

Background: The FabI inhibitor CG400549 is a promising new anti-staphylococcal drug candidate with recently validated human efficacy.
Results: We revealed the molecular determinants conferring S. aureus FabI selectivity to rationally design a compound with an improved antibacterial activity spectrum.
Conclusion: The 4-pyridone PT166 represents a critical step toward Gram-negative and mycobacterial coverage.
Significance: We provide an approach to expand the spectrum of antimicrobial activity.
Keywords: Drug Design, Enzyme Inhibitor, Enzyme Structure, Lipid Synthesis, Protein Drug Interaction, Staphylococcus aureus, CG400549, Enoyl-ACP Reductase, FabI, Target Selectivity
Abstract
Determining the molecular basis for target selectivity is of particular importance in drug discovery. The ideal antibiotic should be active against a broad spectrum of pathogenic organisms with a minimal effect on human targets. CG400549, a Staphylococcus-specific 2-pyridone compound that inhibits the enoyl-acyl carrier protein reductase (FabI), has recently been shown to possess human efficacy for the treatment of methicillin-resistant Staphylococcus aureus infections, which constitute a serious threat to human health. In this study, we solved the structures of three different FabI homologues in complex with several pyridone inhibitors, including CG400549. Based on these structures, we rationalize the 65-fold reduced affinity of CG400549 toward Escherichia coli versus S. aureus FabI and implement concepts to improve the spectrum of antibacterial activity. The identification of different conformational states along the reaction coordinate of the enzymatic hydride transfer provides an elegant visual depiction of the relationship between catalysis and inhibition, which facilitates rational inhibitor design. Ultimately, we developed the novel 4-pyridone-based FabI inhibitor PT166 that retained favorable pharmacokinetics and efficacy in a mouse model of S. aureus infection with extended activity against Gram-negative and mycobacterial organisms.
Introduction
Staphylococcus aureus can cause a variety of bacterial infections ranging from common skin infections to life-threatening pneumonia or bacteremia (1). In particular, methicillin-resistant S. aureus (MRSA)6 poses an imminent risk to immunocompromised patients in healthcare settings all over the world. In addition, the incidence of community-acquired MRSA infections has increased among otherwise healthy individuals (1, 2). The initial occurrence of S. aureus strains resistant to vancomycin, an antibiotic used to treat severe MRSA infections (3), underlines the urgent need for novel anti-staphylococcal drugs.
Isoniazid, a first-line prodrug for the treatment of tuberculosis, inhibits the type II fatty acid biosynthesis pathway of Mycobacterium tuberculosis (4). The clinical success of isoniazid validates the type II fatty acid biosynthesis pathway as an important target for the development of new antibiotics (5). Bacterial fatty acid biosynthesis differs from its mammalian counterpart and is pivotal for the production of several cellular components, such as phospholipids (6, 7). In the last step of the type II fatty acid biosynthesis elongation cycle, the enoyl-acyl carrier protein (ACP) reductase (FabI) catalyzes the reduction of the trans-2-enoyl-ACP double bond (Fig. 1) (8, 9). After the discoveries that activated isoniazid and the antimicrobial agent triclosan inhibit FabI (10–14), this enzyme was investigated as a potential broad spectrum target conserved among many pathogenic organisms (15, 16). The finding that distinct bacteria such as Streptococcus pneumoniae or Vibrio cholerae utilize FabI isoenzymes, including FabK (17), FabL (18), and FabV (19) or can take up exogenous fatty acids from the host blood serum to circumvent the inhibition of FabI (20), has provided some limitations with regards to antibacterial coverage (15). Nevertheless, for several clinically relevant pathogens, such as S. aureus, Staphylococcus epidermidis, Escherichia coli, Haemophilus influenzae, Francisella tularensis, and M. tuberculosis, FabI remains a very promising drug target because these organisms require endogenous fatty acids (4, 6, 21, 22) and contain FabI as the sole enoyl-ACP reductase (23, 24). Accordingly, several drug design programs have been continued, and three S. aureus FabI (saFabI) inhibitors with different scaffolds (Fig. 1) have been advanced to clinical trials (25).
FIGURE 1.

Catalyzed reaction and successful inhibitor classes of S. aureus FabI. FabI catalyzes the reduction of the trans-2-enoyl-ACP substrate via an enolate intermediate (n = 0–8) (78). In the case of saFabI, the hydride (shown in red) is delivered by the reducing agent NADPH (6, 8, 9). During the second step of the reaction, the enolate intermediate is protonated, which leads to the formation of the final acyl-ACP product (the proton is depicted in blue). Examples for the most important FabI inhibitor classes are given in the boxes along with their binding mode in the saFabI active site pocket (PDB codes 4FS3 and 4ALI (6, 23); the CG400549 structure was solved during this study, PDB code 4CV1). For each of these inhibitor scaffolds, one compound is currently in clinical trials (AFN-1252, MUT056399, and CG400549) (25). One common feature of these FabI inhibitors is the formation of a hydrogen bond to Tyr-157 and the cofactor NADP(H). The oxygen atoms involved in this central interaction are colored in magenta. In addition, all depicted inhibitors directly interact with Ala-97 (the interacting inhibitor atoms are highlighted in orange).
Based on the lead compound triclosan, the company Mutabilis (FAB Pharma) developed the diphenyl ether MUT056399 (Fig. 1), which displays activity against S. aureus and several important Gram-negative pathogens (24, 26). In contrast, the pyridone inhibitor CG400549 (Crystal Genomics) as well as the naphthyridinone AFN-1252 (GlaxoSmithKline and Affinium Pharmaceuticals) were shown to be Staphylococcus-specific (Fig. 1) (15, 23, 27–30). Importantly, all three FabI inhibitors have been shown to be efficacious in mouse models of infection (23, 26, 30). Similar to Affinium Pharmaceuticals (AFN-1252), Crystal Genomics announced that the Phase 2a study for CG400549 has been completed and confirmed human efficacy without serious adverse events.
Extensive studies have been conducted on the molecular recognition of diphenyl ethers by saFabI (6, 16, 31). However, little is known about the binding of pyridone inhibitors such as CG400549. These compounds were intended to improve pharmacokinetic properties by replacing the metabolically labile phenol of the diphenyl ether skeleton with a stable pyridone (25). Despite the structural similarity between pyridones and diphenyl ethers, we reveal fundamental differences in the molecular recognition of these two scaffolds by FabI enzymes, which explain their respective narrow and broad spectrum activities. We report the first pyridone-bound crystal structures of saFabI, in addition to a comprehensive kinetic profile of inhibition. In particular, we solved the CG400549-bound saFabI and E. coli FabI (ecFabI) structures, which allowed us to rationalize the selectivity of this compound for the S. aureus homologue. Guided by this information, we sought to develop a compound that combined the pharmacokinetic stability of a pyridone with the broad spectrum characteristics of diphenyl ethers. The novel 4-pyridone inhibitor PT166 represents a significant step toward this goal, exhibiting extended spectrum antimicrobial activity against E. coli, F. tularensis, Proteus mirabilis, and M. tuberculosis, while retaining in vivo efficacy and favorable pharmacokinetics in a murine S. aureus thigh infection model.
EXPERIMENTAL PROCEDURES
Compound Synthesis
The pyridone compounds PT155, PT159, PT166, PT170, PT171, PT172, PT173, PT179, PT191, PT420, and CG400549 were synthesized as described in the supplemental Schemes S1–S5.
Expression and Purification
saFabI was prepared as described previously (6, 32). Briefly, we expressed the safabi gene cloned into a pETM-11 vector in E. coli BL21(DE3), disrupted the cells, and obtained the >95% pure protein in 25 mm Tris-HCl, pH 8.0, and 200 mm NaCl via Ni2+ affinity and size exclusion chromatography. In addition, ecFabI and the M. tuberculosis enoyl-ACP reductase InhA were expressed and purified as described previously (33, 34). Burkholderia pseudomallei FabI (bpFabI) was obtained using a previously described procedure (35) with the final size exclusion chromatography step (Superdex 200 26/60, GE Healthcare/ÄKTA) performed in 20 mm BisTris-HCl, pH 6.5, 500 mm NaCl, 1 mm EDTA.
Crystallization
Prior to concentrating saFabI samples from 2 to 15–19 mg/ml, the protein was incubated for 2 h at 20 °C with a 12-fold molar excess of NADPH and a 20-fold molar excess of inhibitor dissolved in DMSO (CG400549 or PT173, respectively). Diffraction-quality crystals were grown in vapor diffusion experiments with a precipitant solution containing 0.1–0.2 m Li2SO4 and 20–24 w/v % PEG 3350. For CG400549, we obtained crystals of space group P212121 with two different sets of cell parameters (the resulting structures were named CG400549-I and CG400549-II; supplemental Table S1).
Similarly, ecFabI samples at a concentration of 13 mg/ml were incubated for 2 h at 4 °C with a 10-fold molar excess of NADH and a 20-fold molar excess of CG400549 or PT166 (dissolved in DMSO), respectively. Hanging drop vapor diffusion experiments yielded diffraction-quality crystals in drops composed of 1 μl of this protein/ligand mixture and 1 μl of precipitant solution (0.2 m NH4Ac, 0.1 m CAPS, pH 10.5, and 20 w/v % PEG 8000 in the case of CG400549; 0.2 m NH4Ac, 0.1 m sodium citrate, pH 5.6, and 10 w/v % PEG 8000 in the case of PT166).
bpFabI samples at a concentration of 10–30 mg/ml were incubated for 2 h at 20 °C with a 10-fold molar excess of NAD+ and a 20-fold molar excess of PT155 (dissolved in DMSO). Sitting drop vapor diffusion experiments yielded diffraction-quality crystals in drops composed of 0.3 μl of the protein/ligand mixture and 0.3 μl of precipitant solution (20 w/v % PEG 3350 and 200 mm (NH4)2HPO4).
Data Collection and Structure Determination
Prior to flash-freezing in liquid nitrogen, the saFabI ternary complex crystals were successively transferred into solutions composed of mother liquor supplemented with 10 and 25 v/v % ethylene glycol, respectively. Diffraction data were collected at the BESSY II MX beamline 14.1 (36) (λ = 0.918 Å, T = 100 K) equipped with a MarMosaic 225 detector, integrated with Imosflm (CG400549-II and PT173) (37) or XDS (CG400549-I) (38), and further processed using Scala (39). The CG400549 structures were solved by molecular replacement with Phaser (40) using our previously published saFabI structure (PDB code 4ALK; lacking amino acids 196–202) as search model (6). For PT173, the fully refined CG400549-I structure was used as a template for molecular replacement. To avoid model bias, Rfree flags were assigned in thin resolution shells (CG400549-I and -II) or copied from the search model (for PT173). The final structures were obtained by several alternative cycles of model building in Coot (41) and refinement in Refmac 5 (CG400549-bound structures) (42) or Phenix (PT173) (43), respectively (including noncrystallographic symmetry (PT173, CG400549-II) and TLS refinement (44)). Cofactors and inhibitors could be unambiguously assigned based on the 2Fo − Fc and Fo − Fc electron density maps.
Crystals of the CG400549 and PT166 ternary ecFabI complexes were cryo-protected using the corresponding mother liquor supplemented by 25 or 30 v/v % ethylene glycol, respectively. Using a Pilatus 6M detector, diffraction data were collected at MX beamline 14.1 of the BESSY II synchrotron (λ = 0.918 Å, T = 100 K) and at beamline 23-1 of the European Synchrotron Radiation Facility (λ = 1.064 Å, T = 100 K), integrated using XDSAPP (45), and scaled with Scala. Initial phases were determined by molecular replacement in Phaser with our previously published ecFabI structure (PDB code 1QSG) as the search model (13). Model building in Coot and refinement using Refmac 5 (including TLS refinement) yielded the final structure. We did not model the amino acids 193 to 209/211 due to only partial and very weak electron density for the two monomers in the asymmetric unit. In addition, we note that the CG400549 electron density in subunit B was sufficient for model building but inferior compared with monomer A. Dictionaries for the cofactors and inhibitors of the S. aureus and E. coli FabI structures were computed using Grade (46, 47).
bpFabI crystals were cryo-cooled in cryo-protectant containing 25 v/v % glycerol in the mother liquor. Data collection was performed at an in-house x-ray generator (MicroMax-007 HF, Rigaku) at a wavelength of 1.54 Å and recorded with an imaging plate detector (R-Axis HTC, Rigaku). Data were integrated with Imosflm and scaled in Scala. Molecular replacement was performed in Phaser using the PDB entry 3EK2 as a template. For refinement in Refmac and finally Phenix TLS parameters were created using the TLSMD server (44) and a library file supplying restraints for the cofactor and inhibitor was generated by the Prodrg server (48). The structure was refined until convergence (R/Rfree = 14/16%) and validated using the Molprobity server (49).
