

Background: A few enzyme reactions in the mevalonate pathway remain unclear in most archaea.
Results: (R)-Mevalonate 3-phosphate is synthesized by Ta1305 protein from Thermoplasma acidophilum and is converted into isopentenyl diphosphate in cell-free extract from the archaeon.
Conclusion: The mevalonate pathway of T. acidophilum passes through (R)-mevalonate 3-phosphate rather than (R)-mevalonate 5-phosphate.
Significance: A third alternative mevalonate pathway is discovered.
Keywords: Archaea, Biosynthesis, Enzyme, Isoprenoid, Metabolism
Abstract
The lack of a few conserved enzymes in the classical mevalonate pathway and the widespread existence of isopentenyl phosphate kinase suggest the presence of a partly modified mevalonate pathway in most archaea and in some bacteria. In the pathway, (R)-mevalonate 5-phosphate is thought to be metabolized to isopentenyl diphosphate via isopentenyl phosphate. The long anticipated enzyme that catalyzes the reaction from (R)-mevalonate 5-phosphate to isopentenyl phosphate was recently identified in a Cloroflexi bacterium, Roseiflexus castenholzii, and in a halophilic archaeon, Haloferax volcanii. However, our trial to convert the intermediates of the classical and modified mevalonate pathways into isopentenyl diphosphate using cell-free extract from a thermophilic archaeon Thermoplasma acidophilum implied that the branch point intermediate of these known pathways, i.e. (R)-mevalonate 5-phosphate, is unlikely to be the precursor of isoprenoid. Through the process of characterizing the recombinant homologs of mevalonate pathway-related enzymes from the archaeon, a distant homolog of diphosphomevalonate decarboxylase was found to catalyze the phosphorylation of (R)-mevalonate to yield (R)-mevalonate 3-phosphate. The product could be converted into isopentenyl phosphate, probably through (R)-mevalonate 3,5-bisphosphate, by the action of unidentified T. acidophilum enzymes fractionated by anion-exchange chromatography. These findings demonstrate the presence of a third alternative “Thermoplasma-type” mevalonate pathway, which involves (R)-mevalonate 3-phosphotransferase and probably both (R)-mevalonate 3-phosphate 5-phosphotransferase and (R)-mevalonate 3,5-bisphosphate decarboxylase, in addition to isopentenyl phosphate kinase.
Introduction
The mevalonate (MVA)2 pathway is the supply route of active units for isoprenoid biosynthesis, i.e. isopentenyl diphosphate (IPP) and dimethylallyl diphosphate, in archaea, most eukaryotes, and some bacteria, whereas the methylerythritol phosphate pathway replaces it in most bacteria (1). In the classical MVA pathway, (R)-MVA formed from three acetyl-CoA molecules is successively phosphorylated to produce (R)-mevalonate 5-phosphate (MVA-5-P) and (R)-mevalonate 5-diphosphate (MVA-5-PP), and the third phosphorylation step at the 3-hydroxy group gives rise to the concerted elimination of the 3-phosphate group and the carboxyl group to yield IPP (Fig. 1A). IPP is then isomerized to give dimethylallyl diphosphate, which acts as the first priming substrate for prenyl elongation reactions that provide a basis for the numerous structural variations of isoprenoids, which include more than 55,000 compounds (2).
FIGURE 1.

Part of the MVA pathways discovered thus far from the archaea. A, classical pathway identified in S. solfataricus (11). B, modified pathway that was expected thus far and was recently identified in H. volcanii (10) (and in a Cloroflexi bacterium (9)). C, modified pathway identified in T. acidophilum in this study. The conversion from MVA-3-P to IP is likely catalyzed by unidentified enzymes, probably via MVA-3,5-PP.
The MVA pathway in almost all archaea and in some bacteria is considered to be different from the well studied classical pathway in eukaryotes such as yeast and humans (Fig. 1B). Based on the absence of the orthologous genes of two enzymes in the classical MVA pathway, i.e. phosphomevalonate kinase (PMK) and diphosphomevalonate decarboxylase (DMD) (3–5), and on the discovery of a widely conserved enzyme, isopentenyl phosphate kinase (IPK), which catalyzes the phosphorylation of isopentenyl phosphate (IP) (6–8), most archaea are believed to have a modified MVA pathway (Table 1). In the hypothetical pathway, MVA-5-P is decarboxylated to give isopentenyl phosphate (IP), which is then phosphorylated to IPP by the action of IPK. Very recently, the missing link enzyme that converts MVA-5-P into IP was discovered from exceptional lineages of bacteria and archaea, i.e. Chloroflexi and Halobacteria, respectively (9, 10). The organisms share a similar conservation pattern of the orthologs of the MVA pathway-related enzymes; they commonly possess the genes of the orthologs of MVA kinase (MVK), DMD, and IPK but lack that of PMK. The DMD orthologs from a Chloroflexi bacterium, Roseiflexus castenholzii, and from a halophilic archaeon, Haloferax volcanii, were independently identified as the enzyme of interest, i.e. phosphomevalonate decarboxylase (PMD). These findings proved the existence of the modified MVA pathway in the expected shape; however, it should be emphasized that DMD orthologs are absent in other “general” archaea and that no clue has been found as to a pathway that would yield IP in the organisms.
TABLE 1.
Conservation patterns of the orthologs of the MVA pathway-related enzymes
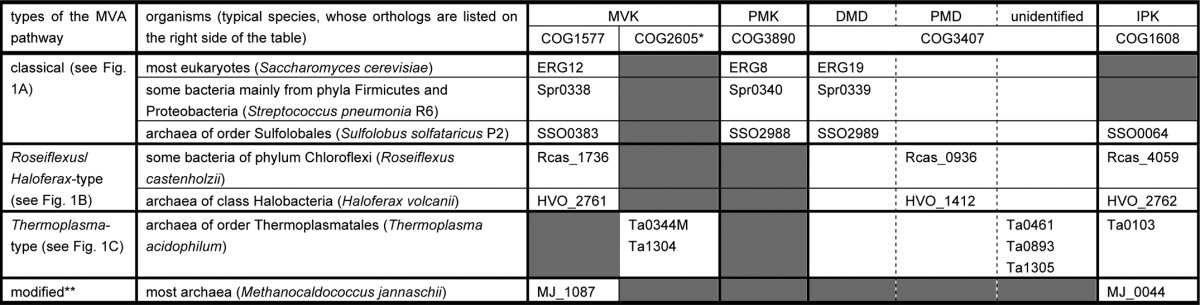
* The proteins of COG2605 have been predicted to be MVK without biochemical evidence.
