

Background: Endorepellin inhibits angiogenesis by simultaneously binding the α2β1 integrin and VEGFR2, attenuating the PI3K/Akt/mTOR and PKC/JNK/AP1 signaling pathways.
Results: Endorepellin evokes autophagy by inducing Beclin 1 and LC3 downstream of VEGFR2 in a Peg3-dependent manner.
Conclusion: Endorepellin causes endothelial cell autophagy through VEGFR2 independent of the α2β1 integrin.
Significance: Endorepellin-evoked endothelial cell autophagy represents a promising strategy for angiostatic-based therapeutics.
Keywords: Angiogenesis, Autophagy, Endothelial Cell, Extracellular Matrix, Extracellular Matrix Proteins, Proteoglycan
Abstract
Endorepellin, the C-terminal fragment of the heparan sulfate proteoglycan perlecan, possesses angiostatic activity via dual receptor antagonism, through concurrent binding to the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2). Here, we discovered that soluble endorepellin induced autophagy in endothelial cells by modulating the expression of Beclin 1, LC3, and p62, three established autophagic markers. Moreover, endorepellin evoked expression of the imprinted tumor suppressor gene Peg3 and its co-localization with Beclin 1 and LC3 in autophagosomes, suggesting a major role for this gene in endothelial cell autophagy. Mechanistically, endorepellin induced autophagy by down-regulating VEGFR2 via the two LG1/2 domains, whereas the C-terminal LG3 domain, the portion responsible for binding the α2β1 integrin, was ineffective. Endorepellin also induced transcriptional activity of the BECN1 promoter in endothelial cells, and the VEGFR2-specific tyrosine kinase inhibitor, SU5416, blocked this effect. Finally, we found a correlation between endorepellin-evoked inhibition of capillary morphogenesis and enhanced autophagy. Thus, we have identified a new role for this endogenous angiostatic fragment in inducing autophagy through a VEGFR2-dependent but α2β1 integrin-independent pathway. This novel mechanism specifically targets endothelial cells and could represent a promising new strategy to potentiate the angiostatic effect of endorepellin and perhaps other angiostatic matrix proteins.
Introduction
Perlecan is a multidomain heparan sulfate proteoglycan encoded by a large gene in both mice and humans (1, 2) with a complex and highly regulated promoter (3–5). The large protein core of ∼500 kDa is endowed with multiple biological functions that have been partly elucidated in the past 20 years. Perlecan is one of the few genes in the human genome that is expressed by both vascular (6–9) and avascular tissues, such as cartilage (10), and is localized at the apical cell surface (11, 12) as well as in the basement membranes of both epithelial and endothelial lined tissues (6, 13, 14). Based on its widespread expression in both embryonic (10) and adult (13) life and its ability to engage various receptor tyrosine kinases (15), perlecan regulates various biological processes, including cell adhesion (16), lipid metabolism (17), thrombosis (18), apoptosis (19), biomechanical properties of blood vessels and cartilage (20, 21), premature rupture of fetal membranes (22), epidermal formation (23), endochondral bone (24) and enamel organ (25) formation, osteophyte formation (26), and corneal structure (27). Moreover, perlecan binds various growth factors (28–32), and its expression is often deregulated in several types of cancer (33–35).
Abnormal expression of proteoglycans (36–38) and generation of processed forms derived from large protein core precursors, as well as deregulated expression of glycolytic enzymes in cancer, have been well documented for over 2 decades (39–42). Perlecan has a dual function in developmental and experimental angiogenesis. Via the N-terminal heparan sulfate chains, perlecan acts primarily as a pro-angiogenic factor (43) by binding and presenting FGF2 and VEGF to its cognate receptors (44–51). Genetically abrogating Hspg2 or preventing Hspg2 expression in early embryogenesis causes cardiovascular defects in mammalians and vertebrates (52–55). In contrast, a C-terminal processed form of perlecan, denoted as endorepellin by signifying the inherent anti-endothelial cell activity (56), inhibits endothelial cell migration, collagen-induced capillary morphogenesis, and blood vessel growth both in vitro and in animal models of squamous and lung carcinomas (57–60). The mechanism of action concerning endorepellin has been partly elucidated by first discovering a major endorepellin receptor expressed by endothelial cells, i.e. the α2β1 integrin (57, 61, 62), a key receptor involved in angiogenesis (63–65). Tumor xenografts generated in mice with a targeted deletion of the α2 integrin fail to respond to systemic delivery of endorepellin, and similarly, α2β1−/− microvascular endothelial cells do not respond to endorepellin (59). Endorepellin triggers the activation of the tyrosine phosphatase SHP-1 via an α2β1 integrin-dependent pathway to dephosphorylate and inactivate various receptor tyrosine kinases, including VEGFR24 (66). Recently, we discovered that endorepellin exerts a dual receptor antagonism by concurrently targeting VEGFR2 and the α2β1 integrin (67). The first two laminin-like globular domains (LG1/2) bind the Ig3–5 domain of VEGFR2, whereas the terminal LG3, liberated in vivo by BMP-1/Tolloid-like metalloproteases (68), binds the α2β1 integrin (69). These two separate branches of endorepellin signaling have a similar outcome by utilizing different mechanisms. Binding to α2β1 integrin causes a signaling cascade that leads to disassembly of actin filaments and focal adhesions that ultimately suppress endothelial cell migration (69, 70). Concurrent binding to VEGFR2 leads to further downstream signaling initiated by dephosphorylation of Tyr1175 by SHP-1 and subsequent downstream transcriptional inhibition of VEGFA (71). This ultimately inhibits VEGFA-induced endothelial cell migration and angiogenesis.
We noticed that a VEGFR2 signaling pathway inhibited by endorepellin included the mammalian target of rapamycin (mTOR), a key inhibitor of autophagy (72). Thus, we hypothesized that endorepellin could evoke autophagy via suppression of VEGFR2-dependent signaling by suppressing the canonical mTOR pathway. In this work, we demonstrate for the first time that endorepellin induces autophagy in endothelial cells through VEGFR2 but independently of the α2β1 integrin. We found that nanomolar concentrations of human recombinant endorepellin induced Beclin 1- and LC3-positive autophagosomes in nutrient-enriched conditions in both human and porcine endothelial cells. Moreover, p62 protein was dynamically modulated by endorepellin and co-localized with LC3 in autophagosomes. Thus, we have discovered a novel mechanism that specifically targets endothelial cells and could provide a promising strategy to potentiate the angiostatic effect of endorepellin and perhaps other proteolytically processed matrix proteins harboring angiostatic activity.
EXPERIMENTAL PROCEDURES
Antibodies, Cells, and Reagents
The mouse anti-rabbit IgG (light chain-specific) was from Cell Signaling. The rabbit antibodies against human Peg3, Beclin 1, LC3-I/II, and mouse monoclonal antibodies (mAb) against Beclin 1 and LC3-I/II were from Abcam. Rabbit mAb against human Vps34 (Vps34, vacuolar protein sorting 34, also known as class III PI3K), p62/SQSTM1, GAPDH, were from Cell Signaling. Rabbit anti-LC3 antibody and mouse mAb against Beclin 1 were procured from Novus Biologicals. Anti-integrin α2 I-domain blocking mAb (1998Z) was from Millipore (Billerica, MA). Secondary HRP-conjugated goat anti-rabbit and anti-mouse antibodies were from Millipore. HRP-conjugated donkey anti-rabbit and sheep anti-mouse were from Millipore. Goat anti-mouse and anti-rabbit (Alexa Fluor-488) and goat anti-mouse and anti-rabbit (Alexa Fluor-594) antibodies were from Invitrogen. Human umbilical vein endothelial cells (HUVEC) were from Lifeline Cell Technology and grown in basal media supplemented with VascuLife EnGS LifeFactors kit (Lifeline Cell Technology) and used within the first five passages. Porcine aortic endothelial cells (PAE) and their transgenic counterparts expressing either VEGFR2 or VEGFR2-GFP-LC3 were described previously (67, 73). PAE cells were stably transfected with a human BECN1 promoter (74) luciferase reporter construct as described previously (73). Dulbecco's phosphate-buffered saline and HBSS were from CellGro. Endorepellin fragments LG1/2 and LG3 were described previously (69). SU5416 was from EMD. Rapamycin was from Sigma. Protein A-Sepharose magnetic beads were from GE Healthcare. SuperSignal West Pico chemiluminescence substrate was from Thermo Fisher Scientific.
Immunoblotting and Immunoprecipitation Assays
Following each treatment, endothelial cells were lysed in RIPA buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mm EDTA/EGTA/sodium vanadate, 10 mm β-glycerophosphate, and the following protease inhibitors: 1 mm phenylmethanesulfonyl fluoride and 10 μg/ml leupeptin/l-1-tosylamido-2-phenylethyl chloromethyl ketone/aprotinin each) for 20 min on ice (75). Insoluble material was removed by centrifugation at 14,000 × g, and protein levels were determined using the DC assay (Bio-Rad). For immunoprecipitation, protein A-Sepharose magnetic beads were absorbed with antibodies for 4 h at 4 °C, and pre-cleared cell lysates were added to the beads for 18 h at 4 °C. After extensive washing, the beads were boiled in reducing buffer, and supernatants were separated by SDS-PAGE. Proteins were then transferred to nitrocellulose membranes (Bio-Rad), probed with the indicated antibodies, developed with enhanced chemiluminescence (Thermo), and detected using ImageQuant LAS-4000 (GE Healthcare).
Immunofluorescence and Confocal Microscopy
Routinely, ∼5 × 104 HUVECs or PAE-VEGFR2 cells were grown on 0.2% gelatin-coated four-chamber slides (Nunc). After each treatment, cells were washed with PBS and fixed for 30 min in 4% paraformaldehyde at 4 °C (76, 77). After blocking in PBS, 5% BSA, cells were incubated with various antibodies for 1 h, washed in PBS, and then incubated with the appropriate secondary antibodies (goat anti-mouse or anti-rabbit IgG Alexa Fluor® 488 or goat anti-mouse or anti-rabbit IgG Alexa Fluor® 594) for 1 h. Nuclei were visualized with DAPI (Vector Laboratories). For immunofluorescence, images were acquired using a Leica DM5500B microscope with Leica Application Suite, Advanced Fluorescence software (version 1.8, Leica Microsystems, Inc., Wetzlar, Germany). A Zeiss LSM-780 confocal laser-scanning microscope was utilized for the acquisition of confocal images with a ×63, 1.3 oil-immersion objective. Merged images represent single optical sections (<0.8 μm), collected with the pinhole set to 1 airy unit for the red channel, and adjusted to give the same optical slice thickness in the green and blue channels. Images were acquired in single confocal planes to determine co-localization using Zeiss LSM-780 software, with filters set at 488/594 nm for dual-channel imaging. All images were analyzed using ImageJ and Adobe Photoshop CS6. Line scanning was performed as described before (71).