To avoid model bias, omit maps were calculated prior to inclusion of cofactors and inhibitors. Data collection and refinement statistics are given in supplemental Table S1 (saFabI) and supplemental Table S2 (ecFabI and bpFabI). Distances and angles were measured for all subunits of the asymmetric unit and are given as mean values ± S.D. Structural figures were prepared using PyMOL (50).
The structure factors and coordinates of the different FabI structures have been deposited in the Protein Data Bank with the PDB entry codes 4CUZ (saFabI-NADPH-PT173), 4CV1 (saFabI-NADPH-CG400549-I), 4CV0 (saFabI-NADPH-CG400549-II), 4CV2 (ecFabI-NADH-CG400549), 4CV3 (ecFabI-NADH-PT166), and 4BKU (bpFabI-NAD+-PT155).
Inhibition Kinetics
Kinetics were performed on a Cary 100 spectrophotometer (Varian) at 20 °C. Reaction velocities were measured by monitoring the oxidation of NAD(P)H to NAD(P)+ at 340 nm (ϵ = 6220 m−1 cm−1). For saFabI, the reaction mixture was identical to that described previously for progress curve experiments (6). For ecFabI, the final reaction mixture contained ecFabI (75 nm), trans-2-butenoyl-CoA (800 μm; Sigma and Advent Bio), NADH (300 μm; Sigma), NAD+ (400 μm; Sigma), and inhibitor (2 v/v % DMSO) in 50 mm potassium phosphate, pH 7.5, 150 mm NaCl, 8 v/v % glycerol. For InhA, the final reaction mixture contained InhA (100 nm), trans-2-octenoyl-CoA (200 μm), NADH (250 μm), NAD+ (200 μm), and inhibitor (2 v/v % DMSO) in 30 mm PIPES, pH 6.8, 150 mm NaCl, 1 mm EDTA, 8 v/v % glycerol. The resulting curves were fit to the Morrison and Walsh integrated rate equation (Equation 1) (51). Kiapp was determined using the standard isotherm equation (Equation 2) or Morrison quadratic equation for tight-binding inhibitors (Equation 3) (52). For the pyridones, the Ki was extracted from Kiapp using Equation 4, where KS and KNAD(P)H values are rationally derived estimates for the real values provided in supplemental Table S3 or (31).
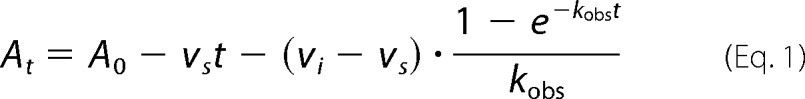 |
At and A0 are the absorbance at time t and time 0; vi and vs are the initial and steady-state velocities, and kobs is the pseudo-first order rate constant for the approach to steady state.
 |
vu is the control, uninhibited velocity, and Kiapp is the IC50 value.
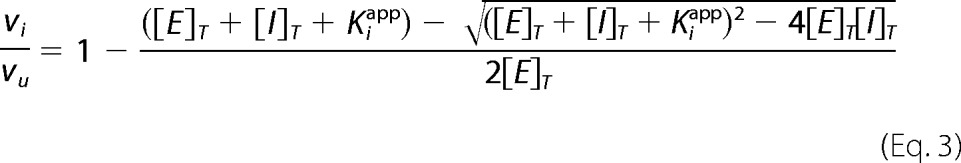 |
[E]T and [I]T are the total enzyme and inhibitor concentrations, respectively.
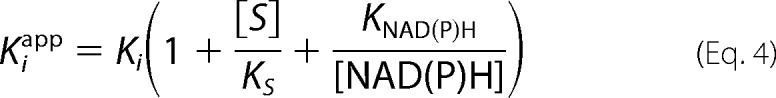 |
KS and KNAD(P)H are the respective dissociation rate constants for the enoyl-CoA substrate and NAD(P)H.
For the jump dilution assay, 10 μm saFabI, 15 μm inhibitor, and 500 μm NADPH were preincubated overnight at room temperature followed by a 1:200 dilution into reaction buffer (50 mm potassium phosphate, pH 7.5, 150 mm NaCl, 1 m potassium glutamate, 8 v/v % glycerol) containing 1.5 mm trans-2-butenoyl-CoA and 350 μm NADPH. The resulting progress curve was fitted to Equation 1. All curve fitting was performed using KaleidaGraph Version 4.1.
Thermal Shift Assay
ThermoFluor experiments were carried out in 96-well plates (Concord) using the CFX96 Real Time PCR Detection System and C1000 Thermal Cycler (Bio-Rad), as described previously (31).
Docking Studies
A computational docking and scoring procedure was used to generate putative binding modes for all pyridone inhibitors investigated in this study. The binding poses were generated with FlexX (BioSolveIT, Sankt Augustin, 2009), version 3.1.4 (53), and rescored with DrugScoreX (G. Neudert and G. Klebe, University of Marburg, 2008), version 0.21, which builds on DrugScore and utilizes the DrugScoreCSD potentials (54–56). To account for the flexibility of the substrate-binding loop, we docked all inhibitors into subunits A and C of the saFabI CG400549-I structure, which represent the two experimentally observed states (for details see “Results”). The selection of the most likely binding pose and receptor was based on the DrugScoreX score combined with visual inspection (supplemental Fig. S1). A comparison with the available experimental binding modes revealed root mean square deviations below 1.1 Å for the 10 best ranked binding poses, with 0.7 Å (CG400549) and 0.8 Å (PT173) for the top ranked pose, respectively (supplemental Fig. S1, A and B). These re-/cross-docking experiments confirm the validity and reliability of our computational approach.
The inhibitors were setup with MOE (Chemical Computing Group, Montreal, 2010), version 2010.10 (57), and energetically minimized (Tripos force field) using SYBYL-X (Tripos, St. Louis, 2009), version 1.0 (58). NADPH was protonated within the saFabI environment using MOE. The saFabI CG400549-I crystal structure was protonated in FlexX, and the binding site region was defined by NADPH and amino acids 93–99, 102, 121, 146–147, 154–157, 160, 164, 190–193, 195, 197–204, and 207. Water molecules within a radius of 6 Å around CG400549 were included during the docking procedure and treated as displaceable particles. FlexX was run in command line mode with a default docking procedure, followed by post-docking optimization. Root mean square deviations were calculated using fconv (G. Neudert and G. Klebe, University of Marburg, 2012), version 1.24 (59).
Determination of MIC Values
MIC values were determined with the microbroth dilution assay according to the Clinical and Laboratory Standards Institute methods for antimicrobial susceptibility tests for aerobically growing bacteria (60).
Selection for Resistance
S. aureus RN4220 was grown at 37 °C in Mueller-Hinton (MH) broth to late log phase (A600 = 1.2). 200 μl of culture was plated on MH agar containing PT166 (2 μg/ml; 5× MIC). After 48 h, five resistant colonies were randomly selected, and their phenotypes were confirmed by re-growth on the same medium containing PT166. The genomic DNA was extracted and purified using the Quick g-DNA Mini Prep kit (ZYMO Research). The S. aureus fabI genes from the PT166-resistant mutants were characterized by double-stranded nucleotide sequencing of PCR products using the following primers: saFabI forward (5′-CTAATTAGGCATATGTTAAATCTTGAAAACAAAACG-3′) and saFabI reverse (5′-GTAAGTGCTCGAGTTATTTAATTGCGTGGAATCC-3′). Sequencing reactions were performed with the ABI PRISM BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems, Foster City, CA), and sequencing data were obtained using an Applied Biosystems 3730 DNA sequencer.
In Vivo Pharmacokinetics
Pharmacokinetic (PK) studies were conducted in female ICR mice via intraperitoneal administration of PT04 (200 mg/kg dose) or PT166 (100 mg/kg dose) in a vehicle of 40 v/v % PEG 400, 40 v/v % EtOH, 20 v/v % H2O. Blood samples were collected from each animal at eight time points (5, 15, and 30 min and 1, 2, 4, 8, and 24 h post-injection). Three mice were sampled per time point. Plasma concentrations for each sample were measured by LC/MS/MS, and PK parameters were calculated with WinNonlin (Pharsight Corp., Mountain View, CA).
In Vivo Efficacy
Antibacterial efficacy of 5-hexyl-2-phenoxyphenol (PT04) and PT166 (Table 1) was evaluated in a neutropenic mouse thigh infection model. Six-week-old male Swiss Webster mice weighing 23–27 g were rendered neutropenic by intraperitoneal injection of cyclophosphamide 4 days (150 mg/kg) and 1 day (100 mg/kg) prior to infection. Previous studies have shown that this can produce severe neutropenia in mice for at least 5 days (61).
TABLE 1.
Thermodynamic parameters for inhibitors of saFabIa
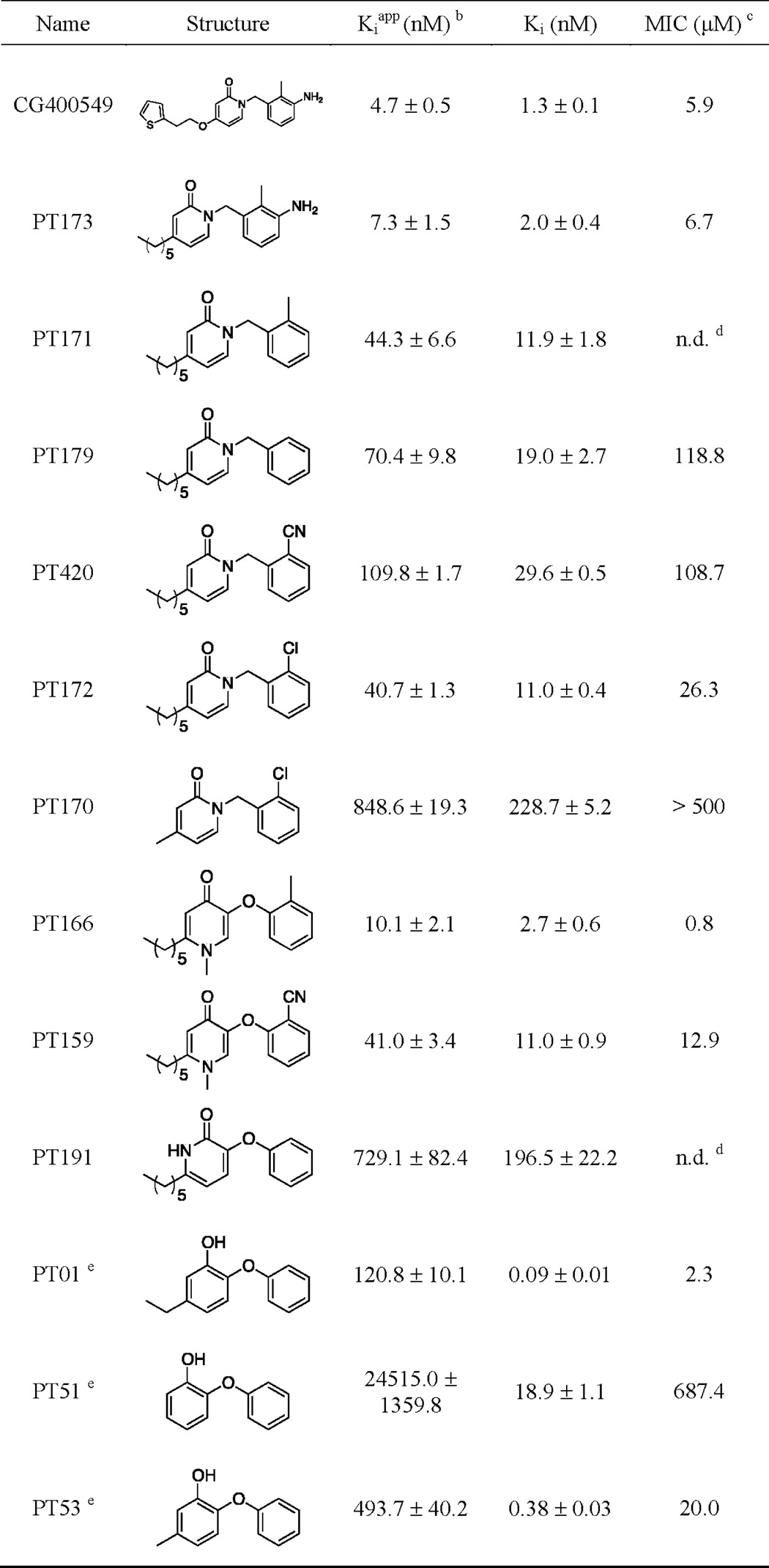
a Curve fitting errors are reported for each value in the table.
b [S]/KS = 2.
c Value was reported against S. aureus RN4220.
d n.d. means not determined.
e Binds to the E-NADP+ binary complex; Ki and Kiapp values were obtained from Ref. 31.