** The modified MVA pathway used by most archaea (and possibly by some bacteria of phylum Bacteroides (3)) is considered to be identical with the Roseiflexus/Haloferax-type MVA pathway, although the enzyme corresponding to PMD has yet to be identified.
By whole-genome comparison, a few other exceptional conservation patterns of the orthologs of MVA pathway enzymes have been found in some limited lineages of archaea (Table 1) (3). Archaea of the genera Sulfolobus and their relatives possess the orthologs of all the enzymes of the classical MVA pathway. The existence of the classical MVA pathway, which is very exceptional in archaeal lineages, was recently proved by expected enzyme activities in Sulfolobus solfataricus (11). Archaea of the order Thermoplasmatales lack the ortholog of PMK. Although the conservation pattern seems similar to that found in Cloroflexi and Halobacteria, it has a few distinctive features. The Thermoplasmatales proteins that have been assigned as MVK homologs, such as Ta0344M and Ta1304 from Thermoplasma acidophilum, are distant from the usual MVKs of eukaryotes, bacteria, and other archaea (4). They are classified in a different protein family (COG2605) from that of the usual MVKs (COG1577). In addition, the Thermoplasmatales archaea have multiple orthologs of DMD; for example, T. acidophilum possesses two probable orthologs of DMD (Ta0461 and Ta0893) in addition to a relatively distant homolog (Ta1305). The phylogenetic tree of the MVA pathway-related enzymes shows that the Thermoplasmatales orthologs occupy unique positions (Fig. 2). In this study, we demonstrate that the mevalonate pathway in T. acidophilum is distinct from other known archaeal pathways (Fig. 1C). In this pathway, MVA is directly phosphorylated to (R)-mevalonate 3-phosphate (MVA-3-P), rather than to MVA-5-P, by the DMD homolog Ta1305. MVA-3-P is converted into IP, probably via (R)-mevalonate 3,5-bisphosphate (MVA-3,5-PP), by unknown enzymes from T. acidophilum. Because IPK has been identified from the archaeon (12), these findings suggest that, in T. acidophilum, a major part of IPP is synthesized via this somewhat convoluted route.
FIGURE 2.

Phylogenetic tree of the orthologs of the MVA pathway-related enzymes listed in Table 1. GHMP means galactokinase/homoserine kinase/mevalonate kinase/phosphomevalonate kinase. AAK means amino acid kinase.
EXPERIMENTAL PROCEDURES
Materials
(All-E) farnesyl diphosphate (FPP) was donated by Dr. Chikara Ohto, Toyota Motor Co., Japan. Partisil K6F normal-phase and LKC-18 reversed-phase precoated TLC plates were purchased from GE Healthcare. [2-14C]MVA-5-P (55 Ci/mol) and [1-14C]IPP (55 Ci/mol) were purchased from American Radiolabeled Chemicals. All other chemicals were of analytical grade.
General Procedures
Restriction enzyme digestions, transformations, and other standard molecular biological techniques were carried out as described by Sambrook et al. (13).
Cultivation of the Microorganism
T. acidophilum (JCM 9062T) was provided by the RIKEN BRC through the Natural Bio-Resource Project of the MEXT, Japan. The organism was cultured in a JCM 180 medium at 60 °C and harvested during the early stationary phase.
Preparation of the Isotope-labeled Substrates
[2-14C]MVA and [2-14C]MVA-5-PP were enzymatically synthesized from [2-14C]MVA-5-P using Antarctic phosphatase (New England Biolabs) and recombinant S. solfataricus PMK, respectively, as described in our previous work (11). [1-14C]IP was synthesized from [1-14C]IPP using Sulfolobus acidocaldarius exo-pyrophosphatase as described in Ref. 11. [U-13C]Mevalonolactone was prepared as described elsewhere (14).
Cell-free Conversion Assay Using the Radiolabeled Substrates
0.95 g of wet T. acidophilum cells were dissolved with 5 ml of 100 mm sodium phosphate buffer, pH 7.5, and were disrupted by sonication with a UP200S ultrasonic homogenizer (Hielscher Ultrasonics, Germany). The homogenate was centrifuged at 15,000 rpm for 30 min, and the supernatant filtered with a 0.45-μm membrane filter was used as the cell-free extract. In a 100-μl volume, 728 pmol (1,480 Bq) of the radiolabeled substrate, i.e. [2-14C]MVA, [2-14C]MVA-5-P, [2-14C]MVA-5-PP, [1-14C]IP, [1-14C]IPP, or later, the 14C-labeled Ta1305 product prepared as described below, was reacted with the cell-free extract of T. acidophilum containing 800 μg of protein, 800 nmol of ATP, 1 nmol of FPP, and an appropriate amount of S. acidocaldarius geranylgeranyl diphosphate (GGPP) synthase that was prepared as described elsewhere (15), in 10 mm sodium phosphate buffer, pH 7.5, containing 10 mm MgCl2 and 50 mm NaF, at 60 °C for 1 h. After the reaction, 200 μl of saturated saline was added to the mixture, and the hydrophobic product GGPP was extracted with 600 μl of 1-butanol that had been saturated with the saline. After treatment with potato acid phosphatase (Sigma), according to a method described by Fujii et al. (16), the alcohol from GGPP was extracted with n-pentane. The radioactivity in the pentane layer was measured with an LSC-5100 liquid scintillation counter (Aloka, Japan). The pentane-extracted alcohol was analyzed by reversed-phase TLC using a precoated plate, LKC-18, developed with acetone/H2O (9:1). The distribution of radioactivity on the TLC plate was detected using a Typhoon FLA7000 image analyzer (GE healthcare).
