV DNA undetectable after 48 wk (wait-listed and post transplantation, respectively) | Overall 1-yr patient survival rates were 86% and 91% (wait-listed and post transplantation, respectively) | [87] |
| HCV-related cirrhosis | ||||
| Group 1: PegIFN-2a + RBV2 Group 2: PegIFN-2b + RBV3 Group 3: PegIFN-2a + placebo4 | 453 in group 1, 444 in group 2 and 224 in group 3 | 69%, 52% and 59% achieve SVR1 after 24 wk (group 1, group 2 and group 3, respectively) | Not analyzed | [81] |
| Treated: IFN5 Untreated: placebo | 72 patients in both treated group and untreated group | Not analyzed | 5-yr overall survival is 50% and 39% in treated and untreated group, respectively | [89] |
| Non-LT and LT cirrhotic patients are all treated for PEG-IFN a2a or a2b plus RBV6 | 43 HCV non-LT cirrhotic patients and 17 LT HCV related-cirrhotic patients | 69.8% and 47.1% achieve EVR and 41.9% and 29.4% achieve SVR (non-LT group and LT group, respectively) | None of the non-LT cirrhotic patients died; LT cirrhotic patients with survival rates of 87% at 1 yr and of 76% at 3 and 5 yr after the treatment | [97] |
Undetectable HCV RNA in serum after 24 wk of post-treatment follow-up;
Once-weekly injections of 180 mcg of PegIFN-α2a plus RBV (1000 mg/d);
Thrice-weekly injections of 3 million units of PegIFN-α2b plus RBV (1000 mg/d);
Once-weekly injections of 180 mcg of PegIFN-α2a plus daily placebo;
Treatment started with 1 mega unit three times weekly for three months then increased every three months to 3, 6, and 9 mega units;
Peg-IFN-α2a or -α2b: at the dose of 180 mcg/wk or 1.5 mcg/kg per week respectively; and RBV 800-1200 mg, based on body weight. HBV: Hepatitis B virus; ETV: Entecavir; TBV: Telbivudine; LAM: Lamivudine; ETV: Entecavir; ADV: Adefovir; TDF: Tenofovir disoproxil fumarate; FTC: Emtricitabine; HCV: Hepatitis C virus; SVR: Sustained virological response; PegIFN: Peginterferon; RBV: Ribavirin; CHB: Chronic hepatitis B; IFN: Interferon; LT: Liver transplanted; EVR: Early virological response.
Antiviral therapy using newer NAs could delay or obviate liver transplantation in some patients[76]. Some clinical guidelines suggest that the clinical improvement in some wait-listed patients with antiviral therapy could lead to their retreat from the transplant list[93,94]. However, if the decompensation is caused by superimposed viral infection, the effect of anti-HBV therapy would be limited. In this situation, liver transplantation should be the most immediate option. Standard of therapy for patients with HBV-related decompensated cirrhosis in accordance with their clinical manifestations, including control of ascites, infection or encephalopathy, should be instituted quickly and sufficiently[93]. Monitoring of HCC and timely transferring consultation for liver transplantation are also mandatory[93,94].
Antiviral treatment of HCV-related cirrhosis
The antiviral treatment of HCV-related cirrhosis poses much greater challenges. IFN remains an essential element of HCV antiviral treatment, but has reduced efficacy and significant toxicity at this stage of cirrhosis[79]. The current standard of care for HCV patients is the therapy with pegylated interferon (Peg-IFN) and ribavirin (RBV), leading to 45% of cure for genotype 1 HCV patients and approximately 80% for patients infected with HCV genotypes 2 and 3[95]. The current standard of care for chronic hepatitis C is the combination of PEG-IFNα and RBV[96]. A recent study provides solid evidence that anti-HCV treatment using recombinant or pegylated IFN plus RBV is equally effective in compensated cirrhosis both before and after liver transplantation[97]. Several clinical trials have demonstrated that the new direct acting antivirals, such as Sofosbuvir, Daclastavir, Asunaprevir, ABT-450, Faldaprevir, Simeprevir, and Deleobuvir, interact with several of vital components of HCV (NS3/4, NS5A, NS5B polymerase, etc.) and have a chance of viral eradication of 80%-90%, with a few negligible side effects[98]. The combination of vaniprevir (a NS3/4A protease inhibitor) with Peg-IFN plus RBV significantly increase rates of sustained virologic response (SVR) among treatment-experienced patients with chronic HCV genotype 1 infection, compared to re-treatment with Peg-IFN plus RBV alone. Vaniprevir is generally well-tolerated for up to 48 wk in those with compensated cirrhosis[99]. Eltrombopag (an oral, nonpeptide, thrombopoietin receptor agonist) can significantly increase platelet numbers in thrombocytopenic patients with HCV-induced cirrhosis, allowing otherwise ineligible or marginal patients to begin and maintain antiviral therapy and leading to significantly increased rates of SVR[100]. It has been well-established that the CC genotype of the genetic polymorphism rs12979860 located at 3 kilobases upstream of the IL28B gene, encoding IFN-lambda-3, is associated with spontaneous clearance of HCV infection and an approximately 2-fold change in response to treatment with Peg-IFN plus RBV[101,102]. The frequency of C allele of rs12979860 is 80.3% in subjects of European ancestry and 56.2% in those of African ancestry[101], which might be one of reasons why the European population is more apt to eradicate HCV than the African population. The frequency of rs12979860 CC genotype is 84.1% in Chinese HCV-positive patients[11], indicating that HCV-related cirrhosis in Chinese HCV carriers should not be as common as in HCV carriers of African ancestry. Subsequent studies have also demonstrated that the IL-28B rs8099917 genotype TT significantly predict SVR in patients chronically infected with HCV genotype 1 to PEG-IFN-RBV therapy[96,103]. The prediction of nonresponse to the treatment is mandatory to avoid side effects and reduce costs[104]. Genotyping the IL-28B rs12979860 and/or rs8099917 should be considered before the treatment of HCV-related cirrhosis. The baseline mean model of end stage-liver disease (MELD) score predicts the risk of hepatic decompensation during antiviral therapy[105], and should be considered for the treatment of HCV-related cirrhosis with peg-IFN and RBV. Thus, antiviral treatment with PEG-IFN+RBV and/or DAAs is recommended to prevent the progression of HCV-related cirrhosis in patients with the CC genotype of rs12979860 and/or the TT genotype of rs8099917 polymorphisms.
