

Background: Glutamate-induced excitotoxicity is a poorly understood but major cause of neuronal degeneration.
Results: Glutamate increased membrane translocation of Annexin A2 (AnxA2), thereby increasing plasmin generation.
Conclusion: Glutamate-induced membrane translocation of AnxA2 is an important mediator of excitotoxicity.
Significance: Our study provides a mechanistic insight into glutamate-induced excitotoxicity and signifies AnxA2 as a potential target in neurodegenerative diseases.
Keywords: Annexin, Calcium, Cell Surface Protein, Glutamate, Plasmin, Proteolysis
Abstract
Glutamate-induced elevation in intracellular Ca2+ has been implicated in excitotoxic cell death. Neurons respond to increased glutamate levels by activating an extracellular proteolytic cascade involving the components of the plasmin-plasminogen system. AnxA2 is a Ca2+-dependent phospholipid binding protein and serves as an extracellular proteolytic center by recruiting the tissue plasminogen activator and plasminogen and mediating the localized generation of plasmin. Ratiometric Ca2+ imaging and time-lapse confocal microscopy demonstrated glutamate-induced Ca2+ influx. We showed that glutamate translocated both endogenous and AnxA2-GFP to the cell surface in a process dependent on the activity of the NMDA receptor. Glutamate-induced translocation of AnxA2 is dependent on the phosphorylation of tyrosine 23 at the N terminus, and mutation of tyrosine 23 to a non-phosphomimetic variant inhibits the translocation process. The cell surface-translocated AnxA2 forms an active plasmin-generating complex, and this activity can be neutralized by a hexapeptide directed against the N terminus. These results suggest an involvement of AnxA2 in potentiating glutamate-induced cell death processes.
Introduction
Excessive accumulation of excitatory amino acids such as glutamate has been implicated in the pathogenesis of several neurodegenerative diseases (1). The hypothesis of glutamate excitotoxicity has been proposed to explain the molecular mechanisms of glutamate-induced neuronal dysfunction and degeneration (2, 3). Glutamate-induced intracellular increase in Ca2+ levels leads to the hyperactivation of several normal Ca2+-mediated physiological processes, including the activation of intracellular kinases, phosphatases, phospholipases, and proteases that contribute to the degeneration of the neurons (4). Despite intensive research, the mechanisms that contribute to glutamate-induced cellular loss are yet to be elucidated. In neurodegenerative diseases like age-related macular degeneration and retinitis pigmentosa, glutamate excitotoxicity has been linked to successive loss of photoreceptor cells (5). To have a better understanding of the mechanistic events of glutamate excitotoxicity that lead to retinal degeneration, we investigated the glutamate-induced, Ca2+-mediated signaling events in a cone-photoreceptor cell line, 661W. 661W has been shown to express several markers of photoreceptor cells and to be sensitive to oxidative stress, similar to observations recorded in primary retinal photoreceptor cells (6). Thus, understanding the molecular mechanisms of glutamate-induced retinal degeneration can lead to the development of better therapeutic approaches for neurodegenerative diseases, including age-related macular degeneration and retinitis pigmentosa. Our study provides new insights into one of the mechanisms that might contribute to glutamate-induced loss of photoreceptors in the retina.
Recent evidence suggests that the components of the plasmin-plasminogen system are involved in retinal degeneration after ischemic and excitotoxic injury (7). The plasmin-plasminogen system is composed of a proteolytic cascade comprising the two plasminogen activators, tissue plasminogen activator (tPA)2 and urokinase plasminogen activator, that culminate in the conversion of the inactive zymogen substrate plasminogen to active plasmin (8). Plasmin, a potent trypsin-like endopeptidase, cleaves the components of the extracellular matrix, like laminin, and contributes to the detachment-induced apoptosis or anoikis (8, 9). The culminating step in the plasmin-plasminogen proteolytic cascade is the generation of cell surface plasmin (8). This is a highly regulated process mediated by several cell surface receptors that act as binding sites for both the substrate plasminogen and its activator, tPA (10).
AnxA2 has been identified as a common receptor with distinct domains for the binding of tPA and its substrate, plasminogen (11). AnxA2-mediated coassembly of tPA and plasminogen accelerates the catalytic efficiency of tPA-mediated plasmin generation 60-fold (11). The mechanisms that regulate the plasmin-generating activity of AnxA2 could have potential contributions in the pathogenesis of diseases in which the plasmin-plasminogen system is implicated.
AnxA2 is a member of the family of Ca2+-dependent anionic phospholipid-binding proteins involved in mediating several intracellular actions of Ca2+ (12). The anionic phospholipid- and Ca2+-binding properties of AnxA2 are imparted by the conserved carboxyl terminal core domain, whereas the variable N terminus is the site for posttranslational modifications and interactions with other proteins (13). The Ca2+-binding residues in the conserved domain of AnxA2 comprise four repeats of a 70-amino acid sequence called annexin repeats, and they confer the ability of AnxA2 to interact reversibly with the cellular membranes in a regulated manner (14). The N terminus possesses phosphorylation sites on tyrosine 23, which is a substrate for phosphorylation by Src kinase (12, 15), serine 25, which has been reported to be phosphorylated by protein kinase C in vitro and in vivo (16), and serine 11, which can only be phosphorylated in vitro (17). Studies have also indicated that AnxA2 is initially targeted to the phospholipid component of the plasma membrane and that this binding mediates the phosphorylation of tyrosine 23 by Src kinase (18). Our previous studies have shown that phosphorylation of AnxA2 at tyrosine 23 significantly promotes the transport of AnxA2 to the plasma membrane (19).
Most members of the annexin family, including AnxA2, are recruited to cellular membranes in response to several stimuli that induce intracellular Ca2+ mobilization (20). Interaction of AnxA2 at the N terminus with p11/S100A10 or by phosphorylation has been proposed to play a regulatory role in the Ca2+-dependent association to the cell membranes (21). Here we used a glutamate-induced Ca2+ influx model in 661W cells to examine the Ca2+-induced translocation of AnxA2 and its N-terminal phosphorylation mutants to the outer surface of the plasma membrane. Our data indicate that glutamate induces the cell surface translocation of AnxA2 and that phosphorylation of tyrosine 23 at the N terminus of AnxA2 is an essential prerequisite for the translocation process. We also show that glutamate-induced cell surface translocation of AnxA2 is associated with a concomitant increase in the cell surface generation of plasmin, which can be inhibited by a peptide directed against the N terminus of AnxA2. Taken together, the data shown here suggest that glutamate induces the cell surface translocation of AnxA2, which can potentiate the glutamate-induced degeneration of photoreceptor cells.
MATERIALS AND METHODS
Cell Culture and Glutamate Treatment
Initially, all work was done on a supposed retinal ganglion cell line, RGC-5, but later on it was found to be the transformed mouse photoreceptor cell line 661W (22). 661W was cultured in low-glucose DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen), 100 units/ml penicillin, and 100 μg/ml streptomycin (Invitrogen) in a humidified atmosphere of 95% air and 5% CO2 at 37 °C. Differentiation of the 661W cells was achieved by supplementing the conditioned medium derived from human non-pigmented ciliary epithelial cells according to methods published previously (23). l-Glutamate (Glu) (Sigma-Aldrich) concentrations of up to 500 μm were used on the basis of literature published previously (24–27).
Plasmid Constructs and Transient Transfection
For the construction of a plasmid expressing AnxA2-GFP fusion protein (AnxA2-GFP), full-length AnxA2 was cloned into the pEGFP-N1 vector (Clontech). We used site-directed mutagenesis kits (Clontech) to generate both single and double phosphorylation and non-phosphorylation mutants of AnxA2 at tyrosine 23 and serine 25. The protein products of the mutants will be subsequently called AnxA2Y23E-GFP, AnxA2Y23F-GFP, AnxA2S25E-GFP, AnxA2S5A-GFP, AnxA2S11A-GFP, AnxA2S11E-GFP, AnxA2Y23ES25E-GFP, AnxA2Y23FS25E, AnxA2Y23ES25A, and AnxA2Y23FS25A. Transient transfection was performed according to protocols published previously (28).
Elution of Cell Surface AnxA2
Ca2+-binding proteins on the cell surface were eluted by a procedure described previously (29). Briefly, confluent 661W cells were washed with ice-cold PBS and incubated in the presence of EDTA(Invitrogen) for 20 min at 37 °C. The EDTA washed from the cells is henceforth referred to as EDTA eluates. The EDTA eluates were centrifuged and concentrated using NANOSEP Omega 10K filters (Pall Corp.) and subjected to SDS-PAGE and Western blot analysis. The EDTA eluates were checked for lack of cytosolic proteins by immunoblotting with anti-3-phosphoglycerate kinase (PGK) antibodies.