Cloning, Recombinant Expression, and Purification of T. acidophilum Proteins
The homolog genes of MVK (ta0344 and ta1304) and DMD (ta0461, ta0893, and ta1305) from T. acidophilum were amplified by KOD DNA polymerase (Toyobo, Japan), using genomic DNA of T. acidophilum as a template and each pair of primers as follows: 5′-aaaaacatatgataatcactcgaacccca-3′ and 5′-aaagtcgactcacctgtcctgatagtagattatgc-3′ for ta0344m; 5′-aaaacatatggtgtacatatcatacagccctc-3′ and 5′-aaagtcgacctaatggccgatcatcc-3′ for ta1304; 5′-aaaaacatatggccccgcatc-3′ and 5′-aaactcgagtcaggtgtagcggtcg-3′ for ta0461; 5′-aaaaacatatgttggatacagccggaatg-3′ and 5′-aaactcgagtcacaggcgatcctcttcg-3′ for ta0893; and 5′-aaaacatatgcctacagatcaataggatcaac-3′ and 5′-aaactcgagtcactctggtcttctatgcca-3′ for ta1305. Each of the amplified DNA fragments was digested with restriction enzymes NdeI and XhoI (NdeI and SalI for ta0344m and ta1304), which recognize the underlined sequences in the paired primers, respectively, and then was ligated with the pET-15b vector (Merck) that had been digested with NdeI and XhoI (NdeI and SalI for cloning ta0344m or ta1304). The Escherichia coli Rosetta (DE3) strain transformed with each of the plasmids was cultivated at 37 °C in an LB medium supplemented with 100 mg/liter ampicillin and 30 mg/liter chloramphenicol. After ∼20 h of cultivation with no induction, cells were harvested. For the expressions of Ta1304 and Ta1305, cells were cultured at 30 and 22 °C, respectively.
The IPK gene from T. acidophilum (ta0103) was amplified with a pair of primers, 5′-cgcgcggcagccatatgatgatactgaagataggcggaag-3′ and 5′-ggatcctcgagcatatcatcttatcaccgtacctatgaatgattc-3′, and the amplified DNA was inserted into pET-15b digested with NdeI using an In-Fusion Advantage PCR cloning kit (TaKaRa, Japan) following the manufacturer's instructions. E. coli BL21(DE3) transformed with the plasmid was cultivated in an LB medium supplemented with 100 mg/liter ampicillin at 37 °C, and 1 mm isopropyl β-d-1-thiogalactopyranoside was added for the induction of enzyme expression when the A600 of the culture reached 0.6. After additional cultivation at 30 °C for 5 h, the cells were harvested.
The harvested cells were disrupted by sonication using a UP200S ultrasonic homogenizer (Hielscher Ultrasonics) in a HisTrap binding buffer containing 20 mm sodium phosphate at pH 7.4, 0.5 m NaCl, and 20 mm imidazole. The homogenates were centrifuged at 15,000 rpm for 30 min to recover the supernatants. Each recombinant enzyme was purified from the supernatant by heat treatment at 60 °C for 30 min and then using a HisTrap FF crude column (GE Healthcare) according to the manufacturer's protocol. The heat treatment process was omitted only for the purification of Ta0461 because the enzyme was denatured by heat treatment. The level of purification was determined by 12% SDS-PAGE.
Analysis of the Reaction Products of the Recombinant Enzymes
In a 25-μl volume, 91 pmol of the 14C-labeled substrate prepared as described above was reacted with 25 pmol of the purified recombinant T. acidophilum proteins, i.e. Ta0344M, Ta1304, Ta0461, or Ta1305, and 100 nmol of ATP, in 40 mm sodium phosphate buffer, pH 7.5, containing 5 mm MgCl2, at 60 °C for 1 h. 10 μl of the solution was spotted on a Partisil K6F normal-phase TLC plate and then developed with CHCl3/pyridine/formate/H2O (12:28:6:4). As necessary, an appropriate amount of S. solfataricus DMD, prepared as described in our previous study (11), was reacted with the post-reaction mixture to check the formation of MVA-5-PP by its conversion into IPP.
Preparation of the 14C-Labeled Ta1305 Product
In a 50-μl volume, 7.28 nmol of [2-14C]MVA-5-P was reacted with 15 units of Antarctic phosphatase (New England Biolabs) at 37 °C for 24 h according to the manufacturer's protocol. After the reaction, the mixture was treated at 65 °C for 15 min to inactivate the phosphatase. The heat-treated mixture was then reacted with 2 nmol of Ta1305 and 1.6 μmol of ATP at 60 °C for 3 h in a 400-μl volume of 40 mm sodium phosphate buffer, pH 7.5, containing 5 mm MgCl2. After filtration with Vivaspin 500 (10 kDa molecular mass cutoff, Funakoshi, Japan), the filtrate was used as the 14C-labeled Ta1305 product.
Analysis of the Ta1305 Product from MVA Using 32P-Labeled ATP
65 mg of (R)-mevalonolactone was reacted with 3 ml of 0.2 n KOH at 37 °C for 1 h. After the reaction, the mixture was neutralized to pH 7.2 with 0.1 n HCl and diluted with water up to 5 ml. The solution was used as 0.1 m (R)-MVA. In a 25-μl volume, 10 nmol of (R)-MVA and 400 pmol of either [γ-32P]ATP or [α-32P]ATP was reacted with 210 pmol of Ta1305 at 60 °C for 1 h in 40 mm sodium phosphate buffer, pH 7.5, containing 5 mm MgCl2. When [γ-32P]ATP was used, the radio-TLC analysis of the product was performed with the same protocol used for the 14C-labeling assay described above. When [α-32P]ATP was used, a Silica Gel 60 TLC plate was developed with propanol-1, methanol, 25% ammonium water (45:15:40). S. solfataricus MVK, prepared as described elsewhere (11), was used for experimental control to produce MVA-5-P and ADP.
MS Analysis of the Ta1305 Product
In a 150-μl volume, 600 nmol of (R)-MVA was reacted with 1.5 nmol of Ta1305 at 60 °C for 1 h in 0.1 m ammonium acetate buffer, pH 6.9, containing 3 μmol of ATP. After filtration with Vivaspin 500 (10 kDa molecular mass cutoff), the filtrate was mixed with the same volume of acetonitrile and analyzed by negative ion electrospray ionization-mass spectrometry with an Esquire 3000 (Bruker) by direct infusion.