RNAi-mediated Gene Silencing
Transient knockdown of PEG3 and VEGFR2 in HUVEC was achieved via transfection of three separate and validated siRNA oligonucleotides specific for PEG3 (sc-97350) from Santa Cruz Biotechnology (Santa Cruz, CA) or targeting VEGFR2 (s7823) from Invitrogen. Scrambled siRNA (siScr, sc-37007) from Santa Cruz Biotechnology served as a negative control for all siRNA experiments presented herein. The following protocol was utilized prior to total RNA isolation (via TRIzol reagent, Invitrogen) or for immunoblotting following lysis in RIPA. As such, six-well plates were seeded with ∼2 × 105 HUVECs followed by overnight incubation at 37 °C, 5% CO2 until cultures were ∼70% confluent. Targeting or scrambled siRNA duplex (80 and 20 pm, respectively) was added to dilute Lipofectamine 2000 RNAiMAX (Invitrogen) in 0.5% serum-supplemented HUVEC media (LifeCell). The transfection mixture was applied for 6 h at 37 °C, 5% CO2, whereupon additional full serum (2% serum-supplemented HUVEC media) media were added overnight. The following day, media were changed, and the transfection was carried out for an additional 48 h, inclusive of any condition as required per experimental setting. Verification of RNAi-mediated knockdown of target mRNA or protein was determined via qPCR-utilizing gene-specific primers or via immunoblotting and reaction with a specific primary antibody, respectively.
Complementary DNA (cDNA) Library Synthesis and Quantitative Real Time-PCR (qPCR) Analysis
Gene expression analysis by qPCR was carried out on confluent (>90%) six-well plates seeded with ∼2 × 105 HUVECs treated variably in accordance with the prevailing experimental parameters in 2% serum-supplemented HUVEC media. Cells were lysed in 1 ml of TRIzol reagent (Invitrogen) for total RNA extraction. Thus, for gene expression analyses, ∼1 μg of total RNA was annealed with oligo(dT18–20) primers, and cDNA was synthesized with SuperScript Reverse Transcriptase II (SSRT II, Invitrogen). PCR amplicons representing target genes (PEG3, VEGFR2, BECN1, and MAPLC3A) and the endogenous housekeeping gene, ACTB, were amplified in quadruplicate independent reactions with the Brilliant SYBR Green Master Mix II reagent (Agilent Technologies, Cedar Creek, TX). All samples were run on the LightCycler 480-II Real Time PCR platform (Roche Applied Science), and the cycle number (Ct) was recorded for each independent reaction. Fold change determinations were made utilizing the comparative Ct method. ΔCt values represent normalized gene expression levels to ACTB. ΔΔCt values were calculated and represent the experimental cDNA minus the corresponding gene levels (ΔCt values) of the calibrator sample (i.e. control). Fold change determinations were calculated using the double ΔCt (ΔΔCt) method (2−ΔΔCt) ± S.E. The data presented herein represent at least three independent trials run in quadruplicate replicates for each gene of interest.
Luciferase Assay
Cells were washed with PBS, and 150 μl of lysis buffer (50 mm potassium phosphate buffer, 2% Triton X-100, 20% glycerol, 4 mm DTT) was added to each well. Cells were then lysed at 25 °C for 10 min, centrifuged at 2000 × g for 2 min, and ∼100 μl of cleared cell lysate was dispensed into a 96-well ELISA plate, together with 100 μl of luciferase assay buffer (100 mm potassium phosphate, 2 mm DTT, 8 mm MgSO4, 175 μm coenzyme A, 750 μm ATP) and 0.5 mm d-luciferin. For light measurement, a plate luminometer (PerkinElmer Life Sciences) was used, and all data were normalized to total cell protein.
Capillary Morphogenesis Assays
Four-well slides (Thermo Scientific) were initially coated with poly-l-lysine (20 μg/ml) (Sigma) and then left to set in the incubator at 37 °C for 1 h before placing the slide under UV light for 15 min. The wells were then coated with 400 μl of Matrigel (BD Biosciences) and incubated for 1 h at 37 °C. Approximately 104 PAE-GFP-LC3 cells were resuspended in warm media either in the presence or absence of endorepellin (200 nm) for 30 min and then placed onto the Matrigel. After 6 h, the wells were washed with PBS and fixed in cold formalin. An inverted epifluorescence microscope (Olympus CKX-41) equipped with an X-CITE 120Q microscope light source system was used for imaging. Analysis of the images was then completed in ImageJ by converting the images to Rainbow RGB after splitting the channels and then making a surface plot. This allowed the fluorescence intensity of the GFP-LC3 to be visualized and quantified.
Statistical Analysis
Immunoblots were quantified by scanning densitometry using ImageJ or Odyssey software for the infrared-labeled secondary antibodies. All data were expressed as mean ± S.E. and statistically analyzed with the Student's t test or paired t test using the Sigma-Stat software 11.0 (SPSS Inc); p values <0.05 were considered statistically significant.
RESULTS
Endorepellin Induces Endothelial Cell Autophagy
To test whether endorepellin could induce autophagy in endothelial cells, we treated HUVEC and PAE-VEGFR2 cells for 6 h with human recombinant endorepellin (200 nm), rapamycin (40 nm), an established mTOR inhibitor and inducer of autophagy, or HBSS to mimic nutrient deprivation. Using differential interference contrast (DIC) microscopy, we found in both endothelial cells that endorepellin induced the formation of large autophagosome-like structures identical to those observed in rapamycin and HBSS treatments (white arrows, Fig. 1A). To confirm the nature of these large cytoplasmic structures, we utilized transgenic and immunological approaches targeting two established autophagic markers, Beclin 1 and LC3 (78). To specifically label autophagosomes, we stably transfected PAE-VEGFR2 cells with GFP-LC3. The lipidated form of LC3, known as LC3-II, re-locates from the cytosol to autophagosome membranes during autophagy (78). The GFP moiety is located at the N terminus of LC3, thereby allowing lipidation that occurs at its C terminus (78). Upon endorepellin treatment, there was a marked re-distribution of GFP-LC3 into large autophagosomes (Fig. 1B). Unlike vehicle-treated controls, Beclin 1 co-localized with GFP-LC3 into large autophagosomes following endorepellin treatment (Fig. 1C). The endorepellin-evoked co-distribution of LC3 and Beclin 1 was further corroborated using confocal laser microscopy and concurrent line scanning, an imaging strategy that measures the pixels along a defined axis (i.e. the distance between the white arrows in Fig. 1, D and E) and establishes co-localization of the differentially labeled fluorophores (79, 80). The extent of overlap between two fluorophores occupying the same pixel is used as a qualitative assessment of a proximity-dependent localization (81). The results showed a clear distinction in the co-localization of both autophagy markers between vehicle-treated (Fig. 1D) and endorepellin-treated (Fig. 1E) endothelial cells. Endorepellin evoked a near complete overlap of the fluorescent profiles for LC3 and Beclin (right panel, Fig. 1E), indicating co-localization.
FIGURE 1.

Endorepellin causes autophagy in endothelial cells inducing the expression and co-localization of LC3 and Beclin 1. A, DIC/fluorescence images of HUVEC or PAE-VEGFR2 cells as indicated and treated with endorepellin (200 nm), rapamycin (40 nm), or HBSS for 6 h showing formation of autophagosomes in the cytoplasm of the cells. Nuclei (blue) were stained with DAPI. B and C, representative fluorescence micrographs of PAE-VEGFR2 cells stably transfected with GFP-LC3 and treated for 6 h with endorepellin (200 nm) or vehicle as indicated. D and E, confocal images of Beclin 1 and LC3 cellular co-localization in HUVECs ± a 6-h treatment with endorepellin. The line-scanned profiles are next to each confocal image.
Endorepellin Induces Autophagy in a Dose-dependent Manner and Promotes LC3 Interaction with Beclin 1
Next, we performed dose-response experiments to evaluate endorepellin activity on LC3-positive autophagosomes using HUVEC and DIC. We discovered that there was a progressive increase of autophagosome formation with increasing amounts of endorepellin. Notably, autophagosomes were already evident at 25 nm endorepellin (Fig. 2B) and became increasingly prominent up to 250 nm (Fig. 2, C–F). Quantification of three independent experiments showed a significant increase in autophagosomes/cell reaching nearly 18-fold over controls at 250 nm endorepellin (p < 0.001, Fig. 2G). To further validate these findings, we performed confocal microscopy and compared the activity of endorepellin vis à vis that of rapamycin or nutrient deprivation. We found that HUVEC treated for 6 h with endorepellin (200 nm) under nutrient-rich conditions contained a large number of LC3-positive autophagosomes (Fig. 2I) similar to those induced by rapamycin (Fig. 2J) or HBSS (Fig. 2K). Quantification of LC3-positive autophagosomes showed a significant increase in the average number of puncta/cell for all three conditions (p < 0.001, Fig. 2L).
FIGURE 2.