Potential stem cell treatment for decompensated cirrhosis
Liver transplantation is the currently last option for the treatment of virus-related decompensated cirrhosis patients who fail to respond to antiviral treatments. Due to the lack of donors, surgical complications, rejection reactions, and high cost for liver transplantation, other strategies have been considered for the treatment of decompensated cirrhosis. Of those, stem cell-based treatments can be expected to be an alternative for patients with liver failure or decompensated cirrhosis because it may improve scarring and supplement hepatocytes[106,107]. Repopulation induced hepatic stem cells (iHepSCs) can become hepatocyte-like cells in the injured liver of fumarylacetoacetate hydrolase (Fah)-deficient mice[108]. Mesenchymal stem cells (MSCs) produce inhibitory cytokines or induce the development of regulatory T cells in the inflammatory and fibrotic processes, therefore they play an immunomodulatory role in this process although many details remain unknown[109]. Interestingly, MSC therapy seems to be effective in regulating the immune response in liver injury, transplantation, and autoimmunity in both patients in clinical trials and animal models of liver disease[110]. MSCs can directly suppress the activation of the main cell source of ECM, HSCs, via MSC-derived IL-10 and TNF-α, and may also induce HSC apoptosis via the Fas/FasL pathway[111]. Therefore, MSCs are considered to work through multiple mechanisms to harmonize a dynamic, integrated response to liver inflammation and fibrosis, which prevents the progressive distortion of hepatic architecture.
Another actual objective of MSC treatment is to substitute impaired hepatocytes in patients with liver failure or decompensated cirrhosis with exogenous functional hepatocytes[106]. For this reason, induced pluripotent stem (iPS) cells and embryonic stem (ES) cells have been shown to be the most competent, producing large numbers of functional hepatocyte-like cells (HLCs) in both animal models and humans. However, ethical issues and indeterminacy about their reaction in vivo in a proper homoeostatic manner have limited their clinical applications[112]. It is currently unknown whether MSC therapy could induce side effects such as hepatic artery dissection, fibrogenesis, and even tumorigenesis. The long-term clinical significance and safety of stem cell-based therapies should be confirmed in large-scale randomized controlled trials. Thus, the co-transplantation of iPS/ES-derived MSCs and HLCs may offer the potency for a series of new therapeutic interventions for liver diseases[106]. It will be highly important to tailor future stem cell therapies to specific patient types due to the mutable feature of different stem cells (ES, iPS, and MSCs).
Regenerative therapies have the potential to provide minimally invasive procedures with few complications. The potential for stem cells in bone marrow (BM) to differentiate into hepatocytes and intestinal cells was confirmed through detection of Y chromosome-containing cells in samples from female recipients of BM cells (BMCs) from male donors[113-115]. Recent studies showed that use of whole bone marrow as a cell therapy in a rodent model with chronic liver disease led to the evolution of hepatic fibrosis[116]. These studies suggest that BMCs are effective sources for regenerative liver therapy. A study group have found that targeting androgen receptor, which is a key factor in male sexual phenotype in bone marrow mesenchymal stem cells (BM-MSCs), can improve the therapeutic efficacy of transplantation for liver fibrosis[117]. Autologous BMC infusion (ABMI) in patients with cirrhosis is one of regenerative therapies. Serum albumin levels and Child-Pugh scores significantly improved after ABMI therapy, and the most important is lack of adverse effects[118]. Thus, ABMI therapy should be developed as a hopeful option for the treatment of decompensated cirrhosis.
CONCLUSION
The process of virus-inducted cirrhosis is a dynamic, multifaceted network. Inflammation provides a suitable microenvironment for the evolution of viral mutation-selection-adaptation, which in turn causes disease-specific viral mutation pattern. The repeated liver damage and tissue repair eventually progress to fibrosis and cirrhosis. In this process, both HSCs and macrophages are important for the fibrogenesis via excessive accumulation of ECM, and macrophages also promote the formation of fibrosis. Oral NAs can prevent viral replication efficiently in viral-related decompensated cirrhosis, cause the stabilization or improvement of liver function, and improve survival. Antiviral treatment should be started as early as the diagnosis has been confirmed. Targeting key signaling pathway should be effective in halting the progression of cirrhosis. Moreover, stem cell-based treatments could be an option for patients with liver failure or decompensated cirrhosis. Future studies should focus more on insight into the cross-link between the mechanisms and therapeutic options.
