

Background: Leishmania donovani adopts several defense mechanisms to become resistant to antileishmanial agents.
Results: Laboratory-grown flavone (baicalein)-resistant parasites exhibit efflux of drug by LdABCC2 transporter. Inside the host macrophage, this parasite up-regulates host MRP2 transporter by an Nrf2-dependent pathway.
Conclusion: Resistant Leishmania parasite exploits ABC transporter in the parasite and inside the host.
Significance: The mechanism of drug resistance in clinical isolates can be probed.
Keywords: Drug Resistance, Flavonoids, Infectious Diseases, Leishmania, Multidrug Transporters
Abstract
In parasites, ATP-binding cassette (ABC) transporters represent an important family of proteins related to drug resistance and other biological activities. Resistance of leishmanial parasites to therapeutic drugs continues to escalate in developing countries, and in many instances, it is due to overexpressed ABC efflux pumps. Progressively adapted baicalein (BLN)-resistant parasites (pB25R) show overexpression of a novel ABC transporter, which was classified as ABCC2 or Leishmania donovani multidrug resistance protein 2 (LdMRP2). The protein is primarily localized in the flagellar pocket region and in internal vesicles. Overexpressed LdABCC2 confers substantial BLN resistance to the parasites by rapid drug efflux. The BLN-resistant promastigotes when transformed into amastigotes in macrophage cells cannot be cured by treatment of macrophages with BLN. Amastigote resistance is concomitant with the overexpression of macrophage MRP2 transporter. Reporter analysis and site-directed mutagenesis assays demonstrated that antioxidant response element 1 is activated upon infection. The expression of this phase II detoxifying gene is regulated by NFE2-related factor 2 (Nrf2)-mediated antioxidant response element activation. In view of the fact that the signaling pathway of phosphoinositol 3-kinase controls microfilament rearrangement and translocation of actin-associated proteins, the current study correlates with the intricate pathway of phosphoinositol 3-kinase-mediated nuclear translocation of Nrf2, which activates MRP2 expression in macrophages upon infection by the parasites. In contrast, phalloidin, an agent that prevents depolymerization of actin filaments, inhibits Nrf2 translocation and Mrp2 gene activation by pB25R infection. Taken together, these results provide insight into the mechanisms by which resistant clinical isolates of L. donovani induce intracellular events relevant to drug resistance.
Introduction
Leishmaniasis is the most serious form of parasitic diseases caused by the protozoan flagellates of the genus Leishmania. Leishmaniasis is transmitted by the phlebotomine sandfly and presents a spectrum of clinical manifestations (1) ranging from ulcerative skin lesions to destructive visceral infection (2). Disability-adjusted life years lost due to several forms of leishmaniasis is close to 1.5–2.0 million affecting almost 88 countries including 13 of the least developed countries. More than 350 million people are at risk for the infection and disease globally, and the disease causes 70,000 deaths each year (3). The key control measures are mainly based on early detection and chemotherapy, which has been hampered by toxicity, side effects, and emergence of drug-resistant parasites. For the last six decades, organic pentavalent antimonials (Sb(V)) have been the first line drugs for the treatment of this disease, and emergence of isolates that are clinically resistant to these drugs poses a serious obstacle for disease control and treatment (4). Therefore, a better understanding of the resistance mechanisms is a priority for the long term use of a drug to sustain its efficacy for the future.
Flavonols are widely distributed in plants and are present in considerable amounts in fruits and vegetables. In addition to their antioxidant effect, flavonols interfere with a large number of biochemical signaling pathway. Among the flavonols, baicalein (BLN)3 has proved to be one of the major antileishmanial agents and induces apoptotic cell death, targeting topoisomerase IB (5). Various studies in cancer cell lines have revealed that the development of resistance to topoisomerase inhibitors is a multifactorial event including altered drug transport, modified drug metabolism and detoxification, and change in drug-target interaction (6). Hence, developing BLN-resistant Leishmania would represent an important strategic tool for a better understanding of the various mechanisms of development of resistance. The first line of defense for a cell against a drug is altered influx or enhanced efflux, leading to lower accumulation of drug molecules inside cells. The enumerable transporters present in Leishmania alter the influx and efflux rates of a drug and manifest a resistant phenotype (7). The ATP-binding cassette (ABC) transporters play a major role in membrane-associated drug resistance by translocating a wide variety of substrates across extra- and intracellular membranes.
ABC proteins are one of the largest families of transmembrane proteins. The strongly conserved nucleotide binding domain, which is composed of three major motifs, is the major characteristic of these transporters. Along with the Walker A and B motifs, the nucleotide binding domain is composed of a characteristic ABC signature “C” motif located just upstream of the Walker B site (8). Eukaryotic ABC proteins can be divided into eight different subfamilies (ABCA–ABCH) on the basis of gene structure and nucleotide binding domain sequence homologies. A previous survey indicated the presence of 42 ABC protein-coding genes in the genomes of Leishmania major and Leishmania infantum (9). Resistance to metalloids in Leishmania requires multiple steps in which Sb(V) is reduced to the trivalent form Sb(III), and the latter is conjugated to trypanothione, a bisglutathione-spermidine conjugate (10). The complex is then transported inside an intracellular detoxification organelle by the ABC transporter MRPA/ABCC3 (11) or extruded outside the cell by an ATP-dependent efflux pump of unknown identity (12, 13). Overexpressed ABCB4 transporter exhibits resistance to vinblastine and daunomycin (14), whereas ABCA transporter governs phospholipid trafficking (15). More recently, an overexpressed ABCG4 transporter has been shown to be involved in miltefosine resistance (16). It has recently been shown that resistance to camptothecin is manifested by overexpression of the ABCG6 efflux pump (17). Several ABCC proteins have been shown to localize to the plasma membrane in many cell types, and it is intriguing that every Leishmania ABCC protein is located intracellularly (18). The ABCC proteins are often implicated in secretion of toxic metabolites and in cellular detoxification. These proteins could be advantageous for an intracellular parasite like Leishmania to transport toxic compounds and waste metabolites inside intracellular compartments instead of excreting them in the phagolysosome of the parasitized macrophages of the mammalian host.