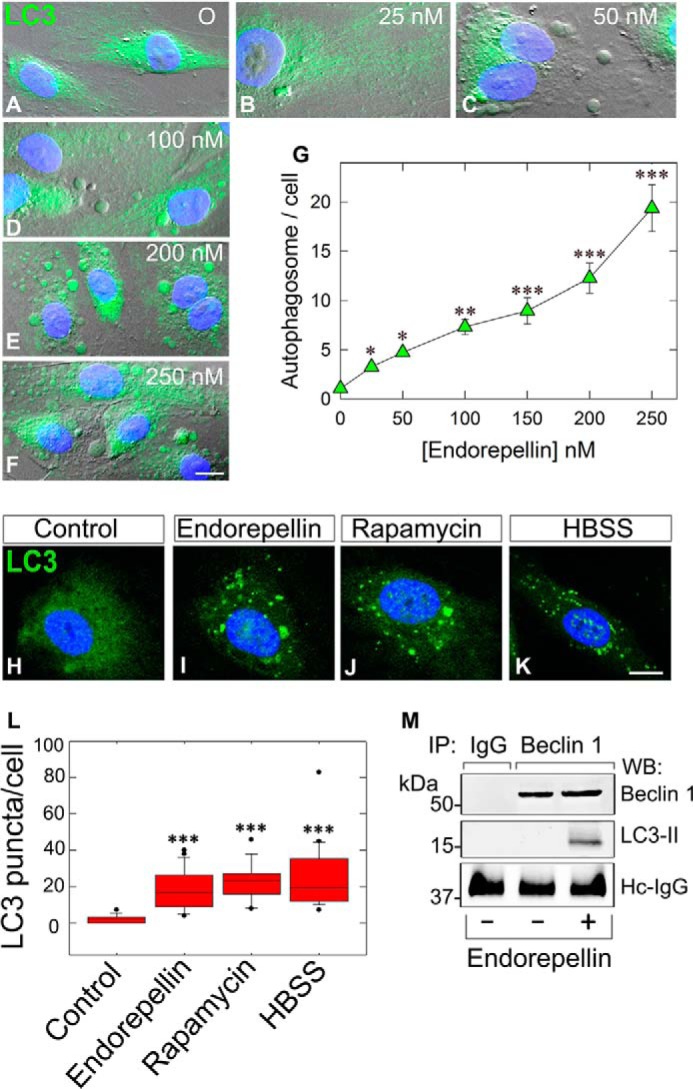
Endorepellin induces endothelial cell autophagy in a dose-dependent manner and promotes LC3-II interaction with Beclin 1. A–F, representative DIC immunofluorescence images of HUVEC exposed to the designated concentrations of endorepellin for 6 h. Notice the progressive formation of autophagic vesicles labeled by anti-LC3 antibody (green) and counterstained with DAPI (blue nuclei). Bar, 10 μm. G, quantification of autophagosomes/HUVEC from three independent experiments. Data represent mean ± S.E. from 52–75 cells/experimental condition; *, p < 0.05; **, p < 0.01; ***, p < 0.001. H–K, representative confocal images of HUVEC treated with endorepellin (200 nm), rapamycin (40 nm), or HBSS for 6 h showing increased expression of LC3 (green) and its relocalization to autophagosomes. L, quantification of the number of LC3-containing vacuoles per cell showing a significant increase in the amount of LC3-positive vacuoles upon treatment. Means ± S.E. (n = 3); ***, p < 0.001. M, representative co-immunoprecipitation of HUVEC stimulated with endorepellin (200 nm) for 6 h. The cells were lysed, immunoprecipitated (IP) with an anti-Beclin 1 antibody, and subjected to Western blot (WB) with anti-Beclin 1 and anti-LC3 as indicated. Nonspecific IgG served as negative control. The experiments were repeated three times with comparable results. Hc-IgG, heavy chain IgG.
Next, we determined whether endorepellin-induced Beclin 1 would physically interact with LC3-II. Using co-immunoprecipitation with anti-Beclin 1 antibodies and immunoblotting with anti-Beclin 1 or LC3, we found that endorepellin-evoked Beclin 1 physically interacted with LC3-II (Fig. 2M), when compared with the negative rabbit IgG control. Thus, these data firmly strengthen the imaging studies.
Peg3 Plays a Key Role in Endorepellin-evoked Autophagy
We have recently discovered that Peg3 acts as a master regulator of autophagy by recruiting Beclin 1 and LC3 upon decorin treatment, as well as canonical autophagy inducers such as rapamycin and nutrient deprivation (73). Thus, we investigated whether Peg3 would also play a crucial role in endorepellin-mediated autophagy in endothelial cells. As in the case of Beclin 1, endorepellin induced Peg3 protein within 6 h and caused a dynamic re-location from a finely granular cytoplasmic distribution onto large vacuoles (Fig. 3A). A comparable increase in Peg3 and redistribution into autophagosomes were evoked by rapamycin and nutrient deprivation (Fig. 3A). Notably, endorepellin-induced Peg3 physically interacted with Beclin 1, as demonstrated by co-immunoprecipitation (Fig. 3B), in contrast to the negative IgG control. Confocal microscopy and line scanning showed co-localization of Peg3 with both Beclin 1 (Fig. 3, C and D) and LC3 (Fig. 3, E and F), respectively, upon endorepellin treatment. We conclude that Peg3 is directly involved in endorepellin-evoked autophagy.
FIGURE 3.

Peg3 expression is affected by endorepellin promoting its interaction and co-localization with the autophagic markers Beclin 1 and LC3 in HUVEC. A, immunofluorescence images showing increasing expression of Peg3 upon endorepellin treatment (200 nm) in HUVEC. HBSS and rapamycin (40 nm) served as positive controls. B, representative co-immunoprecipitation of HUVEC stimulated with endorepellin (ER) (200 nm) for 6 h. The cells were lysed, immunoprecipitated (IP) with an anti-Peg3 antibody, and subjected to Western blot (WB) with anti-Peg3 anti-Beclin 1 as indicated. Nonspecific IgG served as negative control. The experiments were repeated three times with comparable results. Hc-IgG, heavy chain IgG. C–F, confocal images of Peg3 and Beclin 1 or Peg3 and LC3 co-localization in HUVEC upon a 6-h treatment with or without endorepellin (200 nm). The line-scanned profiles are next to each confocal image. The white arrows in the confocal images represent the distance within which the scans were generated. Bar, 10 μm. G, representative Western blot of various proteins, as indicated on the right margin, in HUVEC following time course treatment with soluble endorepellin (200 nm). H, quantification of three independent experiments as in G. Data represent the mean ± S.E.
Endorepellin Enhances Intracellular Levels of Beclin 1 and LC3 and Induces an Autophagic Transcriptional Program via PEG3 and VEGFR2
To corroborate the findings reported above, we investigated by immunoblotting the temporal effects of endorepellin on Peg3, Beclin 1, and LC3 protein levels. Endorepellin caused a time-dependent induction of Peg3 and Beclin 1 (Fig. 3G), both of which peaked at 6 h and remained significantly elevated for up to 8 h (Fig. 3H). Concurrently, there was a marked induction of LC3-II with similar kinetics (Fig. 3, G and H). Thus, endorepellin evokes autophagy concurrent with the induction of Peg3 and of two key autophagic proteins, Beclin 1 and LC3-II, in endothelial cells.
Next, we investigated the ability of endorepellin to induce the expression of autophagy genes in HUVEC. Endorepellin, under nutrient-rich conditions, evoked induction of PEG3 (Fig. 4A), BECN1 (Fig. 4B), and MAPLC3A (Fig. 4C) in a manner analogous to rapamycin (Fig. 4, B and C). Importantly, rapamycin also evoked PEG3 mRNA by ∼4800-fold in HUVEC (data not shown), thus reinforcing PEG3 as a master regulator of endothelial cell autophagy (73).
FIGURE 4.

Endorepellin regulates an autophagy transcriptional program via PEG3 and VEGFR2. A–C, expression of autophagy genes in HUVEC via qPCR under various experimental conditions involving LG1/2 (150 nm, 6 h), LG3 (150 nm, 6 h), and pre-incubation with the α2β1 blocking mAb (10 μg/ml, 30 min) in the presence of endorepellin (200 nm, 6 h) or via RNAi-mediated silencing of PEG3 (siPEG3, 80 pm) alone or in combination with endorepellin (200 nm, 6 h) or in the absence of VEGFR2 (siVEGFR2, 80 pm) following stimulation with endorepellin (200 nm, 6 h) relative to siScr (20 pm) controls for B, PEG3, C, BECN1, or D, MAPLC3A. Rapamycin (40 nm, 2 h) served as a positive control. D, verification of PEG3 (siPEG3, 80 pm) and VEGFR2 (siVEGFR2, 80 pm) RNAi-mediated knockdown relative to siScramble (siScr, 20 pm) controls in the presence or absence of endorepellin (200 nm, 6 h) via quantitative real time-PCR analysis in HUVEC. N.S., not significant. E, qPCR analysis of PEG3, BECN1, and MAPLC3A following endorepellin alone (200 nm, 6 h) or via pre-incubation (30 min) with the VEGFR2 small tyrosine kinase inhibitor SU5416 (30 μm) followed by treatment with endorepellin (200 nm, 6 h) in HUVEC. F, representative immunoblot of Peg3, Beclin 1, and LC3-II following incubation with either endorepellin (200 nm, 6 h), LG1/2 (150 nm, 6 h), or LG3 (150 nm, 6 h). The blocking mAb anti-α2β1 (10 μg/ml) was pre-incubated for 30 min prior to endorepellin stimulation (200 nm, 6 h). Rapamycin (40 nm, 2 h) served as a positive control. All gene expression changes were first normalized to the endogenous housekeeping gene, ACTB, calculated via the ΔΔCt method and reported as fold changes ±S.E. For immunoblotting, GAPDH served as a loading control. All data are representative of three independent trials run in quadruplicate replicates. **, p < 0.01; ***, p < 0.001.
Endorepellin exhibits a unique dual receptor antagonism as the basis for endothelial cell specificity and signal transduction via ligation of VEGFR2 and α2β1 integrin (67). This unique antagonism is a direct consequence of the structure of endorepellin. Endorepellin has a symmetrically modular architecture with three LG domains and intervening EGF repeats. This permits a single LG domain at the N and C termini (LG1 and LG3, respectively), thus placing LG2 in the middle (60). Based on recent molecular dissection of endorepellin (69), we determined which fragment of endorepellin is responsible for modulating autophagy-related (Atg) gene expression. Treatment of HUVEC with LG1/2 induced PEG3 (Fig. 4A), BECN1 (Fig. 4B), and MAPLC3A (Fig. 4C) similar to endorepellin alone. Importantly, LG3 significantly suppressed PEG3 (Fig. 4A), BECN1 (Fig. 4B), and MAPLC3A (Fig. 4C) relative to control. These data indicate that VEGFR2, and not the α2β1 integrin, is the primary signaling receptor for endorepellin-evoked endothelial cell autophagy. This concept was further validated by experiments using anti-α2β1 blocking mAb. Incubation with anti-α2β1 did not abrogate endorepellin-mediated induction of PEG3 (Fig. 4A), BECN1 (Fig. 4B), or MAPLC3A (Fig. 4C). However, there was attenuation for all markers, as maximal expression elicited by endorepellin alone was not attained.