Cell Surface Biotinylation
661W cells treated with glutamate were washed three times with ice-cold PBS and biotinylated with 0.5 mg/ml of sulfo-N-hydroxysulfosuccinimide (NHS) biotin (Pierce) for 30 min at room temperature according to protocols published previously (30). After a series of washes, the cells were lysed in the presence of Triton-X-100 lysis buffer, and the surface -labeled proteins were purified by incubation with avidin-conjugated Sepharose (Sigma) overnight at 4 °C. After another series of washes, Laemmli sample buffer was added to release the surface proteins, and they were separated by SDS-PAGE (4–12%), transferred onto nitrocellulose membranes, and immunoblotted with anti-AnxA2 antibody (BD Transduction Laboratories) to detect endogenous AnxA2 or anti-GFP antibody (Cell Signaling Technology) to detect AnxA2-GFP fusion protein. pAnxA2 Tyr-23 antibody that specifically recognizes the phosphorylated tyrosine residue at the 23rd position of AnxA2 was obtained from Santa Cruz Biotechnologies.
Total Internal Reflection (TIRF) Microscopy
For TIRF microscopy (31), cells were grown on 22-mm glass coverslips (VWR International) and fixed in 2% ice-cold paraformaldehyde in PBS for 10 min at 4 °C. The cells were treated with anti-AnxA2 antibody (BD Transduction Laboratories, 1:500 dilution) for 3 h, washed three times with PBS, and later treated with appropriate Alexa Fluor-conjugated secondary antibody (Molecular Probes, 1:1000 dilution). The cells were observed under an Olympus IX71 microscope equipped with a commercial TIRF attachment using an Olympus ×60 numerical aperture 1.45 PlanApo oil objective and a Hamamatsu C4742-95 high-resolution digital camera utilizing a progressive scan interline transfer charge-coupled device chip with no mechanical shutter and Peltier cooling. Images were acquired with identical image acquisition parameters to monitor differences in the fluorescence intensity between treated and untreated cells. The TIRF microscope (Olympus IX 71with an Olympus TIRF attachment) was set into TIRF mode by looking at a 100× dilution of suspension of 0.1 μm of fluorescent microspheres (Molecular Probes, catalog no. F8800, FluoSpheres® carboxylate-modified microspheres, 0.1 μm, orange fluorescent, 2% solids). The TIRF mirror was adjusted until microspheres begun to flicker. That is, they would appear briefly and disappear rapidly from the field of view. This indicated that the TIFR angle was adjusted correctly. That is, spheres briefly entered the TIRF volume and rapidly diffused away from it. If the angle was too large, nothing could be seen. If the angle was too small, the TIRF was not defined, and many spheres could be seen with low contrast and no flicker.
Live Cell Imaging of Glutamate-induced Intracellular Ca2+ Dynamics
661W cells were grown on sterile glass coverslips and loaded with 3 μm Ca2+ indicator dye, Fluo-3 (Molecular Probes), in Ca2+-free, HEPES-buffered saline solution for 30 min at 37 °C. Ca2+-dependent fluorescence responses of Fluo-3 were monitored in real time by live cell confocal imaging under a Zeiss LSM 410 microscope using a Zeiss ×40, 1.2 numerical aperture C-Apochromat water immersion objective. After the addition of 500 μm glutamate, cells were imaged at an interval 10 s using LSM 4 software (Carl Zeiss) at 488-nm excitation and 526-nm emission wavelengths. To assess the extent of photobleaching of Fluo-3, the loaded cells were exposed to laser illumination continuously for 5 min.
Intracellular Ca2+ ([Ca2+]i) Measurement
661W cells cultured in Ca2+-free medium were loaded with 3 μm Fura-2/AM (Molecular Probes) incubated with 1 μm thapsigargin, followed by treatment with 500 μm glutamate or 5 μm A23187. Ca2+ mobilization in 661W cells was measured using a ratiometric Ca2+ imaging technique with Fura-2/AM Ca2+ indicator dye at 340- and 380-nm excitation wavelengths and 510-nm emission wavelength according to protocols published previously (32). Imaging was performed using a Diaphot microscope (Nikon) and Metafluor software (Universal Imaging). The nanomolar concentrations of [Ca2+]i was calculated using the Grynkiewicz equation (33).
Immunoprecipitation and Immunoblotting
661W cells were transiently transfected with the wild-type pAnxA2-GFP plasmid. The cells were treated with glutamate for 4 h and subjected to EDTA cell surface elution and whole cell lysis (Triton X-100 lysis buffer and 10% glycerol). The eluates and the lysates were incubated at 4 °C for 12 h with 2 μg of anti-GFP antibody (Cell Signaling Technology) immobilized on agarose beads (Sigma). The beads were washed in Triton X-100 lysis buffer in the presence of 10% glycerol and resuspended in Laemmli sample buffer. The immunoprecipitates were analyzed by SDS-PAGE and subjected to immunoblotting with anti-phosphotyrosine (Cell Signaling Technology) and anti-phosphoserine (Millipore) antibodies. All blots were exposed to identical exposure times.
Plasmin Generation Assay
We performed a chromogenic plasmin generation assay in the presence of a competitive peptide inhibitor for the binding of tPA to AnxA2. 661W cells grown on 12-well plates were treated with 500 μm glutamate for 4 h. After a few washes with PBS, the cells were treated with 10 nm recombinant tPA (Molecular Innovations) in the presence of either an experimental peptide (LCKLSL) or a control peptide (LGKLSL) or in the absence of both for 1 h at 37 °C. The assay was performed according to protocols published previously (34). The reaction was initiated by the addition of 100 nm Glu-plasminogen (American Diagnostica). The reaction was monitored by taking 100-μl aliquots of the reaction mixture every 4 min in a 96-well plate and adding 100 μl of 1 mm chromogenic plasmin substrate, S-255 (Diapharma), dissolved in Hanks' balanced salt solution. The rates of plasmin generation were monitored by measuring the change in absorbance at 405 nm (Synergy HT-BioTek). The data are represented as a fold change in plasmin generation by normalizing the relative fluorescence units per minute of the untreated controls to the treated samples.
Image Analysis
Quantification of AnxA2 cell surface fluorescence was performed using TIRF microscopy by using the “Analyze Particle” plugin in ImageJ (http://rsbweb.nih.gov/ij) set at a fixed threshold. The ImageJ “Measure” function was used to measure the integrated fluorescence density of the selected cell. For background correction, the integrated fluorescence of the background was measured. The corrected total cellular intensity was estimated by multiplying the area of the selected cell and the background fluorescence and subtracting them from the integrated cellular density. For comparison purposes, all images were acquired with identical image acquisition parameters and subjected to background correction by subtracting the mean fluorescence intensity of the area void of cells from that of individual cells. Mean values of fluorescence intensity ± S.E. are given.
Statistical Analysis
Results were expressed as mean ± S.E., and statistical analysis was performed using GraphPad Prism 4.02 software. One sample Student's t test was performed, and p < 0.05 was considered significant.
RESULTS
AnxA2 Is Bound to the Extracellular Surface of 661W Cells in a Ca2+-dependent Manner
One of the most important characteristics of AnxA2 is its ability to interact with the plasma membrane in a Ca2+-dependent manner. Substantial evidence indicates that an elevation in the intracellular Ca2+ concentration is a major stimulus for the translocation of AnxA2 to the plasma membrane (12). We first studied the Ca2+-dependent membrane association of AnxA2 in 661W cells by treatment with mild concentrations of a Ca2+ ionophore, A23187 (5 μm). To elute cell surface AnxA2, we used EDTA as stated previously (29). In the presence of mild concentrations (5 μm) of a Ca2+ ionophore, A23187, the cell surface levels of AnxA2 in the EDTA eluates were found to be elevated 1.8-fold (n = 3) when compared with the dimethyl sulfoxide controls (Fig. 1A). We also analyzed the cell surface levels of AnxA2 after Ca2+ ionophore treatment by conjugating all cell surface proteins with a water-soluble and cell-impermeable amine reactive biotin and immobilized them with streptavidin. Western blot analysis of biotin-conjugated and streptavidin-immobilized cell surface extracts revealed that Ca2+ ionophore A23187 treatment resulted in a 2-fold increase in the cell surface levels of AnxA2 (n = 3) compared with the dimethyl sulfoxide control (Fig. 1B). Coomassie staining and immunoblotting with anti-Na,K-ATPase antibody were used as loading controls for EDTA eluates and cell surface biotinylated extracts, respectively.