In this study, we report for the first time the involvement of a novel ABC transporter in rapid BLN efflux-mediated chemoresistance of Leishmania. BLN-resistant Leishmania parasites were developed through progressive adaptation to graded doses of BLN. RT-PCR analysis of ABCC2 (LdMRP2) transporter showed an overexpression pattern in the BLN-resistant parasites. Antibody raised against LdMRP2 showed its localization mainly in the intracellular locations and flagellar pocket region. To our surprise, the BLN-resistant axenic amastigotes failed to show a concomitant increase in MRP2 transporter in promastigotes. This led us to investigate the mechanism of resistance of BLN-resistant amastigotes inside macrophages. Upon infection, resistant amastigotes stimulate macrophage MRP2 transporter and thus become incurable with BLN. These resistant parasites stimulated reorganization of actin filaments in macrophages, and nuclear translocation of Nrf2 is dependent on actin rearrangement, which ultimately activates the ARE1 promoter to produce Mrp2 transcript. This study will help to identify newer therapeutic targets and will provide a better knowledge of the efflux pumps of Leishmania for acquiring resistance against a drug.
EXPERIMENTAL PROCEDURES
Strains and Parasite Culture
Promastigotes of Leishmania donovani (AG83) were cultured in M199 medium (pH 7.4) supplemented with 40 mm HEPES and 10% FBS and maintained at 22 °C. All the resistant cell lines were maintained under drug pressure. Axenic amastigotes were cultured in modified M199 medium (pH 5.3) supplemented with 10 mm sodium succinate, 40 mm HEPES, and 25% FBS and maintained at 37 °C with 5% CO2 (19). Leishmania tarentolae promastigotes were maintained like L. donovani cells with the addition of nourseothricin and phleomycin antibiotics. The transformation of promastigotes to axenic amastigotes was confirmed by microscopy (Nikon A1R confocal microscope).
Generation of BLN-resistant Parasites
Parasites were progressively adapted to BLN by culturing them in medium containing BLN at 0.1, 0.2, 0.5, 1, 2, 5, 10, 15, 20, and 25 μm concentrations. Parasites were cultured two passages up to 5 μm and thereafter three passages up to 25 μm. When 25 μm concentrations were reached, the culture was diluted to ∼10 cells/ml and separately cultured to generate a clonal population of the parasites. The BLN-resistant promastigotes and axenic amastigotes were termed pB25R and aB25R. For L. tarentolae promastigotes, the parasites were exposed to BLN as above, and the resistant strain was named LT.B25R.
Determination of Resistance Levels and Stability
The pB25R parasites were exposed to increasing concentrations (1–150 μm) of BLN with each concentration in triplicate in M199 medium without phenol red. The growth was monitored over a period of 24 h. L. donovani AG83 or pB25R promastigotes were incubated for different times (2, 4, 6, 8, 10, 12, 16, and 24 h) after which the viability of promastigotes was measured using Alamar Blue dye, which is a cell-permeable, nontoxic, nonfluorescent active ingredient (blue) that uses the natural reducing power of viable cells to convert resazurin to the fluorescent molecule resorufin (very bright red). Metabolically active cells convert resazurin to resorufin, thereby generating a quantitative measure of viability and cytotoxicity. The percentage of growth over control was plotted over each drug concentration. The ED50 and ED90 values for each cell type were thereby obtained and tabulated. The pB25R parasites were grown in drug-free medium for 1, 2, 3, 4, 6, and 8 weeks. The ED50 values were obtained as above after the respective time periods to determine the stability of resistance (17). In some experiments, cells were treated with several pharmacological inhibitors such as verapamil (5 μm) (20) or probenecid (4 mm) (21) for 1 h prior to treatment with BLN.
Cross-resistance Studies
Both wild type and BLN-resistant parasites were harvested in exponential growth phase, diluted to a concentration of 1 × 106 parasites/ml, and incubated with either general antileishmanial agents or with topoisomerase inhibitors. The concentration of sodium antimony gluconate (SAG) ranged from 0.5 to 30 μm, and that of miltefosine was 0.5–50 μm. Triplicate values of the percentage of live promastigotes over that of the control were taken, and the ED50 values were calculated.
Energy Depletion and Protein Modification Experiments
The resistant parasites were washed twice in HEPES-buffered saline (HBS; 21 mm HEPES, 137 mm NaCl, 5 mm KCl, 0.7 mm NaH2PO4, and 20 mm glucose adjusted to pH 7.4) and resuspended in the same buffer at 2 × 107 parasites/ml. For energy depletion, parasites were preincubated with 20 μm NaN3 for 30 min in HBS buffer without glucose. For protein modification, parasites were treated with 1 mm N-ethylmaleimide (NEM) for 30 min. Cells were centrifuged and resuspended in fresh HBS containing 25 μm BLN (HBS without glucose for NaN3 experiments) and incubated at 22 °C for the promastigotes (22). A similar set was incubated in ice. After 4 h of incubation, the amount of accumulated BLN in terms of mean fluorescence units at 540 nm was obtained (5). The resistant parasites, which were not pretreated with NaN3 or NEM and not incubated in ice, served as the respective controls. The percentage of accumulated BLN was obtained from the equation
 |
where the values of accumulated BLN are in fluorescence units at 540 nm.
Cloning of LdABCC2 and Transfection
The ABCC2 transporter in Leishmania represents an ORF, LmjF.23.0220, in the L. major genome database. Sense primer 5′-ATAAGAATGCGGCCGCATGCAGTACGAGGACCCGCG-3′ and antisense primer 5′-GGACTAGTTTAAGCCATCGCCACCTGAATCA-3′ were used to amplify the LdABCC2 gene by PCR from the cDNA pool of L. donovani AG83 strain. It was cloned in the NotI and SpeI sites of the vector pXG-GFP2+/. The GFP-tagged LdABCC2-like transporter cloned in pXG-GFP2+/ (a kind gift from Dr. S. M. Beverley) and the empty vector were next electroporated into Leishmania wild type cells using a low voltage protocol, and after 24 h, transfectants were selected using G418 solution. The transfectants were termed WT+G-Abcc2 and WT+G, respectively. Once stable transfectants were obtained, the cultures were grown in low pH M199 medium as stated earlier for their transformation into axenic amastigotes.
Antisense Constructs
The L. tarentolae T7.TR strain was procured from Jena Bioscience. It has the T7 RNA polymerase and Tet repressor genes integrated in the small ribosomal subunit locus, and both genes are under the control of the antibiotics nourseothricin and hygromycin (100 μg/ml each). The vector pLew82v4 (a kind gift from Dr. George A. M. Cross) has a T7 promoter and a Tet operator sequence. The antisense construct of LdABCC2 (MRP2) was generated by PCR amplification of the nucleotide region 1801–2131 using the primers 5′-CGGGATCCGCTGTGCTGGAGAAGGTCGAC-3′ and5′-CCCAAGCTTCACAGACACGTCGCGCAGCAG-3′ andcloned in reverse orientation in the HindIII and BamHI sites of the pLew82v4 vector. The antisense construct pLew82v4/aMRP2 was electroporated separately into the L. tarentolae T7.TR promastigotes and plated onto semisolid M199 agar plates containing nourseothricin (100 μg/ml), hygromycin (100 μg/ml), and phleomycin (25 μg/ml) (23). Positive clones were obtained after 2 weeks, and the transfectant promastigotes harboring pLew82v4/aMRP2 were termed “LTB25R/aMRP2.”