Next, we determined the requirement of PEG3 in the expression of Atg targets stimulated by endorepellin. We verified RNAi-mediated gene silencing of PEG3 in HUVEC in the presence or absence of endorepellin (Fig. 4D). Importantly, we recapitulated the necessity of PEG3 in maintaining basal BECN1 levels but not MAPLC3A (Fig. 4, B and C) (73). Crucially, depletion of PEG3 utterly abrogated the proclivity of endorepellin to induce BECN1 (Fig. 4B) and MAPLC3A (Fig. 4C). These data establish a necessity of PEG3 for endorepellin-evoked autophagic gene induction. Moreover, these data further underscore the central role of PEG3 in initiating and regulating a pro-autophagic gene program in response to soluble matrix molecules.
Next, we depleted VEGFR2 (Fig. 4D) from HUVEC and interrogated the effect on autophagy gene induction. Under steady-state conditions, endorepellin did not affect VEGFR2 mRNA (N.S., Fig. 4D). Consistent with the ability of LG1/2 to be sufficient for PEG3, BECN1, and MAPLC3A induction, loss of VEGFR2 completely blocked PEG3 expression (p < 0.001, Fig. 4A), BECN1 (p < 0.001, Fig. 4B), or MAPLC3A (p < 0.001, Fig. 4C) in HUVEC. Moreover, similar results were obtained following pre-incubation with the VEGFR2-specific kinase small molecule inhibitor SU5416 (82). Abrogating signaling prevented endorepellin induction of PEG3, BECN1, and MAPLC3A in HUVEC (Fig. 4E). Thus, VEGFR2 and associated downstream signaling are required for endorepellin-evoked Atg regulation.
Next, we corroborated the regulation of Peg3, Beclin 1, and LC3 at the protein level following stimulation with the endorepellin fragments as well as with the α2β1-blocking antibody. Notably, LG1/2 induced Peg3, Beclin 1, and LC3 above control levels (Fig. 4F). However, similar to the transcriptional regulation evoked by endorepellin and rapamycin, LG1/2 did not induce these autophagy markers to the same extent (Fig. 4F). Moreover, the distal fragment of endorepellin, LG3, had no effect on Beclin 1 or LC3-II (Fig. 4F). Importantly, LG3 alone mediated Peg3 suppression (Fig. 4F) analogous to the transcriptional repression reported above (cf. Fig. 4A). Finally, pre-incubation with the anti-α2β1 blocking mAb attenuated the ability of endorepellin to induce Peg3, Beclin 1, and LC3 (Fig. 4F) in accordance with the transcriptional analysis reported above. Therefore, these results indicate a complex signaling mechanism, mediated primarily by the N-terminal fragment (encompassing LG1/2) physically engaging VEGFR2. Moreover, the role of the α2β1 integrin may not be necessary for the inherent bioactivity of the LG1/2 fragment of endorepellin, but it may be required for transducing signals from intact endorepellin for regulating Atg gene expression. Binding evokes the rapid expression of key autophagy regulators in endothelial cells in a strict Peg3-dependent manner
Endorepellin Induces Redistribution of Vps34 into Beclin 1-positive Autophagosomes in a VEGFR2-dependent Manner
The finding that Beclin 1 and LC3 co-localized upon endorepellin treatment and the involvement of another upstream regulator of autophagy, Peg3, led us to study another key component of newly formed autophagosomes, Vps34, a class III PI3K involved in the initial formation of the phagophore (83). We found a dynamic redistribution of Vps34 from the plasma membrane onto large intracytoplasmic vacuoles (Fig. 5A), which were Beclin 1-positive (Fig. 5B). We note that the protein levels of Vps34 were not changed upon endorepellin treatment (data not shown).
FIGURE 5.

Endorepellin induces redistribution of Vps34 into Beclin 1-positive autophagosomes in a VEGFR2-dependent but α2β1 integrin-independent manner. A, immunofluorescence images of HUVEC treated with endorepellin (200 nm) for 6 h showing a redistribution of Vps34 from the membrane to the cytoplasm of the cells. Nuclei were stained with DAPI (blue). B, immunofluorescence images of Beclin 1 and Vps34 co-localization in HUVEC upon a 6-h treatment with endorepellin (200 nm). Notice the complete block of VEGFR2 small molecule inhibitor SU5416 (30 μm). C, immunofluorescence images of Beclin 1 and Peg3 cellular co-localization in HUVECs upon a 6-h treatment with endorepellin (200 nm), LG1/2 (150 nm), or LG3 (150 nm). Notice that only LG1/2 was capable of inducing autophagy. D, immunofluorescence images of LC3 (green) in HUVEC upon a 6-h treatment with endorepellin (200 nm), and either the α2β1 integrin blocking mAb (10 μg/ml) or in combination with endorepellin as indicated. E, immunofluorescence images of LC3 (green) in HUVEC upon a 6-h treatment with LG1/2 (150 nm) and either the α2β1 integrin blocking mAb (10 μg/ml) or in combination with LG1/2 as indicated. Bars, 12 μm.
We have recently shown that endorepellin attenuates two major signaling branches of VEGFR2 as follows: the PI3K/PDK1/Akt/mTOR and the PKC/JNK/AP1 pathways (71). We hypothesized that endorepellin could release the mTOR-mediated repression of autophagy through mTOR suppression evoked by antagonizing VEGFR2. Thus, we pre-incubated HUVEC with SU5416 (30 μm) for 30 min before co-incubation with endorepellin (200 nm) for 6 h. Importantly, the translocation of Vps34/Beclin 1 into autophagosomes was completely blocked by SU5416 (Fig. 5B), indicating that the tyrosine kinase activity of VEGFR2 is required for autophagic induction following endorepellin treatment. These findings further support the signaling requirement for Atg gene expression established above (cf. Fig. 4E).
Next, we investigated the bioactive endorepellin fragments (discussed above) for competent autophagic induction in endothelial cells. Unlike LG1/2, LG3 was unable to induce autophagy in endothelial cells as shown by the failure to form Peg3/Beclin 1-positive autophagosomes (Fig. 5C). Thus, endorepellin-evoked autophagy is an intrinsic activity of the two LG1/2 domains, and these effects are neither dependent on nor mediated by the α2β1 integrin. Collectively, our findings provide a novel mechanism evoked by endorepellin and suggest that Peg3 could play a crucial role in the early stages of phagophore formation by recruiting the Beclin 1-Vps34 complex to the isolation membrane. Once formed, the Peg3-Beclin 1-Vps34 complex permits recruitment of lipidated LC3-II for mature autophagosome formation.
Endorepellin Induces Autophagy through a VEGFR2-dependent but α2β1 Integrin-independent Manner
To further investigate the differential activity of the α2β1 integrin and VEGFR2, we performed image analysis using a mAb blocking the α2β1 integrin. Pre-incubation for 30 min with the saturable amounts of α2β1 integrin blocking mAb (10 μg/ml), followed by a subsequent 4-h co-incubation with endorepellin (200 nm), did not affect the pro-autophagic activity of endorepellin. Specifically, when treated with endorepellin, both in the absence and presence of the α2β1 integrin blocking mAb, LC3 localized to large autophagosomes (Fig. 5D). Similar effects were also observed when LG1/LG2 was used (Fig. 5E).
Next, we corroborated the above evidence by observing an increase in protein levels of Beclin 1 after a 6-h treatment with endorepellin. This increase was blocked by SU5416 (Fig. 6A). Additional evidence of the importance of VEGFR2 in endorepellin-evoked autophagy was through siRNA-mediated depletion of the receptor. Treatment with siRNA for VEGFR2 blocked Beclin 1 and Peg3 induction by endorepellin (Fig. 6A).
FIGURE 6.
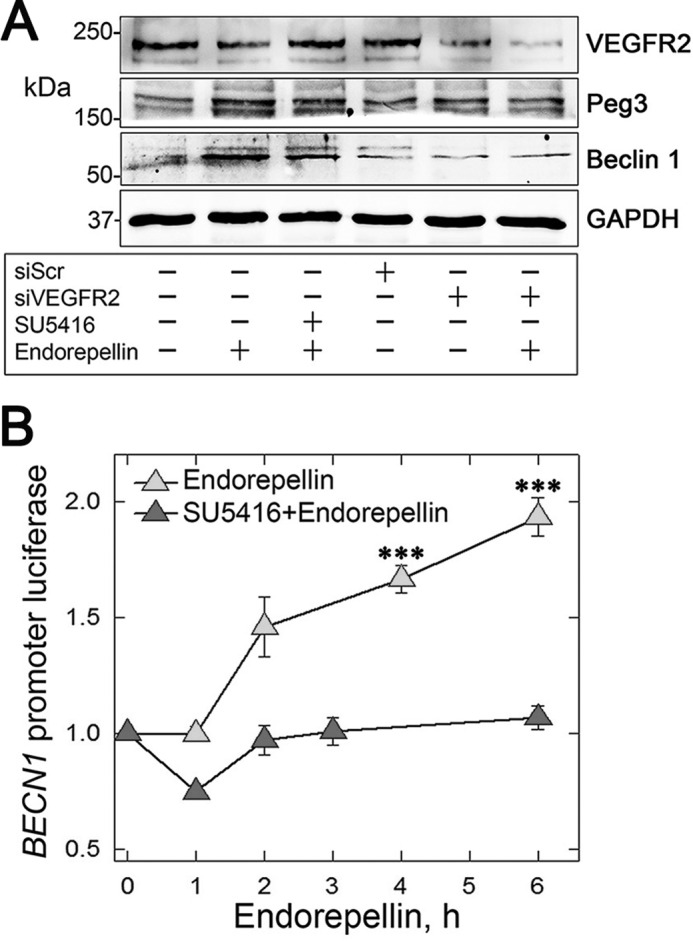
Endorepellin induces autophagy through a VEGFR2-dependent pathway and evokes transcription of BECN1 gene. A, representative immunoblotting following a 6-h treatment with endorepellin (200 nm) of HUVEC treated with scrambled siRNA (siScr), siRNA targeting the VEGFR2 (siVEGFR2), or SU5416 as indicated. The blotting is representative of three independent experiments with similar results. B, representative luciferase reporter assays of PAE-VEGFR2 BECN1-Luc cells treated with soluble endorepellin (200 nm) for the designated times, in the presence or absence of SU5416 (30 μm). Data are mean ± S.E., normalized to total cell protein. The values at 4 and 6 h are statistically significant (***, p < 0.001) as compared with time 0 and the SU5416-treated samples.