FIGURE 1.

AnxA2 translocates to the cell surface in response to increased concentrations of intracellular Ca2+, whereas glutamate mobilizes intracellular Ca2+ in 661W cells. A, representative Western blot analysis of EDTA cell surface eluates from 661W cells treated with the Ca2+ ionophore A23187. Following a 4-h treatment with A23187, the 661W monolayers were incubated with EDTA (Invitrogen) for 20 min at 37 °C. The cell surface eluates were concentrated and immunoblotted for AnxA2 using a mouse monoclonal anti-AnxA2 antibody (left panel, top row). To normalize for loading, SDS-PAGE was performed with identical concentrations of protein, as in the left panel, top row, and the gel was stained with Coomassie Blue (left panel, bottom row). An ∼50-kD, Coomassie-stained band whose levels seemed invariant with the treatment conditions was used to account for differences in protein loading. DMSO, dimethyl sulfoxide. ***, p < 0.001. B, for cell surface biotinylation, 661W cells treated with A23187 were incubated for 30 min in a cell-impermeable sulfo-N-hydroxysulfosuccinimide biotin reagent. After cell lysis, the biotinylated proteins were precipitated with streptavidin-conjugated agarose beads and eluted with Laemmli buffer. The extracts were immunoblotted with anti-AnxA2 antibody (left panel, top row). As a loading control, the blot was probed with anti-Na,K-ATPase antibody (left panel, bottom row). A and B, fold change in A23187 treatment compared with dimethyl sulfoxide treatment is plotted. **, p < 0.01, and ***, p < 0.001. C, 661W cells preloaded with 3 μm Fluo-3 for 30 min were imaged under a Zeiss LSM confocal microscope for 700 s after treatment with 500 μm glutamate at a rate of one image taken every 10 s. The wavelengths were set to 488 nm excitation and 526 nm emission, and the images were taken at identical image acquisition parameters under a ×20 objective. A significant increase in Fluo-3 intensity was observed starting at 80 s, and levels were sustained even after 570 s. D, quantification of [Ca2+]i after thapsigargin and glutamate treatment was performed by converting the 340/380 ratio of Fura-2 fluorescence intensity to nanomolar concentrations of intracellular Ca2+ using the Grynkiewicz equation, as described previously (33). Fura-2 fluorescence ratios are shown in supplemental Fig. 1B.
Glutamate Induces Mobilization of Intracellular Ca2+ in 661W Cells
We also studied glutamate-induced intracellular Ca2+ mobilization by live cell confocal microscopy. In 661W cells preloaded with 3 μm of Fluo-3, changes in fluorescence intensity were observed starting 120 s after the addition of glutamate, and the elevated levels were sustained even after 570 s (Fig. 1C). To specifically demonstrate that it is the intracellular Ca2+ reserves that are responsible for membrane translocation of AnxA2, cells cultured in Ca2+-free medium were loaded with Fura-2/AM and incubated with 1 μm thapsigargin, followed by treatment with 5 μm A23187 (supplemental Fig. 1A) or 500 μm glutamate (supplemental Fig. 1B).
The 340/380 ratio of Fura-2 fluorescence intensity from supplemental Fig. 1B was converted to nanomolar concentrations of intracellular Ca2+ using the Grynkiewicz equation (Fig. 1D) (33). The addition of thapsigargin (1 μm) induced an initial increase in [Ca2+]i (∼400 nm), followed by a recovery to steady levels. The addition of glutamate induced [Ca2+]i to nearly 750 nm.
These studies reveal that glutamate, at a concentration of 500 μm, leads to a mobilization of intracellular Ca2+ from intracellular Ca2+ reserves. Each experiment was repeated at least three times.
Glutamate-induced Intracellular Ca2+ Mobilization Stimulates the Translocation of Both Endogenous and AnxA2-GFP to the Cell Surface in 661W Cells
We studied the effect of glutamate exposure on the translocation of AnxA2 to the plasma membrane. In 661W cells, glutamate, at a concentration of 500 μm, increased the cell surface levels of endogenous AnxA2 in a time-dependent manner, starting 10 min after the addition of glutamate. The surface levels of AnxA2, 4 h after treatment with glutamate, remained elevated by 71 ± 3.2% (mean ± S.D., n = 3) compared with the levels 0 min after treatment with glutamate (Fig. 2A). Coomassie-stained bands were used as a loading control for the EDTA eluates. Further, to directly show that the glutamate induced membrane-translocated AnxA2 is predominantly tyrosine 23-phosphorylated, we used a mouse monoclonal antibody that specifically recognizes the phosphorylated tyrosine residue at the 23rd position of AnxA2. Treatment with glutamate showed a significant increase (almost 1.8-fold) in pAnxA2 Tyr-23 when compared with the vehicle control. (Fig. 2B).
FIGURE 2.

Glutamate induces cell surface translocation of endogenous and AnxA2-GFP in 661W cells. A, Western blot analysis of EDTA eluates from 661W cells treated with 500 μm glutamate for the indicated time periods. Coomassie staining of the EDTA eluates was used as a loading control B, EDTA eluates from 661W cells in the presence or absence of 500 μm glutamate for 4 h were collected and immunoblotted with pAnxA2 Tyr-23 antibody. Na,K-ATPase served as a loading control. Right panel, quantitation. **, p < 0.01. The fold change in pAnxA2 Y23 level with respect to vehicle control (1 m HCl) is shown. C, representative images of TIRF microscopy performed on vehicle control-treated (1 m HCl) and glutamate-treated (250 μm and 500 μm) 661W cells according to the procedures described above. The images were acquired at identical acquisition settings. A significant increase in the cell surface-associated levels of AnxA2 was observed in glutamate-treated 661W cells compared with the untreated controls. A graphical representation of mean relative fluorescence units (relative fluorescence units) of cell surface AnxA2 immunostaining was plotted after normalizing for the background intensity (right panel). D, 661W cells were transiently transfected with either the pEGFP-N1 empty plasmid vector or the AnxA2-GFP plasmid vector and treated with glutamate (250 μm and 500 μm) for 4 h. EDTA eluates and cell surface biotinylated extracts were collected. Western blotting with anti-GFP antibody revealed a 62-kD band representative of the AnxA2-GFP fusion protein, whose levels were up-regulated on treatment with glutamate (500 μm) in both the EDTA eluates (left panel, top row) and cell surface biotinylated extracts (Biot.) (right panel, top row). The fusion protein was not detected in the empty vector-transfected cells. Coomassie-stained bands and Na,K-ATPase were used as loading controls for EDTA eluates and biotinylated extracts, respectively. Each experiment was repeated three times (n = 3).
To visualize the cell surface-associated AnxA2 exclusively, 661W cells treated with glutamate for 4 h were immunostained with anti-AnxA2 antibody and examined by TIRF microscopy (Fig. 2C, left panel). The evanescent field produced by total internally reflected light excites the fluorescent molecules at the cell-substrate interface, which is ideal for the visualization of fluorescent molecules at the cell surface. Because TIRF microscopy selectively excites the fluorophores present at the plasma membrane, cell surface-associated AnxA2 was observed as punctate spots on the membrane. On quantification of the intensity of cell surface AnxA2, we observed a significant difference in the expression levels of cell surface AnxA2 in glutamate-treated cells compared with the vehicle control (Fig. 2C, right panel). These results suggest that glutamate-treated 661W cells exhibit increased levels of cell surface-associated AnxA2.
To further confirm glutamate-induced cell surface translocation of AnxA2, we used a plasmid vector containing AnxA2 fused with GFP at its carboxyl terminus. We examined the cell surface translocation of the AnxA2-GFP fusion protein on treatment with glutamate. Western blotting of the EDTA eluates (Fig. 2D, left panel) and cell surface-biotinylated extracts (Fig. 2D, right panel) demonstrated an increase in the expression levels of AnxA2-GFP fusion protein in glutamate-treated (250 and 500 μm) 661W cells. These results suggest that glutamate-mediated elevation in the intracellular levels of Ca2+ mobilizes AnxA2-GFP fusion protein to the plasma membrane in a Ca2+-dependent manner. Coomassie staining and immunoblotting with anti-Na,K-ATPase antibody were used as loading controls for EDTA elution and cell surface biotinylation, respectively.