NMR Analysis of the Ta1305 Product
20 mg of [U-13C]mevalonolactone was hydrolyzed with 192 μl of 1 m KOH at 37 °C for 1 h. After neutralization and dilution, the solution was used as a [U-13C]MVA solution. A 400-μl sample containing 1.95 μmol of [U-13C]MVA, 10 μmol of ATP, 50 μmol of ammonium acetate buffer, pH 6.9, and 12.5% D2O was prepared for a 13C NMR analysis of [U-13C]MVA using an AVANCE III HD 600 NMR spectrometer equipped with a cryoprobe (Bruker). After the analysis of the substrate, 5 nmol of Ta1305 in 100 mm ammonium acetate buffer, pH 6.9, was added to the sample mixture for enzyme reaction at 60 °C for 2 h. The mixture was filtered with Vivaspin 500 (10 kDa molecular mass cutoff), and the 13C NMR spectrum of the filtrate was analyzed.
Anion-exchange Chromatography
The cell-free extract from 0.9 g of wet T. acidophilum cells was prepared as described above, with the exception of the use of 50 mm sodium phosphate buffer, pH 6.5, for cell disruption. Two HiTrap Q HP columns (1 ml, GE Healthcare) connected in tandem were equilibrated with 20 ml of 50 mm sodium phosphate buffer, pH 6.5, using an FPLC system (Pharmacia, Sweden). The crude extract from T. acidophilum was applied to the column. Proteins were eluted with the following NaCl gradient in the buffer at a flow rate of 1 ml/min: 0–20 min, 0 m; 20–40 min, linear gradient from 0 to 1 m; and 40–60 min, 1 m. The eluent was fractionated into the first 20-ml fraction (at a retention time of 0–20 min), subsequent 2-ml fractions (20–40 min, 2 min each), and a final 20-ml fraction (40–60 min).
Conversion of Radiolabeled Substrates with Fractionated T. acidophilum Enzymes
Radio-TLC assay using fractionated enzymes was carried out basically with the same protocols for the assay with purified enzymes. In a 25-μl volume, 91 pmol of the 14C-labeled Ta1305 product ([2-14C]MVA-3-P) was reacted with 15 μl each of T. acidophilum enzyme fractions and 100 nmol of ATP, in 40 mm sodium phosphate buffer, pH 7.5, containing 5 mm MgCl2, at 60 °C for 2 h. After the reaction, 10 μl of the reaction mixture was analyzed by TLC. For the enzymatic analyses of the unidentified products, an appropriate amount of S. solfataricus DMD or T. acidophilum IPK was added to the pre-reaction mixture as needed.
Phylogenetic Analysis
The phylogenetic analysis of the MVA pathway-related enzymes was performed with the program Clustal Omega (17). The phylogenetic tree was depicted using a program NJplot (18).
RESULTS
Because IPK was discovered from T. acidophilum (12), the archaeon was expected to use the modified MVA pathway that passes through MVA-5-P and IP (Fig. 1B). To determine whether the enzymatic activity was associated with the classical or the modified MVA pathway in the cells of T. acidophilum, the cell-free extract of the archaeon was reacted with the radiolabeled intermediates of the pathways, i.e. [2-14C]MVA, [2-14C]MVA-5-P, [2-14C]MVA-5-PP, or [1-14C]IP, and the activity to synthesize IPP was measured using S. acidocaldarius GGPP synthase (19). Because most of the later reactions of the known MVA pathways are ATP-dependent, ATP and Mg2+ were added. NaF was added as an inhibitor for phosphatases in the extract. If the radiolabeled substrates were converted into IPP, it would be condensed with FPP, which was added as an allylic substrate for the prenyl elongation reaction, to form radiolabeled GGPP. The hydrophobic product was extracted with 1-butanol and then hydrolyzed by phosphatase treatment. Its alcoholic form, geranylgeraniol, was again extracted with n-pentane for TLC-autoradiography analysis (Fig. 3). The result from this preliminary experiment was surprising. MVA and IP were efficiently converted into IPP, although MVA-5-P and MVA-5-PP were not. The interpretation of this result was not simple because the crude extract from T. acidophilum, which might contain enzymes decomposing the intermediates, was used for the experiment; however, the result suggested the existence of an undiscovered biosynthetic route to IPP, which passes through MVA and IP, rather than through MVA-5-P and MVA-5-PP.
FIGURE 3.
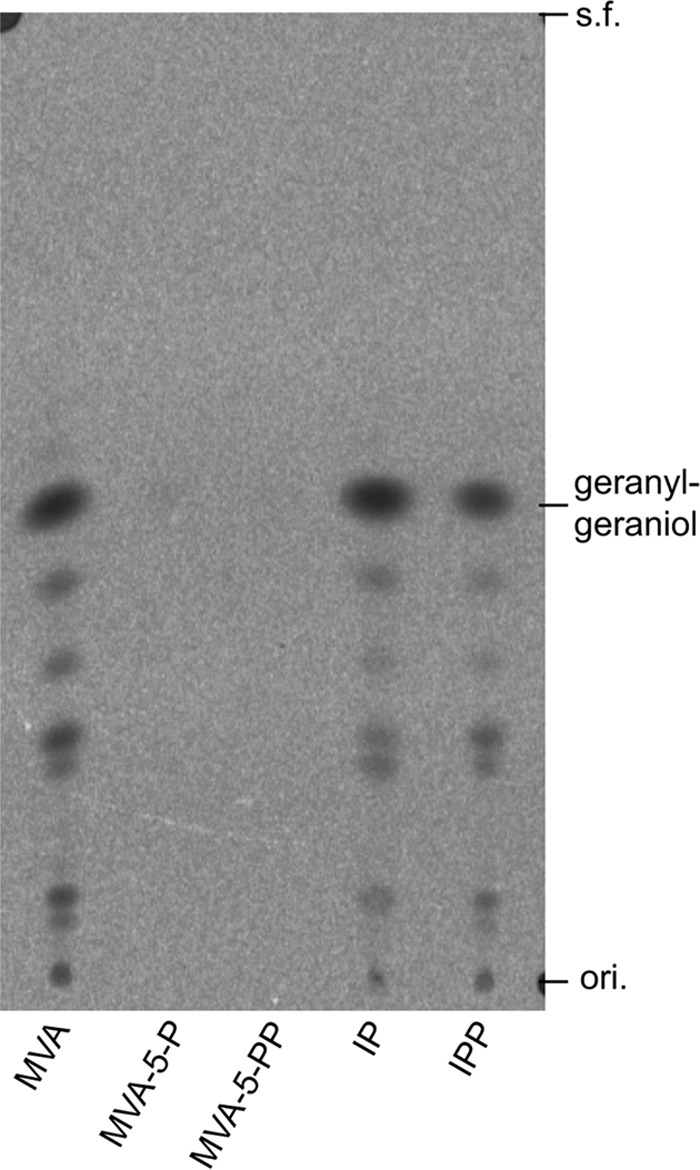
Radio-TLC analysis of products from the cell-free conversion assay. Formation of IPP from 14C-labeled substrates through conversion by T. acidophilum cell-free extract was confirmed by its condensation with FPP by the action of GGPP synthase. The product GGPP was hydrolyzed by phosphatase to geranylgeraniol and then detected by reversed-phase TLC developed with acetone/H2O (9:1) and followed by autoradiography. ori., origin; s.f., solvent front.