Armed with this compelling evidence at the protein level, we investigated the transcriptional activity of the BECN1 promoter through luciferase reporter assays of PAE-VEGFR2BECN1-Luc cells. We found a significant time-dependent increase in BECN1 promoter activity, which peaked at 6 h, upon endorepellin treatment (p < 0.001 (Fig. 6B)). Interestingly, co-incubation with SU5416 blocked endorepellin-evoked BECN1 promoter activity (Fig. 6B). This indicates that VEGFR2 is required for the downstream signaling of endorepellin and transcriptional regulation of BECN1.
Endorepellin Causes p62 Induction and Its Relocalization into LC3-positive Autophagosomes
Another key player in the autophagic degradation of many proteins is p62/SQSTM1 (p62 hereafter), a specific substrate that is cleared through the autophagy-lysosomal pathway (84, 85). This degradation is accompanied by interactions with lipidated LC3-II into the phagophore/isolation membrane and remains associated with the completed autophagosome. Having established that endorepellin and its LG1/2 domain induce the interaction and co-localization of Beclin 1-Vps34 complex with LC3, we examined the temporal expression of p62 in HUVEC. We discovered that p62 was induced in a time-dependent manner by endorepellin and LG1/2, reaching maximal levels at 6 h, and then subsequently declining to basal levels at 12 h (Fig. 7, A and B). Endorepellin triggered co-localization of p62 with LC3-positive structures (Fig. 7C), suggesting that p62 is degraded by endorepellin-induced autophagy through direct interaction with LC3. This assumption was confirmed by the discovery that the LG1/2 was able to evoke co-distribution of p62 and LC3 in autophagosomes, in contrast to LG3 domain that failed to induce significant changes in HUVEC (Fig. 7D).
FIGURE 7.

p62 expression is affected by endorepellin promoting its relocalization to LC3-positive autophagosomes in HUVEC. A, representative immunoblotting showing a time course induction of p62 upon endorepellin treatment (200 nm), with GAPDH as control. B, quantification of p62 levels evoked by endorepellin (200 nm) treatment for various intervals as indicated. The values represent the mean ± S.E. of six independent experiments; *, p < 0.05; **, p < 0.01. C, immunofluorescence images of HUVEC treated with endorepellin (200 nm) for 6 h showing a relocalization of p62 (red) into LC3-positive autophagosomes. Nuclei are stained with DAPI (blue). Bar, 10 μm. D, immunofluorescence images of p62 (red) and LC3 (green) co-localization in HUVEC upon a 6-h treatment with vehicle (control), LG1/2 (150 nm), or LG3 (150 nm). Nuclei are stained with DAPI. Bar, 10 μm.
Endorepellin Inhibits Capillary Morphogenesis and Concurrently Stimulates Autophagy
We were interested in trying to link autophagy and angiostasis in the same assay. We achieved this by undertaking a Matrigel-based angiogenesis assay using PAE-VEGFR2 cells stably transfected with GFP-LC3. We found that in the vehicle-treated samples, clear branching morphogenesis and tube-like formation occurred within 6 h (Fig. 8, A and B). In stark contrast, incubation with endorepellin (200 nm) completely blocked capillary morphogenesis (Fig. 8, C and D). Concurrent with inhibition of tube formation, endorepellin evoked an increase in fluorescence intensity, i.e. enhanced GFP-LC3, as visualized by surface plot analysis (Fig. 8, compare D with B). We note that all images were captured with the same exposure, gain, and intensity. Quantification of established angiogenic parameters, including the number of branch points, individual tube length, and tube number, was significantly suppressed by endorepellin (p < 0.001, Fig. 8E). Thus, inhibition of branching morphogenesis correlates with enhanced autophagy evoked by endorepellin.
FIGURE 8.

Endorepellin inhibits capillary tube formation in tandem with autophagy induction. A and C, representative fluorescence images of 6-h vehicle-treated (control) or endorepellin-treated (200 nm) PAE-VEGFR2 cells stably expressing GFP-LC3. Approximately 104 cells were seeded on Matrigel previously gelled in a four-chamber slide. All images were taken with the same exposure and gain. Bar, 50 μm. B and D, three-dimensional surface plots processed on ImageJ representing the fluorescence intensity of the corresponding images. E, quantification of various angiogenic (capillary morphogenesis) parameters as indicated in the y axis from images similar to those shown in A and C, respectively. The data were acquired using the tube formation analysis tool WimTube offered by Wimasis Image Analysis, GmbH. ***, p < 0.001.
DISCUSSION
During the past decade, interest in defining the basic cellular mechanisms of autophagy and its roles in human health and disease has become widespread. Autophagy is a cellular degradation pathway for the clearance of damaged or superfluous proteins and organelles (78). In addition to eliminating protein aggregates and damaged organelles, autophagy promotes cellular senescence and cell surface antigen presentation. Moreover, autophagy protects against genome instability and prevents necrosis, thereby counteracting diseases such as cancer, neurodegeneration, cardiomyopathy, diabetes, liver disease, autoimmune diseases, and infections (83). Autophagy induction in response to stress and starvation also has a crucial role in normal cells. For example, Atg5-deficient mice fail to live through the neonatal survival period, during which tissues show signs of amino acid depletion and metabolic insufficiency. These findings support a pro-survival role for autophagy in both normal tissues and in response to metabolic stress (78).
A working model summarizing our most recent findings concerning the mechanism of action for endorepellin-evoked endothelial cell autophagy and anti-angiogenesis is provided in Fig. 8. Accordingly, we propose that endorepellin engages VEGFR2 via its two proximal LG domains and acts as a partial agonist. This signaling emanated by VEGFR2 evokes the simultaneous formation of Peg3-, Beclin 1-, and LC3-positive complexes and suppression of the anti-autophagic PI3K/Akt/mTOR pathway (Fig. 9). Transcriptional induction of PEG3 is also required for downstream activation of BECN1 and MAPLC3A. Accumulated Peg3, Beclin 1, and lipidated LC3 promote autophagosome nucleation and maturation with concomitant dissolution of the actin cytoskeleton for angiogenic inhibition mediated by interaction of LG3 with the α2β1 integrin (Fig. 9).
FIGURE 9.

Working model elucidating the mechanism in which endorepellin induces autophagy through the VEGFR2 α2β1 integrin-independent manner. Please, see the text for detailed information.
We hypothesized that endorepellin could remove the repression of autophagy induced by mTOR, a downstream component of the PI3K pathway, through the binding to the VEGFR2 in endothelial cells and consequent inhibition of the PI3K/PDK1/AKT/mTOR pathway (71). Using DIC microscopy, we found that in both endothelial cell types, endorepellin induced the formation of characteristic autophagosomes comparable with those evoked by rapamycin or nutrient deprivation. This observation was further corroborated by the finding that endorepellin induced expression of the Atg proteins Beclin 1, LC3, and p62 and promoted their co-localization and physical interaction in HUVEC. Importantly, endorepellin-evoked autophagy was analogous to traditional autophagic stimuli such as rapamycin and nutrient deprivation.
The Atg proteins are required for activation of autophagy, formation of autophagosomes, sequestration of intracellular constituents, and targeting and fusion of autophagosomes to lysosomes where the contents are degraded and recycled (83). Beclin 1 is part of the class III PI3K-Vps34 complex that is also required for autophagy. ATG8 (also known as LC3) is another ubiquitin-like protein that is cleaved and lipidated and becomes a component of the autophagosomal membrane. This membrane translocation event is commonly used to monitor autophagy (72). Notably, endorepellin induced a re-distribution of Vps34 in the cytoplasm of HUVEC and a subsequent co-localization with Beclin 1, suggesting that inhibiting mTOR precedes and evokes the formation of Beclin 1/Vps34 pre-autophagic signaling complexes.
We have recently discovered that Peg3, a decorin-induced gene (86), is a master regulator of autophagy through the recruitment and transcriptional induction of Beclin 1 and LC3 (73). Peg3 was originally identified as a paternally imprinted gene and is a relatively large protein (∼180 kDa) that contains an N-terminal SCAN protein-protein interaction domain and 12 Kruppel-type zinc fingers (87, 88). Notably, Peg3−/− mice show growth retardation and impaired maternal nurturing (89). Peg3 is involved in TNF-NFκB signaling (90) as well as p53-mediated apoptosis (91) and Wnt signaling (92). We found transcriptional induction of PEG3 upon endorepellin treatment with a dynamic co-distribution of Peg3 with both Beclin 1 and LC3, supporting a role for this gene in endorepellin-induced autophagy. In accordance with the finding that Peg3 binds Beclin 1 following decorin stimulation in HUVEC (73), we found that endorepellin also promotes Peg3-Beclin 1 interaction. This finding suggests that a general feature of soluble matrix constituents with anti-angiogenic activities is to promote Peg3/Beclin 1 association consummate with autophagic induction. Moreover, the role of Peg3 in relaying endorepellin-evoked Atg gene expression was confirmed as silencing of Peg3 prevented BECN1 and MAPLC3A expression. These data support a role for Peg3 as a master autophagic regulator responsible for coordinating downstream responses from matrix signaling.
We have discovered that endorepellin specifically targets endothelial cells via a dual receptor antagonism, acting as a molecular bridge, ligating both VEGFR2 and α2β1 integrin (67, 71). Following engagement, endorepellin seemingly attenuates VEGFR2 phosphorylation at Tyr1175, a key residue from which the phospholipase C-γ signaling cascade originates (93). However, endorepellin requires positive VEGFR2-dependent signaling for sustained autophagic induction concurrent with PEG3, BECN1, and MAPLC3A transcriptional induction as SU5416 abrogated this response. Furthermore, a similar outcome was obtained by RNAi-mediated silencing of VEGFR2. Together, these results support the importance of the major pro-angiogenic VEGFR2 tyrosine kinase in endorepellin-induced autophagy.