MRSA strain BAA1762 was cultured in MH broth to mid-log phase (A600 = 0.4; 3 × 108 cfu/ml) and harvested by centrifugation. Cell pellets were resuspended in freshly sterilized heart infusion (BHI) broth to a final inoculum of 1 × 107 cfu/ml. 50 μl of this suspension (5 × 105 cfu) was injected intramuscularly into the left thigh of each mouse, and 50 μl of BHI broth was injected into the right thigh as control. The drug was administered by subcutaneous injection at 1 and 12 h post-infection (100 mg/kg dose per injection). Mice were euthanized 24 h post-infection. Each thigh muscle was subsequently collected and homogenized in saline containing 10 w/v % BHI broth. The bacterial load was determined by counting colony-forming units of serial dilutions plated on MH sheep blood agar.
For in vivo efficacy and PK studies, all animals were maintained in accordance with criteria set by the American Association for Accreditation of Laboratory Animal Care. This study was approved by the Institutional Animal Care and Use Committee (IACUC) at Stony Brook University.
RESULTS
CG400549 and Related 2-Pyridones Selectively Inhibit Growth of S. aureus
The 2-pyridone CG400549 exhibits potent activity against S. aureus in contrast to many other bacteria such as E. coli, Listeria monocytogenes, Helicobacter pylori, and Pseudomonas aeruginosa (29). We confirmed this disparity by comparing growth inhibition of S. aureus and E. coli with various 2-pyridone inhibitors (Tables 1 and 2). In contrast to the Staphylococcus-specific 2-pyridones, diphenyl ethers are active against a broader spectrum of microorganisms. It is therefore important to elucidate whether the narrow spectrum behavior of 2-pyridones can be attributed to FabI-specific differences between species and why these compounds behave differently compared with the structurally similar diphenyl ethers.
TABLE 2.
Thermodynamic parameters for inhibitors of ecFabIa
a Curve fitting errors are reported for each value in the table.
b [S]/KS = 0.19.
c Value is reported against E. coli MG1655 ΔacrAB, a strain with a knockout of the specific efflux pump AcrAB.
d n.d. means not determined.
e The MIC value against E. coli MG1655 with the intact AcrAB efflux pump is >425 μm.
f This binds to the E-NAD+ binary complex. Ki and Kiapp values were determined via single point progress curve analysis, as described in Ref. 31. Rationally derived kinetic parameters for each step in the mechanistic model depicted in Fig. 2B are listed in supplemental Table S3.
g MIC values against E. coli MG1655 with the intact AcrAB efflux pump were 10–30-fold higher.
In Contrast to the Diphenyl Ethers, 2-Pyridones Bind to the E-NADPH Complex and Exhibit Fast-on Kinetics
Very recently, we found that diphenyl ethers bind exclusively to the E-NADP+ enzyme-product complex generated via catalysis (31). Despite the obvious structural similarity between pyridone and diphenyl ether inhibitors (Fig. 1), thermal shift assays revealed preferable binding of pyridones to the E-NADPH complex (Fig. 2A and Table 1). Thus, these inhibitors are uncompetitive with respect to NADPH but competitive with respect to the acyl substrate (Fig. 2B). Our kinetic studies confirmed this pattern of saFabI inhibition, showing enhanced inhibition with increasing concentrations of NADPH and decreasing concentrations of the substrate trans-2-octenoyl-CoA used in the assay (data not shown). Moreover, we were able to co-crystallize pyridones with saFabI using the reduced form of the cofactor. Accordingly, the saFabI target can be productively inhibited at two different stages of the catalytic cycle, as exemplified by pyridones and diphenyl ethers (Fig. 2B). This finding is of particular importance for the development of improved FabI inhibitors because most known scaffolds bind adjacent to the nicotinamide ring of the cofactor (16). The absence or presence of a buried positive charge in the E-NADPH versus E-NADP+ binary complex likely will have a critical influence on the true affinity of potential inhibitors (62). However, the relative potencies of inhibitors with different modes of action are highly dependent on the experimental assay condition (see below). Because NADPH binds with higher affinity to the enzyme compared with NADP+, the E-NADPH binary complex exists at a higher population for most experimental assay conditions (8, 31). Thus, even though CG400549 exhibits weaker true thermodynamic affinity (Ki) than most diphenyl ethers, its apparent inhibition constant (Kiapp) is much more potent in our standard enzyme assay (Table 1), requiring classical tight binding analysis (Fig. 2C) (31).
FIGURE 2.

Different mechanisms of saFabI inhibition. A, thermal shift analyses of saFabI bound to NADPH, NADP+, and/or inhibitor (CG400549). The measurement variability is approximately ±0.2 °C. B, distinct mechanisms of saFabI inhibition. Based on our recent kinetic and structural results, we reasoned that the enzyme binds NADPH first followed by the substrate (31). In contrast to diphenyl ethers (highlighted in red), which bind to the enzyme-product complex generated via catalysis (E-NADP+, depicted in blue) (31), pyridone compounds (A), PT166 (supplemental Fig. S2) and AFN-1252 (71) (all shown in red), preferentially inhibit saFabI at the enzyme-substrate complex state (E-NADPH, depicted in blue). C, representative plot of fractional velocity (v/v0) as a function of inhibitor concentration for a potent pyridone (CG400549, in this example). The shape is characteristic of tight-binding inhibition. The best fit curve to the Morrison quadratic equation (Equation 3) yields Kiapp = 4.73 ± 0.50 nm and [E]T = 92.73 ± 2.52 nm (R2 = 0.99). D, representative set of forward progress curves with pyridone-based inhibitors of saFabI. The plot depicts rapid-onset inhibition at different PT173 concentrations. E, as a reference, this plot illustrates the slow-onset inhibition of saFabI by the diphenyl ether PT04 (31). Note the clear observation of curvature that is absent in D. F, jump dilution curve for CG400549 (black) following preincubation with NADPH and saFabI. The jump dilution curve for the slow off diphenyl ether inhibitor PT52 (gray) following preincubation with NADP+ and saFabI is shown as reference (full recovery of activity; tR = 30 min, where tR is the residence time) (31). The lack of curvature for CG400549 is consistent with rapid off kinetics.
Pyridone and diphenyl ether saFabI inhibitors also differ with respect to their apparent association and dissociation kinetics. Diphenyl ethers exhibit slow binding kinetics and bind with long residence times to their target (Fig. 2, E and F) (6, 31). In contrast, progress curves of saFabI in the presence of pyridones are linear, displaying apparent rapid-onset kinetics (Fig. 2D). This is likely attributed to the higher population of E-NADPH compared with E-NADP+. In fact, an estimate of the actual association rate constant yields a value in the same range as for the diphenyl ethers (31). Moreover, the rapid appearance of activity following jump dilution is highly indicative of fast off kinetics (Fig. 2F). This is consistent with the weaker true thermodynamic affinity (Ki) of pyridones compared with diphenyl ethers. In other words, differences in koff are driven by Ki rather than kon.
The mode of action can have significant implications for cell growth inhibition. In open systems, substrate accumulation may eventually diminish the effect of competitive inhibitors (63). Therefore, it is important to consider the relationship between substrate concentration and Kiapp (Fig. 3A). In Fig. 3B, we observe a linear correlation in the double logarithmic plot of Ki versus MIC for both 2-pyridones and diphenyl ethers, consistent with on-target effects. However, 100-fold higher overall ternary complex affinity (Ki × Kd, NADP(H)) is needed for 2-pyridones to obtain a similar cellular potency as diphenyl ethers. Although this could be due to differences in cell permeability, this observation may also arise from substrate accumulation. The MIC is a thermodynamic parameter that is essentially equivalent to a physiological apparent inhibition constant (Kiapp). For competitive inhibitors, such as the pyridone compounds, the presence of high substrate concentrations may weaken the apparent affinity. Alternatively, because the rate of substrate reduction (kcat = 40 min−1) will increase as substrate concentration increases (KS = 0.75 mm), a higher proportion of E-NADP+ is formed, which can bind to the uncompetitive diphenyl ether compounds. At a substrate concentration range of 40–80 times KS, the relative Kiapp values are very predictive of the pattern of MIC values for both 2-pyridones and diphenyl ethers (Fig. 3C). This allows us to translate Ki to Kiapp values that are readily compared despite different modes of action. The true level of substrate accumulation in the cell is likely to be lower than predicted by this calculation. The cell contains longer chain substrates with faster turnover rates (6), which will increase the apparent affinity of diphenyl ethers. For instance, if we assume an average FabI turnover rate of 400 min−1, the predicted substrate concentration range would be 4–8 times KS, which is a much more reasonable estimate.
FIGURE 3.

Rationalizing the in vitro cellular potency of competitive and uncompetitive FabI inhibitors. A, relationship between acyl substrate concentration as a multiple of KS ([S]/KS) and the apparent affinity (Kiapp) of pyridones (PT170, PT172, and CG400549) and diphenyl ethers (PT51, PT53, and PT01) is illustrated. The plots are simulated based on Ki values against saFabI (Table 1) (31), the mechanism of inhibition shown in Fig. 2B, and the kinetic model described in Ref. 31. The range of substrate concentration that best correlates relative Kiapp to relative MIC for both classes of compounds is shaded in gray. B, double logarithmic plot depicts a strong linear correlation between Ki of the overall ternary complex (Ki × Kd, NADP(H)) and MIC for the 2-pyridone series (●) and diphenyl ethers (○). Points corresponding to 4-pyridones (×) are superimposed. Note that in this plot, the MIC for PT170 was assumed to be 550 μm, which is a lower limit estimate. C, this double logarithmic plot of Kiapp and MIC illustrates how 2-pyridones and diphenyl ethers can lie on the same linear correlation at the estimated substrate concentration [S] in the cell. Data points correspond to inhibitor Kiapp values at [S]/KS of 1 (○), 10 (×) and 100 (♦). The linear correlation for points corresponding to [S]/KS = 100 is depicted.
The utility of a scaffold for lead optimization is related, in part, to its intrinsic potency. We define this as the potency of a relatively unmodified scaffold, i.e. the starting point that determines how much affinity optimization is needed. For instance, PT170 and PT53 represent relatively unmodified 2-pyridone and diphenyl ether scaffolds, respectively (Table 1). The accumulation of substrate likely weakens the intrinsic potency of the competitive 2-pyridone scaffold relative to the uncompetitive diphenyl ether scaffold by more than 20-fold (Fig. 3A). Thus, further optimization of 2-pyridones is needed to achieve potent cellular activity. As we demonstrate below, such optimization is much more readily attained in the case of saFabI compared with other FabI homologues, providing the rationale for the narrow spectrum activity of CG400549 and related 2-pyridone compounds.
Clinical Candidate CG400549 Interacts Tightly with saFabI
CG400549 binds with high affinity (Ki = 1.27 nm) to saFabI (Table 1). To provide insight into the underlying molecular interactions, we solved two different saFabI-NADPH-CG400549 ternary complex structures (CG400549-I and CG400549-II, respectively; unless stated otherwise, the CG400549-I structure was used for the following analyses; see also supplemental Table S1). Based on the associated 2Fo − Fc omit maps, we unambiguously reveal the binding mode of CG400549 (Fig. 4A), which enables the formation of two central hydrogen bonds of the pyridone carbonyl oxygen with the Tyr-157 hydroxyl and the NADPH nicotinamide ribose 2′-OH at distances of 2.74 ± 0.07 and 2.69 ± 0.10 Å, respectively (Fig. 4B). Although reminiscent of interactions found for other inhibitor scaffolds (Fig. 1), these hydrogen bonds are 0.15 Å longer for pyridones in comparison with diphenyl ethers. This supports our hypothesis that diphenyl ethers bind to saFabI in their deprotonated form (31) leading to shorter charge-assisted hydrogen bonds. Additional long range hydrogen bonds are formed between the 3′-amino group of CG400549 (Fig. 1) and the main chain oxygen and nitrogen of Ala-97 at distances of 3.56 ± 0.06 and 3.32 ± 0.09 Å, respectively (Fig. 4B). Moreover, a water molecule is frequently bound between this B-ring NH2 group and Ala-95 at 3.36 ± 0.18 Å (Fig. 4B). Interestingly, Ala-97 is engaged in direct interactions to several potent saFabI inhibitors (Fig. 1). Triclosan and AFN-1252 form halogen and hydrogen bonds with Ala-97, respectively, and the amide group of MUT056399 was similarly suggested to interact with this residue (Fig. 1) (6, 23, 24).