Reporter Dye Uptake Assay
The nonfluorescent calcein ester prodrug calcein AM has been shown to be a substrate for MRP1 and MRP2, and Rhodamine 123 (Rh123) is a substrate for P-glycoprotein (24). Rh123, a fluorescent cationic dye used as a tracer dye to determine the rate and direction of flow and transport, is an established substrate for P-glycoprotein. Conversely, calcein AM in its native form is nonfluorescent but upon entering the cell is cleaved by esterases and becomes nonpermeable and fluorescent. As both these probes are selectively pumped out via active protein pumps, it is understood that a cell with a higher activity of transporters would have lesser fluorescence. Therefore, Rh123 and calcein AM serve as useful tools for the study of transport proteins. Treated or untreated parasites in 24-well plates were incubated with 10 μm calcein AM for 4 h either alone or in the presence of probenecid or verapamil. Extracellular medium was removed, and the cells were washed twice with 1 ml of ice-cold phosphate-buffered saline. Cells were then lysed by addition of 500 μl of 0.4% Triton X-100 and 20 mm Tris-HCl (pH 9.0). An aliquot of 200 μl of the cell lysate was then transferred to an opaque 96-well plate. Fluorescence was measured with an excitation of 470 nm and an emission of 580 nm for Rh123 dye and an excitation of 490 nm and an emission of 515 nm for calcein.
Measurements of Endocytosis
Uptake of the endocytosis marker FM 4-64 was measured on PerkinElmer Life Sciences LS55 luminescence spectrometer. A total of 4 × 106 parasites/ml were incubated with 10 μm FM 4-64 in HBS plus 0.5% BSA at 22 °C or on ice for 30 min. Thereafter, the parasites were either washed one time with cold phosphate-buffered saline (PBS) to remove the label bound to the plasma membrane and flagellar pocket and then resuspended in PBS or directly analyzed by measuring their cellular fluorescence as described above by scanning the emission between 506 and 750 nm (25).
Secreted Acid Phosphatase (SAP) Activity Assay
SAP activity was assayed as described (26). Briefly, Leishmania parasites (2 × 108 cells/ml) were incubated with 25 μm BLN. After every 20 min of incubation, 200 μl of supernatants from Leishmania cultures were lysed with 1% Triton X-100 and incubated with p-nitrophenyl phosphate (Sigma) as a substrate. The results were expressed in nanomoles of substrate hydrolyzed/2 × 108 promastigotes for 30 min (extinction coefficient, 17.8 mm−1) (27).
Gene Expression Analysis
Total RNA was prepared from wild type, BLN-resistant, SAG-resistant (GE1), DIM-resistant, and BLN-treated BLN-resistant promastigotes using the Total RNA Isolation kit (Roche Applied Science). cDNA was synthesized from 100 ng of total RNA using Superscript IITM RNase H− reverse transcriptase (Invitrogen) and oligo(dT)12–18 primers (Invitrogen) following the manufacturer's instructions. Semiquantitative PCR was performed in a 25-μl volume using 20 pmol each of sense and antisense primers corresponding to LdABCC1, LdABCC2, LdTOP1L, and LdGAPDH using the following profile: initial denaturation at 95 °C for 5 min followed by 25 cycles with denaturation at 95 °C for 1 min, annealing at 57 °C for 40 s, and extension at 68 °C for 40 s and a final extension of 3 min. The primers were designed such that each set amplified a 220–250-bp fragment. Real time PCR was performed with cDNAs of genes showing altered expression in semiquantitative PCR. Three separate reactions were carried out using three different RNA preparations in a 25-μl volume using SYBR Green Super Mix (Applied Biosystem) and the same primer sets in a 7300 Real Time PCR System (Applied Biosystem). Reactions were carried out using the following profile: initial denaturation at 95 °C for 5 min followed by 35 cycles with denaturation at 95 °C for 40 s, annealing at 60 °C for 40 s, and extension at 65 °C for 40 s. The PCR was followed by a melt curve analysis to ascertain whether the expected products were amplified (28). The relative amounts of PCR products generated were obtained from the threshold cycle (Ct) value, and amplification efficiencies were normalized by dividing the values by the relative amount of the GAPDH gene used as a control.
Cloning of N-terminal Fragment of LdABCC2, Overexpression, and Antibody Purification
Rabbit anti-LdABCC2 antibodies were produced against a recombinant protein corresponding to positions 1–130 of the N terminus of LdABCC2. The 390 bp were cloned in the NdeI and BamHI sites of pET16b vector, which was transformed into the Escherichia coli Rosetta strain. Thereafter, one of the culture was grown to an A600 of 0.5 and induced with 0.25 mm isopropyl β-d-thiogalactopyranoside for 6 h at 23 °C. The protein was purified from the lysate by Ni2+-nitrilotriacetic acid column chromatography. The polyclonal antiserum was obtained by several subcutaneous immunizations of a New Zealand White rabbit with 150 μg of purified peptide (29).
Cell Death Detection
Cell death of BLN-resistant L. tarentolae T7.TR promastigotes (LTB25R) and aMRP2-transfected LTB25R parasites (LTB25R/aMRP2) in the presence of 20 μm BLN was tested by binding of FITC-annexin V and propidium iodide using an annexin V staining kit (Invitrogen). Flow cytometry was carried out for treated and untreated parasites (30). The gating was done so that the FL-1 channel denotes the mean intensity of FITC-annexin V, whereas the FL-2 channel denotes the mean intensity of propidium iodide. The data represented here are a mean of three experiments.
DNA Fragmentation Assay
An estimate of the extent of DNA fragmentation after drug treatments was carried out using a cell death detection ELISA kit (Roche Applied Science). Promastigote samples (5 × 106 cells/ml) were collected at 2-h intervals, and the histone-associated DNA fragments (mononucleosome and oligonucleosome) were detected using the manufacturer's protocol. DNA fragmentation was estimated by spectrophotometric measurement of microtiter plates in a Thermo Multiskan EX plate reader at 405 nm.