We then attempted to find the enzyme that catalyzes the conversion of MVA from T. acidophilum. The prime candidates were the homologs of the enzymes of the classical MVA pathway as follows: MVK homologs Ta0344M and Ta1304; and DMD homologs Ta0461, Ta0893, and Ta1305. The homologs were recombinantly expressed in E. coli and purified using the N-terminally fused polyhistidine tag (data not shown). Only Ta0893 could not be purified because it was expressed as an inclusion body, although the molecular weight of the expressed protein seemed smaller than that expected from the amino acid sequence of Ta0893. Purified Ta0344M and Ta0461 showed no activity toward all the radiolabeled substrates (data not shown). Ta1304 exhibited obvious MVK activity, as expected from its sequential homology (Fig. 4A). However, even though it is a DMD homolog, Ta1305 almost completely converted MVA into a compound with an Rf value that was similar to, but slightly larger than, that of MVA-5-P based on TLC analysis (Fig. 4B). The enzyme showed no detectable DMD activity. More curiously, the enzyme exhibited very weak PMK activity and converted a slight portion of MVA-5-P into MVA-5-PP. The formation of MVA-5-PP was confirmed by its conversion into IPP through the reaction with S. solfataricus DMD (data not shown) (11). This inconsistent result suggested that the Ta1305 reaction product from MVA was not MVA-5-P, because it was not converted further into MVA-5-PP.
FIGURE 4.

Radio-TLC analysis of products from reactions with Ta1304 (A) and Ta1305 (B). Radiolabeled substrates, i.e. [2-14C]MVA, [2-14C]MVA-5-P, [2-14C]MVA-5-PP, and [1-14C]IP, were reacted with purified enzymes in the presence of ATP and Mg2+. The products and [1-14C]IPP as an authentic compound were analyzed by normal-phase TLC developed with CHCl3/pyridine/formate/H2O (12:28:6:4) and followed by autoradiography. ori., origin; s.f., solvent front.
We therefore attempted to determine the structure of the Ta1305 product that had formed from MVA. Without ATP, the product was not formed (data not shown). Using [γ-32P]ATP and [α-32P]ATP, we confirmed the incorporation of the γ-phosphate group into the product and the formation of ADP through the Ta1305 reaction, respectively (Fig. 5). This result suggests that the γ-phosphate group from ATP was transferred to MVA to yield the product. This expectation was verified by negative electron spray ionization-MS analysis of the compounds formed after the Ta1305 reaction (Fig. 6A). Because we noticed that the reaction of Ta1305 was insensitive to the addition of EDTA (data not shown), MgCl2 was omitted from the reaction thereafter. An ion with an m/z of 226.7, which was remarkably detected in the ion spectrum, corresponded either with MVA-5-P or its isomer. The formation of ADP was also confirmed by the appearance of an ion with an m/z of 425.8, although those with values of m/z at 146.7 and 505.8 were likely derived from unreacted MVA and ATP, respectively. The MS/MS analysis of an ion with an m/z value of 226.7 gave fragmentation ions with m/z values of 211.7, 208.7, 182.7, and 128.7, which corresponded with [M − CH3]−, [M − H2O]−, [M − CO2]−, and [M − H3PO4]−, respectively (Fig. 6B). Next, we performed NMR analysis of the Ta1305 product using [U-13C]MVA. In the 13C NMR spectrum of the reaction mixture, from which the enzyme had been removed, the U-13C-labeled product gave strong signals with characteristic 13C-13C coupling, which is usually not detected under the conditions of natural isotopic abundance (Table 2 and Fig. 7). Compared with the spectrum of [U-13C]MVA, only the signal that was assigned to C3 of MVA shifted after the reaction with Ta1305, suggesting that the hydroxyl group at C3 is the site for phosphorylation. Moreover, after the reaction, only the signal of C3 got an additional hyperfine coupling of 8 Hz, which probably arose from the two-bond 13C-31P coupling (Fig. 7B). In addition, MS analysis of the Ta1305 product from [U-13C]MVA gave an ion with an m/z value of 232.7, which was 6 units larger than that observed in the analysis of the nonlabeled product (Fig. 6C). These results clearly showed that the product was MVA-3-P and that Ta1305 is ATP:(R)-MVA 3-phosphotransferase.
FIGURE 5.

Radio-TLC analysis of the Ta1305 product from MVA and 32P-labeled ATP. A, nonlabeled MVA and [γ-32P]ATP were reacted with purified Ta1305 or S. solfataricus MVK. The products were analyzed by normal-phase TLC developed with CHCl3/pyridine/formate/H2O (12:28:6:4) and followed by autoradiography. B, nonlabeled MVA and [α-32P]ATP were reacted with purified Ta1305 or S. solfataricus MVK. The products were analyzed by normal-phase TLC developed with propanol-1, methanol, 25% ammonium water (45:15:40). ori., origin; s.f., solvent front.