FIGURE 4.

Molecular interactions of saFabI with the pyridone inhibitors CG400549 and PT173. A, 2Fo − Fc omit map for CG400549 bound to saFabI. According to the omit map (shown as mesh at 1σ), CG400549 (cyan) unambiguously binds to the hydrophobic saFabI active site pocket. An intersection of the CG400549-I structure (subunit G, depicted in gray surface representation) provides insight into this cavity. B, binding mode of CG400549 (cyan) in complex with saFabI and NADPH. Interactions between inhibitor, cofactor, and protein are highlighted by yellow dashed lines. Selected residues of the saFabI binding pocket are shown as gray sticks (CG400549-I structure, subunit G). C, NCS-averaged 2Fo − Fc omit map for PT173. The omit map is shown at 1σ and reveals the presence of PT173 (shown in green). Subunit F of the PT173 structure is depicted in gray schematic representation. D, experimental binding geometry of PT173 (green). Selected residues of the saFabI-NADPH-PT173 structure (gray, subunit F) and the central hydrogen bonding network (yellow dashed lines) are depicted.
The unique 5-substituent of CG400549 (atoms of the diphenyl ether and pyridone scaffolds are numbered as indicated in the two lower boxes of Fig. 1) binds to a hydrophobic pocket with the thiophene moiety trapped between the three aromatic amino acids Tyr-147, Tyr-157, and Phe-204 (Fig. 4, A and B). The side-on π-stacking interaction between Tyr-157 of the catalytic triad and the thiophene ring may contribute to the high affinity of CG400549 toward saFabI (Table 1). In particular, the thiophene sulfur interacts with the edge of Tyr-157 (Cδ) at a distance of 3.97 ± 0.11 Å (62). A similar interaction is found between this sulfur atom and Phe-204 (Cδ) at 4.15 ± 0.16 Å (Fig. 4B). Remarkably, among 13 CG400549-resistant S. aureus strains, 10 were characterized by a single F204L mutation, whereas the residual three had no mutations in the fabi gene (30). In contrast, multiple resistance mutations are known for diphenyl ethers (8, 9, 26, 64, 65). Accordingly, the Phe-204-thiophene interaction seems to be critical for the activity of CG400549.
In addition to the CG400549 structures, we solved an saFabI structure in complex with NADPH and PT173, which displays a similar affinity toward saFabI as CG400549 (Ki = 1.97 nm versus 1.27 nm) and contains a 5-hexyl group that mimics the natural enoyl-ACP substrate (Fig. 1) (31). Although the resolution was much lower (supplemental Table S1), clear density was observed for the cofactor and inhibitor (Fig. 4C). The binding mode of PT173 in the saFabI active site pocket is similar to CG400549, and we observed the same central hydrogen bonding network between the inhibitor, cofactor, and Tyr-157 (Fig. 4D).
Despite the relative success of CG400549, little is known about pyridone FabI inhibitors and their structure-activity relationships (SAR) (66–68). Thus, we synthesized a series of pyridone compounds and investigated their ability to inhibit saFabI (Table 1). To rationalize the SAR results, we generated putative binding modes for all investigated inhibitors using a validated docking procedure, which could reproduce the CG400549 and PT173 binding geometries with low root mean square deviations (0.71 and 0.83 Å, respectively) (supplemental Fig. S1). Because of the lipophilic environment, bulky and hydrophobic substituents are preferred at the 5-position. Hence, PT170 is the least potent compound of our pyridone series. Replacing the 5-methyl group by a 5-hexyl group (PT172) leads to a 21-fold affinity enhancement, which is underlined by the favorable scores for the additional carbon atoms (supplemental Fig. S1). Similar to the 2′-chloro substituent of PT172, the 2′-methyl group of PT171, which is also present in CG400549, increases affinity by a factor of 2 compared with the unsubstituted analogue PT179. In comparison with the 2′-Cl and 2′-Me groups, a 2′-CN substituent leads to decreased potency. Interestingly, the SAR at this position is different from diphenyl ethers for which 2′-cyano is the best substituent (31). Pyridone 2′-substituents are predicted to bind in a similar orientation (supplemental Fig. S1) as observed for diphenyl ethers. The introduction of a 3′-amino group as present in CG400549 and PT173 further enhances the affinity of PT171 by a factor of 6, which highlights the energetically favorable character of the observed long range hydrogen bonds between those inhibitors and Ala-97 (Fig. 4B). Replacement of the PT173 5-hexyl group with the 5-substituent of CG400549 further improves the affinity of the drug candidate 1.5-fold and might be explained by the additional aromatic interactions we observed.
saFabI Conformational States Differ between Pyridone and Diphenyl Ether Ternary Complex Structures
In accordance with the distinct kinetic behavior of pyridone and diphenyl ether inhibitors, we observed considerable structural differences between the corresponding ternary complex structures. A per-residue root mean square deviation plot reveals variations in mainly three regions of the protein (Fig. 5A), the two substrate-binding loops (SBL and SBL-2; residues 194–204 and 94–108) and the phosphate recognition helix α2 (residues 40–54), which confers the unique NADPH specificity to saFabI (6). Complete closure of the SBL has been proposed to constitute the rate-limiting step of slow binding FabI inhibition by diphenyl ethers (16). Indeed, we found more “open” SBL states in the saFabI-NADPH-CG400549 structures compared with the triclosan-bound structure (Fig. 5B). Most likely the observation of the open state can partially be attributed to steric interference between the thiophene moiety of CG400549 and the side chain of Val-201 in the SBL if it would adopt the completely “closed” state (Fig. 5B, gray). Hence, Val-201 is shifted by 2.9 ± 0.1 Å into the more open CG400549-bound structure (Fig. 5B, cyan). Interestingly, some subunits of the CG400549-I structure reveal a second, even more open SBL state; the root mean square deviation increases from 1.6 to 2.0 Å compared with the triclosan-bound reference structure, with an additional shift in helix α7, which we previously identified to be very flexible prior to ligand binding (Fig. 5B, yellow) (6). Both SBL conformations in the saFabI-NADPH-CG400549 complex seem to be energetically equally favorable due to the observation of subunits with both states in equilibrium. The more closed state is stabilized by a sulfate ion, which is bound to backbone amides of the SBL; this state differs from the completely closed triclosan-bound structure by a Lys-199 to Gly-200 backbone flip (Fig. 5B, right inset; ψK199 and ϕG200 change by 156° and 159°, respectively), which is observed for seven of the eight CG400549-I monomers and might also be responsible for the opening movement. An additional shift of residues 202–208 with extensive variations for Gly-202 and Gly-203 leads to the most open form (Fig. 5B, left inset), which is related to the conformation observed for the AFN-1252 structure (23).
FIGURE 5.
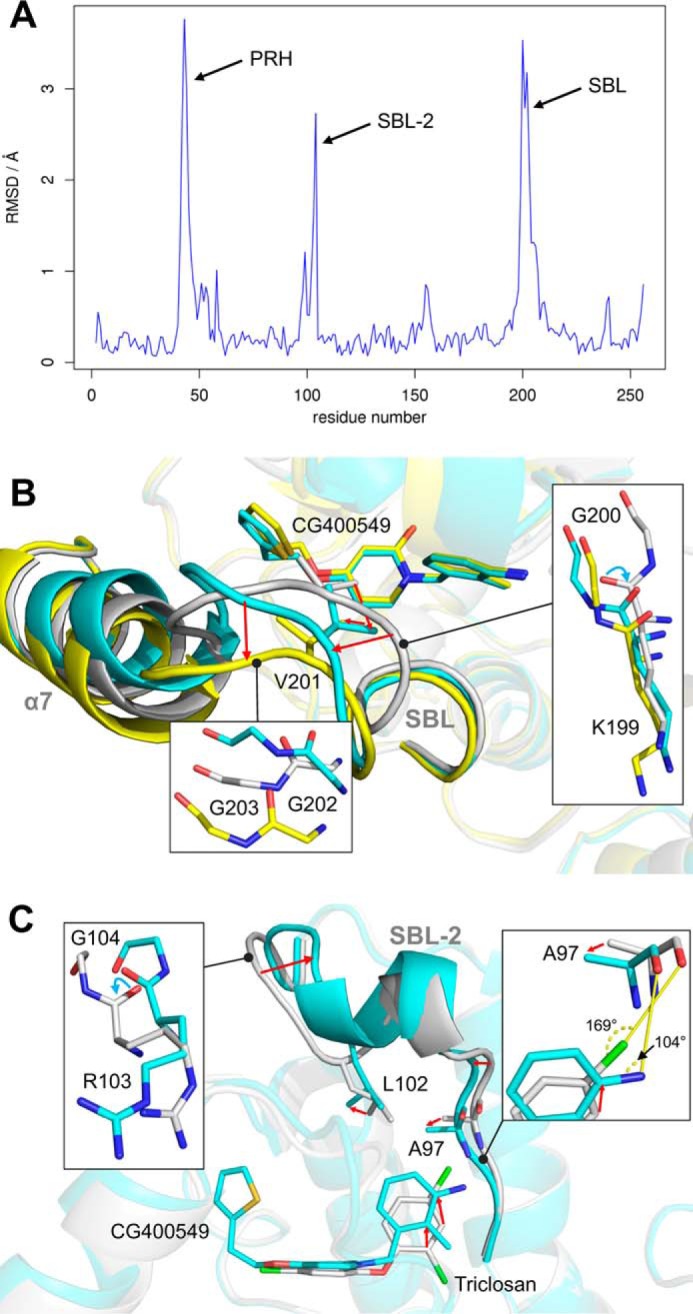
Structural variations between pyridone and diphenyl ether ternary complex structures. A, structural differences between diphenyl ether and pyridone ternary complexes. Per residue root mean square deviation (RMSD) values between our triclosan-bound (PDB code 4ALI, subunit H) and CG400549-bound (CG400549-I, subunit C) structures were calculated using Theseus (79) and are plotted against the residue number. The inhibitor-bound structures of these two scaffolds differ considerably in three regions of the protein (PRH = phosphate recognition helix α2; SBL-2 = substrate-binding loop 2; SBL = substrate-binding loop). B, conformational states of the SBL. The different subunits of our CG400549-I structures reveal two distinct states of the SBL and the attached helix α7 (shown in cyan and yellow for subunits A and C, respectively; residual parts of the protein are displayed in transparent colors for clarity). Compared with these conformations, the SBL is more closed in our triclosan-bound structure (shown in gray; PDB code 4ALI, subunit H). Detailed views of the three different conformations are displayed in the insets. Red arrows indicate the conformational changes from the closed to the open substrate-binding loop states. Cyan arrows highlight backbone flips. C, conformational states of the SBL-2. Selected residues, the inhibitors, and the SBL-2 are shown for the CG400549-I (cyan; subunit A) and triclosan-bound structures (gray; PDB code 4ALI, subunit H).
Similar to the SBL, a second loop that also contributes to the substrate binding pocket (SBL-2) was almost exclusively found to be in an open conformation in the saFabI-NADPH-CG400549 structures (Fig. 5C, cyan). In contrast, we observed alternative closed and open SBL-2 conformations for the diphenyl ether-bound structures (6). An Arg-103 to Gly-104 backbone flip (Fig. 5C, left inset; ψR103 and ϕG104 change by 180° and 144°, respectively) clearly differentiates the two states. This flip is induced by (or induces) a 1.5 ± 0.5 Å shift of Leu-102 out of the binding pocket, which in turn might be caused by (or causes) a considerable movement of the CG400549 B-ring toward this residue (Fig. 5C). The resulting differences between the pyridone and diphenyl ether binding poses (Fig. 5C) rationalize the varying SAR profiles at the 2′-position for both scaffolds and the success of the 3′-amino substituent in the case of pyridones. In particular, the 2′- and 3′-carbon atoms are relocated by 1.3 ± 0.1 and 1.6 ± 0.1 Å, respectively, which places the CG400549 3′-carbon at the position of the triclosan 4′-carbon. Consequently, 3′-pyridone and 4′-diphenyl ether substituents are ideally oriented to interact with the important anchor residue Ala-97. For instance, the triclosan 4′-Cl is halogen-bonded to the free electron pair of the Ala-97 carbonyl oxygen via the favorable linear geometry (Fig. 5C, right inset) (6, 62). To enable an equally favorable angular geometry for a hydrogen bond between the CG400549 3′-NH2 group and Ala-97 along one amino hydrogen atom (62), the Ala-97 carbonyl oxygen is shifted by 0.8 ± 0.1 Å toward the B-ring of the inhibitor, which in turn approaches Ala-97 to reduce the interaction distance (Fig. 5C, right inset). As exemplified by these considerations, the exact knowledge of the different conformations that can be attained by a protein target is pivotal for the design of improved inhibitors.