In Vitro Mf Culture
BALB/c mice originally obtained from The Jackson Laboratories (Bar Harbor, ME) and reared in the institute animal facilities were used for experimental purposes with prior approval of the animal ethics committee. Macrophages were isolated from mice 36–48 h postinjection (intraperitoneal) with 2% (w/v) hydrolyzed starch by peritoneal lavage with ice-cold phosphate-buffered saline. In vitro infection with AG83 and pB25R promastigotes was carried out as described (31). BLN was added at different concentrations (ranging from 1 to 25 μm) to infected macrophages and left for another 24-h period. Cells were then fixed in methanol and stained with 2% Giemsa. Percentages of infected cells and total numbers of intracellular parasites were determined by manual counting in at least 250 cells using a light microscope.
FACS Analysis
The levels of expression of Mf-MRP2 on normal, AG83-infected, and pB25R-infected macrophages were determined by immunostaining followed by flow cytometry (BD Biosciences) as described (32). The appropriate isotype control was used for each individual case. Because these antibodies may cross-react with nonspecific parasite proteins, all stainings were performed 18–24 h after washing the unpermeabilized infected cells in the cold to specifically check the expression of MRP2 on the host cell surface. To detect the percent population of LdMRP2-positive cells in resistant pB25R parasites, the parasites were fixed prior to incubation with LdMRP2 antibody raised in rabbit. All the data were analyzed using BD FACScan software.
Microscopy
Promastigotes were harvested, washed three times in cold PBS (130 mm NaCl, 2.6 mm KCl, 1.2 mm KH2PO4, and 8.1 mm Na2HPO4 (pH 7.4)), and settled onto slides. Fixation was allowed to proceed sequentially at −20 °C in ethanol for 5 min and in acetone for 8 min. The slides were then incubated with anti-LdABCC2 antibodies (dilution, 1:500) or preimmune serum for 1 h at 37 °C. After three washes in PBS with 0.5% bovine serum albumin, the slides were further incubated with Atto 488-conjugated goat anti-rabbit IgG (Sigma) for 1 h at 37 °C and washed as above. All the above steps were performed in a humidified chamber. After mounting, images were acquired with Nikon A1R confocal microscope.
Plasmid Constructs for Promoter Analysis and Mutagenesis
The 5′-flanking region of the mouse Mrp2 promoter was amplified by PCR from genomic DNA isolated from murine peritoneal macrophage cells with high fidelity Taq polymerase (Roche Applied Science). The isolated PCR product was ligated into KpnI-XhoI sites of the pGL3 Basic vector (Promega, Madison, WI). The forward primer for the full-length construct was GGTAACCTAGGGTCTGGGCTTGATTCT (FARE1895). Three deletion constructs (−60 to +99, −120 to +99, and −854 to +99) were generated. The primers used for amplification were as follows: GGTAACACTATTAAGTTGTCAGGATGAAAGG (FARE60), GGTAACAGTCACATGTCTGCTCACTGGA (FARE120), GGTAACAAAGATACAATAGAAAATAA (FARE854), and CATCGTGATTTCCAGGGCACTGAGCTC (RUni; reverse primer for all constructs). ARE1 in the Mrp2 promoter was mutated using the QuikChange mutagenesis kit (Stratagene). For mutation of ARE1, an antisense primer (5′-TTGCTAGATGAATGcGTcTGaCTCCCAGTGAGCAG-3′) and a sense primer (5′-CTGCTCACTGGGAGtCAgAcGCATTCATCTAGCAA-3′) (mutated bases are indicated in lowercase type) were used in a PCR according to the manufacturer's directions with the 0.2-kb plasmid generated above as template. PCR products were digested with DpnI for 6 h to cleave the wild type promoter.
Transient Transfections
RAW264.7 cells were grown in complete medium, and 24 h before transfection, the cells were seeded on 12-well plates at a density of 1 × 105 cells/well. Cells were transfected with 10 μg of DNA, 3.5 μl of LipofectamineTM, and 6 μl of Lipofectamine Plus reagent (Invitrogen) as described previously (33). Total DNA included 7.5 μg of luciferase pGL3-reporter construct together with 2.5 μg of SV40-Renilla pGL3-reporter construct (Promega) as an internal control. After 4 h of incubation, the transfection medium was replaced with complete medium and kept for another 12 h. Following successful transfection, cells were infected either with AG83 promastigotes or with pB25R promastigotes or left uninfected. Infected and uninfected cells were harvested after 12 h, and cell extracts were assayed for enzyme-specific activity of the reporter gene used. Luciferase and Renilla luciferase activity were measured using the Dual-Luciferase assay kit according to the manufacturer's (Promega) instructions and normalized for transfection efficiency with the luminescence of the Renilla luciferase activity. LUC activities were normalized with Renilla internal control, and the activities were measured by the Dual-Luciferase Reporter Assay system (Promega) with a luminometer (EG&G Wallac, Gaithersburg, MD).
Preparation of Nuclear and Cytoplasmic Fractions
Nuclear and cytoplasmic protein extracts were prepared as described (34). Cells were collected by scraping after the required infection or treatment by centrifugation at 800 × g for 5 min at 4 °C, resuspended in 500 μl of ice-cold hypotonic buffer (10 mm HEPES (pH 7.9), 10 mm KCl, 0.1 mm EDTA, 2 mm DTT, 0.5 mm PMSF, and protease inhibitor mixture), and allowed to swell. After 30 min of incubation on ice, Nonidet P-40 was added to a final concentration of 0.6%, and the cells were mixed and centrifuged for 3 min at 16,000 × g at 4 °C. Supernatants were used as cytoplasmic fractions for assay of Nrf2, and immunoblots were stored at −70 °C until use.
Pellets containing crude nuclei were further washed with swelling buffer without Nonidet P-40 and finally resuspended in 100 μl of extraction buffer containing 20 mm HEPES (pH 7.9), 400 mm NaCl, 1 mm EDTA, 10 mm DTT, 1 mm PMSF, and protease inhibitor mixture (Sigma-Aldrich) and then incubated for 30 min in ice on a rocking platform. The protein concentration was determined using the Bradford assay (Bio-Rad). Nuclear fractions were stored at −70 °C until use.
Immunoblot Analysis
SDS-polyacrylamide gel electrophoresis and immunoblot analyses were performed as described (35). The samples were fractionated by 7.5 (for MRP2) or 12% (actin and Nrf2) gel electrophoresis and electrophoretically transferred to nitrocellulose paper. The nitrocellulose paper was incubated with polyclonal rabbit anti-Nrf2 antibody (1:1000) (Santa Cruz Biotechnology, Santa Cruz, CA) or anti-actin antibody (Cell Signaling Technology) (1:1000) followed by incubation with alkaline horseradish peroxidase-conjugated secondary antibodies and developed using an enhanced chemiluminescence detection kit. The loading control was measured using β-tubulin or H2B (Cell Signaling Technology).