Footnotes
P- Reviewers: Amarapurkar DN, Chuang WL, Ido A, Lin ZY, Schemmer P, Tasci I S- Editor: Ma YJ L- Editor: Wang TQ E- Editor: Zhang DN
References
- 1.Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006;45:529–538. doi: 10.1016/j.jhep.2006.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Zhang HW, Yin JH, Li YT, Li CZ, Ren H, Gu CY, Wu HY, Liang XS, Zhang P, Zhao JF, et al. Risk factors for acute hepatitis B and its progression to chronic hepatitis in Shanghai, China. Gut. 2008;57:1713–1720. doi: 10.1136/gut.2008.157149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin J, Zhang H, He Y, Xie J, Liu S, Chang W, Tan X, Gu C, Lu W, Wang H, et al. Distribution and hepatocellular carcinoma-related viral properties of hepatitis B virus genotypes in Mainland China: a community-based study. Cancer Epidemiol Biomarkers Prev. 2010;19:777–786. doi: 10.1158/1055-9965.EPI-09-1001. [DOI] [PubMed] [Google Scholar]
- 4.Yin J, Zhang H, Li C, Gao C, He Y, Zhai Y, Zhang P, Xu L, Tan X, Chen J, et al. Role of hepatitis B virus genotype mixture, subgenotypes C2 and B2 on hepatocellular carcinoma: compared with chronic hepatitis B and asymptomatic carrier state in the same area. Carcinogenesis. 2008;29:1685–1691. doi: 10.1093/carcin/bgm301. [DOI] [PubMed] [Google Scholar]
- 5.Chan HL, Wong GL, Tse CH, Chim AM, Yiu KK, Chan HY, Sung JJ, Wong VW. Hepatitis B virus genotype C is associated with more severe liver fibrosis than genotype B. Clin Gastroenterol Hepatol. 2009;7:1361–1366. doi: 10.1016/j.cgh.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 6.Yin J, Xie J, Zhang H, Shen Q, Han L, Lu W, Han Y, Li C, Ni W, Wang H, et al. Significant association of different preS mutations with hepatitis B-related cirrhosis or hepatocellular carcinoma. J Gastroenterol. 2010;45:1063–1071. doi: 10.1007/s00535-010-0253-1. [DOI] [PubMed] [Google Scholar]
- 7.Lok AS, McMahon BJ. Chronic hepatitis B. Hepatology. 2007;45:507–539. doi: 10.1002/hep.21513. [DOI] [PubMed] [Google Scholar]
- 8.Simmonds P, Bukh J, Combet C, Deléage G, Enomoto N, Feinstone S, Halfon P, Inchauspé G, Kuiken C, Maertens G, et al. Consensus proposals for a unified system of nomenclature of hepatitis C virus genotypes. Hepatology. 2005;42:962–973. doi: 10.1002/hep.20819. [DOI] [PubMed] [Google Scholar]
- 9.Bruno S, Crosignani A, Maisonneuve P, Rossi S, Silini E, Mondelli MU. Hepatitis C virus genotype 1b as a major risk factor associated with hepatocellular carcinoma in patients with cirrhosis: a seventeen-year prospective cohort study. Hepatology. 2007;46:1350–1356. doi: 10.1002/hep.21826. [DOI] [PubMed] [Google Scholar]
- 10.Lee MH, Yang HI, Lu SN, Jen CL, Yeh SH, Liu CJ, Chen PJ, You SL, Wang LY, Chen WJ, et al. Hepatitis C virus seromarkers and subsequent risk of hepatocellular carcinoma: long-term predictors from a community-based cohort study. J Clin Oncol. 2010;28:4587–4593. doi: 10.1200/JCO.2010.29.1500. [DOI] [PubMed] [Google Scholar]
- 11.Rao H, Wei L, Lopez-Talavera JC, Shang J, Chen H, Li J, Xie Q, Gao Z, Wang L, Wei J, et al. Distribution and clinical correlates of viral and host genotypes in Chinese patients with chronic hepatitis C virus infection. J Gastroenterol Hepatol. 2014;29:545–553. doi: 10.1111/jgh.12398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng Y, Du Y, Zhang Q, Han X, Cao G. Human cytidine deaminases facilitate hepatitis B virus evolution and link inflammation and hepatocellular carcinoma. Cancer Lett. 2014;343:161–171. doi: 10.1016/j.canlet.2013.09.041. [DOI] [PubMed] [Google Scholar]
- 13.Lee J, Wu CC, Lee KJ, Chuang TH, Katakura K, Liu YT, Chan M, Tawatao R, Chung M, Shen C, et al. Activation of anti-hepatitis C virus responses via Toll-like receptor 7. Proc Natl Acad Sci USA. 2006;103:1828–1833. doi: 10.1073/pnas.0510801103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 15.Chang J, Block TM, Guo JT. The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antiviral Res. 2012;96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 16.Karimi-Googheri M, Daneshvar H, Nosratabadi R, Zare-Bidaki M, Hassanshahi G, Ebrahim M, Arababadi MK, Kennedy D. Important roles played by TGF-β in hepatitis B infection. J Med Virol. 2014;86:102–108. doi: 10.1002/jmv.23727. [DOI] [PubMed] [Google Scholar]
- 17.Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 2007;13:1324–1332. doi: 10.1038/nm1663. [DOI] [PubMed] [Google Scholar]
- 18.Watanabe A, Hashmi A, Gomes DA, Town T, Badou A, Flavell RA, Mehal WZ. Apoptotic hepatocyte DNA inhibits hepatic stellate cell chemotaxis via toll-like receptor 9. Hepatology. 2007;46:1509–1518. doi: 10.1002/hep.21867. [DOI] [PubMed] [Google Scholar]
- 19.García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: a lesson in détente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- 20.Sadler AJ, Williams BR. Interferon-inducible antiviral effectors. Nat Rev Immunol. 2008;8:559–568. doi: 10.1038/nri2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 22.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 23.Bode JG, Brenndörfer ED, Häussinger D. Hepatitis C virus (HCV) employs multiple strategies to subvert the host innate antiviral response. Biol Chem. 2008;389:1283–1298. doi: 10.1515/BC.2008.147. [DOI] [PubMed] [Google Scholar]