Recently, we have dissected the bioactivity of endorepellin and its two main fragments (LG1/2 and LG3) on VEGFR2 and α2β1 integrin (69), respectively. Together with the present results, our findings suggest a major role for VEGFR2 in binding endorepellin and inducing autophagy in an α2β1 integrin-independent manner. This assumption was confirmed by the discovery that, unlike LG3, LG1/2 promotes the formation of large autophagosomes and mediates transcriptional regulation of Atg genes comparable with those induced by endorepellin, mTOR inhibition, or nutrient deprivation. In contrast, LG3 signaling via α2β1 integrin results in autophagic gene suppression. Moreover, the necessity of VEGFR2 and dispensability of the α2β1 integrin are underscored by the finding that utilization of an α2β1 integrin-blocking mAb has no effect on autophagic induction. The N-terminal LG1/2 domains are inherently pro-autophagic and require VEGFR2. Concomitant with endorepellin-evoked autophagic induction, branching morphogenesis is also abrogated but with a concurrent increase in GFP-LC3 fluorescence. Thus, deciphering whether autophagy via Peg3 is required for suppressing capillary morphogenesis is an important goal of our future research.
The intrinsic nature of endorepellin abrogating tumor angiogenesis and metabolism, by selectively targeting the tumor vasculature, has been verified in vivo (58). As endorepellin activates endothelial cell autophagy downstream of VEGFR2 signaling, it is plausible that autophagic elimination of critical organelles might inhibit neovascularization, thereby impeding tumorigenesis.
Autophagic induction is emerging as a more common theme among matrix constituents. Endostatin, for example, is a well characterized angiogenesis inhibitor generated by C-terminal proteolysis of the heparan sulfate proteoglycan, collagen XVIII (94). In addition to apoptosis, endostatin also activates autophagy by Beclin 1 and β-catenin levels in HUVEC (95) and induces autophagic cell death in the EAhy926 human endothelial cell line (96). Moreover, Kringle 5, an antiangiogenic factor derived from human plasminogen, induces both autophagy and apoptotic cell death in HUVEC (97). Thus, proteolytically processed fragments of larger parent molecules can evoke autophagy, suggesting the potential involvement of a common signaling pathway for angiostatic proteins (98). Moreover, autophagic regulation via matrix components might be a highly dynamic process. For example, CD47, the receptor for the anti-angiogenic effector thrombospondin 1 (99) and for regulating cardiovascular function and responses to stress (100), suppresses autophagy (101). Specifically, lack of CD47 permits autophagic activation in Jurkat T-cells and HUVEC (101), which ultimately confers a radioprotective state for cells and tissues.
In conclusion, we propose that endorepellin, the C-terminal cleavage product of perlecan, functions not only as an endogenous anti-angiogenic cue but also as a potent pro-autophagic effector for endothelial cells while residing in a nutrient-rich microenvironment. Endorepellin, by specifically binding VEGFR2, induces the formation of the Peg3-Vps34-Beclin 1 autophagic complexes via inhibition of the PI3K/Akt/mTOR pathway. Importantly, endorepellin-evoked autophagy requires the recently discovered master regulator Peg3 for initiation. Therefore, this autophagic response may be a novel target for enhancing the therapeutic efficacy of angiogenesis inhibitors.
Acknowledgments
We thank S. Ramakrishnan for providing the GFP-LC3 plasmid, L. Claesson-Welsh for providing the PAE-VEGFR2 cells, F. Demarchi for providing the BECN1 promoter-luciferase construct, and A. Shellard and A. Torres for valuable help in the initial stages of this work.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 CA3948, RO1 CA47282, and RO1 CA164462 (to R. V. I.).
- VEGFR2
- VEGF receptor 2
- LG
- laminin-like globular domain
- HUVEC
- human umbilical vein endothelial cells
- PAE
- porcine aortic endothelial cells
- DIC
- differential interference contrast microscopy
- mTOR
- mammalian target of rapamycin
- Atg
- autophagy-related
- LC3
- microtubule associated light chain 3
- qPCR
- quantitative PCR
- HBSS
- Hanks' balanced salt solution.
REFERENCES
- 1. Noonan D. M., Fulle A., Valente P., Cai S., Horigan E., Sasaki M., Yamada Y., Hassell J. R. (1991) The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J. Biol. Chem. 266, 22939–22947 [PubMed] [Google Scholar]
- 2. Murdoch A. D., Dodge G. R., Cohen I., Tuan R. S., Iozzo R. V. (1992) Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 267, 8544–8557 [PubMed] [Google Scholar]
- 3. Cohen I. R., Grässel S., Murdoch A. D., Iozzo R. V. (1993) Structural characterization of the complete human perlecan gene and its promoter. Proc. Natl. Acad. Sci. U.S.A. 90, 10404–10408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Iozzo R. V., Pillarisetti J., Sharma B., Murdoch A. D., Danielson K. G., Uitto J., Mauviel A. (1997) Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming factor-β via a nuclear factor 1-binding element. J. Biol. Chem. 272, 5219–5228 [DOI] [PubMed] [Google Scholar]
- 5. Sharma B., Iozzo R. V. (1998) Transcriptional silencing of perlecan gene expression by interferon-γ. J. Biol. Chem. 273, 4642–4646 [DOI] [PubMed] [Google Scholar]
- 6. Carson D. D., Tang J.-P., Julian J. (1993) Heparan sulfate proteoglycan (perlecan) expression by mouse embryos during acquisition of attachment competence. Dev. Biol. 155, 97–106 [DOI] [PubMed] [Google Scholar]
- 7. Iozzo R. V., Cohen I. R., Grässel S., Murdoch A. D. (1994) The biology of perlecan: the multifaceted heparan sulphate proteoglycan of basement membranes and pericellular matrices. Biochem. J. 302, 625–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Farach-Carson M. C., Carson D. D. (2007) Perlecan–a multifunctional extracellular proteoglycan scaffold. Glycobiology 17, 897–905 [DOI] [PubMed] [Google Scholar]
- 9. Farach-Carson M. C., Warren C. R., Harrington D. A., Carson D. D. (2014) Border patrol: insights into the unique role of perlecan/heparan sulfate proteoglycan 2 at cell and tissue borders. Matrix Biol. 34, 64–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Handler M., Yurchenco P. D., Iozzo R. V. (1997) Developmental expression of perlecan during murine embryogenesis. Dev. Dyn. 210, 130–145 [DOI] [PubMed] [Google Scholar]
- 11. Iozzo R. V. (1984) Biosynthesis of heparan sulfate proteoglycan by human colon carcinoma cells and its localization at the cell surface. J. Cell Biol. 99, 403–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iozzo R. V. (1987) Turnover of heparan sulfate proteoglycan in human colon carcinoma cells. A quantitative biochemical and autoradiographic study. J. Biol. Chem. 262, 1888–1900 [PubMed] [Google Scholar]
- 13. Murdoch A. D., Liu B., Schwarting R., Tuan R. S., Iozzo R. V. (1994) Widespread expression of perlecan proteoglycan in basement membranes and extracellular matrices of human tissues as detected by a novel monoclonal antibody against domain III and by in situ hybridization. J. Histochem. Cytochem. 42, 239–249 [DOI] [PubMed] [Google Scholar]
- 14. Couchman J. R., Ljubimov A. V., Sthanam M., Horchar T., Hassell J. R. (1995) Antibody mapping and tissue localization of globular and cysteine-rich regions of perlecan domain III. J. Histochem. Cytochem. 43, 955–963 [DOI] [PubMed] [Google Scholar]
- 15. Whitelock J. M., Melrose J., Iozzo R. V. (2008) Diverse cell signaling events modulated by perlecan. Biochemistry 47, 11174–11183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Whitelock J. M., Graham L. D., Melrose J., Murdoch A. D., Iozzo R. V., Underwood P. A. (1999) Human perlecan immunopurified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 18, 163–178 [DOI] [PubMed] [Google Scholar]
- 17. Fuki I. V., Iozzo R. V., Williams K. J. (2000) Perlecan heparan sulfate proteoglycan. A novel receptor that mediates a distinct pathway for ligand catabolism. J. Biol. Chem. 275, 25742–25750 [DOI] [PubMed] [Google Scholar]
- 18. Nugent M. A., Nugent H. M., Iozzo R. V., Sanchack K., Edelman E. R. (2000) Perlecan is required to inhibit thrombosis after deep vascular injury and contributes to endothelial cell-mediated inhibition of intimal hyperplasia. Proc. Natl. Acad. Sci. U.S.A. 97, 6722–6727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Laplante P., Raymond M.-A., Labelle A., Abe J., Iozzo R. V., Hebért M.-J. (2006) Perlecan proteolysis induces α2β1 integrin and Src-family kinases dependent anti-apoptotic pathway in fibroblasts in the absence of focal adhesion kinase activation. J. Biol. Chem. 281, 30383–30392 [DOI] [PubMed] [Google Scholar]
- 20. Baker A. B., Ettenson D. S., Jonas M., Nugent M. A., Iozzo R. V., Edelman E. R. (2008) Endothelial cells provide feedback control for vascular remodeling through a mechanosensitive autocrine TGF-β signaling pathway. Circ. Res. 103, 289–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wilusz R. E., Defrate L. E., Guilak F. (2012) A biomechanical role for perlecan in the pericellular matrix of articular cartilage. Matrix Biol. 31, 320–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Thadikkaran L., Crettaz D., Siegenthaler M. A., Gallot D., Sapin V., Iozzo R. V., Queloz P. A., Schneider P., Tissot J. D. (2005) The role of proteomics in the assessment of premature rupture of fetal membranes. Clin. Chim. Acta 360, 27–36 [DOI] [PubMed] [Google Scholar]