Coimmunoprecipitation
To detect actin-bound Nrf2 transcription factor, either the nuclear fraction or total cell lysate (100 μg in 300 μl each) was incubated with polyclonal rabbit anti-Nrf2 antibody (2 μg) (Santa Cruz Biotechnology) for 8 h at 4 °C on a shaking platform. The antigen-antibody complex was immunoprecipitated after overnight incubation at 4 °C with protein G-agarose. Immune complexes were solubilized in 50 μl of 2× Laemmli buffer and boiled for 10 min at 100 °C. The boiled fractions were centrifuged. The supernatant was allowed to cool to room temperature. Samples were analyzed using 12% SDS-PAGE and then transferred to nitrocellulose membranes. The samples were then immunoblotted with anti-actin antibody (36). For some experiments, anti-actin antibody was used for immunoprecipitation followed by blotting with anti-Nrf2 antibody. Blots were developed using an ECL chemiluminescence detection kit for immunostaining.
Staining of Actin with Fluorescein-labeled Phalloidin
To selectively stain filamentous polymerized actin (F-actin) in the cytoskeleton of the cytoplasm, the plated cells (on a Lab-TekTM II Chamber SlideTM) were infected with AG83 or pB25R promastigotes or left uninfected. The cells were washed with PBS and fixed for 10 min in 3.7% formaldehyde solution in PBS. Cells were then washed extensively in PBS, dehydrated with acetone, and permeabilized with 0.5% Nonidet P-40 in PBS. Cells were stained with a 50 μg/ml fluorescent phalloidin conjugate (phalloidin-Atto 565) solution in PBS (containing 1% DMSO from the original stock solution) for 40 min at room temperature in a humidified chamber according to the manufacturer's instructions.
Colocalization of Nrf2 and actin in the nucleus in pB25R-infected macrophages was determined using the combination of two fluorescent stainings. Cells were then fixed for 10 min in 3.7% formaldehyde solution in PBS, washed extensively in PBS, dehydrated with acetone, and permeabilized with 0.1% Nonidet P-40 in PBS. Actin was labeled inside the nucleus with anti-actin antibody (Sigma-Aldrich) followed by Atto 594-conjugated anti-rabbit IgG antibody, whereas Nrf2 was stained with rabbit anti-Nrf2 antibody followed by Alexa Fluor 488-conjugated anti-rabbit IgG antibody (Molecular Probes). Stained cells were washed twice with PBS and examined using a laser-scanning confocal microscope (Nikon A1R).
Measurement of G-actin/F-actin Ratio
Determination of the amount F-actin content compared with free G-actin content was performed using a G-actin/F-actin in vivo assay kit (Cytoskeleton Inc., Denver, CO) according to the manufacturer's instructions. Briefly, experimental cell samples (RAW264.7) were homogenized in cell lysis and F-actin stabilization buffer (50 mm PIPES, 50 mm NaCl, 5 mm MgCl2, 5 mm EGTA, 5% (v/v) glyceral, 0.1% (v/v) Nonidet P-40, 0.1% (v/v) Triton X-100, 0.1% (v/v) Tween 20, 0.1% (v/v) 2-mercaptoethanol, 0.001% (v/v) antifoam, and a protease inhibitor mixture followed by centrifugation for 1 h at 100,000 × g to separate the F-actin from G-actin pool. Supernatants of the protein extracts were collected at 30 °C. Pellets were resuspended in ice-cold distilled H2O plus 1 μm cytochalasin D and then incubated on ice for 1 h to dissociate F-actin. The resuspended pellets were gently mixed every 15 min. Equal amounts of both the supernatants (G-actin) and the resuspended pellets (F-actin) were subjected to immunoblot analysis with actin antibody (Cytoskeleton Inc.). F-actin and G-actin levels were quantified from three individual Western blots using Bio-Rad Quantity One software and are represented as means.
Immunocytochemistry of Nrf2
Peritoneal mouse macrophage cells were grown on Lab-Tek Chamber Slides (Nalgene Nunc International Corp., Rochester, NY) and infected either with AG83 or pB25R promastigotes or left uninfected. For immunostaining, the cells were fixed in 100% methanol for 30 min and washed three times with PBS. After blocking in 5% bovine serum albumin in PBS for 1 h at room temperature, the cells were incubated overnight with polyclonal rabbit anti-Nrf2 antibody (1:100) in PBS containing 0.5% bovine serum albumin. The cells were incubated with Atto 594-conjugated anti-rabbit IgG antibody (1:500) after serial washings with PBS. UltraCruzTM mounting medium (Santa Cruz Biotechnology) was used for counterstaining with DAPI as well as for mounting purposes and verification of the location and integrity of nuclei. Stained cells were examined using a laser-scanning confocal microscope (Nikon A1R).
EMSA
A double-stranded DNA probe containing the ARE1 or the corresponding ARE1 mutant oligonucleotide sequences (ARE1M) were end-labeled with [γ-32P]ATP using T4 polynucleotide kinase and used for gel shift analysis (37). The DNA probes were 5′-ACTGGGATGACATAGCATTCATC-3′ (ARE1 sense) and 5′-GATGAATGCTATGTCATCCCAGT-3′ (ARE1 antisense) where boldface indicates the ARE core sequence or 5′-ACTGGGAGTCAGACGCATTCATC-3′ (ARE1M sense) where underlining indicates the ARE core mutation and 5′-GATGAATGCGTCTGACTCCCAGT-3′ (ARE1M antisense). The sense and corresponding antisense single-stranded DNA were annealed in a stoichiometric ratio of 1:1 at 25 °C for 1 h. Five micrograms of nuclear protein extract and the dsDNA probes (2 ng; 32P-labeled) were incubated in the binding buffer (10 mm HEPES (pH 7.9), 60 mm KCl, 10% (v/v) glycerol, 1 mm EDTA, 10 mm Na3PO4, 1 mm DTT, 3 mm MgCl2, 1 μg of poly(dI-dC), and 1 mg of sonicated salmon sperm single-stranded DNA) for 40 min at 20 °C. The samples were electrophoresed on a 6% polyacrylamide gel in 0.25× Tris borate-EDTA buffer at 100 V for 200 min.
For the supershift assay, an anti-Nrf2 antibody (sc-722X, Santa Cruz Biotechnology) was added after 4 h of the binding reaction at 4 °C. DNA-protein interactions were detected by electrophoresis on a nondenaturing 6% polyacrylamide gel in 0.25× Tris borate-EDTA buffer. Gels were dried, and DNA-protein complexes were visualized by autoradiography.