FIGURE 6.

Negative electron spray ionization-MS analysis of the Ta1305 product from MVA. A, ion spectrum of components in the post-reaction mixture of Ta1305 with nonlabeled MVA and ATP, obtained by a direct infusion method. B, MS/MS analysis of the ion of m/z 226.7. C, ion spectrum of components in the post-reaction mixture of Ta1305 with [U-13C]MVA and ATP.
TABLE 2.
13C NMR data for [U-13C]MVA and the Ta1305 product from [U-13C]MVA
| Compound | Carbon no. | Chemical shift | Coupling patterna |
|---|---|---|---|
| [U-13C]MVA | 1 | 180.0 ppm | d |
| 2 | 47.7 | dd | |
| 3 | 70.9 | q | |
| 4 | 42.6 | t | |
| 5 | 57.9 | d | |
| 6 | 26.0 | d | |
| Ta1305 product | 1 | 178.9 | d |
| 2 | 48.2 | dd | |
| 3 | 78.5 | qd | |
| 4 | 42.2 | t | |
| 5 | 57.8 | d | |
| 6 | 25.3 | d |
a The abbreviations used are as follows: d, doublet; t, triplet; q, quartet; dd, doublet doublet; qd, quartet doublet.
FIGURE 7.

13C NMR analysis of the Ta1305 reaction product. A, signal at 70.9 ppm in the NMR spectrum of a pre-reaction mixture is assigned to the C3 of [U-13C]MVA. It exhibits an apparent quartet coupling pattern (J = ∼38 Hz). Smaller signals around it probably come from other components in the reaction mixture. B, corresponding signal at 78.5 ppm in the spectrum of a post-reaction mixture has acquired an additional hyperfine two-bond 13C-31P coupling (J = 8 Hz), which results in a quartet-doublet coupling pattern.
At that point, however, we noticed that a recently disclosed patent application made by Global Bioenergies, France, and Scientist of Fortune S.A., Luxembourg, had identified Ta1305 as a DMD homolog that can convert MVA into MVA-3-P (20). The inventors determined the product structure based on the result of MS analysis and on the fact that the product can be converted into isopentenol by the action of known DMDs and their orthologs. They even conducted kinetic analysis of Ta1305 and got acceptable constants (Km for (RS)-MVA of 0.32 mm and kcat of 1.5 s−1). However, they regarded the enzyme simply as a tool to produce isopentenol, which can be applied for industrial uses, and did not consider the physiological role of Ta1305, except for presenting the possibility that the enzyme “actually represents a mevalonate-5-monophosphate decarboxylase” (20). Thus, we tried to determine whether MVA-3-P would be metabolized to IPP in the cells of T. acidophilum, what intermediate the metabolic pathway passes through, and what would be the percentage of total IPP production in T. acidophilum from the new pathway.
Therefore, we again performed the conversion assay of radiolabeled substrates, including MVA-3-P using T. acidophilum cell-free extract. GGPP was extracted from the reaction mixture with 1-butanol, and the radioactivity extracted with n-pentane after phosphatase treatment of the butanol extract was used to quantify the conversion efficiency of the substrates (Fig. 8A) because part of the radioactivity from unreacted substrates, such as spontaneously lactonized MVA, can also be extracted in the 1-butanol layer. A TLC-autoradiography analysis proved that a major part of radioactivity in the pentane layer was derived from geranylgeraniol (Fig. 8B). The conversion efficiency of MVA-3-P was ∼25%, which was comparable with those of IPP and IP. As shown in Fig. 3, the conversion efficiency of MVA was also high (∼15%), whereas those of MVA-5-P and MVA-5-PP were significantly lower. It seems conceivable that the efficiency reached a plateau at around 20% of conversion efficiency, possibly due to a shortage of FPP; however, that result strongly suggests that MVA-3-P, like MVA and IP, is an intermediate of the major MVA pathway in T. acidophilum, which yields IPP as an active unit for isoprenoid biosynthesis. The Thermoplasma-type modified MVA pathway is distinct from the modified pathway recently discovered from R. castenholzii and H. volcanii and, of course, from the classical MVA pathway, and it does not pass through MVA-5-P as well as MVA-5-PP.
FIGURE 8.

Conversion efficiency of intermediates of the MVA pathways in the reaction with T. acidophilum cell-free extract. Radioactive intermediates including [2-14C]MVA-3-P were used for the assay. Their conversion into IPP was confirmed by the use of a prenyl-elongation reaction with S. acidocaldarius GGPP synthase. Conversion efficiency was calculated by dividing the radioactivity extracted in an n-pentane layer by that of an added intermediate. ori., origin; s.f., solvent front.