Comparison of ecFabI and saFabI Inhibition by 2-Pyridones
To determine whether the narrow spectrum behavior of CG400549 can be partly attributed to target-specific differences between species, we obtained and compared the structures and inhibition kinetics of CG400549 with respect to E. coli and S. aureus FabI. Indeed, CG400549 shows a 65-fold reduced affinity to ecFabI in comparison with saFabI (Tables 1 and 2), which translates into >64-fold lower antibacterial activity (Table 3). Nevertheless, the inhibitor clearly bound to the binary ecFabI-NADH complex with a similar binding geometry as observed for saFabI (Fig. 6, A and B). However, in contrast to the related saFabI ternary complex, the substrate-binding loop and the attached helix α7, including residues 193–214, were found to be disordered in this ecFabI structure (Fig. 6A). Based on a comparison of these structures, we propose that the saFabI residues Val-201 and Ile-207, which are located in this region of the protein, contribute to the specificity of CG400549 toward saFabI. The more bulky Ile-200 and Met-206 ecFabI residues (corresponding to saFabI positions 201 and 207, respectively) restrict the available space for the large 5-substituents as present in CG400549 (Fig. 6B). Accordingly, the elongation of the 5-substituent in 5-ethyl-2-phenoxyphenol (PT01) to 5-hexyl-2-phenoxyphenol (PT04) does not enhance the affinity toward ecFabI (Table 2), whereas the affinity increases ∼10-fold for saFabI (31). We have recently shown that these inhibitors are transition state analogues with the 5-substituent alkyl chain extending toward the fatty acyl binding channel (31). Although the kcat increases for longer enoyl substrates in the case of saFabI (31), it is similar among the different substrate chain lengths for E. coli and F. tularensis FabI, which both carry the V201I and I207M substitutions and are inhibited by PT01 and PT04 with similar potency (6, 33, 69, 70). Thus, the enlarged binding pocket of saFabI might partially explain the specific action of the comparatively bulky CG400549 and AFN-1252 clinical candidates (Fig. 1). Interestingly, the C terminus of the ecFabI monomer located on the opposite side of the homo-tetrameric protein (in particular, Met-256′) seals this acyl-binding cavity and likely restricts the side chain mobility of Met-206 (Fig. 6B). The resulting steric interference between the substrate-binding loop residues Met-206 and Ile-200 and the thiophene moiety of CG400549 presumably results in the experimentally observed enhanced mobility of this loop. In contrast, the C terminus of saFabI is shorter and lacks a residue corresponding to Met-256′, thus enabling Ile-207 to move away from bulky 5-substituents. Consequently, important contacts between this inhibitor and the substrate-binding loop (e.g. with Phe204; see also Fig. 4B) are more readily attained, partially explaining the selectivity of CG400549 toward the saFabI homologue.
TABLE 3.
Spectrum of antibacterial activity for different classes of FabI inhibitors
| Organism | Enoyl-reductase isoforma | MIC (μm) PT01 | MIC (μm) CG400549 | MIC (μm) PT166 |
|---|---|---|---|---|
| S. aureus RN4220 | FabI | 2.3 | 5.9 | 0.8 |
| E. coli MG1655 | FabI | 18.7 | >375 | >425 |
| E. coli MG1655 ΔacrABb | FabI | 0.6 | >375 | 6.7 |
| F. tularensis LVS | FabI | 0.1c | 5.9 | 0.4 |
| P. mirabilis ATCC35659 | FabI | NDd | >750 | 213.8 |
| M. tuberculosis H37Rv | FabI | 72.9 | >300 | 10.5 |
| B. pseudomallei Bp400b | FabI and FabV | 326.7e | 376.0 | 213.8 |
FIGURE 6.

Rationalizing the Staphylococcus-specific activity of CG400549 and designing PT166 with an extended activity spectrum. A, CG400549 (lilac) in its complex with ecFabI and NADH. The 2Fo − Fc omit map is depicted for CG400549 (shown as mesh at 1σ) and clearly reveals the presence of this molecule, although the SBL region was found to be disordered in this structure (the disordered region starts after residue 192 (colored in red) as highlighted by a red arrow; subunit A of the ecFabI-NADH-CG400549 structure is shown in yellow schematic representation). As a reference, the SBL of a superimposed ecFabI-NAD+-triclosan structure is shown in light green (PDB code 1QSG) (13). Together with the SBL, the loop comprising Ala-95 defines a portal toward the solvent. B, comparison between the CG400549-bound S. aureus (inhibitor in cyan, for clarity only present in the inset, amino acids in gray) and ecFabI (inhibitor in lilac, amino acids in yellow) ternary complexes. The inset contains a comparison between the CG400549 binding modes, which differ in their B-ring conformations via changes in the torsion angles between the two aromatic rings (indicated by red arrowheads). Met-256′ of the subunit on the opposite side of the ecFabI homo-tetramer is depicted in pink. Residues Ile-200 and Met-206 of the 1QSG structure (ecFabI-NAD+-triclosan) are shown as reference in green. C, putative binding mode of PT166 (yellow sticks) within the saFabI pocket (CG400549-I structure, subunit A). PT166 was docked into the saFabI binding cavity using the validated approach described under “Experimental Procedures.” The radii of the blue (red) spheres indicate the values of the favorable (unfavorable) score for each individual atom, as determined with DrugScoreX. Our bpFabI-NAD+-PT155 structure (light green sticks) confirms this binding mode and suggests a rotation of Phe-204 to avoid the steric interference with the N-methyl group of 4-pyridone inhibitors (indicated by the red sphere). Moreover, the putative binding mode is in accordance with our PT166-bound ecFabI structure (orange sticks, subunit A). The docking results for the residual pyridone inhibitors of Table 1 are summarized in supplemental Fig. S1. D, 2Fo − Fc omit map (shown as mesh at 1 σ) for PT166 (orange sticks) bound to ecFabI. An intersection of the ecFabI-NADH-PT166 structure (subunit A, depicted in yellow surface representation) is shown. The SBL, which usually covers the binding site (in front of the cavity), is disordered.
In line with our assumption that ecFabI is a good model for all FabIs insensitive to CG400549, an alignment of the FabI sequences from clinically relevant pathogens, which can be sensitive to FabI-specific inhibitors, reveals that staphylococcal FabIs differ fundamentally from classical FabI proteins such as ecFabI (Fig. 7). Most strikingly, all nonstaphylococcal FabIs included in this comparison contain extended C termini. A comparison of the available corresponding structures clearly shows that the four amino acids following the C-terminal Lys-256 of saFabI occlude the acyl-binding cavity for classical FabIs and thus restrict the available space for bulky 5-substituents (red box in Fig. 7). In particular, the additional large hydrophobic residues at position 257 (256 for ecFabI) or 259 (Met, Leu, and Ile) will most likely interfere with bulky residues located at position 207, which thus cannot avoid the interference with large 5-substituents without the opening of the SBL (Fig. 6B). The length of the C terminus might therefore be an ideal indicator whether or not the corresponding FabI is sensitive to compounds such as CG400549 or AFN-1252. The presence of Val-201 instead of an isoleucine is an additional unique characteristic of FabIs sensitive to the more voluminous compounds and may facilitate the production of bulky branched-chain fatty acids abundant in S. aureus (Figs. 6B and 7) (6).
FIGURE 7.

Alignment of FabI sequences from clinically relevant pathogens. The sequences of FabI from S. aureus (saFabI), S. epidermidis (seFabI), E. coli (ecFabI), B. pseudomallei (bpFabI), F. tularensis (ftFabI), H. influenzae (hiFabI), Neisseria gonorrhoeae (ngFabI), Neisseria meningitidis (nmFabI), Moraxella catarrhalis (mcFabI), Proteus mirabilis (pmFabI), H. pylori (hpFabI) and M. tuberculosis (InhA) were aligned using ClustalW (80). These organisms are known to contain FabI as the sole enoyl-ACP reductase (exception: FabV is additionally present in B. pseudomallei) (23, 24, 29, 81). Residues that are likely responsible for the unique NADPH specificity, enhanced mobility (6), and enlarged binding pocket of saFabI are indicated by magenta, black, and red stars, respectively. The C-terminal regions occluding the acyl-binding cavity and the RKXXS motif conferring NADPH specificity are highlighted with red and magenta boxes, respectively. The blue star indicates the location of Met-99. This figure was prepared using ESPript (82).
In addition to the tolerance for bulky 5-substituents, hydrogen bonding interactions with Ala-97 seem to be particularly favorable in the case of saFabI and are exploited by the three drug candidates, as indicated by crystallographic and computational studies (Fig. 1) (23, 24). In the case of CG400549, the more rigid Pro-96 of ecFabI (Asn-98 in the case of saFabI) slightly changes the orientation of the Ala-95 carbonyl (Ala-97 for saFabI) (Fig. 6B). This might explain the observed rotation in the B-ring of CG400549, which seems to be required for the maintenance of the hydrogen bond to this residue. Interestingly, this interaction is solvent-exposed (Fig. 6A) and less buried in ecFabI due to the presence of Gly-97, which corresponds to Met-99 in saFabI (Fig. 6B). This observation rationalizes why the 3′-amino substituent leads to a decrease in affinity in the case of ecFabI (Table 2) because solvent-exposed interactions tend to be energetically less favorable (62). Thus, other classical FabI proteins harbor a more hydrophilic residue at this position compared with saFabI (Fig. 7). Furthermore, treatment of S. aureus with AFN-1252, which also binds to the saFabI-NADPH complex and forms a hydrogen bond with Ala-97 (Fig. 1), predominantly selects for an M99T mutation, and it was recently suggested that this residue confers S. aureus selectivity to AFN-1252 (23, 71). Thus, the buried hydrogen bonding interaction with Ala-97 likely plays an important role in the selective affinity of CG400549 and AFN-1252 toward saFabI.
4-Pyridone PT166 Is a Potent FabI Inhibitor with Extended Spectrum in Vitro Activity and in Vivo Efficacy against S. aureus
In contrast to saFabI, strategies to optimize the binding affinity of 2-pyridones for ecFabI and related homologues may be limited due to the more constricted space in the binding crevice combined with the relatively low intrinsic potency of 2-pyridones compared with the diphenyl ether scaffold. Instead, to attain broad spectrum activity, a modified scaffold should be designed that possesses a higher intrinsic potency for all FabI homologues. One possibility is the replacement of the methylene bridge with an ether linkage, thereby changing the conformational preference prior to binding. The biologically active Ar-X-Ar conformation (Ar = aromatic ring, X = CH2 or O, Figs. 4 and 6) is more readily available for the bisaryl ether system thus leading to an entropic advantage upon binding (72). This was implemented in the design of C-substituted 2-pyridone (in contrast, 2-pyridones such as CG400549 are substituted at the nitrogen atom) and 4-pyridone analogues. As expected, both prefer to bind in a ternary complex with NADPH (supplemental Fig. S2), thus preserving the 2-pyridone mode of action. Compared with the analogous N-substituted 2-pyridone PT179, the C-substituted 2-pyridone PT191 binds 10-fold less tightly to saFabI (Table 1). Although the enol form of PT191 displays a similar putative binding geometry as PT179 (supplemental Fig. S1) and more closely resembles the potent diphenyl ether PT04 (31, 66), the increased desolvation costs for this more polar compound are likely responsible for the decreased affinity. In contrast, the 4-pyridones PT166 and PT159 have 3–4-fold enhanced affinities for saFabI with respect to the analogous 2-pyridones PT171 and PT420 (Table 1), consistent with a higher intrinsic potency of this scaffold. PT166 also potently inhibited ecFabI (Table 2), suggesting that the higher intrinsic potency may indeed translate to broad spectrum activity. Thus, substituting the methylene bridge of the initially reported 4-pyridones (67) with an ether linkage (73) seems to be a successful strategy to further improve FabI pyridone inhibitors.