RESULTS
Development of Baicalein-resistant Promastigotes and Stability of Resistance
The wild type promastigote cultures were exposed to baicalein (0.2–25 μm) through various passages. The parasites were grown stably in the medium in the presence of 25 μm BLN and were termed pB25R. The parasites were then selected using a clonal selection method. The sensitivity of the clonally selected parasites was then determined by growing them in increasing concentrations of BLN, and ED50 and ED90 values were obtained (Table 1). One single cell clone was used for all the subsequent experiments after confirming the drug resistance. To compare susceptibility to BLN, clonally selected parasites were exposed to increasing concentrations of the drug. The growth of pB25R parasites was reduced to 50% by treatment with 50 μm concentrations and was hardly affected by a lower dosage of BLN, whereas the growth of wild type (WT) parasites was reduced to 50% at 4.31 μm concentration (Fig. 1A). This result clearly established the development of resistance in pB25R to circumvent the cytotoxic effect of BLN.
TABLE 1.
ED50 and ED90 values of WT and BLN-resistant (Res) parasites obtained from Fig. 1A
| Parasite name | ED50 | ED90 |
|---|---|---|
| μm | μm | |
| AG83 (WT) | 4.31 ± 1.41 | 42.12 ± 6.17 |
| pB25R (Res) | 32.41 ± 4.23 | >100 |
FIGURE 1.

A, cytotoxicity of BLN-resistant cell lines. L. donovani WT and BLN-resistant (pB25R) parasites were grown in the presence of increasing concentrations of BLN (1–100 μm) at 22 °C. Aliquots were taken at intervals, and the percentage of viable promastigotes was measured by Alamar Blue reagent. Proliferation of wild type and resistant parasites in the absence of any drug was considered as the maximal growth control. Parasite growth in the presence of the drugs was expressed as percentage of growth over control. Results (percentage over control ±S.E.) were performed in triplicate and averaged. Graphical presentation was performed with GraphPad Prism version 5.00 (GraphPad Software, San Diego, CA), and the fitted lines (asymmetric) from these data points (n = 3) have R2 values of 0.9012 and 0.9471, respectively. B, enhanced efflux of BLN in resistant pB25R Leishmania promastigotes. Wild type AG83 and pB25R parasites were loaded under conditions that yielded similar amounts of intracellular drug (2 h for pB25R and wild type AG83), and the amount of BLN retained in parasites maintained after that in drug-free culture medium was measured at different time points up to 150 min. C, SAP activity in WT and pB25R Leishmania. Bars represent the SAP activity in supernatants from Leishmania cultures incubated in the presence of BLN (20 μm) for different time intervals as indicated on the x axis. The values are presented as nmol of p-nitrophenyl phosphate hydrolyzed in 30 min/2 × 108 promastigotes. D, analysis of the stability of BLN resistance. The resistant parasites (pB25R) were grown in respective drug-free medium for 8 weeks. The sensitivity of the parasites to BLN was observed in the 1st, 2nd, 3rd, 4th, 6th, and 8th weeks. Their respective ED50 and ED90 values were calculated and plotted. Error bars represent S.E.
pB25R promastigotes showed nearly similar uptake for BLN from the medium. When parasites were grown in culture containing 25 μm BLN, ∼10% of the BLN was taken up within 45 min by wild type or resistant cells, and saturation was reached in about 2 h for both the parasites. As control, WT AG83 and pB25R cells were incubated on ice, a condition that blocks endocytosis but allows a residual translocation activity. BLN accumulation was only 5–12% higher by wild type parasites than resistant parasites at different time points (data not shown). This reflects that BLN uptake is similar in both type of parasites. Accumulation of FM 4-64 further confirmed that there was no significant difference in the endocytosis process. FM 4-64 has been used previously as an endocytosis marker in Leishmania parasites. FM 4-64 primarily binds to plasma membrane of the parasites and to the flagellar pocket from which it is endocytosed to a multivesicular network and lysosomes. The uptake of FM 4-64 was similar for both the parasites (data not shown).
To study whether the resistance mechanism was due to active efflux pumps following accumulation of BLN after 2 h, wild type AG83 and pB25R parasites were loaded with 25 μm BLN under conditions where drug accumulation was similar. The amount of BLN retained in parasites maintained in drug-free culture medium was measured at different time points. The efflux of BLN was about 60% faster for resistant parasites (measured every 20 min after drug removal for 150 min).The results show a logarithmic decrease in efflux pattern in pB25R parasites and an almost linear pattern of efflux of BLN in wild type parasites (Fig. 1B). This study emphasizes that the differences in the levels of BLN accumulation are not due to a defect in uptake activity but are due to increased drug efflux.
To follow whether the resistant promastigotes utilize higher exocytosis, SAP activity, a marker for exocytosis activity, was assayed. Parasites resistant to BLN showed a significantly (p < 0.005) higher secreted SAP activity with respect to wild type parasites (Fig. 1C). The SAP activity was highest after 2 h of drug internalization. This result suggests that resistant promastigotes induced an increase in the exocytic activity.
After drug resistance was established, drug pressure was removed from pB25R cell types, and the parasites were allowed to grow in drug-free medium for 10 weeks. The sensitivity of the parasites to BLN was observed in the 1st, 2nd, 3rd, 4th, 6th, and 8th weeks, and their respective ED50 and ED90 values were calculated and plotted (Fig. 1D). Upon prolonged incubation without BLN, the resistivity of the parasites fell appreciably throughout the days, and after 8 weeks postincubation, the resistance was reversed.
The extent of cross-resistance of the pB25R parasites was determined by growing the parasites in the presence of general antileishmanial compounds as well as other topoisomerase inhibitors. The general antileishmanial compounds included miltefosine, amphotericin B, and paromomycin, and topoisomerase inhibitors include dihydrobetulinic acid, camptothecin, diindolylmethane, luteolin, and quercetin. The parasites were found to be sensitive to all the antileishmanial compounds. Of the topoisomerase inhibitors, CPT, DIM, and quercetin all showed potent antileishmanial activity against the resistant parasites, whereas the parasites were cross-resistant to luteolin. Manifestation of CPT resistance occurs through overexpression of ABCG6 transporter (17), which causes decreased uptake, whereas resistance to DIM causes mutation in LdTOP1LS, which escapes the trapping of DNA-LdTOP1LS covalent complex inside parasites. To our surprise, pB25R becomes cross-resistant to luteolin, which may be due to a similar kind of inhibitory action or molecule associated with resistance (Table 2).
TABLE 2.