Because IP is unlikely to be the intermediate proximally downstream from MVA-3-P, next we searched enzyme activities that can convert MVA-3-P into downstream intermediates. Such activities were sought from T. acidophilum enzymes fractionated by anion-exchange column chromatography. An enzyme fraction at a retention time of 30–32 min (and also the fractions before and after it, although weakly) converted MVA-3-P into a compound with an Rf value identical to that of IP in TLC analysis (Fig. 9A). A faint, tailed spot with an Rf value similar to that of IPP was also observed. The addition of purified T. acidophilum IPK in the reaction resulted in a fading of the spot of the IP-like compound and in increase in the IPP-like compound instead (Fig. 9B), suggesting that the 30–32-min fraction could convert MVA-3-P mainly into IP. Conversely, earlier eluted enzyme fractions at 0–30 min converted MVA-3-P into a compound with an Rf value similar to that of MVA-5-PP (Fig. 9A). However, the addition of purified S. solfataricus DMD converted the compound into an IP-like compound (Fig. 9C). The product from DMD reaction with MVA-5-PP, namely IPP, was not produced. This result strongly suggests that the compound is mevalonate 3,5-bisphosphate (MVA-3,5-PP), because the patent application mentioned above has shown that the treatment of MVA-3-P with various DMDs yields isopentenol (20). A similar decarboxylation reaction catalyzed by DMD is expected to synthesize IP from MVA-3,5-PP. Because the biosynthetic pathway from MVA-3-P to IP via MVA-3,5-PP seems quite reasonable, we synthesized the compound that was believed to be MVA-3,5-PP from MVA-3-P using the 22–24-min fraction and, after filtration, reacted it with T. acidophilum cell-free extract or the 30–32-min fraction. Both reactions yielded IP, as expected (Fig. 9D). These results support the presence of a third alternative MVA pathway in T. acidophilum (Fig. 1C). In the modified pathway, MVA is phosphorylated to MVA-3-P by the action of Ta1305, namely ATP:(R)-MVA 3-phosphotransferase. MVA-3-P is presumably converted into IP via MVA-3,5-PP by unidentified enzymes, probably (R)-MVA-3-P 5-phosphotransferase and (R)-MVA-3,5-PP decarboxylase. Then IP is phosphorylated by IPK to yield IPP. The absence, or at least a negligible presence, of the known MVA pathways that pass through MVA-5-P (Fig. 1, A and B) in T. acidophilum was also supported by a conversion assay using the same enzyme fractions and radiolabeled MVA-5-P, in which none of the fractions converted MVA-5-P (data not shown).
FIGURE 9.

Conversion of MVA-3-P with fractionated T. acidophilum enzymes. A, [2-14C]MVA-3-P was reacted with each fraction of anion-exchange column chromatography of T. acidophilum cell-free extract in the presence of ATP and Mg2+. The reaction mixtures were analyzed by normal-phase TLC. B, radio-TLC analysis of the products from the reaction with the 30–32-min fraction and T. acidophilum IPK. C, analysis of the products from the reaction with the 22–24-min fraction. S. solfataricus DMD was added during the reaction, and the reaction mixture was analyzed by normal-phase TLC. D, radio-TLC analysis of the products obtained from MVA-3-P by consecutive reactions with different fractions. After conversion of MVA-3-P using the 22–24-min fraction, the reaction mixture was filtered and then reacted either with T. acidophilum cell-free extract or the 30–32 min fraction. ori., origin; s.f., solvent front.
DISCUSSION
Ta1305, which was identified as ATP:(R)-MVA 3-phosphotransferase, should be a key enzyme of the third alternative MVA pathway that we discovered from T. acidophilum in this study. Distribution of its orthologs is very limited among archaea in the class Thermoplasmata. This means that the Thermoplasma-type MVA pathway is likely distributed also among the limited archaeal species. In archaea other than Halobacteria and Thermoplasmata (and the relatives of S. solfataricus that possess the classical MVA pathway), however, no research has elucidated the biosynthetic route that supplies IP for IPK. Such general archaea possess neither the ortholog of Ta1305 nor that of PMD. Therefore, it remains unclear how IP is synthesized in most archaea. Although they possess an MVK homolog gene, and MVK activity has been identified in a few cases (21, 22), it is also unclear which of the two modified MVA pathways, i.e. the Roseiflexus/Haloferax-type pathway via MVA-5-P and the Thermoplasma-type pathway via MVA-3-P, is actually used by them. Of course, there remains the possibility of another undiscovered route.
Ta1305, PMD, and DMD commonly catalyze the phosphorylation of the 3-hydroxyl group of the MVA moiety of their substrates, i.e. MVA, MVA-5-P, and MVA-5-PP, respectively. However, Ta1305 is distinct from the others because it does not catalyze the subsequent elimination of the 3-phosphate group that accompanies decarboxylation. The difference is presumably due to the structural properties of the enzymes. Ta1305 lacks some residue that was conserved among DMDs and PMDs. For example, a highly conserved aspartate residue in DMD/PMD, which corresponds to Asp-283 of Staphylococcus epidermidis DMD, is not conserved in Ta1305 and its orthologs (data not shown). Based on the results from a previous mutagenic study (23) and on the ternary crystal structures of S. epidermidis DMD, Barta et al. (24) proposed that the aspartate residue is the active site residue that abstracts 3-hydroxy proton to promote phosphotransfer from ATP. Considering that a phosphotransfer reaction occurs also in Ta1305, the enzyme likely has a substituting residue at a different position. However, it is more fascinating to think that the main role of the aspartate residue is to aid in the elimination of the 3-phosphate group and that its absence causes a functional difference between Ta1305 and DMD/PMD. Mutagenic and/or structural studies on Ta1305 would provide clues to solve this problem.
Considering the existence of the multiple homologs of MVK and DMD in T. acidophilum, it is tempting to imagine that one of the MVK homologs catalyzes the 5-phosphorylation of MVA-3-P and that one of the DMD homologs promotes the decarboxylation of MVA-3,5-PP to form IP. The distance between the T. acidophilum MVK homologs, i.e. Ta0344M and Ta1304, and usual MVKs suggests their different preference to the substrates, whereas Ta1304 has MVK activity. In a similar manner, the DMD homologs other than Ta1305, i.e. Ta0461 and Ta0893, are distinguishable from standard DMDs. In a recent paper announcing the first discovery of PMD, Dellas et al. (9) reported the difference in the amino acid sequences between DMDs and the orthologs of PMD. The difference presumably endows PMD with a preference to MVA-5-P over MVA-5-PP. They also reported that Ta0461 and Ta0893 share such sequential properties of PMD, although it is unlikely that MVA-5-P is metabolized in T. acidophilum cells, as we have shown here. This information implies that the enzymes might have a preference for a substrate that possesses a mono-phosphate group at C5, such as MVA-3,5-PP. Our group is presently attempting to prove this hypothesis.
Acknowledgment
We thank Kazushi Koga, Nagoya University, for help with the NMR analysis.
This work was supported by Japan Society for the Promotion of Science KAKENHI Grants 23580111, 23108531, and 25108712 (to H. H.).
This article was selected as a Paper of the Week.