Glutamate-induced Cell Surface Translocation of AnxA2 is NMDA Receptor-mediated
Glutamate-induced excitotoxicity is mediated by the hyperstimulation of the NMDA-type glutamate receptors (35). In the presence of MK-801 (10 μm), a selective antagonist of the NMDA receptor, glutamate-induced elevation in the cell surface levels of annexin A2 was reduced ∼9-fold at 2 h and 5-fold at 4 h compared with no MK-801 treatment (Fig. 3A). 2,3-Dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), a non-NMDA receptor antagonist, failed to result in a similar effect (Fig. 3B), suggesting that the activity of the NMDA receptor is essential for glutamate to exert its effects on AnxA2 translocation. Treatment with a Ca2+ channel blocker, nifedipine (50 μΜ), did not influence the glutamate-mediated cell surface translocation of AnxA2 (Fig. 3C).
FIGURE 3.

Glutamate-induced AnxA2 cell surface translocation is NMDA receptor-mediated. A, Western blot analysis (left panel) of the cell surface biotinylated extracts from 661W cells treated for different times with MK-801 (10 μm), a NMDA receptor antagonist, in the presence of glutamate. Right panel, quantitation. **, p < 0.01. B, the effect of an inhibitor for the non-NMDA receptor, NBQX (25 μm), on glutamate-induced cell surface translocation of AnxA2 was tested in a time dependent manner. A Western blot analysis (left panel) for the cell surface biotinylated extracts is shown. Right panel, quantitation. C, Western blot analysis (left panel) for the cell surface biotinylated extract showing the effect of calcium channel blocker nifedipine on the glutamate-induced translocation of AnxA2 to the membrane in a time-dependent manner. Right panel, quantitation. Anti-Na,K-ATPase antibody served as a loading control for A–C. The fold change to the respective vehicle (dimethyl sulfoxide) control treatments is shown. For comparison, we also checked the cell surface levels of AnxA2 in untreated and glutamate-treated cells in the absence of MK-801, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX), and nifedipine for the indicated time. Each experiment was repeated three times (n = 3).
Phosphorylation of AnxA2 at Tyrosine 23 Is Critical for Glutamate-induced AnxA2 Cell Surface Translocation
Because the endogenous and wild-type AnxA2-GFP-fused protein responded similarly to glutamate treatment, as shown before, we next asked whether the phosphorylation status of AnxA2 is important in the translocation process. Because AnxA2 possesses three potential phosphorylation sites, tyrosine 23, serine 25, and serine 11, at its N terminus, we tested the involvement of these three sites in the translocation process. For this purpose, we constructed single point mutants with a phosphomimetic (AnxA2Y23E-GFP, AnxA2S25E-GFP, and AnxA2S11E-GFP) and a non-phosphomimetic variant (AnxA2Y23F-GFP, AnxA2S25A-GFP, and AnxA2S11A-GFP) at tyrosine 23, serine 25, or serine 11. On transient transfection of 661W cells and glutamate treatment (500 μm for 4 h), we studied the extent to which the mutant variants were translocated to the cell surface using EDTA cell surface elution and cell surface biotinylation. In the case of the non-phosphomimetic tyrosine 23 mutant AnxA2Y23F-GFP (Fig. 4A), we could not detect any AnxA2-GFP that translocated to the membrane in the presence or absence of glutamate. On the other hand, in the case of the phosphomimetic mutant AnxA2Y23E-GFP (Fig. 4B), we saw a significant increase in AnxA2-GFP levels in glutamate-treated cells when compared with non-treated cells, both in EDTA eluates (2-fold) and biotinylated extracts (3-fold) (quantification in supplemental Fig. 2A). The other variants at serine 25, AnxA2S25A-GFP and AnxA2S25E-GFP (Fig. 4, C and D, respectively) had a similar or increased cell surface distribution with glutamate treatment compared with no glutamate treatment. Furthermore, the serine 11 phosphorylation mutants AnxA2S11E-GFP and AnxA2S11A-GFP did not influence the cell surface translocation of AnxA2 (data not shown). The intracellular levels of AnxA2-GFP did not seem to be affected on glutamate treatment in the cell lysates of all phosphorylation mutants.
FIGURE 4.
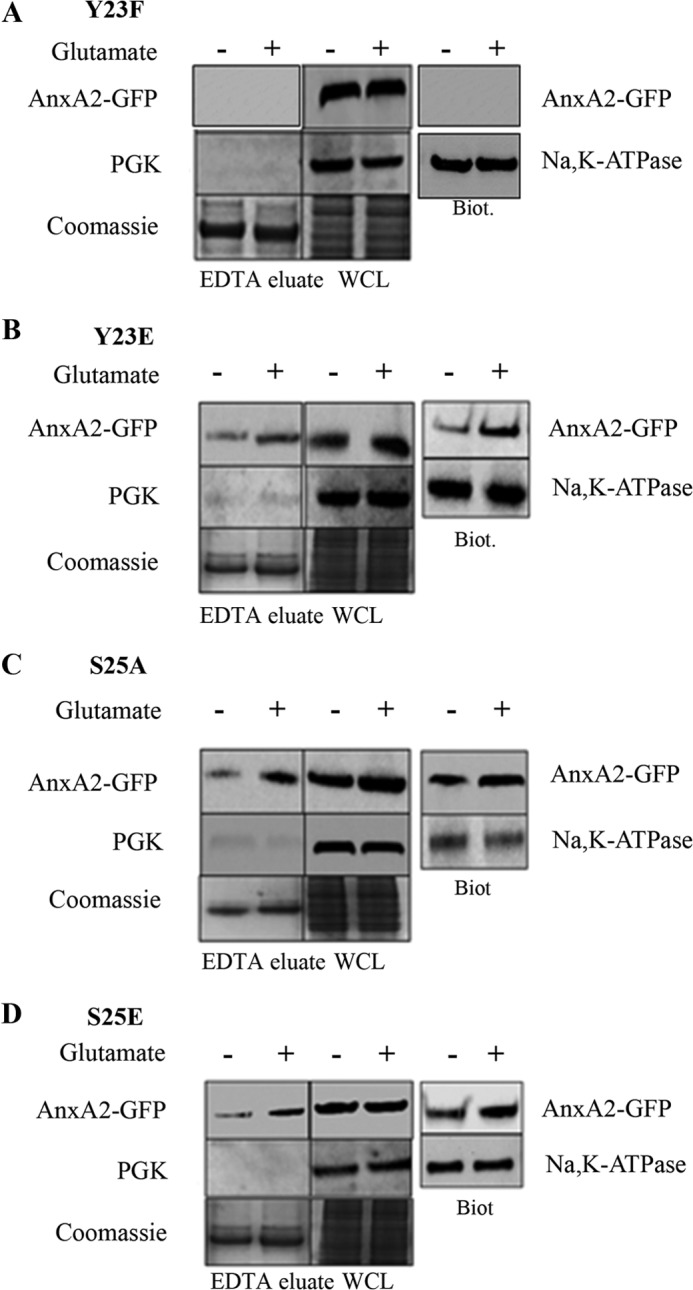
Cell surface translocation of single phosphorylation mutants at tyrosine 23 and serine 25 on glutamate treatment. A, 661W cells were transfected with a plasmid vector expressing a non-phosphomimetic mutant at tyrosine 23 (AnxA2Y23F-GFP) and subjected to glutamate treatment for 4 h. The EDTA eluates and cell lysates were collected. SDS-PAGE and Western blotting was performed with anti-GFP antibody (top row). To determine the purity of the EDTA eluates, Western blotting was performed with PGK (center row). As a control for loading, the gel was stained with Coomassie (bottom row). The cell surface AnxA2 was extracted by conjugation with a hydrophilic, cell-impermeable biotin analog and precipitated with streptavidin. Western blotting of the cell surface biotinylated extracts (Biot.) was performed with anti-GFP antibody (right column, top). As a control for loading, the blot was probed with anti-Na,K-ATPase antibody (right column, bottom). WCL, whole cell lysate. B, 661W cells were transfected with a plasmid vector expressing the phosphomimetic mutant at tyrosine 23 (AnxA2Y23E-GFP) and treated with glutamate for 4 h. The EDTA eluates and cell lysates were collected and subjected to Western blotting with anti-GFP antibody (top row). Immunoblotting with PGK (center row) and Coomassie staining of the gel (bottom row) were performed as described above. The cell surface biotinylated extracts were collected, and Western immunoblotting was performed with anti-GFP antibody (right column). Na,K-ATPase was used to control for loading. C and D, 661W cells were transfected with a plasmid vector expressing a non-phosphomimetic mutant (AnxA2S25A-GFP) and a phosphomimetic mutant at serine 25 (AnxA2S25E-GFP), respectively, treated with glutamate, and subjected to Western blotting as described above.