- 24.Thimme R, Wieland S, Steiger C, Ghrayeb J, Reimann KA, Purcell RH, Chisari FV. CD8(+) T cells mediate viral clearance and disease pathogenesis during acute hepatitis B virus infection. J Virol. 2003;77:68–76. doi: 10.1128/JVI.77.1.68-76.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunn C, Brunetto M, Reynolds G, Christophides T, Kennedy PT, Lampertico P, Das A, Lopes AR, Borrow P, Williams K, et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J Exp Med. 2007;204:667–680. doi: 10.1084/jem.20061287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shin EC, Seifert U, Kato T, Rice CM, Feinstone SM, Kloetzel PM, Rehermann B. Virus-induced type I IFN stimulates generation of immunoproteasomes at the site of infection. J Clin Invest. 2006;116:3006–3014. doi: 10.1172/JCI29832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gremion C, Grabscheid B, Wölk B, Moradpour D, Reichen J, Pichler W, Cerny A. Cytotoxic T lymphocytes derived from patients with chronic hepatitis C virus infection kill bystander cells via Fas-FasL interaction. J Virol. 2004;78:2152–2157. doi: 10.1128/JVI.78.4.2152-2157.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larrubia JR, Benito-Martínez S, Calvino M, Sanz-de-Villalobos E, Parra-Cid T. Role of chemokines and their receptors in viral persistence and liver damage during chronic hepatitis C virus infection. World J Gastroenterol. 2008;14:7149–7159. doi: 10.3748/wjg.14.7149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nattermann J, Schneiders AM, Leifeld L, Langhans B, Schulz M, Inchauspé G, Matz B, Brackmann HH, Houghton M, Sauerbruch T, et al. Serum antibodies against the hepatitis C virus E2 protein mediate antibody-dependent cellular cytotoxicity (ADCC) J Hepatol. 2005;42:499–504. doi: 10.1016/j.jhep.2004.12.018. [DOI] [PubMed] [Google Scholar]
- 30.Yan ZH, Fan Y, Wang XH, Mao Q, Deng GH, Wang YM. Relationship between HLA-DR gene polymorphisms and outcomes of hepatitis B viral infections: a meta-analysis. World J Gastroenterol. 2012;18:3119–3128. doi: 10.3748/wjg.v18.i24.3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cangussu LO, Teixeira R, Campos EF, Rampim GF, Mingoti SA, Martins-Filho OA, Gerbase-DeLima M. HLA class II alleles and chronic hepatitis C virus infection. Scand J Immunol. 2011;74:282–287. doi: 10.1111/j.1365-3083.2011.02568.x. [DOI] [PubMed] [Google Scholar]
- 32.Lazarevic I, Djordjevic J, Cupic M, Karalic D, Delic D, Svirtlih N, Simonovic J, Svorcan P, Milic N, Jovanovic T. The influence of single and combined IL28B polymorphisms on response to treatment of chronic hepatitis C. J Clin Virol. 2013;58:254–257. doi: 10.1016/j.jcv.2013.06.014. [DOI] [PubMed] [Google Scholar]
- 33.Ezzikouri S, Alaoui R, Rebbani K, Brahim I, Fakhir FZ, Nadir S, Diepolder H, Khakoo SI, Thursz M, Benjelloun S. Genetic variation in the interleukin-28B gene is associated with spontaneous clearance and progression of hepatitis C virus in Moroccan patients. PLoS One. 2013;8:e54793. doi: 10.1371/journal.pone.0054793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarobe P, Lasarte JJ, Casares N, López-Díaz de Cerio A, Baixeras E, Labarga P, García N, Borrás-Cuesta F, Prieto J. Abnormal priming of CD4(+) T cells by dendritic cells expressing hepatitis C virus core and E1 proteins. J Virol. 2002;76:5062–5070. doi: 10.1128/JVI.76.10.5062-5070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Radziewicz H, Ibegbu CC, Fernandez ML, Workowski KA, Obideen K, Wehbi M, Hanson HL, Steinberg JP, Masopust D, Wherry EJ, et al. Liver-infiltrating lymphocytes in chronic human hepatitis C virus infection display an exhausted phenotype with high levels of PD-1 and low levels of CD127 expression. J Virol. 2007;81:2545–2553. doi: 10.1128/JVI.02021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Golden-Mason L, Palmer B, Klarquist J, Mengshol JA, Castelblanco N, Rosen HR. Upregulation of PD-1 expression on circulating and intrahepatic hepatitis C virus-specific CD8+ T cells associated with reversible immune dysfunction. J Virol. 2007;81:9249–9258. doi: 10.1128/JVI.00409-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Radziewicz H, Ibegbu CC, Hon H, Osborn MK, Obideen K, Wehbi M, Freeman GJ, Lennox JL, Workowski KA, Hanson HL, et al. Impaired hepatitis C virus (HCV)-specific effector CD8+ T cells undergo massive apoptosis in the peripheral blood during acute HCV infection and in the liver during the chronic phase of infection. J Virol. 2008;82:9808–9822. doi: 10.1128/JVI.01075-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yao ZQ, Eisen-Vandervelde A, Waggoner SN, Cale EM, Hahn YS. Direct binding of hepatitis C virus core to gC1qR on CD4+ and CD8+ T cells leads to impaired activation of Lck and Akt. J Virol. 2004;78:6409–6419. doi: 10.1128/JVI.78.12.6409-6419.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yao ZQ, Shata MT, Tricoche N, Shan MM, Brotman B, Pfahler W, Hahn YS, Prince AM. gC1qR expression in chimpanzees with resolved and chronic infection: potential role of HCV core/gC1qR-mediated T cell suppression in the outcome of HCV infection. Virology. 2006;346:324–337. doi: 10.1016/j.virol.2005.11.020. [DOI] [PubMed] [Google Scholar]