- MVA
- mevalonate
- DMD
- diphosphomevalonate decarboxylase
- FPP
- farnesyl diphosphate
- GGPP
- geranylgeranyl diphosphate
- IP
- isopentenyl phosphate
- IPK
- isopentenyl phosphate kinase
- IPP
- isopentenyl diphosphate
- MVA-3-P
- mevalonate 3-phosphate
- MVA-3,5-PP
- mevalonate 3,5-bisphosphate
- MVA-5-P
- mevalonate 5-phosphate
- MVA-5-PP
- mevalonate 5-diphosphate
- MVK
- mevalonate kinase
- PMD
- phosphomevalonate decarboxylase
- PMK
- phosphomevalonate kinase.
REFERENCES
- 1. Kuzuyama T., Hemmi H., Takahashi S. (2010) in Comprehensive Natural Products II Chemistry and Biology (Mander L., Liu H.-W., eds) pp. 493–516, Elsevier, Oxford, UK [Google Scholar]
- 2. Thulasiram H. V., Erickson H. K., Poulter C. D. (2007) Chimeras of two isoprenoid synthases catalyze all four coupling reactions in isoprenoid biosynthesis. Science 316, 73–76 [DOI] [PubMed] [Google Scholar]
- 3. Lombard J., Moreira D. (2011) Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 28, 87–99 [DOI] [PubMed] [Google Scholar]
- 4. Matsumi R., Atomi H., Driessen A. J., van der Oost J. (2011) Isoprenoid biosynthesis in Archaea–biochemical and evolutionary implications. Res. Microbiol. 162, 39–52 [DOI] [PubMed] [Google Scholar]
- 5. Smit A., Mushegian A. (2000) Biosynthesis of isoprenoids via mevalonate in Archaea: the lost pathway. Genome Res. 10, 1468–1484 [DOI] [PubMed] [Google Scholar]
- 6. Dellas N., Noel J. P. (2010) Mutation of archaeal isopentenyl phosphate kinase highlights mechanism and guides phosphorylation of additional isoprenoid monophosphates. ACS Chem. Biol. 5, 589–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Grochowski L. L., Xu H., White R. H. (2006) Methanocaldococcus jannaschii uses a modified mevalonate pathway for biosynthesis of isopentenyl diphosphate. J. Bacteriol. 188, 3192–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mabanglo M. F., Schubert H. L., Chen M., Hill C. P., Poulter C. D. (2010) X-ray structures of isopentenyl phosphate kinase. ACS Chem. Biol. 5, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dellas N., Thomas S. T., Manning G., Noel J. P. (2013) Discovery of a metabolic alternative to the classical mevalonate pathway. eLife 2, e00672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vannice J. C., Skaff D. A., Keightley A., Addo J. K., Wyckoff G. J., Miziorko H. M. (2014) Identification in Haloferax volcanii of phosphomevalonate decarboxylase and isopentenyl phosphate kinase as catalysts of the terminal enzymatic reactions in an archaeal alternate mevalonate pathway. J. Bacteriol. 196, 1055–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nishimura H., Azami Y., Miyagawa M., Hashimoto C., Yoshimura T., Hemmi H. (2013) Biochemical evidence supporting the presence of the classical mevalonate pathway in the thermoacidophilic archaeon Sulfolobus solfataricus. J. Biochem. 153, 415–420 [DOI] [PubMed] [Google Scholar]
- 12. Chen M., Poulter C. D. (2010) Characterization of thermophilic archaeal isopentenyl phosphate kinases. Biochemistry 49, 207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sambrook J., Fritsch E. F., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 14. Sugai Y., Miyazaki S., Mukai S., Yumoto I., Natsume M., Kawaide H. (2011) Enzymatic total synthesis of gibberellin A4 from acetate. Biosci. Biotechnol. Biochem. 75, 128–135 [DOI] [PubMed] [Google Scholar]
- 15. Hemmi H., Shibuya K., Takahashi Y., Nakayama T., Nishino T. (2004) (S)-2,3-Di-O-geranylgeranylglyceryl phosphate synthase from the thermoacidophilic archaeon Sulfolobus solfataricus. Molecular cloning and characterization of a membrane-intrinsic prenyltransferase involved in the biosynthesis of archaeal ether-linked membrane lipids. J. Biol. Chem. 279, 50197–50203 [DOI] [PubMed] [Google Scholar]
- 16. Fujii H., Koyama T., Ogura K. (1982) Efficient enzymatic hydrolysis of polyprenyl pyrophosphates. Biochim. Biophys. Acta 712, 716–718 [PubMed] [Google Scholar]
- 17. Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G. (2011) Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal omega. Mol. Syst. Biol. 7, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Perrière G., Gouy M. (1996) WWW-Query: An on-line retrieval system for biological sequence banks. Biochimie 78, 364–369 [DOI] [PubMed] [Google Scholar]
- 19. Ohnuma S., Suzuki M., Nishino T. (1994) Archaebacterial ether-linked lipid biosynthetic gene. Expression cloning, sequencing, and characterization of geranylgeranyl-diphosphate synthase. J. Biol. Chem. 269, 14792–14797 [PubMed] [Google Scholar]
- 20. Delcourt M., Anissimova M., Marliere P. (October 10, 2013) International Patent Application PCT/EP2013/057108
- 21. Huang K. X., Scott A. I., Bennett G. N. (1999) Overexpression, purification, and characterization of the thermostable mevalonate kinase from Methanococcus jannaschii. Protein Expr. Purif. 17, 33–40 [DOI] [PubMed] [Google Scholar]
- 22. Primak Y. A., Du M., Miller M. C., Wells D. H., Nielsen A. T., Weyler W., Beck Z. Q. (2011) Characterization of a feedback-resistant mevalonate kinase from the archaeon Methanosarcina mazei. Appl. Environ. Microbiol. 77, 7772–7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Krepkiy D., Miziorko H. M. (2004) Identification of active site residues in mevalonate diphosphate decarboxylase: implications for a family of phosphotransferases. Protein Sci. 13, 1875–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Barta M. L., McWhorter W. J., Miziorko H. M., Geisbrecht B. V. (2012) Structural basis for nucleotide binding and reaction catalysis in mevalonate diphosphate decarboxylase. Biochemistry 51, 5611–5621 [DOI] [PMC free article] [PubMed] [Google Scholar]