Cross-resistance study of other antileishmanial compounds
| Antileishmanial compounds | AG83 ED50 | pB25R ED50 |
|---|---|---|
| μm | μm | |
| Miltefosine | 16.3 ± 2.5 | 15.7 ± 3.4 |
| Amphotericin B | 0.125 ± 0.014 | 0.131 ± 0.011 |
| Paromomycin | 52.24 ± 8.64 | 55.31 ± 14.07 |
| Dihydrobetulinic acid | 3.1 ± 0.24 | 2.9 ± 0.17 |
| Camptothecin | 1.33 ± 0.21 | 1.35 ± 0.11 |
| 3,3′-Diindolylmethane | 1.52 ± 0.17 | 1.61 ± 0.24 |
| Luteolin | 15.27 ± 2.31 | 74.91 ± 1.51 |
| Quercetin | 51.34 ± 8.41 | 49.26 ± 9.17 |
| Baicalein | 4.31 ± 1.41 | 42.12 ± 4.23 |
Resistant Promastigotes Overexpress Classical MRP Transporter Activity
Next we studied the energy and protein dependence of baicalein resistance in promastigotes of pB25R parasites. To deplete the intracellular ATP pool, the parasites were pretreated with 20 mm NaN3 and thereafter resuspended in fresh medium containing BLN, and BLN accumulation was measured. The percentage of BLN accumulation increased up to 62% for the NaN3-treated parasites with respect to untreated resistant parasites (Fig. 2A). Whereas in wild type parasites, no significant difference was seen in NaN3-treated parasites. NEM is known to modify sulfhydryl residues of protein and thus leads to protein inactivation. Resistant parasites pretreated with 1 mm NEM followed by resuspension in the fresh medium containing BLN were measured for intracellular BLN accumulation and apoptotic cell death. Pretreatment with NEM followed by baicalein treatment enhanced BLN accumulation by 45% compared with that of untreated pB25R promastigotes. Incubation of the promastigotes at 4 instead of 22 °C also increased the BLN accumulation to 80% of the control (Fig. 2A). Thus, it can be envisaged that increased BLN accumulation may be due to inactivation of protein-mediated active processes that lead to defective efflux of baicalein from the cells.
FIGURE 2.

A, effect of energy depletion, protein inactivation, and temperature dependence (Temp) of BLN efflux in resistant promastigotes (pB25R). Resistant parasites were preincubated with 20 μm NaN3 or 1 mm NEM and resuspended in BLN-containing fresh medium. One set was incubated in ice. BLN accumulation was measured after 2 h of incubation, and the percentage of BLN accumulation was obtained. The data shown are means of three independent experiments each performed in triplicate. B, effect of probenecid on pB25R Leishmania. The exponentially growing cells at 106 promastigotes/ml in drug-free medium were pretreated with 4 mm probenecid for 2 h and then resuspended in fresh medium containing BLN at 20 μm. Next, at each 2-h interval, aliquots are taken and stained using trypan blue, and a promastigote count was taken under a light microscope. The fitted lines (asymmetric) from these data points (n = 3) have R2 values of 0.9936, 0.9983, and 0.9977, respectively. C, effect of verapamil on pB25R Leishmania. The exponentially growing cells at 106 promastigotes/ml in drug-free medium were pretreated with 5 μm verapamil for 2 h and then resuspended in fresh medium containing BLN at 20 μm. Next, at each 2-h interval, aliquots were taken and stained using trypan blue, and a promastigote count was taken under a light microscope. The fitted lines (asymmetric) from these data points (n = 3) have R2 values of 0.9581, 0.9373, and 0.9872, respectively. D, the accumulation of Rh123 reporter dye was measured after 20 min of incubation of the cells (WT, pB25R, and probenecid-pretreated pB25R) with the substrate Rh123. The mean fluorescence intensity at 515 nm was plotted against time. The negative control group was treated with medium alone. The fitted lines (asymmetric) from these data points (n = 3) have R2 values of 0.9211, 0.9445, and 0.9634, respectively. Error bars represent S.D.
To check the specificity of this active efflux system, specific blockers were used, viz. probenecid (blocker of MRP) and verapamil (blocker of MDR). The chemosensitivity toward baicalein was reverted back to almost wild type when incubated in the presence of probenecid (Fig. 2B). In the presence of verapamil, pB25R parasites showed no effect to BLN (Fig. 2C). This confirms that the active efflux pump is MRP-related and probenecid-sensitive.
To understand whether MRP is involved in drug resistance, we studied the status of drug efflux pumps. Rh123, a fluorescent cationic dye, was used to determine the rate and direction of flow and transport of P-glycoprotein. Conversely, calcein AM, a nonfluorescent dye in its native form, upon entering the cell is cleaved by esterases and becomes nonpermeable and fluorescent. As both these probes are selectively pumped out via active protein pumps, it is understood that a cell with a higher activity of transporters would have lesser fluorescence. The classical blockers probenecid and verapamil had no effect on Rh123 accumulation in both wild type and resistant promastigotes. To study whether MRP pumps were present in the resistant promastigotes, we used calcein AM as the substrate. Accumulation of calcein in wild type promastigotes showed a 4 times higher fluorescence than did pB25R promastigotes (Fig. 2D), indicating that a lower accumulation was associated with the resistance phenomenon. Pretreatment of probenecid prior to BLN treatment steeply enhanced calcein accumulation in resistant promastigotes, corroborating the fact that MRP pumps are operative in resistant parasites.
Involvement of LdMRP2-like Transporter Protein in Baicalein Resistance
In this study, the active protein that was associated with drug efflux inside the promastigotes was probenecid-sensitive, which may be due to overexpression of MRP-like protein inside cells. It has been suggested by Leprohon et al. (18) that MRP-like protein transporters are localized inside cells but are absent in eukaryotic counterparts. In pB25R promastigotes and axenic amastigotes, there was a significant amount of MRP2 protein expression compared with wild type parasites. Semiquantitative PCR revealed that the levels of MRP1 transporter (Fig. 3A) were 1.4- and 1.1-fold in BLN-resistant promastigotes and axenic amastigotes compared with wild type parasites and amastigotes, respectively, whereas the levels of MRP2 were 22- and 2.7-fold, respectively, compared with wild type parasites (Fig. 3B). Analysis of overexpression of MRP2 in other resistant parasites (SAG-resistant GE1, diindolylmethane-resistant DIMR, and camptothecin-resistant CPTR) failed to show any up-regulation of MRP2 gene (Fig. 3C). Incubation of pB25R parasites in BLN-free medium for a prolonged time period gradually reverted parasites back to the wild type phenomenon.
FIGURE 3.