- 40.Brenndörfer ED, Brass A, Söderholm J, Frelin L, Aleman S, Bode JG, Sällberg M. Hepatitis C virus non-structural 3/4A protein interferes with intrahepatic interferon-γ production. Gut. 2012;61:589–596. doi: 10.1136/gut.2010.232116. [DOI] [PubMed] [Google Scholar]
- 41.Riezu-Boj JI, Larrea E, Aldabe R, Guembe L, Casares N, Galeano E, Echeverria I, Sarobe P, Herrero I, Sangro B, et al. Hepatitis C virus induces the expression of CCL17 and CCL22 chemokines that attract regulatory T cells to the site of infection. J Hepatol. 2011;54:422–431. doi: 10.1016/j.jhep.2010.07.014. [DOI] [PubMed] [Google Scholar]
- 42.Belkaid Y. Regulatory T cells and infection: a dangerous necessity. Nat Rev Immunol. 2007;7:875–888. doi: 10.1038/nri2189. [DOI] [PubMed] [Google Scholar]
- 43.Chen L, Zhang Q, Chang W, Du Y, Zhang H, Cao G. Viral and host inflammation-related factors that can predict the prognosis of hepatocellular carcinoma. Eur J Cancer. 2012;48:1977–1987. doi: 10.1016/j.ejca.2012.01.015. [DOI] [PubMed] [Google Scholar]
- 44.Han YF, Zhao J, Ma LY, Yin JH, Chang WJ, Zhang HW, Cao GW. Factors predicting occurrence and prognosis of hepatitis-B-virus-related hepatocellular carcinoma. World J Gastroenterol. 2011;17:4258–4270. doi: 10.3748/wjg.v17.i38.4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bièche I, Asselah T, Laurendeau I, Vidaud D, Degot C, Paradis V, Bedossa P, Valla DC, Marcellin P, Vidaud M. Molecular profiling of early stage liver fibrosis in patients with chronic hepatitis C virus infection. Virology. 2005;332:130–144. doi: 10.1016/j.virol.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 46.Takahara Y, Takahashi M, Zhang QW, Wagatsuma H, Mori M, Tamori A, Shiomi S, Nishiguchi S. Serial changes in expression of functionally clustered genes in progression of liver fibrosis in hepatitis C patients. World J Gastroenterol. 2008;14:2010–2022. doi: 10.3748/wjg.14.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Utsunomiya T, Okamoto M, Hashimoto M, Yoshinaga K, Shiraishi T, Tanaka F, Mimori K, Inoue H, Watanabe G, Barnard GF, et al. A gene-expression signature can quantify the degree of hepatic fibrosis in the rat. J Hepatol. 2004;41:399–406. doi: 10.1016/j.jhep.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 48.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 49.Hösel M, Quasdorff M, Wiegmann K, Webb D, Zedler U, Broxtermann M, Tedjokusumo R, Esser K, Arzberger S, Kirschning CJ, et al. Not interferon, but interleukin-6 controls early gene expression in hepatitis B virus infection. Hepatology. 2009;50:1773–1782. doi: 10.1002/hep.23226. [DOI] [PubMed] [Google Scholar]
- 50.Borkham-Kamphorst E, Stoll D, Gressner AM, Weiskirchen R. Antisense strategy against PDGF B-chain proves effective in preventing experimental liver fibrogenesis. Biochem Biophys Res Commun. 2004;321:413–423. doi: 10.1016/j.bbrc.2004.06.153. [DOI] [PubMed] [Google Scholar]
- 51.Wietzke-Braun P, Manhardt LB, Rosenberger A, Uy A, Ramadori G, Mihm S. Spontaneous elimination of hepatitis C virus infection: a retrospective study on demographic, clinical, and serological correlates. World J Gastroenterol. 2007;13:4224–4229. doi: 10.3748/wjg.v13.i31.4224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bowen DG, Walker CM. Mutational escape from CD8+ T cell immunity: HCV evolution, from chimpanzees to man. J Exp Med. 2005;201:1709–1714. doi: 10.1084/jem.20050808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernandez J, Taylor D, Morhardt DR, Mihalik K, Puig M, Rice CM, Feinstone SM, Major ME. Long-term persistence of infection in chimpanzees inoculated with an infectious hepatitis C virus clone is associated with a decrease in the viral amino acid substitution rate and low levels of heterogeneity. J Virol. 2004;78:9782–9789. doi: 10.1128/JVI.78.18.9782-9789.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Osiowy C, Giles E, Tanaka Y, Mizokami M, Minuk GY. Molecular evolution of hepatitis B virus over 25 years. J Virol. 2006;80:10307–10314. doi: 10.1128/JVI.00996-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Peng ZG, Zhao ZY, Li YP, Wang YP, Hao LH, Fan B, Li YH, Wang YM, Shan YQ, Han YX, et al. Host apolipoprotein B messenger RNA-editing enzyme catalytic polypeptide-like 3G is an innate defensive factor and drug target against hepatitis C virus. Hepatology. 2011;53:1080–1089. doi: 10.1002/hep.24160. [DOI] [PubMed] [Google Scholar]
- 56.Vartanian JP, Henry M, Marchio A, Suspène R, Aynaud MM, Guétard D, Cervantes-Gonzalez M, Battiston C, Mazzaferro V, Pineau P, et al. Massive APOBEC3 editing of hepatitis B viral DNA in cirrhosis. PLoS Pathog. 2010;6:e1000928. doi: 10.1371/journal.ppat.1000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yin J, Xie J, Liu S, Zhang H, Han L, Lu W, Shen Q, Xu G, Dong H, Shen J, et al. Association between the various mutations in viral core promoter region to different stages of hepatitis B, ranging of asymptomatic carrier state to hepatocellular carcinoma. Am J Gastroenterol. 2011;106:81–92. doi: 10.1038/ajg.2010.399. [DOI] [PubMed] [Google Scholar]