Based on our docking studies, we suggest a binding mode for the 4-pyridones similar to those observed for 2-pyridones (Fig. 6C). However, despite the enhanced affinity of PT166 with respect to PT171, the additional 4-methyl group is characterized by an unfavorable score. Thus, we hypothesized that Phe-204 will change its conformational state upon 4-pyridone binding to avoid the steric interference with the N-methyl group. Because 4-pyridones did not co-crystallize with saFabI, we initially used PT155, which carries an additional 4′-NH2 group compared with PT166, and we determined a ternary complex structure with B. pseudomallei FabI (bpFabI). Indeed, this structure confirmed the predicted 4-pyridone binding mode, and Phe-203 (corresponding to the saFabI residue Phe-204) was found to adopt a different conformation presenting one of its π-faces to the N-methyl group of PT155 (Fig. 6C). Finally, we were able to solve an ecFabI-NADH-PT166 structure (Fig. 6D), which further validated the proposed PT166-binding mode (Fig. 6C). Similar to the ecFabI-NADH-CG400549 structure, the substrate-binding loop was found to be disordered or in a very open conformation in these ecFabI and bpFabI structures, respectively. Interestingly, an open SBL conformation was very recently also observed in the structure of PT155 bound to InhA (74).
For the 4-pyridone analogues, in vitro MIC measurements against S. aureus RN4220 lie near the 2-pyridone linear correlation, consistent with their similar mode of action (Fig. 3B). To confirm that the main target of PT166 in vivo is indeed saFabI, we performed selection experiments, which resulted in mutations of the fabI gene in 4 of 5 sampled colonies, including the previously characterized A95V variant (Table 4) (8). Relative to CG400549, PT166 had a >50-fold lower MIC against an E. coli MG1655 strain lacking the efflux pump AcrAB, consistent with its more potent inhibition of ecFabI (Table 2). Gratifyingly, relative to CG400549, PT166 also exhibited improved activity against the Gram-negative pathogens F. tularensis and P. mirabilis (Table 3). Thus, we rationally extended the spectrum of antibacterial activity by using ecFabI as the paradigm for Gram-negative FabI homologues. Furthermore, similar optimization principles were applicable for the inhibition of mycobacterial FabI proteins. PT166 bound more potently than CG400549 to the M. tuberculosis enoyl-ACP reductase InhA with Ki values of 22 and 582 nm, respectively, and translated to significantly enhanced in vitro anti-mycobacterial activity (MIC = 10.5 μm; Table 3).
TABLE 4.
Selection for S. aureus RN4220 resistance to PT166
| Strain | Nucleotide change | Amino acid change | MIC |
|---|---|---|---|
| μm | |||
| RN4220 | 0.8 | ||
| 166R.1 | GCA → GTA | A95V | 8.8 |
| 166R.2 | GCA → GTA | A95V | 8.8 |
| 166R.3 | GCA → GTA | A95V | 8.8 |
| TTC → TTG | F252L | ||
| 166R.4 | GCA → GTA | A95V | 8.8 |
| GAA → GAT | E71D | ||
| 166R.5 | No change | No change | 17.6 |
Pyridones are a metabolically stable alternative to diphenyl ethers, which contain a hydroxyl group that can be susceptible to glucuronidation and sulfonation (75). This PK advantage may be key to the success of the clinical candidates CG400549 and AFN-1252. Importantly, PT166 maintained a superior PK profile compared with the diphenyl ether PT04 (Table 5). Cmax and AUC0–24 h (where AUC0–24 h is the area under the plasma concentration-time curve over 24 h) of PT166 are 9- and 5-fold higher than that of PT04, respectively, despite the fact that the dose of PT04 was double that of PT166. We also tested the efficacy of PT166 in a neutropenic mouse model of MRSA infection. As a control, no bacteria were observed in the right thigh muscle of both treated and untreated mice, confirming the lack of significant migration of bacteria. In the infected thigh, however, significant bacterial burden was observed in the different treatment groups. As expected, oxacillin, a clinical antibiotic similar to methicillin, exhibited no in vivo antibacterial efficacy (Fig. 8). However, a 100 mg/kg intramuscular dose of PT166 significantly decreased the bacterial burden in the infected thigh by 2.8 log cfu/g tissue. In comparison, the same dose of the diphenyl ether PT04 only decreased the bacterial burden by 0.9 log cfu/g tissue.
TABLE 5.
In vivo pharmacokinetic profile for PT166 and PT04
| PK parameter | PT04 | PT166 |
|---|---|---|
| Dosage (mg/kg) | 200 | 100 |
| AUC0–24 (μg h/ml)a | 11.8 | 53.0 |
| t½ (h)b | 4.5 | 2.7 |
| tmax (h)c | 1.0 | 0.25 |
| Cmax (μg/ml)d | 5.1 | 45.9 |
a AUC0–24 is the area under the plasma concentration-time curve over 24 h.
b t½ is the time taken for plasma concentration to fall to 50% of its original value.
c tmax is the time when Cmax occurs.
d Cmax is the maximum plasma concentration of the drug.
FIGURE 8.

In vivo efficacy. The efficacy of selected compounds against MRSA strain BAA1762 in a neutropenic mouse thigh infection model is shown. Error bars represent the standard deviation for replicate data (n = 5 in each group).
DISCUSSION
Pyridones constitute a very promising and relatively new class of FabI inhibitors (16). Compared with diphenyl ethers, CG400549 has superior pharmacokinetic properties and proven clinical efficacy against S. aureus infections. However, it also has lower activity against many other important pathogens. Understanding the molecular basis for such selectivity can guide the development of pyridone-based FabI inhibitors with broad spectrum potential.
Despite the structural similarity of the pyridone and diphenyl ether scaffolds, we observed differences with respect to their mode of inhibition, which have significant implications for the spectrum of activity. Pyridones bind predominantly to the E-NADPH complex, whereas diphenyl ethers exclusively interact with E-NADP+ generated via catalysis (Fig. 2, A and B) (31). We recently proposed that diphenyl ethers bind in a deprotonated state to their target (31). In such a scenario, the positively charged and thus electron-deficient oxidized nicotinamide ring of the cofactor forms a charge-assisted π-π stacking interaction with the electron-rich phenolate A-ring (Fig. 1). The inability of the much less acidic pyridones to form this ionic interaction might explain their reduced affinity for E-NADP+.
The diphenyl ether and pyridone-bound saFabI ternary complexes likely reflect different stages during the hydride transfer step of the enzymatic reaction (Fig. 9A). We have recently shown that diphenyl ethers are analogues of a late enolate-like transition state (31). In contrast, the pyridone structure more closely resembles the enoyl-ACP substrate (Fig. 9A) and thus binds preferably in a ternary complex with the reduced cofactor, which is present prior to the hydride transfer. We envision an incremental closure of the SBL during substrate binding and hydride transfer in which the loop is fully closed at the transition state to minimize its energy (Fig. 9). In this respect, the different conformations of the SBL presented in this study define important structural snapshots along the reaction coordinate of enzyme catalysis (Fig. 5B). The observation of an opened substrate-binding site for pyridones confirms our hypothesis that these inhibitors are more substrate-like compared with the diphenyl ethers. In the light of the two alternatively ordered SBL conformations versus the disordered loop in the apoenzyme (Fig. 9B, states 1 and 2) (6, 32), it could be argued that the pyridone ternary complexes represent a state between the substrate complex and the transition state for enolate formation (Fig. 9C). The electron-donating effect of the pyridone nitrogen, which leads to a phenolate-like resonance structure, might thereby mimic the transfer of the negatively charged hydride ion (Fig. 9A).
FIGURE 9.

Conformational states sampled along the reaction coordinates of inhibitor binding and substrate turnover. A, pyridone and diphenyl ether inhibitors resemble different species along the enzymatic reaction coordinate. In contrast to the enolate-like diphenyl ethers which are transition state analogues (31), pyridones are more substrate-like. The corresponding moieties of inhibitors and species along the reaction coordinate are highlighted in green. B, conformational states sampled by the SBL and the attached helix α7 of saFabI. Red arrows indicate the conformational changes we propose to occur during the enzymatic reaction (see also C). Prior to the binding of cofactor and substrate or inhibitor, the SBL is disordered, and helix α7 attains a very open conformation (state 1 colored in pink = PDB code 4ALM, subunit B; state 2 colored in lilac = PDB code 4ALM, subunit C; further details about the conformational changes upon ligand binding are provided in our previous report (6)). The more substrate-like pyridone inhibitors likely induce a conformational state between the ternary E-NADPH-S complex and the transition state of the hydride transfer (state 3 colored in yellow = CG400549-I structure, subunit C; state 4 colored in cyan = CG400549-I structure, subunit A; see also Fig. 5B). In contrast, the transition state analogue triclosan and several other diphenyl ethers (6, 31) induce the likely fully closed state of the SBL (state 5 colored in gray = PDB code 4ALI, subunit H). C, qualitative energy diagram for substrate turnover by saFabI. The numbers 1–5 indicate the conformational states (colors according to B), which are likely sampled along the reaction coordinate of the enzymatic reaction (see also B). D, approximate energy diagrams for saFabI in complex with NADPH and pyridone (black) or NADP+ and diphenyl ether inhibitor (red). The overall affinities of both ternary complexes are assumed to be identical. By shifting stabilization from cofactor to inhibitor, the residence time of the overall complex is increased. This rationalizes the difference in off-rate kinetics between the diphenyl ethers and pyridones. Note that, technically, koff for the pyridone complex (E-NADPH-I) is equal to k−2 because the E-NADPH complex is catalytically active. C is defined as a constant with a value greater than 1. The numbers 1–5 indicate the conformational states (colors according to B), which are likely sampled along the reaction coordinate of inhibitor binding (see also B).
As the hydride transfer reaction proceeds, the increasing positive charge on the nicotinamide ring and the closure of the substrate-binding loop shifts the balance of ternary complex stabilization more toward the fatty acyl relative to the cofactor component, which is reflected in the relative affinities of the investigated inhibitors and cofactor forms. The transition state mimicking diphenyl ethers display affinities that are 3 orders of magnitude higher compared with analogous substrate-like 2-pyridones, whereas NADP+ binds ∼1000-fold less tightly to saFabI with respect to NADPH (Fig. 9D) (31). Accordingly, the cumulative ternary complex affinities (Ki × Kd, NADP(H)), which also integrate cofactor affinity, of comparable E-NADP+-diphenyl ether and E-NADPH-pyridone complexes are calculated to be very similar. There is only a 2-fold difference in the cumulative affinity of the E-NADP+-PT04 complex compared with the analogous E-NADPH-PT179 complex. Diphenyl ethers are stabilized to a greater extent partly because of the closure of the SBL that occurs farther along the reaction coordinate. At this stage, NADP+ exists in the ternary complex, which also enhances the binding affinity of deprotonated diphenyl ethers. However, in comparison with E-NADPH, the steady-state concentration of E-NADP+ is very small due to the fast dissociation of the oxidized cofactor generated via catalysis (31). Hence, the resulting apparent inhibitor association rate (kon·[E-NADP+]·[I]) of diphenyl ethers is slow explaining the observed slow binding phenomenon, although the actual association rate constants kon of pyridones and diphenyl ethers are very similar. Thus, in saFabI the ordering of the substrate-binding loop is likely correlated rather than causative with respect to the observation of slow-onset kinetics with diphenyl ethers. The slow-off kinetics observed with diphenyl ethers is likely the consequence of its potent thermodynamic affinity.
Among the three saFabI inhibitors investigated in clinical trials, CG400549 and AFN-1252 have been shown to be Staphylococcus-specific (23, 28–30), whereas the diphenyl ether MUT056399 is also active against several Gram-negative pathogens (24). As depicted in Fig. 3, this is likely attributed to different modes of inhibition. Substrate accumulation weakens the intrinsic potency of competitive inhibitors in contrast to uncompetitive inhibitors. Thus, relatively unmodified diphenyl ethers are already able to potently inhibit cell growth. In contrast, further optimization of binding affinity is necessary for pyridones to achieve cellular efficacy, particularly in the presence of active efflux pumps. This argument likely extends to the naphthyridinones, such as AFN-1252, which was recently crystallized in complex with saFabI and 3′-NADPH (Fig. 1) (23).