To further investigate whether phosphorylation at tyrosine 23 occurs in conjunction with phosphorylation at serine 25 to facilitate the cell surface translocation of AnxA2, we constructed phospho-mimicking and non-phospho-mimicking double mutants of AnxA2 at tyrosine 23 and serine 25. Both cell surface biotinylation and EDTA elution in AnxA2Y23FS25A-GFP and AnxA2Y23FS25E-GFP double non-phosphomimetic mutants detected no AnxA2-GFP, indicating that phosphorylation of the tyrosine 23 site is required for cell surface translocation of AnxA2. (Fig. 5, A and B). The other two mutants, AnxA2Y23ES25A-GFP and AnxA2Y23ES25E-GFP, were observed to have a similar cell surface distribution as that of the single mutants, with elevated cell surface levels on glutamate treatment and invariant cytosolic levels (Fig. 5, C and D, respectively) (quantification in supplemental Fig. 3, A and B). For all mutants, Coomassie staining and immunoblotting with anti-Na,K-ATPase were used as loading controls for EDTA eluates and biotinylated extracts, respectively. The purity of the EDTA eluates was tested in each case by immunoblotting with PGK. Taken together, these results indicate that tyrosine 23 at the N terminus of AnxA2 plays a predominant role in the cell surface translocation of AnxA2.
FIGURE 5.
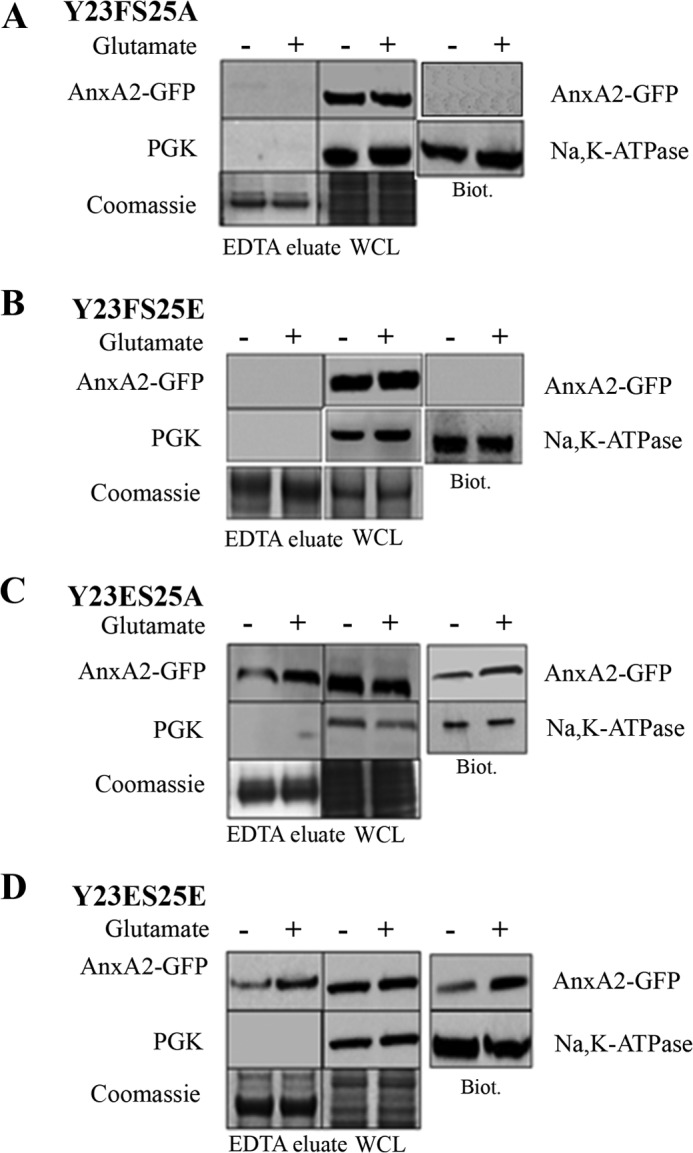
Distribution of tyrosine 23 and serine 25 double phosphorylation mutants of AnxA2 in response to glutamate treatment. A–D, 661W cells were transiently transfected with plasmid vectors expressing phosphomimetic and non-phosphomimetic double mutants at tyrosine 23 and serine 25 (AnxA2Y23FS25A-GFP, AnxA2Y23FS25E-GFP, AnxA2Y23ES25A-GFP, and AnxA2Y23ES25E-GFP). The cells were treated with 500 μm glutamate for 4 h. The EDTA eluates and cell surface biotinylated extracts were collected as mentioned previously and immunoblotted with anti-GFP antibody. The EDTA eluates were tested for their purity by immunoblotting with anti-PGK antibody. A Coomassie-stained gel was used as a loading control for the EDTA eluates. The biotinylated extracts were probed with anti-Na,K-ATPase antibody to determine equal loading. WCL, whole cell lysate; Biot., biotinylated extracts. All the blots were exposed to identical exposure times.
Glutamate-induced Cell Surface AnxA2 Is Phosphorylated Predominantly at Tyrosine 23
EDTA eluates of 661W cells transfected with AnxA2-GFP fusion construct were immunoprecipitated with anti-GFP antibody. On Western blotting with anti-AnxA2 antibody, a time-dependent increase in the cell surface levels of AnxA2-GFP fusion protein was observed in the EDTA eluates (Fig. 6A). Next, we investigated the phosphorylation status of glutamate-induced cell surface AnxA2. For this purpose, the immunoprecipitates were immunoblotted with anti-phosphotyrosine and anti-phosphoserine antibodies. It was observed that the cell surface-associated AnxA2-GFP fusion protein was tyrosine-phosphorylated and that its levels were elevated on treatment with 500 μm glutamate (Fig. 6B). In contrast, we did not detect any serine-phosphorylated AnxA2-GFP in the EDTA eluates of glutamate-treated and untreated cells (Fig. 6C). From these data, it appears that glutamate-induced mobilization of intracellular Ca2+ leads to the translocation of tyrosine-phosphorylated AnxA2 to the cell surface. On the other hand, serine-phosphorylated AnxA2 does not seem to have a significant contribution to the cell surface pool of AnxA2. As a control for loading the light chain of the immunoprecipitated antibody (IgG LC) was used. The fold change compared with GFP antibody treatment is shown.
FIGURE 6.
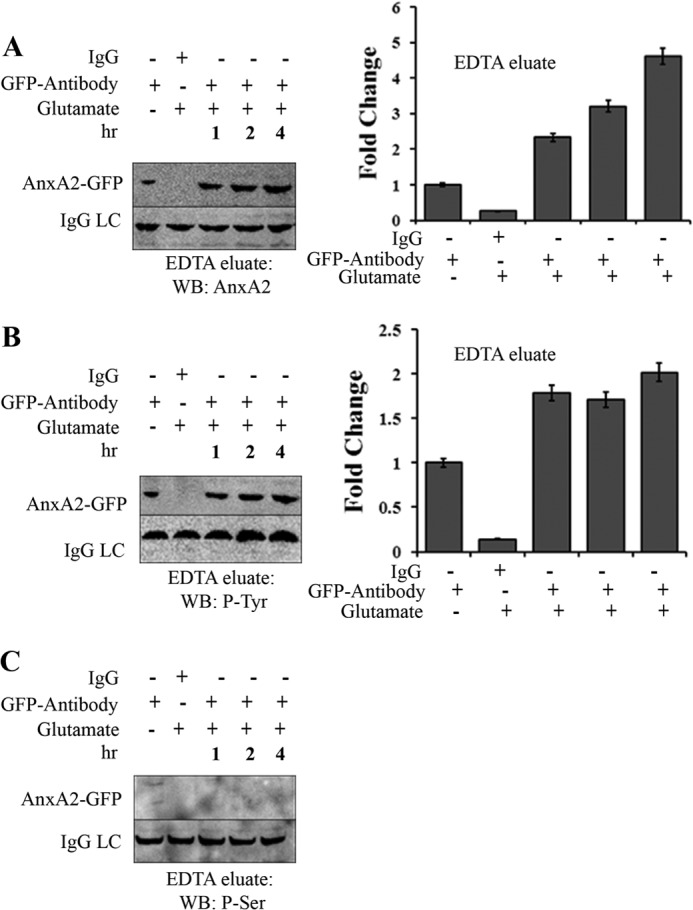
Glutamate-induced cell surface AnxA2 is tyrosine-phosphorylated. A, 661W cells transfected with a plasmid vector expressing wild-type AnxA2-GFP were treated with 500 μm glutamate for 1, 2, and 4 h. The cells were incubated in EDTA (Invitrogen) for 20 min. The EDTA eluates were immunoprecipitated with anti-GFP antibody, subjected to SDS-PAGE, and analyzed by Western blotting (WB) with anti-AnxA2 antibody. The eluates were incubated with nonspecific anti-mouse IgG as negative controls. LC, light chain. B and C, immunoprecipitates of the EDTA eluates collected as mentioned above were immunoblotted with anti-phosphotyrosine and anti-phosphoserine antibodies. The IgG light chain of the antibody used for immunoprecipitation was used as a control for loading. All blots were exposed to identical exposure times. The fold change to the respective GFP antibody treatments is shown. C, because we could not detect any serine-phosphorylated (P-Ser) AnxA2-GFP in the EDTA eluates of glutamate-treated and untreated cells, it could not be quantified. P-Tyr, tyrosine-phosphorylated.