- 23. Sher I., Zisman-Rozen S., Eliahu L., Whitelock J. M., Maas-Szabowski N., Yamada Y., Breitkreutz D., Fusenig N. E., Arikawa-Hirasawa E., Iozzo R. V., Bergman R., Ron D. (2006) Targeting perlecan in human keratinocytes reveals novel roles for perlecan in epidermal formation. J. Biol. Chem. 281, 5178–5187 [DOI] [PubMed] [Google Scholar]
- 24. Ishijima M., Suzuki N., Hozumi K., Matsunobu T., Kosaki K., Kaneko H., Hassell J. R., Arikawa-Hirasawa E., Yamada Y. (2012) Perlecan modulates VEGF signaling and is essential for vascularization in endochondral bone formation. Matrix Biol. 31, 234–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ida-Yonemochi H., Satokata I., Ohshima H., Sato T., Yokoyama M., Yamada Y., Saku T. (2011) Morphogenetic roles of perlecan in the tooth enamel organ: An analysis of overexpression using transgenic mice. Matrix Biol. 30, 379–388 [DOI] [PubMed] [Google Scholar]
- 26. Kaneko H., Ishijima M., Futami I., Tomikawa-Ichikawa N., Kosaki K., Sadatsuki R., Yamada Y., Kurosawa H., Kaneko K., Arikawa-Hirasawa E. (2013) Synovial perlecan is required for osteophyte formation in knee osteoarthritis. Matrix Biol. 32, 178–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Inomata T., Ebihara N., Funaki T., Matsuda A., Wantanabe Y., Ning L., Xu Z., Murakami A., Arikawa-Hirasawa E. (2012) Perlecan-deficient mutation impairs corneal epithelial structure. Invest. Ophthalmol. Vis. Sci. 53, 1277–1284 [DOI] [PubMed] [Google Scholar]
- 28. Mongiat M., Taylor K., Otto J., Aho S., Uitto J., Whitelock J. M., Iozzo R. V. (2000) The protein core of the proteoglycan perlecan binds specifically to fibroblast growth factor-7. J. Biol. Chem. 275, 7095–7100 [DOI] [PubMed] [Google Scholar]
- 29. Mongiat M., Otto J., Oldershaw R., Ferrer F., Sato J. D., Iozzo R. V. (2001) Fibroblast growth factor-binding protein is a novel partner for perlecan protein core. J. Biol. Chem. 276, 10263–10271 [DOI] [PubMed] [Google Scholar]
- 30. Mongiat M., Fu J., Oldershaw R., Greenhalgh R., Gown A. M., Iozzo R. V. (2003) Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), a glycoprotein involved in bone formation and angiogenesis. J. Biol. Chem. 278, 17491–17499 [DOI] [PubMed] [Google Scholar]
- 31. Gonzalez E. M., Mongiat M., Slater S. J., Baffa R., Iozzo R. V. (2003) A novel interaction between perlecan protein core and progranulin: potential effects on tumor growth. J. Biol. Chem. 278, 38113–38116 [DOI] [PubMed] [Google Scholar]
- 32. Whitelock J. M., Iozzo R. V. (2005) Heparan sulfate: a complex polymer charged with biological activity. Chem. Rev. 105, 2745–2764 [DOI] [PubMed] [Google Scholar]
- 33. Cohen I. R., Murdoch A. D., Naso M. F., Marchetti D., Berd D., Iozzo R. V. (1994) Abnormal expression of perlecan proteoglycan in metastatic melanomas. Cancer Res. 54, 5771–5774 [PubMed] [Google Scholar]
- 34. Mathiak M., Yenisey C., Grant D. S., Sharma B., Iozzo R. V. (1997) A role for perlecan in the suppression of growth and invasion in fibrosarcoma cells. Cancer Res. 57, 2130–2136 [PubMed] [Google Scholar]
- 35. Datta S., Pierce M., Datta M. W. (2006) Perlecan signaling: helping hedgehog stimulate prostate cancer growth. Int. J. Biochem. Cell Biol. 38, 1855–1861 [DOI] [PubMed] [Google Scholar]
- 36. Iozzo R. V., Cohen I. (1993) Altered proteoglycan gene expression and the tumor stroma. Experientia 49, 447–455 [DOI] [PubMed] [Google Scholar]
- 37. Iozzo R. V. (1988) Proteoglycans and neoplasia. Cancer Metastasis Rev. 7, 39–50 [DOI] [PubMed] [Google Scholar]
- 38. Iozzo R. V. (1998) Matrix proteoglycans: from molecular design to cellular function. Annu. Rev. Biochem. 67, 609–652 [DOI] [PubMed] [Google Scholar]
- 39. Sanderson R. D. (2001) Heparan sulfate proteoglycans in invasion and metastasis. Semin. Cell Dev. Biol. 12, 89–98 [DOI] [PubMed] [Google Scholar]
- 40. Sanderson R. D., Yang Y., Suva L. J., Kelly T. (2004) Heparan sulfate proteoglycans and heparanase-partners in osteolytic tumor growth and metastasis. Matrix Biol. 23, 341–352 [DOI] [PubMed] [Google Scholar]
- 41. Sanderson R. D., Iozzo R. V. (2012) Targeting heparanase for cancer therapy at the tumor-matrix interface. Matrix Biol. 31, 283–284 [DOI] [PubMed] [Google Scholar]
- 42. Jung M., Lord M. S., Cheng B., Lyons J. G., Alkhouri H., Hughes J. M., McCarthy S. J., Iozzo R. V., Whitelock J. M. (2013) Mast cells produce novel shorter forms of perlecan that contain functional endorepellin: A role in angiogenesis and wound healing. J. Biol. Chem. 288, 3289–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Iozzo R. V. (2005) Basement membrane proteoglycans: from cellar to ceiling. Nat. Rev. Mol. Cell Biol. 6, 646–656 [DOI] [PubMed] [Google Scholar]
- 44. Aviezer D., Hecht D., Safran M., Eisinger M., David G., Yayon A. (1994) Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell 79, 1005–1013 [DOI] [PubMed] [Google Scholar]
- 45. Sharma B., Handler M., Eichstetter I., Whitelock J. M., Nugent M. A., Iozzo R. V. (1998) Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Invest. 102, 1599–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aviezer D., Iozzo R. V., Noonan D. M., Yayon A. (1997) Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA. Mol. Cell. Biol. 17, 1938–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zoeller J. J., Whitelock J. M., Iozzo R. V. (2009) Perlecan regulates developmental angiogenesis by modulating the VEGF-VEGFR2 axis. Matrix Biol. 28, 284–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Iozzo R. V., San Antonio J. D. (2001) Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J. Clin. Invest. 108, 349–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chuang C. Y., Lord M. S., Melrose J., Rees M. D., Knox S. M., Freeman C., Iozzo R. V., Whitelock J. (2010) Heparan sulfate-dependent signaling of fibroblast growth factor 18 by chondrocyte-derived perlecan. Biochemistry 49, 5524–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Howell A., Landberg G., Bergh J. (2009) Breast tumor stroma is a prognostic indicator and a target for therapy. Breast Cancer Res. 11, S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Iozzo R. V., Sanderson R. D. (2011) Proteoglycans in cancer biology, tumour microenvironment and angiogenesis. J. Cell. Mol. Med. 15, 1013–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Costell M., Carmona R., Gustafsson E., González-Iriarte M., Fässler R., Muñoz-Chápuli R. (2002) Hyperplastic conotruncal endocardial cushions and transposition of great arteries in perlecan-null mice. Circ. Res. 91, 158–164 [DOI] [PubMed] [Google Scholar]
- 53. Zoeller J. J., McQuillan A., Whitelock J., Ho S.-Y., Iozzo R. V. (2008) A central function for perlecan in skeletal muscle and cardiovascular development. J. Cell Biol. 181, 381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. González-Iriarte M., Carmona R., Pérez-Pomares J. M., Macías D., Costell M., Muñoz-Chápuli R. (2003) Development of the coronary arteries in a murine model of transposition of great arteries. J. Mol. Cell. Cardiol. 35, 795–802 [DOI] [PubMed] [Google Scholar]
- 55. Gustafsson E., Almonte-Becerril M., Bloch W., Costell M. (2013) Perlecan maintains microvessel integrity in vivo and modulates their formation in vitro. PLoS One 8, e53715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mongiat M., Sweeney S. M., San Antonio J. D., Fu J., Iozzo R. V. (2003) Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 278, 4238–4249 [DOI] [PubMed] [Google Scholar]
- 57. Bix G., Fu J., Gonzalez E. M., Macro L., Barker A., Campbell S., Zutter M. M., Santoro S. A., Kim J. K., Höök M., Reed C. C., Iozzo R. V. (2004) Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through the α2β1 integrin. J. Cell Biol. 166, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bix G., Castello R., Burrows M., Zoeller J. J., Weech M., Iozzo R. A., Cardi C., Thakur M. L., Barker C. A., Camphausen K., Iozzo R. V. (2006) Endorepellin in vivo: targeting the tumor vasculature and retarding cancer growth and metabolism. J. Natl. Cancer Inst. 98, 1634–1646 [DOI] [PubMed] [Google Scholar]
- 59. Woodall B. P., Nyström A., Iozzo R. A., Eble J. A., Niland S., Krieg T., Eckes B., Pozzi A., Iozzo R. V. (2008) Integrin α2β1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 283, 2335–2343 [DOI] [PubMed] [Google Scholar]
- 60. Willis C. D., Schaefer L., Iozzo R. V. (2012) in Extracellular Matrix: Pathobiology and Signaling (Karamanos N. K., ed) pp. 171–184, Walter de Gruyter GmbH & Co. KG, Berlin [Google Scholar]
- 61. San Antonio J. D., Zoeller J. J., Habursky K., Turner K., Pimtong W., Burrows M., Choi S., Basra S., Bennett J. S., DeGrado W. F., Iozzo R. V. (2009) A key role for the integrin α2β1 in experimental and developmental angiogenesis. Am. J. Pathol. 175, 1338–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Iozzo R. V., Zoeller J. J., Nyström A. (2009) Basement membrane proteoglycans: modulators par excellence of cancer growth and angiogenesis. Mol. Cells 27, 503–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Senger D. R., Claffey K. P., Benes J. E., Perruzzi C. A., Sergiou A. P., Detmar M. (1997) Angiogenesis promoted by vascular endothelial growth factor: regulation through α1β1 and α2β1 integrins. Proc. Natl. Acad. Sci. U.S.A. 94, 13612–13617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Senger D. R., Perruzzi C. A., Streit M., Koteliansky V. E., de Fougerolles A. R., Detmar M. (2002) The α1β1 and α2β1 integrins provide critical support for vascular endothelial growth factor signaling, endothelial cell migration, and tumor angiogenesis. Am. J. Pathol. 160, 195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sweeney S. M., DiLullo G., Slater S. J., Martinez J., Iozzo R. V., Lauer-Fields J. L., Fields G. B., San Antonio J. D. (2003) Angiogenesis in collagen I requires α2β1 ligation of a GFP*GER sequence and possible p38 MAPK activation and focal adhesion disassembly. J. Biol. Chem. 278, 30516–30524 [DOI] [PubMed] [Google Scholar]