Our structural data rationalize the pyridone SAR profile and clearly reveal the ability of the CG400549 5-, 2′-, and 3′-substituents to enhance its affinity toward saFabI (Fig. 4 and Table 1). In particular, the hydrogen bond formed between the CG400549 and PT173 3′-amino group with Ala-97 leads to a 6-fold increase in affinity (Fig. 4B). In the case of ecFabI, this hydrogen bond is not protected from solvent exposure due to an M99G substitution (Fig. 6B), thereby reducing the affinity of such compounds and increasing the selectivity of CG400549 for saFabI (Table 2). Because of the amino acid residues Ile-200 and Met-206 and an elongated C terminus, including Met-256′, ecFabI also harbors a smaller binding pocket compared with saFabI (Fig. 6B). The wider acyl cavity of saFabI facilitates the accommodation of branched and longer acyl substrates or, analogously, bulky diphenyl ether 5-substituents (6, 31, 33, 70). Importantly, CG400549 and AFN-1252 contain large moieties at this position, which might further contribute to the Staphylococcus-specific spectrum of these two clinical trial inhibitors (Fig. 1). In fact, bulky pyridone 5-substituents interfere with the ecFabI and bpFabI SBL, although it is in a relatively closed state for saFabI with bound AFN-1252 (23) and CG400549 (Figs. 5B and 6A). This important difference might be further explained by the presence of the additional C-terminal extension in typical FabIs such as ecFabI and bpFabI, which occludes their substrate-binding sites (Fig. 7). In contrast, saFabI contains a significantly shorter C terminus compared with most other structurally characterized FabI proteins. The wider acyl cavity and thus the enhanced affinity of CG400549 can be related to the ability of saFabI to efficiently utilize bulky branched-chain fatty acyl substrates (6).
Based on the SAR profile of ecFabI, the ability to optimize binding affinity via substituents on the scaffold is very limited. A pyridone-based compound with broad spectrum activity must necessarily have higher intrinsic potency than the 2-pyridones. To achieve this, we used a 4-pyridone scaffold that retains the bridging oxygen of diphenyl ethers, thus providing an entropic advantage upon binding to FabI. Although the 4-pyridone inhibitor PT166 shares features with both the 2-pyridone and diphenyl ether scaffolds, its inhibition mechanism is the same as observed for 2-pyridones (Fig. 2B). Using the thermal shift assay, we could clearly show that this compound inhibits saFabI at the E-NADPH stage (supplemental Fig. S2), just like CG400549 (Fig. 2A). The 2-pyridone/substrate-like behavior of PT166 is further confirmed by a comparison of our ecFabI structures. In contrast to the triclosan-bound structure (13), where the SBL is in a closed state, this loop is disordered in the complexes with PT166 and CG400549 (Fig. 6). Furthermore, the structure of InhA in complex with the 4-pyridone PT155 shares a very similar open SBL conformation with the substrate-bound form of this enzyme (74, 76). As expected based on these considerations, the SAR and predicted binding mode for the 4-pyridones is highly reminiscent of the 2-pyridone series, but 4-pyridones possess superior potency at the enzymatic and cellular levels (Tables 1 and 2). Importantly, the higher intrinsic potency also translates into an extended spectrum of activity for the promising lead compound PT166 (against S. aureus, E. coli, F. tularensis, P. mirabilis, and M. tuberculosis; Table 3). As with CG400549 (77), replacement of the metabolically labile hydroxyl group with a carbonyl successfully improved the pharmacokinetic profile of PT166 compared with the diphenyl ether PT04 (Table 5). Additionally, this compound primarily acts on target (Table 4), and it significantly reduced bacterial burden in a murine model of MRSA infection (Fig. 8), validating its potential as a drug lead for future optimization and development. Further in vivo studies are needed to more precisely compare the pharmacokinetics and efficacy of the diverse FabI inhibitor scaffolds.
In summary, we have elucidated the structural and mechanistic basis for selective saFabI inhibition by pyridones, including the clinical candidate CG400549. Our study has yielded an insightful glimpse into conformational states along the coordinate of the saFabI enzymatic reaction, providing yet another reminder of the elegant connection between catalysis and inhibition. Importantly, we were able to rationally design the promising lead compound PT166, which merges the pharmacokinetic advantages of a pyridone with the potential for an extended spectrum of antibacterial activity. ecFabI and InhA served as paradigms for enoyl-ACP reductase homologues in Gram-negative and mycobacterial organisms, respectively. A similar approach can be applied toward the development of much needed narrow and broad spectrum antibiotics against novel targets.
Supplementary Material
Acknowledgments
We thank the staff at the beamline 14.1 (BESSY II) operated by the Helmholtz-Zentrum Berlin and at the European Synchrotron Radiation Facility beamline ID 23-1 for technical support. Furthermore, we thank the staff at the Translational Experimental Therapeutics Laboratory at Stony Brook. We also thank National Institutes of Health for the shared Instrumentation Grant 1 S10 RR023680-1 from NCRR.
This work was supported, in whole or in part, by National Institutes of Health Grants GM102864, AI044639, and AI070383 (to P. J. T.) This work was also supported by Deutsche Forschungsgemeinschaft Grants SFB630 (to C. A. S. and C. K.) and Forschungszentrum Grant FZ82 (to C. K.).

- MRSA
- methicillin-resistant S. aureus
- ACP
- acyl carrier protein
- FabI
- trans-2-enoyl-ACP reductase
- saFabI
- S. aureus FabI
- ecFabI
- E. coli FabI
- PK
- pharmacokinetics
- bpFabI
- B. pseudomallei FabI
- MIC
- minimal inhibitory concentration
- SAR
- structure-activity relationship
- SBL
- substrate-binding loop
- PDB
- Protein Data Bank
- CAPS
- 3-(cyclohexylamino)propanesulfonic acid
- BHI
- brain heart infusion
- MH
- Mueller-Hinton
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol.
REFERENCES
- 1. Pantosti A., Venditti M. (2009) What is MRSA? Eur. Respir. J. 34, 1190–1196 [DOI] [PubMed] [Google Scholar]
- 2. Naimi T. S., LeDell K. H., Como-Sabetti K., Borchardt S. M., Boxrud D. J., Etienne J., Johnson S. K., Vandenesch F., Fridkin S., O'Boyle C., Danila R. N., Lynfield R. (2003) Comparison of community- and health care-associated methicillin-resistant Staphylococcus aureus infection. JAMA 290, 2976–2984 [DOI] [PubMed] [Google Scholar]
- 3. Sievert D. M., Rudrik J. T., Patel J. B., McDonald L. C., Wilkins M. J., Hageman J. C. (2008) Vancomycin-resistant Staphylococcus aureus in the United States, 2002–2006. Clin. Infect. Dis. 46, 668–674 [DOI] [PubMed] [Google Scholar]
- 4. Marrakchi H., Lanéelle G., Quémard A. (2000) InhA, a target of the antituberculous drug isoniazid, is involved in a mycobacterial fatty acid elongation system, FAS-II. Microbiology 146, 289–296 [DOI] [PubMed] [Google Scholar]
- 5. Zhang Y. M., White S. W., Rock C. O. (2006) Inhibiting bacterial fatty acid synthesis. J. Biol. Chem. 281, 17541–17544 [DOI] [PubMed] [Google Scholar]
- 6. Schiebel J., Chang A., Lu H., Baxter M. V., Tonge P. J., Kisker C. (2012) Staphylococcus aureus FabI: inhibition, substrate recognition, and potential implications for in vivo essentiality. Structure 20, 802–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Payne D. J., Miller W. H., Berry V., Brosky J., Burgess W. J., Chen E., DeWolf Jr. W. E., Jr., Fosberry A. P., Greenwood R., Head M. S., Heerding D. A., Janson C. A., Jaworski D. D., Keller P. M., Manley P. J., Moore T. D., Newlander K. A., Pearson S., Polizzi B. J., Qiu X., Rittenhouse S. F., Slater-Radosti C., Salyers K. L., Seefeld M. A., Smyth M. G., Takata D. T., Uzinskas I. N., Vaidya K., Wallis N. G., Winram S. B., Yuan C. C., Huffman W. F. (2002) Discovery of a novel and potent class of FabI-directed antibacterial agents. Antimicrob. Agents Chemother. 46, 3118–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Xu H., Sullivan T. J., Sekiguchi J., Kirikae T., Ojima I., Stratton C. F., Mao W., Rock F. L., Alley M. R., Johnson F., Walker S. G., Tonge P. J. (2008) Mechanism and inhibition of saFabI, the enoyl reductase from Staphylococcus aureus. Biochemistry 47, 4228–4236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heath R. J., Li J., Roland G. E., Rock C. O. (2000) Inhibition of the Staphylococcus aureus NADPH-dependent enoyl-acyl carrier protein reductase by triclosan and hexachlorophene. J. Biol. Chem. 275, 4654–4659 [DOI] [PubMed] [Google Scholar]
- 10. Banerjee A., Dubnau E., Quemard A., Balasubramanian V., Um K. S., Wilson T., Collins D., de Lisle G., Jacobs W. R., Jr. (1994) inhA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 263, 227–230 [DOI] [PubMed] [Google Scholar]
- 11. Dessen A., Quémard A., Blanchard J. S., Jacobs W. R., Jr., Sacchettini J. C. (1995) Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 267, 1638–1641 [DOI] [PubMed] [Google Scholar]
- 12. Levy C. W., Roujeinikova A., Sedelnikova S., Baker P. J., Stuitje A. R., Slabas A. R., Rice D. W., Rafferty J. B. (1999) Molecular basis of triclosan activity. Nature 398, 383–384 [DOI] [PubMed] [Google Scholar]
- 13. Stewart M. J., Parikh S., Xiao G., Tonge P. J., Kisker C. (1999) Structural basis and mechanism of enoyl reductase inhibition by triclosan. J. Mol. Biol. 290, 859–865 [DOI] [PubMed] [Google Scholar]
- 14. Heath R. J., Rubin J. R., Holland D. R., Zhang E., Snow M. E., Rock C. O. (1999) Mechanism of triclosan inhibition of bacterial fatty acid synthesis. J. Biol. Chem. 274, 11110–11114 [DOI] [PubMed] [Google Scholar]
- 15. Payne D. J., Gwynn M. N., Holmes D. J., Pompliano D. L. (2007) Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug. Discov. 6, 29–40 [DOI] [PubMed] [Google Scholar]
- 16. Lu H., Tonge P. J. (2008) Inhibitors of FabI, an enzyme drug target in the bacterial fatty acid biosynthesis pathway. Acc. Chem. Res. 41, 11–20 [DOI] [PubMed] [Google Scholar]
- 17. Heath R. J., Rock C. O. (2000) A triclosan-resistant bacterial enzyme. Nature 406, 145–146 [DOI] [PubMed] [Google Scholar]
- 18. Heath R. J., Su N., Murphy C. K., Rock C. O. (2000) The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilis. J. Biol. Chem. 275, 40128–40133 [DOI] [PubMed] [Google Scholar]
- 19. Massengo-Tiassé R. P., Cronan J. E. (2008) Vibrio cholerae FabV defines a new class of enoyl-acyl carrier protein reductase. J. Biol. Chem. 283, 1308–1316 [DOI] [PubMed] [Google Scholar]
- 20. Brinster S., Lamberet G., Staels B., Trieu-Cuot P., Gruss A., Poyart C. (2009) Type II fatty acid synthesis is not a suitable antibiotic target for Gram-positive pathogens. Nature 458, 83–86 [DOI] [PubMed] [Google Scholar]
- 21. Parsons J. B., Frank M. W., Subramanian C., Saenkham P., Rock C. O. (2011) Metabolic basis for the differential susceptibility of Gram-positive pathogens to fatty acid synthesis inhibitors. Proc. Natl. Acad. Sci. U.S.A. 108, 15378–15383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balemans W., Lounis N., Gilissen R., Guillemont J., Simmen K., Andries K., Koul A. (2010) Essentiality of FASII pathway for Staphylococcus aureus. Nature 463, E3. [DOI] [PubMed] [Google Scholar]
- 23. Kaplan N., Albert M., Awrey D., Bardouniotis E., Berman J., Clarke T., Dorsey M., Hafkin B., Ramnauth J., Romanov V., Schmid M. B., Thalakada R., Yethon J., Pauls H. W. (2012) Mode of action, in vitro activity, and in vivo efficacy of AFN-1252, a selective antistaphylococcal FabI inhibitor. Antimicrob. Agents Chemother. 56, 5865–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gerusz V., Denis A., Faivre F., Bonvin Y., Oxoby M., Briet S., LeFralliec G., Oliveira C., Desroy N., Raymond C., Peltier L., Moreau F., Escaich S., Vongsouthi V., Floquet S., Drocourt E., Walton A., Prouvensier L., Saccomani M., Durant L., Genevard J. M., Sam-Sambo V., Soulama-Mouze C. (2012) From triclosan toward the clinic: discovery of nonbiocidal, potent FabI inhibitors for the treatment of resistant bacteria. J. Med. Chem. 55, 9914–9928 [DOI] [PubMed] [Google Scholar]