Because we detected that cell surface-associated AnxA2 is predominantly tyrosine-phosphorylated, we wanted to check whether it is the tyrosine 23 residue that is responsible for the membrane translocation of AnxA2. For this, we tested whether inhibition of the phosphorylation event could prevent the cell surface translocation of AnxA2 by using a specific antibody against pAnxA2 Y23. EDTA eluates were collected, concentrated, and immunoblotted with pAnxA2 Tyr-23 antibody. Treatment with sodium orthovanadate, a tyrosine phosphatase inhibitor, augmented the cell surface levels of pAnxA2 Tyr-23 in both untreated (2.5-fold increase) and glutamate-treated cells (3-fold increase) (n = 4) (Fig. 7A), whereas okadaic acid, a serine/threonine phosphatase inhibitor, had no effect on the cell surface levels of pAnxA2 Tyr-23 (Fig. 7B). In addition, when we treated the cells with genistein, a general tyrosine kinase inhibitor, we observed that the glutamate-induced translocation of AnxA2 was inhibited markedly inhibited in both glutamate-treated (almost 13-fold) and untreated cells (almost 10-fold). (Fig. 7C). Fig. 7C, right panel, shows the quantification of the respective blots. The fold change compared with the respective vehicle treatments is shown. These results confirm that it is predominantly the tyrosine 23 residue that is phosphorylated upon glutamate treatment, leading to its translocation to the membrane.
FIGURE 7.
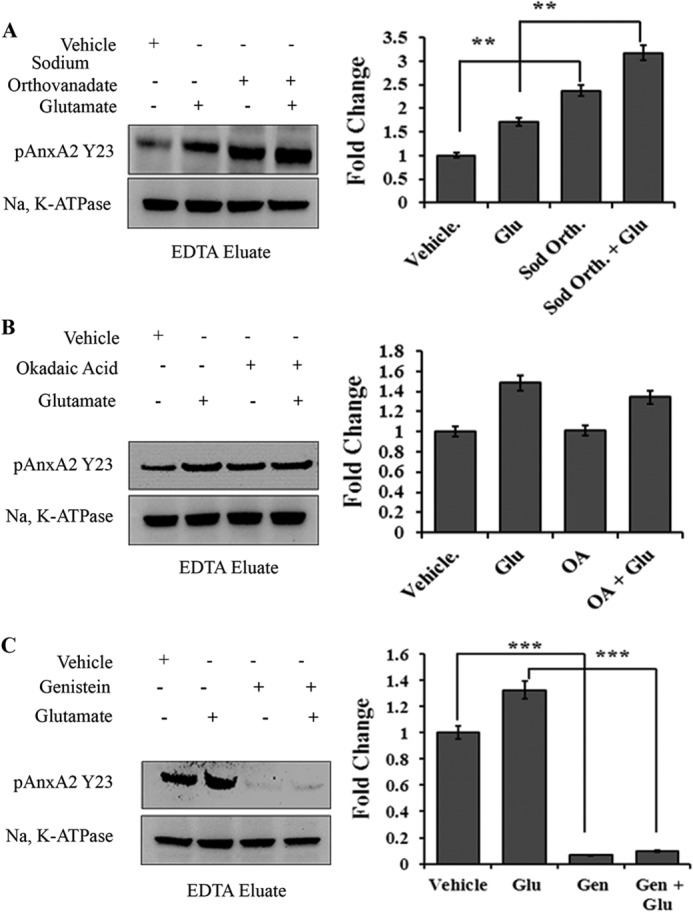
Tyrosine 23 phosphorylation of AnxA2 is critical for its cell surface translocation. 661W cells were incubated with or without glutamate (500 μm, 4 h) in the presence or absence of the respective chemicals. EDTA eluates were collected, concentrated using Nanosep 10KD filters, and immunoblotted with mouse monoclonal antibody that specifically recognizes the phosphorylated tyrosine 23 residue of AnxA2. Na,K-ATPase was used as the loading control. A, Western blot analysis of EDTA eluates after incubation with the tyrosine phosphatase inhibitor sodium orthovanadate (Sod. Orth., 1 mm) (left panel). Right panel, densitometric analysis of the Western blot analyses. Glu, glutamate. **, p < 0.01. B, Western blot analysis of EDTA eluates after incubation with the serine/threonine phosphatase inhibitor okadaic acid (OA, 100 nm) (left panel). Right panel, densitometric analysis of the Western blot analyses. C, Western blot analysis of EDTA eluates after incubation with the general tyrosine kinase inhibitor genistein (Gen, 100 μm) (left panel). Right panel, densitometric analysis of the Western blot analyses (n = 3). ***, p < 0.001. The fold change with respect to vehicle treatment is shown.
Glutamate Enhances AnxA2-mediated Cell Surface Plasmin Generation
Having demonstrated that glutamate induces the cell surface translocation of AnxA2, we examined whether the increased cell surface pool of AnxA2 is associated with a concomitant increase in the AnxA2-mediated generation of plasmin.
To test this, we made use of a competitive peptide inhibitor of tPA binding to AnxA2. Previous reports have identified a hexapeptide, LCKLSL, from regions 7–12 as the minimum sequence required for the binding of tPA to AnxA2 (36). The competitive peptide was assayed for its ability to inhibit the binding of tPA to AnxA2 both in vitro and in vivo, and it has been observed that the LCKLSL peptide resulted in a 40–60% reduction in the binding of tPA to AnxA2 (37). It was has also been observed that peptides with a replacement of the cysteine residue at position 8 with glycine (LGKLSL) did not possess any inhibitory effects (36).
As shown in Fig. 8, the hexapeptide LCKLSL, at a concentration of 250 μm, resulted in a 1.4 ± 0.2-fold (mean ± S.E., n = 6) reduction in plasmin generation on glutamate treatment compared with the control peptide, LGKLSL. The peptide seemed to have only moderate effects on the baseline levels of plasmin generation in untreated cells (data not shown). Taken together, these results suggest that the glutamate-induced increase in the cell surface levels of AnxA2 directly contributes to a corresponding increase in the AnxA2-mediated cell surface generation of plasmin.
FIGURE 8.
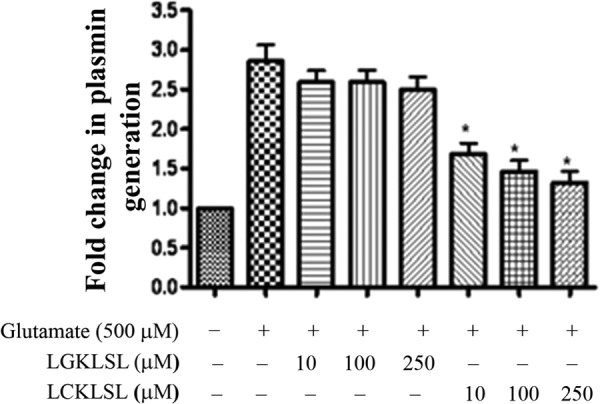
Glutamate enhances AnxA2-mediated cell surface plasmin generation. A, 661W cells were cultured in monolayers and treated with 500 μm glutamate for 4 h. The cells were washed gently and treated with recombinant tPA (10 nm) in the presence of either the experimental (LCKLSL) or the control peptide (LGKLSL) at the indicated concentrations for 1 h at 37 °C. After three washes, recombinant plasminogen (100 nm) was added to the reaction mixture, and the reaction was monitored by removing aliquots every 4 min and measuring the amount of plasmin generated by cleavage of a chromogenic substrate of plasmin, S-2251. To serve as controls, either tPA or glutamate was omitted from the reaction mixture. The fold change in plasmin generation was obtained by normalizing different treatment groups with the untreated controls, whose value was set to 1.0 (A405 nm = 3.68). *, p ≤ 0.05 (n = 6).