- 66. Nyström A., Shaik Z. P., Gullberg D., Krieg T., Eckes B., Zent R., Pozzi A., Iozzo R. V. (2009) Role of tyrosine phosphatase SHP-1 in the mechanism of endorepellin angiostatic activity. Blood 114, 4897–4906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Goyal A., Pal N., Concannon M., Paul M., Doran M., Poluzzi C., Sekiguchi K., Whitelock J. M., Neill T., Iozzo R. V. (2011) Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2). J. Biol. Chem. 286, 25947–25962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gonzalez E. M., Reed C. C., Bix G., Fu J., Zhang Y., Gopalakrishnan B., Greenspan D. S., Iozzo R. V. (2005) BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 280, 7080–7087 [DOI] [PubMed] [Google Scholar]
- 69. Willis C. D., Poluzzi C., Mongiat M., Iozzo R. V. (2013) Endorepellin laminin-like globular repeat 1/2 domains bind Ig3–5 of vascular endothelial growth factor (VEGF) receptor 2 and block pro-angiogenic signaling by VEGFA in endothelial cells. FEBS J. 280, 2271–2284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bix G., Iozzo R. V. (2005) Matrix revolutions: “tails” of basement-membrane components with angiostatic functions. Trends Cell Biol. 15, 52–60 [DOI] [PubMed] [Google Scholar]
- 71. Goyal A., Poluzzi C., Willis C. D., Smythies J., Shellard A., Neill T., Iozzo R. V. (2012) Endorepellin affects angiogenesis by antagonizing diverse VEGFR2-evoked signaling pathways: transcriptional repression of HIF-1α and VEGFA and concurrent inhibition of NFAT1 activation. J. Biol. Chem. 287, 43543–43556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Choi A. M., Ryter S. W., Levine B. (2013) Autophagy in human health and disease. New Engl. J. Med. 368, 651–662 [DOI] [PubMed] [Google Scholar]
- 73. Buraschi S., Neill T., Goyal A., Poluzzi C., Smythies J., Owens R. T., Schaefer L., Torres A., Iozzo R. V. (2013) Decorin causes autophagy in endothelial cells via Peg3. Proc. Natl. Acad. Sci. U.S.A. 110, E2582–E2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Copetti T., Bertoli C., Dalla E., Demarchi F., Schneider C. (2009) p65/RelA modulates BECN1 transcription and autophagy. Mol. Cell. Biol. 29, 2594–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Iozzo R. V., Chakrani F., Perrotti D., McQuillan D. J., Skorski T., Calabretta B., Eichstetter I. (1999) Cooperative action of germline mutations in decorin and p53 accelerates lymphoma tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 96, 3092–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Rudnicka L., Varga J., Christiano A. M., Iozzo R. V., Jimenez S. A., Uitto J. (1994) Elevated expression of type VII collagen in the skin of patients with systemic sclerosis. J. Clin. Invest. 93, 1709–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ryynänen M., Ryynänen J., Sollberg S., Iozzo R. V., Knowlton R. G., Uitto J. (1992) Genetic linkage of Type VII collagen (COL7A1) to dominant dystrophic epidermolysis bullosa in families with abnormal anchoring fibrils. J. Clin. Invest. 89, 974–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Rubinsztein D. C., Codogno P., Levine B. (2012) Autophagy modulation as a potential therapeutic target for diverse diseases. Nat. Rev. Drug Discov. 11, 709–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Thurston T. L., Wandel M. P., von Muhlinen N., Foeglein A., Randow F. (2012) Galectin 8 targets damaged vesicles for authophagy to defend cells against bacterial invasion. Nature 482, 414–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Sai J., Raman D., Liu Y., Wikswo J., Richmond A. (2008) Parallel phosphatidylinositol 3-kinase (P13K)-dependent and Src-dependent pathways lead to CXCL8-mediated Rac2 activation and chemotaxis. J. Biol. Chem. 283, 26538–26547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Molinaro P., Viggiano D., Nisticò R., Sirabella R., Secondo A., Boscia F., Pannaccione A., Scorziello A., Mehdawy B., Sokolow S., Herchuelz A., Di Renzo G. F., Annunziato L. (2011) NA+-Ca2+ exchanger (NCX3) knock-out mice display an impairment in hippocampal long-term potentiation and spatial learning and memory. J. Neurosci. 31, 7312–7321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fong T. A., Shawver L. K., Sun L., Tang C., App H., Powell T. J., Kim Y. H., Schreck R., Wang X., Risau W., Ullrich A., Hirth K. P., McMahon G. (1999) SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (Flk-1/KDR) that inhibits tyrosine kinase catalysis, tumor vascularization, and growth of multiple tumor types. Cancer Res. 59, 99–106 [PubMed] [Google Scholar]
- 83. Mizushima N., Komatsu M. (2011) Autophagy: renovation of cells and tissues. Cell 147, 728–741 [DOI] [PubMed] [Google Scholar]
- 84. Komatsu M., Ichimura Y. (2010) Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 584, 1374–1378 [DOI] [PubMed] [Google Scholar]
- 85. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Buraschi S., Neill T., Owens R. T., Iniguez L. A., Purkins G., Vadigepalli R., Evans B., Schaefer L., Peiper S. C., Wang Z. X., Iozzo R. V. (2012) Decorin protein core affects the global gene expression profile of the tumor microenvironment in a triple-negative orthotopic breast carcinoma xenograft model. PLoS One 7, e45559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kuroiwa Y., Kaneko-Ishino T., Kagitani F., Kohda T., Li L.-L., Tada M., Suzuki R., Yokoyama M., Shiroishi T., Wakana S., Barton S. C., Ishino F., Surani M. A. (1996) Peg3 imprinted gene on proximal chromosome 7 encodes for a zinc finger protein. Nat. Genet. 12, 186–190 [DOI] [PubMed] [Google Scholar]
- 88. Relaix F., Weng X., Marazzi G., Yang E., Copeland N., Jenkins N., Spence S. E., Sassoon D. (1996) Pw1, a novel zinc finger gene implicated in the myogenic and neuronal lineages. Dev. Biol. 177, 383–396 [DOI] [PubMed] [Google Scholar]
- 89. Li L.-L., Keverne E. B., Aparicio S. A., Ishino F., Barton S. C., Surani M. A. (1999) Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284, 330–333 [DOI] [PubMed] [Google Scholar]
- 90. Relaix F., Wei X. J., Wu X., Sassoon D. A. (1998) Peg3/Pw1 is an imprinted gene involved in the TNF-NFκB signal transduction pathway. Nat. Genet. 18, 287–291 [DOI] [PubMed] [Google Scholar]
- 91. Relaix F., Wei Xj, Li W., Pan J., Lin Y., Bowtell D. D., Sassoon D. A., Wu X. (2000) Pw1/Peg3 is a potential cell death mediator and cooperates with Siah1a in p53-mediated apoptosis. Proc. Natl. Acad. Sci. U.S.A. 97, 2105–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Jiang X., Yu Y., Yang H. W., Agar N. Y., Frado L., Johnson M. D. (2010) The imprinted gene PEG3 inhibits Wnt signaling and regulates glioma growth. J. Biol. Chem. 285, 8472–8480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Olsson A.-K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) VEGF receptor signalling-in control of vascular function. Nat. Rev. Mol. Cell Biol. 7, 359–371 [DOI] [PubMed] [Google Scholar]
- 94. Seppinen L., Pihlajaniemi T. (2011) The multiple functions of collagen XVIII in development and disease. Matrix Biol. 30, 83–92 [DOI] [PubMed] [Google Scholar]
- 95. Nguyen T. M., Subramanian I. V., Xiao X., Ghosh G., Nguyen P., Kelekar A., Ramakrishnan S. (2009) Endostatin induces autophagy in endothelial cells by modulating Beclin 1 and β-catenin levels. J. Cell. Mol. Med. 13, 3687–3698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Chau Y.-P., Lin J.-Y., Chen J. H., Tai M.-H. (2003) Endostatin induces autophagic cell death in EAhy926 human endothelial cells. Histol. Histopathol. 18, 715–726 [DOI] [PubMed] [Google Scholar]
- 97. Nguygen T. M., Subramanian I. V., Kelekar A., Ramakrishnan S. (2007) Kringle 5 of human plasminogen, an angiogenesis inhibitor, induces both autophagy and apoptotic death in endothelial cells. Blood 109, 4793–4802 [DOI] [PubMed] [Google Scholar]
- 98. Ramakrishnan S., Nguygen T. M., Subramanian I. V., Kelekar A. (2007) Autophagy and angiogenesis inhibition. Autophagy 3, 512–515 [DOI] [PubMed] [Google Scholar]
- 99. Murphy-Ullrich J. E., Iozzo R. V. (2012) Thrombospondins in physiology and disease: new tricks for old dogs. Matrix Biol. 31, 152–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Roberts D. D., Miller T. W., Rogers N. M., Yao M., Isenberg J. S. (2012) The matricellular protein thrombospondin-1 globally regulates cardiovascular function and responses to stress via CD47. Matrix Biol. 31, 162–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Soto-Pantoja D. R., Miller T. W., Pendrak M. L., DeGraff W. G., Sullivan C., Ridnour L. A., Abu-Asab M., Wink D. A., Tsokos M., Roberts D. D. (2012) CD47 deficiency confers cell and tissue radioprotection by activation of autophagy. Autophagy 8, 1628–1642 [DOI] [PMC free article] [PubMed] [Google Scholar]