- 25. Gerusz V. (2010) in Annual Reports in Medicinal Chemistry (John E. M., ed) pp. 295–311, Academic Press, New York [Google Scholar]
- 26. Escaich S., Prouvensier L., Saccomani M., Durant L., Oxoby M., Gerusz V., Moreau F., Vongsouthi V., Maher K., Morrissey I., Soulama-Mouze C. (2011) The MUT056399 inhibitor of FabI is a new antistaphylococcal compound. Antimicrob. Agents Chemother. 55, 4692–4697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Seefeld M. A., Miller W. H., Newlander K. A., Burgess W. J., DeWolf W. E., Jr., Elkins P. A., Head M. S., Jakas D. R., Janson C. A., Keller P. M., Manley P. J., Moore T. D., Payne D. J., Pearson S., Polizzi B. J., Qiu X., Rittenhouse S. F., Uzinskas I. N., Wallis N. G., Huffman W. F. (2003) Indole naphthyridinones as inhibitors of bacterial enoyl-ACP reductases FabI and FabK. J. Med. Chem. 46, 1627–1635 [DOI] [PubMed] [Google Scholar]
- 28. Silver L. L. (2011) Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yum J. H., Kim C. K., Yong D., Lee K., Chong Y., Kim C. M., Kim J. M., Ro S., Cho J. M. (2007) In vitro activities of CG400549, a novel FabI inhibitor, against recently isolated clinical staphylococcal strains in Korea. Antimicrob. Agents Chemother. 51, 2591–2593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Park H. S., Yoon Y. M., Jung S. J., Kim C. M., Kim J. M., Kwak J. H. (2007) Antistaphylococcal activities of CG400549, a new bacterial enoyl-acyl carrier protein reductase (FabI) inhibitor. J. Antimicrob. Chemother. 60, 568–574 [DOI] [PubMed] [Google Scholar]
- 31. Chang A., Schiebel J., Yu W., Bommineni G. R., Pan P., Baxter M. V., Khanna A., Sotriffer C. A., Kisker C., Tonge P. J. (2013) Rational optimization of drug-target residence time: insights from inhibitor binding to the Staphylococcus aureus FabI enzyme-product complex. Biochemistry 52, 4217–4228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Priyadarshi A., Kim E. E., Hwang K. Y. (2010) Structural insights into Staphylococcus aureus enoyl-ACP reductase (FabI), in complex with NADP and triclosan. Proteins 78, 480–486 [DOI] [PubMed] [Google Scholar]
- 33. Sivaraman S., Zwahlen J., Bell A. F., Hedstrom L., Tonge P. J. (2003) Structure-activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant FabIs. Biochemistry 42, 4406–4413 [DOI] [PubMed] [Google Scholar]
- 34. Luckner S. R., Liu N., am Ende C. W., Tonge P. J., Kisker C. (2010) A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem. 285, 14330–14337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu N., Cummings J. E., England K., Slayden R. A., Tonge P. J. (2011) Mechanism and inhibition of the FabI enoyl-ACP reductase from Burkholderia pseudomallei. J. Antimicrob. Chemother. 66, 564–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mueller U., Darowski N., Fuchs M. R., Förster R., Hellmig M., Paithankar K. S., Pühringer S., Steffien M., Zocher G., Weiss M. S. (2012) Facilities for macromolecular crystallography at the Helmholtz-Zentrum Berlin. J. Synchrotron Radiat. 19, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leslie A. G. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 + ESF-EAMCB Newsletter on Protein Crystallography, No. 26 [Google Scholar]
- 38. Kabsch W. (1993) Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 26, 795–800 [Google Scholar]
- 39. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 40. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 42. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 43. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Painter J., Merritt E. A. (2006) TLSMD web server for the generation of multi-group TLS models. J. Appl. Crystallogr. 39, 109–111 [Google Scholar]
- 45. Krug M., Weiss M. S., Heinemann U., Mueller U. (2012) XDSAPP: a graphical user interface for the convenient processing of diffraction data using XDS. J. Appl. Crystallogr. 45, 568–572 [Google Scholar]
- 46. Smart O. S., Womack T. O., Sharff A., Flensburg C., Keller P., Paciorek W., Vonrhein C., Bricogne G. (2011) Grade. Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 47. Bruno I. J., Cole J. C., Kessler M., Luo J., Motherwell W. D., Purkis L. H., Smith B. R., Taylor R., Cooper R. I., Harris S. E., Orpen A. G. (2004) Retrieval of crystallographically-derived molecular geometry information. J. Chem. Inf. Comput. Sci. 44, 2133–2144 [DOI] [PubMed] [Google Scholar]
- 48. Schüttelkopf A. W., van Aalten D. M. (2004) PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr. D Biol. Crystallogr. 60, 1355–1363 [DOI] [PubMed] [Google Scholar]
- 49. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. DeLano W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA [Google Scholar]
- 51. Morrison J. F., Walsh C. T. (1988) The behavior and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 201–301 [DOI] [PubMed] [Google Scholar]
- 52. Morrison J. F. (1969) Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta 185, 269–286 [DOI] [PubMed] [Google Scholar]
- 53. Rarey M., Kramer B., Lengauer T., Klebe G. (1996) A fast flexible docking method using an incremental construction algorithm. J. Mol. Biol. 261, 470–489 [DOI] [PubMed] [Google Scholar]
- 54. Velec H. F., Gohlke H., Klebe G. (2005) DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction. J. Med. Chem. 48, 6296–6303 [DOI] [PubMed] [Google Scholar]
- 55. Gohlke H., Hendlich M., Klebe G. (2000) Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol. 295, 337–356 [DOI] [PubMed] [Google Scholar]
- 56. Neudert G., Klebe G. (2011) DSX: a knowledge-based scoring function for the assessment of protein-ligand complexes. J. Chem. Inf. Model. 51, 2731–2745 [DOI] [PubMed] [Google Scholar]
- 57. Chemical Computing Group (2010) Molecular Operating Environment, Version 2010.10 Montreal, Quebec, Canada [Google Scholar]
- 58. Tripos (2009) SYBYL-X, Version 1.0 St. Louis, MO [Google Scholar]
- 59. Neudert G., Klebe G. (2011) fconv: format conversion, manipulation and feature computation of molecular data. Bioinformatics 27, 1021–1022 [DOI] [PubMed] [Google Scholar]
- 60. Clinical and Laboratory Standards Institute (2006) Approved Standard M7-A5, 6th Ed., Clinical and Laboratory Standards Institute, Wayne, PA [Google Scholar]
- 61. Gerber A. U., Vastola A. P., Brandel J., Craig W. A. (1982) Selection of aminoglycoside-resistant variants of Pseudomonas aeruginosa in an in vivo model. J. Infect. Dis. 146, 691–697 [DOI] [PubMed] [Google Scholar]
- 62. Bissantz C., Kuhn B., Stahl M. (2010) A medicinal chemist's guide to molecular interactions. J. Med. Chem. 53, 5061–5084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Westley A. M., Westley J. (1996) Enzyme inhibition in open systems. Superiority of uncompetitive agents. J. Biol. Chem. 271, 5347–5352 [DOI] [PubMed] [Google Scholar]
- 64. Fan F., Yan K., Wallis N. G., Reed S., Moore T. D., Rittenhouse S. F., DeWolf W. E., Jr., Huang J., McDevitt D., Miller W. H., Seefeld M. A., Newlander K. A., Jakas D. R., Head M. S., Payne D. J. (2002) Defining and combating the mechanisms of triclosan resistance in clinical isolates of Staphylococcus aureus. Antimicrob. Agents Chemother. 46, 3343–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Brenwald N. P., Fraise A. P. (2003) Triclosan resistance in methicillin-resistant Staphylococcus aureus (MRSA). J. Hosp. Infect. 55, 141–144 [DOI] [PubMed] [Google Scholar]
- 66. Tipparaju S. K., Joyasawal S., Forrester S., Mulhearn D. C., Pegan S., Johnson M. E., Mesecar A. D., Kozikowski A. P. (2008) Design and synthesis of 2-pyridones as novel inhibitors of the Bacillus anthracis enoyl-ACP reductase. Bioorg. Med. Chem. Lett. 18, 3565–3569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kitagawa H., Kumura K., Takahata S., Iida M., Atsumi K. (2007) 4-Pyridone derivatives as new inhibitors of bacterial enoyl-ACP reductase FabI. Bioorg. Med. Chem. 15, 1106–1116 [DOI] [PubMed] [Google Scholar]
- 68. Takahata S., Iida M., Yoshida T., Kumura K., Kitagawa H., Hoshiko S. (2007) Discovery of 4-pyridone derivatives as specific inhibitors of enoyl-acyl carrier protein reductase (FabI) with antibacterial activity against Staphylococcus aureus. J. Antibiot. 60, 123–128 [DOI] [PubMed] [Google Scholar]
- 69. Lu H., England K., am Ende C., Truglio J. J., Luckner S., Reddy B. G., Marlenee N. L., Knudson S. E., Knudson D. L., Bowen R. A., Kisker C., Slayden R. A., Tonge P. J. (2009) Slow-onset inhibition of the FabI enoyl reductase from Francisella tularensis: residence time and in vivo activity. ACS Chem. Biol. 4, 221–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ward W. H., Holdgate G. A., Rowsell S., McLean E. G., Pauptit R. A., Clayton E., Nichols W. W., Colls J. G., Minshull C. A., Jude D. A., Mistry A., Timms D., Camble R., Hales N. J., Britton C. J., Taylor I. W. (1999) Kinetic and structural characteristics of the inhibition of enoyl (acyl carrier protein) reductase by triclosan. Biochemistry 38, 12514–12525 [DOI] [PubMed] [Google Scholar]
- 71. Yao J., Maxwell J. B., Rock C. O. (2013) Resistance to AFN-1252 arises from missense mutations in Staphylococcus aureus enoyl-acyl carrier protein reductase (FabI). J. Biol. Chem. 288, 36261–36271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Brameld K. A., Kuhn B., Reuter D. C., Stahl M. (2008) Small molecule conformational preferences derived from crystal structure data. A medicinal chemistry focused analysis. J. Chem. Inf. Model. 48, 1–24 [DOI] [PubMed] [Google Scholar]
- 73. Shin K. J., Roh E. J., Chung M. K., Cha H. J., Seo S. H. (February 3, 2011) Korea Patent WO/2011/014008 A2
- 74. Li H.-J., Lai C.-T., Pan P., Yu W., Liu N., Bommineni G. R., Garcia-Diaz M., Simmerling C., Tonge P. J. (2014) A structural and energetic model for the slow-onset inhibition of the Mycobacterium tuberculosis enoyl-ACP reductase InhA. ACS Chem. Biol. 9, 986–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang L. Q., Falany C. N., James M. O. (2004) Triclosan as a substrate and inhibitor of 3′-phosphoadenosine 5′-phosphosulfate-sulfotransferase and UDP-glucuronosyl transferase in human liver fractions. Drug. Metab. Dispos. 32, 1162–1169 [DOI] [PubMed] [Google Scholar]
- 76. Rozwarski D. A., Vilchèze C., Sugantino M., Bittman R., Sacchettini J. C. (1999) Crystal structure of the Mycobacterium tuberculosis enoyl-ACP reductase, InhA, in complex with NAD+ and a C16 fatty acyl substrate. J. Biol. Chem. 274, 15582–15589 [DOI] [PubMed] [Google Scholar]
- 77. Ro S., Son K. H., Kim Y. E., Chang H. J., Park S. B., Choi J. R., Cho J. M. (2009) Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, CA, September 12–15, 2009, American Society for Microbiology, San Francisco [Google Scholar]
- 78. White S. W., Zheng J., Zhang Y. M., Rock (2005) The structural biology of type II fatty acid biosynthesis. Annu. Rev. Biochem. 74, 791–831 [DOI] [PubMed] [Google Scholar]
- 79. Theobald D. L., Wuttke D. S. (2008) Accurate structural correlations from maximum likelihood superpositions. PLoS Comput. Biol. 4, e43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X, Version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 81. Lu H., Tonge P. J. (2010) Mechanism and inhibition of the FabV enoyl-ACP reductase from Burkholderia mallei. Biochemistry 49, 1281–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Gouet P., Courcelle E., Stuart D. I., Métoz F. (1999) ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.