DISCUSSION
In this study, we provide evidence to show that a glutamate-mediated increase in the intracellular levels of Ca2+results in the cell surface translocation of both endogenous and AnxA2-GFP to the cell surface. We also observed that AnxA2 translocates to the cell surface in the presence of a Ca2+ ionophore, A23187, suggesting that elevated intracellular Ca2+ levels are a key stimulus for the translocation process. Furthermore, we show that the Ca2+-dependent cell surface translocation of AnxA2 requires the phosphorylation of tyrosine 23 at the N terminus of AnxA2 and that mutation of this residue significantly inhibits the translocation process. The cell surface-translocated AnxA2-GFP fusion protein was observed to be tyrosine-phosphorylated on glutamate treatment. We confirmed the involvement of the tyrosine 23 by using a specific antibody that recognizes the phosphorylated 23rd tyrosine residue of AnxA2. Further, on use of a tyrosine phosphatase inhibitor, sodium orthovanadate, we saw an increase in the membrane translocation of pAnxA2 upon glutamate treatment, whereas the translocation process was inhibited upon treatment with a tyrosine kinase inhibitor, genistein. These results are in agreement with our previous findings that show the requirement of tyrosine 23 phosphorylation in the cell surface translocation of AnxA2 (19).
Our data on both the single mutants of serine 25 or tyrosine 23 as well as the double mutants at tyrosine 23 and serine 25 suggest that tyrosine 23 is predominantly responsible for the glutamate-induced membrane translocation of AnxA2. We also questioned the involvement of serine 11 in the cell surface translocation of AnxA2, and our data on the phosphomimetic and non-phosphomimetic single mutants at serine 11 suggest that this residue does not influence the translocation process. Furthermore, we also demonstrated that the cell surface AnxA2 is an active plasmin-generating complex and that this activity can be inhibited by a hexapeptide directed against the N terminus of AnxA2. These results indicate that the N terminus of AnxA2 is important not only for its membrane translocation but also for plasmin generation and, thereby, extracellular matrix degradation. During ischemic and excitotoxic damage, photoreceptors and inner retinal cells are vulnerable to elevated levels of extracellular glutamate caused by disruptions in glutamate homeostasis (38). Elevated intracellular concentrations of Ca2+ occur as a result of hyperstimulation of glutamate receptors and entry of extracellular Ca2+ or the release of Ca2+ from the intracellular store-operated Ca2+ channels (39). In addition, cell-mediated proteolysis alters cell-extracellular matrix interactions, which could potentiate the excitotoxic injury. Studies in the CNS have shown that, under conditions of ischemic injury, extracellular tPA and plasmin can exacerbate ischemic neuronal damage by further damaging the blood-brain barrier, activation of the NMDA receptor by cleavage of the NR1 subunit, amplification of intracellular Ca2+ signaling, and activation of matrix-bound metalloproteinases (40, 41). tPA also potentiates neuronal loss, and inhibitors of the tPA/plasmin proteolytic cascade have been found to confer protection to CNS neurons (42). Because the retina is commonly viewed as an extension of the CNS, the mechanism of action of tPA could be similar in the two systems. We propose that, although secreted levels of tPA are not found to be induced on ischemic injury in the retina, under ischemic conditions when there is a leakage of circulating plasminogen, extracellular tPA and plasminogen can bind to their cell surface receptors and potentiate the generation of plasmin, which could contribute to the loss of retinal ganglion cells. Plasmin-plasminogen activators, along with matrix metalloproteinases, have been implicated as key representatives in this process. These receptors for plasmin generation act as docking sites for the binding of circulating plasminogen and tPA and result in not only an increase in the catalytic efficiency of plasmin generation but in the protection of plasmin from being degraded by its physiological inhibitors (43). Inhibiting the activity of the AnxA2 catalytic center using small-molecule hexapeptides can have potential implications on inhibiting the pathological processes associated with enhanced plasmin generation (37). Because cell surface AnxA2 is the major fibrinolytic receptor facilitating the enhanced generation of plasmin, regulation of the cell surface translocation of AnxA2 may be an important process in the generation of plasmin and its downstream effects. It is also known that AnxA2 binds to the membrane in a Ca2+-dependent manner. Elevation of intracellular levels of Ca2+ has been shown previously to be one of the key stimuli that translocate AnxA2 to the cell surface (44).
The data presented here suggest a role for AnxA2 in potentiating the glutamate-induced loss of photoreceptors in the retina. Previous reports have indicated that even a modest increase in the cell surface levels of AnxA2 can have profound implications in the pathogenesis of several diseases. Dysregulation in the cell surface expression levels of AnxA2 has been shown to manifest as bleeding diseases, conferring on the cells the ability to generate increased levels of plasmin (45). Many cancer cells have also been shown to abundantly express cell surface AnxA2, which confers on them the invasive and metastatic behavior by the generation of plasmin or by the plasmin-mediated activation of matrix metalloproteinases (46). Therefore, modulating the activity of a potent plasmin-generating receptor, AnxA2, can serve as a potential therapeutic target for photoreceptor-degenerative diseases like retinitis pigmentosa and age-related macular degeneration.
Supplementary Material
Acknowledgments
We thank the microscope core facility at the Center for Commercialization of Fluorescence Technologies at the University of North Texas Health Science Center for help with confocal and TIRF imaging.
This work was supported by National Institute on Minority Health and Health Disparities Grant 1P20 MD006882 (to J. K. V.).

This article contains supplemental Figs. 1–3.
- tPA
- tissue plasminogen activator
- PGK
- phosphoglycerate kinase
- TIRF
- total internal reflection.
REFERENCES
- 1. Vorwerk C. K., Gorla M. S., Dreyer E. B. (1999) An experimental basis for implicating excitotoxicity in glaucomatous optic neuropathy. Surv. Ophthalmol. 43, S142–S150 [DOI] [PubMed] [Google Scholar]
- 2. Doble A. (1999) The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol. Ther. 81, 163–221 [DOI] [PubMed] [Google Scholar]
- 3. Sattler R., Tymianski M. (2000) Molecular mechanisms of calcium-dependent excitotoxicity. J. Mol. Med. 78, 3–13 [DOI] [PubMed] [Google Scholar]
- 4. Lipton S. A., Rosenberg P. A. (1994) Excitatory amino acids as a final common pathway for neurologic disorders. N. Engl. J. Med. 330, 613–622 [DOI] [PubMed] [Google Scholar]
- 5. Lin Y., Jones B. W., Liu A., Vazquéz-Chona F. R., Lauritzen J. S., Ferrell W. D., Marc R. E. (2012) Rapid glutamate receptor 2 trafficking during retinal degeneration. Mol. Neurodegener. 7, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nixon E., Simpkins J. W. (2012) Neuroprotective effects of nonfeminizing estrogens in retinal photoreceptor neurons. Invest. Ophthalmol. Vis. Sci. 53, 4739–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chintala S. K. (2006) The emerging role of proteases in retinal ganglion cell death. Exp. Eye Res. 82, 5–12 [DOI] [PubMed] [Google Scholar]
- 8. Plow E. F., Herren T., Redlitz A., Miles L. A., Hoover-Plow J. L. (1995) The cell biology of the plasminogen system. FASEB J. 9, 939–945 [DOI] [PubMed] [Google Scholar]
- 9. Syrovets T., Simmet T. (2004) The cell biology of the plasminogen system. Cell Mol. Life Sci. 61, 873–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Longstaff C. (2002) Plasminogen activation on the cell surface. Front. Biosci. 7, d244–d255 [DOI] [PubMed] [Google Scholar]