- 58.Zhang Q, Yin J, Zhang Y, Deng Y, Ji X, Du Y, Pu R, Han Y, Zhao J, Han X, et al. HLA-DP polymorphisms affect the outcomes of chronic hepatitis B virus infections, possibly through interacting with viral mutations. J Virol. 2013;87:12176–12186. doi: 10.1128/JVI.02073-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen CH, Hung CH, Lee CM, Hu TH, Wang JH, Wang JC, Lu SN, Changchien CS. Pre-S deletion and complex mutations of hepatitis B virus related to advanced liver disease in HBeAg-negative patients. Gastroenterology. 2007;133:1466–1474. doi: 10.1053/j.gastro.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 60.Liu S, Zhang H, Gu C, Yin J, He Y, Xie J, Cao G. Associations between hepatitis B virus mutations and the risk of hepatocellular carcinoma: a meta-analysis. J Natl Cancer Inst. 2009;101:1066–1082. doi: 10.1093/jnci/djp180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamori A, Kawada N. HLA class II associated with outcomes of hepatitis B and C infections. World J Gastroenterol. 2013;19:5395–5401. doi: 10.3748/wjg.v19.i33.5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kitab B, Essaid El Feydi A, Afifi R, Trepo C, Benazzouz M, Essamri W, Zoulim F, Chemin I, Alj HS, Ezzikouri S, et al. Variability in the precore and core promoter regions of HBV strains in Morocco: characterization and impact on liver disease progression. PLoS One. 2012;7:e42891. doi: 10.1371/journal.pone.0042891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee JH, Han KH, Lee JM, Park JH, Kim HS. Impact of hepatitis B virus (HBV) x gene mutations on hepatocellular carcinoma development in chronic HBV infection. Clin Vaccine Immunol. 2011;18:914–921. doi: 10.1128/CVI.00474-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brenner DA, Kisseleva T, Scholten D, Paik YH, Iwaisako K, Inokuchi S, Schnabl B, Seki E, De Minicis S, Oesterreicher C, et al. Origin of myofibroblasts in liver fibrosis. Fibrogenesis Tissue Repair. 2012;5 Suppl 1:S17. doi: 10.1186/1755-1536-5-S1-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev. 2008;88:125–172. doi: 10.1152/physrev.00013.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang JX, Mikami K, Shah VH, Torok NJ. Leptin induces phagocytosis of apoptotic bodies by hepatic stellate cells via a Rho guanosine triphosphatase-dependent mechanism. Hepatology. 2008;48:1497–1505. doi: 10.1002/hep.22515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao G, Hatting M, Nevzorova YA, Peng J, Hu W, Boekschoten MV, Roskams T, Muller M, Gassler N, Liedtke C, et al. Jnk1 in murine hepatic stellate cells is a crucial mediator of liver fibrogenesis. Gut. 2013:Epub ahead of print. doi: 10.1136/gutjnl-2013-305507. [DOI] [PubMed] [Google Scholar]
- 68.Kluwe J, Pradere JP, Gwak GY, Mencin A, De Minicis S, Osterreicher CH, Colmenero J, Bataller R, Schwabe RF. Modulation of hepatic fibrosis by c-Jun-N-terminal kinase inhibition. Gastroenterology. 2010;138:347–359. doi: 10.1053/j.gastro.2009.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. J Clin Invest. 2007;117:539–548. doi: 10.1172/JCI30542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Forbes SJ, Parola M. Liver fibrogenic cells. Best Pract Res Clin Gastroenterol. 2011;25:207–217. doi: 10.1016/j.bpg.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 71.Shrivastava S, Mukherjee A, Ray R, Ray RB. Hepatitis C virus induces interleukin-1β (IL-1β)/IL-18 in circulatory and resident liver macrophages. J Virol. 2013;87:12284–12290. doi: 10.1128/JVI.01962-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology. 2014;59:2034–2042. doi: 10.1002/hep.26754. [DOI] [PubMed] [Google Scholar]
- 73.Friedman SL. Cytokines and fibrogenesis. Semin Liver Dis. 1999;19:129–140. doi: 10.1055/s-2007-1007105. [DOI] [PubMed] [Google Scholar]
- 74.Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, Wu S, Lang R, Iredale JP. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest. 2005;115:56–65. doi: 10.1172/JCI22675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen LP, Zhao J, Du Y, Han YF, Su T, Zhang HW, Cao GW. Antiviral treatment to prevent chronic hepatitis B or C-related hepatocellular carcinoma. World J Virol. 2012;1:174–183. doi: 10.5501/wjv.v1.i6.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Papatheodoridis GV, Manolakopoulos S, Archimandritis AJ. Current treatment indications and strategies in chronic hepatitis B virus infection. World J Gastroenterol. 2008;14:6902–6910. doi: 10.3748/wjg.14.6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yuen MF, Lai CL. Treatment of chronic hepatitis B: Evolution over two decades. J Gastroenterol Hepatol. 2011;26 Suppl 1:138–143. doi: 10.1111/j.1440-1746.2010.06545.x. [DOI] [PubMed] [Google Scholar]
- 78.Lok AS, McMahon BJ. Chronic hepatitis B: update 2009. Hepatology. 2009;50:661–662. doi: 10.1002/hep.23190. [DOI] [PubMed] [Google Scholar]
- 79.Mutimer DJ, Lok A. Management of HBV- and HCV-induced end stage liver disease. Gut. 2012;61 Suppl 1:i59–i67. doi: 10.1136/gutjnl-2012-302076. [DOI] [PubMed] [Google Scholar]