- 11. Cesarman G. M., Guevara C. A., Hajjar K. A. (1994) An endothelial cell receptor for plasminogen/tissue plasminogen activator (t-PA): II: annexin II-mediated enhancement of t-PA-dependent plasminogen activation. J. Biol. Chem. 269, 21198–21203 [PubMed] [Google Scholar]
- 12. Gerke V., Creutz C. E., Moss S. E. (2005) Annexins: linking Ca2+ signalling to membrane dynamics. Nat. Rev. Mol. Cell Biol. 6, 449–461 [DOI] [PubMed] [Google Scholar]
- 13. Raynal P, Pollard HB. (1994) Annexins: the problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim. Biophys. Acta 1197, 63–93 [DOI] [PubMed] [Google Scholar]
- 14. Rescher U., Gerke V. (2004) Annexins: unique membrane binding proteins with diverse functions. J. Cell Sci. 117, 2631–2639 [DOI] [PubMed] [Google Scholar]
- 15. Glenney J. R., Jr. (1985) Phosphorylation of p36 in vitro with pp60src: regulation by Ca2+ and phospholipid. FEBS Lett. 192, 79–82 [DOI] [PubMed] [Google Scholar]
- 16. Gould K. L., Woodgett J. R., Isacke C. M., Hunter T. (1986) The protein-tyrosine kinase substrate p36 is also a substrate for protein kinase C in vitro and in vivo. Mol. Cell. Biol. 6, 2738–2744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Regnouf F., Sagot I., Delouche B., Devilliers G., Cartaud J., Henry J. P., Pradel L. A. (1995) “In vitro” phosphorylation of annexin 2 heterotetramer by protein kinase C: comparative properties of the unphosphorylated and phosphorylated annexin 2 on the aggregation and fusion of chromaffin granule membranes. J. Biol. Chem. 270, 27143–27150 [DOI] [PubMed] [Google Scholar]
- 18. Bellagamba C., Hubaishy I., Bjorge J. D., Fitzpatrick S. L., Fujita D. J., Waisman D. M. (1997) Tyrosine phosphorylation of annexin II tetramer is stimulated by membrane binding. J. Biol. Chem. 272, 3195–3199 [DOI] [PubMed] [Google Scholar]
- 19. Valapala M., Vishwanatha J. K. (2011) Lipid raft endocytosis and exosomal transport facilitate extracellular trafficking of annexin A2. J. Biol. Chem. 286, 30911–30925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Blanchard S., Barwise J. L., Gerke V., Goodall A., Vaughan P. F., Walker J. H. (1996) Annexins in the human neuroblastoma SH-SY5Y: demonstration of relocation of annexins II and V to membranes in response to elevation of intracellular calcium by membrane depolarisation and by the calcium ionophore A23187. J. Neurochem. 67, 805–813 [DOI] [PubMed] [Google Scholar]
- 21. He K. L., Deora A. B., Xiong H., Ling Q., Weksler B. B., Niesvizky R., Hajjar K. A. (2008) Endothelial cell annexin A2 regulates polyubiquitination and degradation of its binding partner S100A10/p11. J. Biol. Chem. 283, 19192–19200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Krishnamoorthy R. R., Clark A. F., Daudt D., Vishwanatha J. K., Yorio T. (2013) A forensic path to RGC-5 cell line identification: lessons learned. Invest. Ophthalmol. Vis. Sci. 54, 5712–5719 [DOI] [PubMed] [Google Scholar]
- 23. Tchedre K. T., Yorio T. (2008) σ-1 receptors protect RGC-5 cells from apoptosis by regulating intracellular calcium, Bax levels, and caspase-3 activation. Invest. Ophthalmol. Vis. Sci. 49, 2577–2588 [DOI] [PubMed] [Google Scholar]
- 24. Murphy T. H., Miyamoto M., Sastre A., Schnaar R. L., Coyle J. T. (1989) Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron 2, 1547–1558 [DOI] [PubMed] [Google Scholar]
- 25. Ankarcrona M., Dypbukt J. M., Bonfoco E., Zhivotovsky B., Orrenius S., Lipton S. A., Nicotera P. (1995) Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron 15, 961–973 [DOI] [PubMed] [Google Scholar]
- 26. Cheung N. S., Pascoe C. J., Giardina S. F., John C. A., Beart P. M. (1998) Micromolar l-glutamate induces extensive apoptosis in an apoptotic-necrotic continuum of insult-dependent, excitotoxic injury in cultured cortical neurons. Neuropharmacology 37, 1419–1429 [DOI] [PubMed] [Google Scholar]
- 27. Santos-Carvalho A., Elvas F., Alvaro A. R., Ambrósio A. F., Cavadas C. (2013) Neuropeptide Y receptors activation protects rat retinal neural cells against necrotic and apoptotic cell death induced by glutamate. Cell Death Dis. 4, e636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu J., Vishwanatha J. K. (2007) Regulation of nucleo-cytoplasmic shuttling of human annexin A2: a proposed mechanism. Mol. Cell. Biochem. 303, 211–220 [DOI] [PubMed] [Google Scholar]
- 29. Mai J., Finley R. L., Jr., Waisman D. M., Sloane B. F. (2000) Human procathepsin B interacts with the annexin II tetramer on the surface of tumor cells. J. Biol. Chem. 275, 12806–12812 [DOI] [PubMed] [Google Scholar]
- 30. Jacovina A. T., Zhong F., Khazanova E., Lev E., Deora A. B., Hajjar K. A. (2001) Neuritogenesis and the nerve growth factor-induced differentiation of PC-12 cells requires annexin II-mediated plasmin generation. J. Biol. Chem. 276, 49350–49358 [DOI] [PubMed] [Google Scholar]
- 31. Burghardt T. P., Ajtai K., Borejdo J. (2006) In situ single-molecule imaging with attoliter detection using objective total internal reflection confocal microscopy. Biochemistry 45, 4058–4068 [DOI] [PubMed] [Google Scholar]
- 32. Prasanna G., Krishnamoorthy R., Clark A. F., Wordinger R. J., Yorio T. (2002) Human optic nerve head astrocytes as a target for endothelin-1. Invest. Ophthalmol. Vis. Sci. 43, 2704–2713 [PubMed] [Google Scholar]
- 33. Grynkiewicz G., Poenie M., Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 34. Deshet N., Lupu-Meiri M., Espinoza I., Fili O., Shapira Y., Lupu R., Gershengorn M. C., Oron Y. (2008) Plasminogen-induced aggregation of PANC-1 cells requires conversion to plasmin and is inhibited by endogenous plasminogen activator inhibitor-1. J. Cell. Physiol. 216, 632–639 [DOI] [PubMed] [Google Scholar]
- 35. Shen Y., Liu X. L., Yang X. L. (2006) N-methyl-d-aspartate receptors in the retina. Mol. Neurobiol. 34, 163–179 [DOI] [PubMed] [Google Scholar]
- 36. Roda O., Valero M. L., Peiró S., Andreu D., Real F. X., Navarro P. (2003) New insights into the tPA-annexin A2 interaction: is annexin A2 CYS8 the sole requirement for this association? J. Biol. Chem. 278, 5702–5709 [DOI] [PubMed] [Google Scholar]
- 37. Valapala M., Thamake S. I., Vishwanatha J. K. (2011) A competitive hexapeptide inhibitor of annexin A2 prevents hypoxia-induced angiogenic events. J. Cell Sci. 124, 1453–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Delyfer M. N., Forster V., Neveux N., Picaud S., Léveillard T., Sahel J. A. (2005) Evidence for glutamate-mediated excitotoxic mechanisms during photoreceptor degeneration in the rd1 mouse retina. Mol. Vis. 11, 688–696 [PubMed] [Google Scholar]
- 39. Ferreira I. L., Duarte C. B., Carvalho A. P. (1996) Ca2+ influx through glutamate receptor-associated channels in retina cells correlates with neuronal cell death. Eur. J. Pharmacol. 302, 153–162 [DOI] [PubMed] [Google Scholar]
- 40. Strickland S., Gualandris A., Rogove A. D., Tsirka S. E. (1996) Extracellular proteases in neuronal function and degeneration. Cold Spring Harbor Symp. Quant. Biol. 61, 739–745 [PubMed] [Google Scholar]
- 41. Tsirka S. E., Gualandris A., Amaral D. G., Strickland S. (1995) Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature 377, 340–344 [DOI] [PubMed] [Google Scholar]
- 42. Miles L. A., Hawley S. B., Baik N., Andronicos N. M., Castellino F. J., Parmer R. J. (2005) Plasminogen receptors: the sine qua non of cell surface plasminogen activation. Front. Biosci. 10, 1754–1762 [DOI] [PubMed] [Google Scholar]
- 43. Kim J., Hajjar K. A. (2002) Annexin II: a plasminogen-plasminogen activator co-receptor. Front. Biosci. 7, d341–d348 [DOI] [PubMed] [Google Scholar]
- 44. Hajjar K. A., Acharya S. S. (2000) Annexin II and regulation of cell surface fibrinolysis. Ann. N.Y. Acad. Sci. 902, 265–271 [DOI] [PubMed] [Google Scholar]
- 45. Menell J. S., Cesarman G. M., Jacovina A. T., McLaughlin M. A., Lev E. A., Hajjar K. A. (1999) Annexin II and bleeding in acute promyelocytic leukemia. N. Engl. J. Med. 340, 994–1004 [DOI] [PubMed] [Google Scholar]
- 46. Sharma M. C., Sharma M. (2007) The role of annexin II in angiogenesis and tumor progression: a potential therapeutic target. Curr. Pharm. Des. 13, 3568–3575 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.