- 80.Peng CY, Chien RN, Liaw YF. Hepatitis B virus-related decompensated liver cirrhosis: benefits of antiviral therapy. J Hepatol. 2012;57:442–450. doi: 10.1016/j.jhep.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 81.Roche B, Samuel D. Treatment of patients with HBV-related decompensated cirrhosis and liver transplanted patients. Clin Liver Dis. 2013;17:451–473. doi: 10.1016/j.cld.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 82.Chen YC, Chu CM, Yeh CT, Liaw YF. Natural course following the onset of cirrhosis in patients with chronic hepatitis B: a long-term follow-up study. Hepatol Int. 2007;1:267–273. doi: 10.1007/s12072-007-5001-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dakin H, Fidler C, Harper C. Mixed treatment comparison meta-analysis evaluating the relative efficacy of nucleos(t)ides for treatment of nucleos(t)ide-naive patients with chronic hepatitis B. Value Health. 2010;13:934–945. doi: 10.1111/j.1524-4733.2010.00777.x. [DOI] [PubMed] [Google Scholar]
- 84.Liaw YF, Raptopoulou-Gigi M, Cheinquer H, Sarin SK, Tanwandee T, Leung N, Peng CY, Myers RP, Brown RS, Jeffers L, et al. Efficacy and safety of entecavir versus adefovir in chronic hepatitis B patients with hepatic decompensation: a randomized, open-label study. Hepatology. 2011;54:91–100. doi: 10.1002/hep.24361. [DOI] [PubMed] [Google Scholar]
- 85.Liaw YF, Sheen IS, Lee CM, Akarca US, Papatheodoridis GV, Suet-Hing Wong F, Chang TT, Horban A, Wang C, Kwan P, Buti M, Prieto M, Berg T, Kitrinos K, Peschell K, Mondou E, Frederick D, Rousseau F, Schiff ER. Tenofovir disoproxil fumarate (TDF), emtricitabine/TDF, and entecavir in patients with decompensated chronic hepatitis B liver disease. Hepatology. 2011;53:62–72. doi: 10.1002/hep.23952. [DOI] [PubMed] [Google Scholar]
- 86.Huang Y, Wu H, Wu S, Fu D, Ma Y, Shen X. A meta-analysis of nucleos(t)ide analogues in patients with decompensated cirrhosis due to hepatitis B. Dig Dis Sci. 2013;58:815–823. doi: 10.1007/s10620-012-2414-y. [DOI] [PubMed] [Google Scholar]
- 87.Chan HL, Chen YC, Gane EJ, Sarin SK, Suh DJ, Piratvisuth T, Prabhakar B, Hwang SG, Choudhuri G, Safadi R, et al. Randomized clinical trial: efficacy and safety of telbivudine and lamivudine in treatment-naïve patients with HBV-related decompensated cirrhosis. J Viral Hepat. 2012;19:732–743. doi: 10.1111/j.1365-2893.2012.01600.x. [DOI] [PubMed] [Google Scholar]
- 88.Shim JH, Lee HC, Kim KM, Lim YS, Chung YH, Lee YS, Suh DJ. Efficacy of entecavir in treatment-naïve patients with hepatitis B virus-related decompensated cirrhosis. J Hepatol. 2010;52:176–182. doi: 10.1016/j.jhep.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 89.Schiff E, Lai CL, Hadziyannis S, Neuhaus P, Terrault N, Colombo M, Tillmann H, Samuel D, Zeuzem S, Villeneuve JP, et al. Adefovir dipivoxil for wait-listed and post-liver transplantation patients with lamivudine-resistant hepatitis B: final long-term results. Liver Transpl. 2007;13:349–360. doi: 10.1002/lt.20981. [DOI] [PubMed] [Google Scholar]
- 90.Fontana RJ, Hann HW, Perrillo RP, Vierling JM, Wright T, Rakela J, Anschuetz G, Davis R, Gardner SD, Brown NA. Determinants of early mortality in patients with decompensated chronic hepatitis B treated with antiviral therapy. Gastroenterology. 2002;123:719–727. doi: 10.1053/gast.2002.35352. [DOI] [PubMed] [Google Scholar]
- 91.Lian JS, Zeng LY, Chen JY, Jia HY, Zhang YM, Xiang DR, Yu L, Hu JH, Lu YF, Zheng L, et al. De novo combined lamivudine and adefovir dipivoxil therapy vs entecavir monotherapy for hepatitis B virus-related decompensated cirrhosis. World J Gastroenterol. 2013;19:6278–6283. doi: 10.3748/wjg.v19.i37.6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.European Association For The Study Of The Liver. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J Hepatol. 2012;57:167–185. doi: 10.1016/j.jhep.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 93.Garcia-Tsao G, Lim JK. Management and treatment of patients with cirrhosis and portal hypertension: recommendations from the Department of Veterans Affairs Hepatitis C Resource Center Program and the National Hepatitis C Program. Am J Gastroenterol. 2009;104:1802–1829. doi: 10.1038/ajg.2009.191. [DOI] [PubMed] [Google Scholar]
- 94.Murray KF, Carithers RL. AASLD practice guidelines: Evaluation of the patient for liver transplantation. Hepatology. 2005;41:1407–1432. doi: 10.1002/hep.20704. [DOI] [PubMed] [Google Scholar]
- 95.Fried MW, Shiffman ML, Reddy KR, Smith C, Marinos G, Gonçales FL, Häussinger D, Diago M, Carosi G, Dhumeaux D, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 96.European Association of the Study of the Liver. 2011 European Association of the Study of the Liver hepatitis C virus clinical practice guidelines. Liver Int. 2012;32 Suppl 1:2–8. doi: 10.1111/j.1478-3231.2011.02703.x. [DOI] [PubMed] [Google Scholar]
- 97.Ponziani FR, Annicchiarico EB, Siciliano M, D’Aversa F, Pompili M, Gasbarrini A. Treatment of hepatitis C in compensated cirrhotic patients is equally effective before and after liver transplantation. World J Gastroenterol. 2013;19:3255–3262. doi: 10.3748/wjg.v19.i21.3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marinho RT, Barreira DP. Hepatitis C, stigma and cure. World J Gastroenterol. 2013;19:6703–6709. doi: 10.3748/wjg.v19.i40.6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rodriguez-Torres M, Stoehr A, Gane EJ, Serfaty L, Lawitz E, Zhou A, Bourque M, Bhanja S, Strizki J, Barnard RJ, et al. Combination of Vaniprevir With Peginterferon and Ribavirin Significantly Increases the Rate of SVR in Treatment-Experienced Patients With Chronic HCV Genotype 1 Infection and Cirrhosis. Clin Gastroenterol Hepatol. 2013:Epub ahead of print. doi: 10.1016/j.cgh.2013.09.067. [DOI] [PubMed] [Google Scholar]
- 100.Afdhal NH, Dusheiko GM, Giannini EG, Chen PJ, Han KH, Mohsin A, Rodriguez-Torres M, Rugina S, Bakulin I, Lawitz E, et al. Eltrombopag increases platelet numbers in thrombocytopenic patients with HCV infection and cirrhosis, allowing for effective antiviral therapy. Gastroenterology. 2014;146:442–52.e1. doi: 10.1053/j.gastro.2013.10.012. [DOI] [PubMed] [Google Scholar]
- 101.Thomas DL, Thio CL, Martin MP, Qi Y, Ge D, O’Huigin C, Kidd J, Kidd K, Khakoo SI, Alexander G, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461:798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 103.Luo Y, Jin C, Ling Z, Mou X, Zhang Q, Xiang C. Association study of IL28B: rs12979860 and rs8099917 polymorphisms with SVR in patients infected with chronic HCV genotype 1 to PEG-INF/RBV therapy using systematic meta-analysis. Gene. 2013;513:292–296. doi: 10.1016/j.gene.2012.10.030. [DOI] [PubMed] [Google Scholar]
- 104.Estrabaud E, Vidaud M, Marcellin P, Asselah T. Genomics and HCV infection: progression of fibrosis and treatment response. J Hepatol. 2012;57:1110–1125. doi: 10.1016/j.jhep.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 105.Dultz G, Seelhof M, Herrmann E, Welker MW, Friedrich-Rust M, Teuber G, Kronenberger B, von Wagner M, Vermehren J, Sarrazin C, et al. Baseline MELD score predicts hepatic decompensation during antiviral therapy in patients with chronic hepatitis C and advanced cirrhosis. PLoS One. 2013;8:e71262. doi: 10.1371/journal.pone.0071262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhang Z, Wang FS. Stem cell therapies for liver failure and cirrhosis. J Hepatol. 2013;59:183–185. doi: 10.1016/j.jhep.2013.01.018. [DOI] [PubMed] [Google Scholar]
- 107.Forbes SJ, Newsome PN. New horizons for stem cell therapy in liver disease. J Hepatol. 2012;56:496–499. doi: 10.1016/j.jhep.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 108.Yu B, He ZY, You P, Han QW, Xiang D, Chen F, Wang MJ, Liu CC, Lin XW, Borjigin U, et al. Reprogramming fibroblasts into bipotential hepatic stem cells by defined factors. Cell Stem Cell. 2013;13:328–340. doi: 10.1016/j.stem.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 109.Sun L, Akiyama K, Zhang H, Yamaza T, Hou Y, Zhao S, Xu T, Le A, Shi S. Mesenchymal stem cell transplantation reverses multiorgan dysfunction in systemic lupus erythematosus mice and humans. Stem Cells. 2009;27:1421–1432. doi: 10.1002/stem.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Uccelli A, Moretta L, Pistoia V. Mesenchymal stem cells in health and disease. Nat Rev Immunol. 2008;8:726–736. doi: 10.1038/nri2395. [DOI] [PubMed] [Google Scholar]
- 111.Akiyama K, Chen C, Wang D, Xu X, Qu C, Yamaza T, Cai T, Chen W, Sun L, Shi S. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell. 2012;10:544–555. doi: 10.1016/j.stem.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang P, He Z, Ji S, Sun H, Xiang D, Liu C, Hu Y, Wang X, Hui L. Induction of functional hepatocyte-like cells from mouse fibroblasts by defined factors. Nature. 2011;475:386–389. doi: 10.1038/nature10116. [DOI] [PubMed] [Google Scholar]
- 113.Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Teperman L, Henegariu O, Krause DS. Liver from bone marrow in humans. Hepatology. 2000;32:11–16. doi: 10.1053/jhep.2000.9124. [DOI] [PubMed] [Google Scholar]
- 114.Alison MR, Poulsom R, Jeffery R, Dhillon AP, Quaglia A, Jacob J, Novelli M, Prentice G, Williamson J, Wright NA. Hepatocytes from non-hepatic adult stem cells. Nature. 2000;406:257. doi: 10.1038/35018642. [DOI] [PubMed] [Google Scholar]
- 115.Okamoto R, Yajima T, Yamazaki M, Kanai T, Mukai M, Okamoto S, Ikeda Y, Hibi T, Inazawa J, Watanabe M. Damaged epithelia regenerated by bone marrow-derived cells in the human gastrointestinal tract. Nat Med. 2002;8:1011–1017. doi: 10.1038/nm755. [DOI] [PubMed] [Google Scholar]
- 116.Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon-Walker TT, Hartland S, Ramachandran P, Van Deemter M, Hume DA, Iredale JP, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology. 2011;53:2003–2015. doi: 10.1002/hep.24315. [DOI] [PubMed] [Google Scholar]
- 117.Huang CK, Lee SO, Lai KP, Ma WL, Lin TH, Tsai MY, Luo J, Chang C. Targeting androgen receptor in bone marrow mesenchymal stem cells leads to better transplantation therapy efficacy in liver cirrhosis. Hepatology. 2013;57:1550–1563. doi: 10.1002/hep.26135. [DOI] [PubMed] [Google Scholar]
- 118.Terai S, Ishikawa T, Omori K, Aoyama K, Marumoto Y, Urata Y, Yokoyama Y, Uchida K, Yamasaki T, Fujii Y, et al. Improved liver function in patients with liver cirrhosis after autologous bone marrow cell infusion therapy. Stem Cells. 2006;24:2292–2298. doi: 10.1634/stemcells.2005-0542. [DOI] [PubMed] [Google Scholar]