A, analysis of MRP transporter levels associated with the resistant phenotype. Gene expression of LdABCC2 and LdABCC1 in WT and BLN-resistant promastigotes (Prom) and axenic amastigotes (Amas) was analyzed by RT-PCR. The products were run in a 1.5% agarose gel. B, relative expression of MRP2 gene in resistant parasites. Real time RT-PCR was carried out for 35 cycles using RNA isolated from the above stated parasites as well as 8-week BLN-unexposed parasites. The threshold cycle values were taken, and relative expression was calculated using GAPDH as internal control. The -fold expression of MRP2 transcript in each parasite was calculated using the 2−ΔΔCT method. The -fold expression in promastigotes and axenic amastigotes was plotted as means ± S.D. C, association of MRP2 gene with other resistant Leishmania strains. Gene expression of LdABCC2 in WT, SAG-resistant (GE1), CPT-resistant (CPTR), DIM-resistant (DIMR), BLN-resistant (pB25R), and BLN-treated BLN-resistant (pB25R+BLN) parasites and at the indicated weeks (wk) of BLN-unexposed BLN-resistant parasites was analyzed by RT-PCR. D, baicalein sensitivity of wild type, BLN-resistant, empty vector-transfected, and LdABCC2-transfected promastigotes. Parasites (2 × 106 cells/ml) were grown in M199 medium without phenol red and incubated with 25 μm CPT. Optical density was measured at 600 nm, and the percentage of live promastigotes over that of control was plotted. Results represent the means ± S.D. of experiments performed in triplicate. E, involvement of MRP2 gene in baicalein-resistant phenomenon. Baicalein-resistant (25 μm) L. tarentolae (LT.T7.TR) were developed in the laboratory and termed LTB25R. A 300-bp antisense construct of LdMRP2 was cloned in pLew82v4 vector (pLew/aMRP2). The empty vector (pLew82v4) or antisense construct (pLew/aMRP2) was transfected in LTB25R parasites and selected using phleomycin. The transformed parasites were grown in the presence or absence of BLN (20 μm) for 4 h, and the apoptotic population was quantified using flow cytometry in a BD FACSAria II cytometer and analyzed using BD FACScan software. PI, propidium iodide. F, enhanced DNA fragmentation in MRP2-down-regulated parasites. The above stated parasites were treated with 20 μm BLN for the indicated time periods. As a negative control, LTB25R parasites were also treated with 0.2% DMSO. Values were obtained from the Multiskan EX readings at 405 nm. The percentage was plotted as units of time. Data represent means ± S.D. (n = 3). G, decreased efflux of BLN in antisense construct (aMRP2)-transfected resistant L. tarentolae strains (LTB25R/aMRP2). Wild type L. tarentolae (LT.T7.TR), resistant parasites (LTB25R), and MRP2-down-regulated L. tarentolae (LTB25R/aMRP2) were loaded under conditions that yielded similar amounts of intracellular BLN, and the amount of BLN retained in parasites maintained after that in drug-free culture medium was measured at different time points up to 150 min. The data represent means ± S.D. The fitted lines (asymmetric) from these data points (n = 3) have R2 values of 0.9915, 0.9987, and 0.9841, respectively. Error bars represent S.D.
The overexpressed ABCC2 transporter is annotated as LmjF.23.0220 in the L. major genome database (GeneDB). Using sense and antisense primers, the ORF of ABCC2 was amplified by PCR using an L. donovani cDNA template. The PCR product of 4710 bp encoding a protein of 1570 amino acids with a predicted mass of 173.7 kDa was cloned in the pXG-GFP2+/ vector and termed LdABCC2. The ORF of 4710 bp encoded a protein of 1570 amino acids with a predicted mass of 173.7 kDa. Wild type promastigotes and pB25R, pWT+G, and WT+G-Abcc2 parasites were treated with 25 μm BLN, and cell counts were taken at 2-h intervals. Fig. 3D shows that there was a rapid decrease in the percentage of live pWT and pWT+G promastigotes, but for pB25R and pWT+G-Abcc2 parasites, which overexpress LdABCC2, the percentage of live promastigotes increased over time. Hence, the mutant and overexpressor parasites are resistant to BLN. Thus, it was elucidated that overexpression of LdABCC2 alone could decrease the BLN accumulation in the parasites, thereby conferring BLN resistance.
Baicalein is known to inhibit L. donovani heterodimeric type IB topoisomerase (5). Resistance to baicalein may also be due to overexpression of LdTOP1LS, which may lead to an altered interaction of baicalein and topoisomerase. Therefore, we checked the pattern of expression of LdTOP1L in resistant and wild type parasites. In both promastigotes and axenic amastigotes, there was almost 1.6- and 1.3-fold decreased expression of LdTOP1L (data not shown).
To confirm the role of LdMRP2 in the resistant phenotype, down-regulation of MRP2 gene was carried out by antisense RNA as described (23). Tetracycline-induced production of antisense LdMRP2 RNA in LT.T7.TR/aMRP2 transfectants down-regulated LtMRP2 in vivo. Clonally selected resistant parasites (LTB25R) showed comparable ED50 and ED90 values as pB25R. BLN was added to parasites after 24 h of tetracycline-induced or uninduced pLew82 (empty vector) and LTB25R/aMRP2 transfectants, and the percentage of apoptotic promastigotes was monitored after 8 h of BLN treatment. Treatment of LtMRP2-down-regulated parasites (LTB25R/aMRP2) with BLN induced fast apoptotic death (Fig. 3E) and DNA degradation (Fig. 3F), whereas BLN treatment of LTB25R parasites produced no significant change. Therefore, the absence of MRP2 transporter in LTB25R/aMRP2 parasites generated a negative feedback response that led to the accumulation of a higher amount of baicalein in the parasites, resulting in DNA degradation and programmed cell death. Hence, the involvement of LdMRP2 in BLN-induced resistance of Leishmania was conclusively elucidated.
We further checked the SAP activity for the efflux mechanism in LTB25R and LTB25R/aMRP2 promastigotes. SAP activity of LT.T7.TR promastigotes was drastically reduced to almost that of LTB25R/aMRP2 promastigotes (Fig. 3G). Reduced efflux thus corroborates the higher accumulation of BLN inside the cells.
LdMRP2 Transporter Localizes in the Intracellular Compartment and Flagellar Pocket of Resistant Promastigotes
To characterize the level of up-regulation and localization of LdMRP2 in the resistant promastigotes, 390 bp from the N terminus were chosen because the N-terminal region was found to be exposed, and the transmembrane regions cannot interact with the antibody. Following isopropyl β-d-thiogalactopyranoside induction of transformed bacteria carrying the pET16b-ΔLdMRP2 construct, an additional 14-kDa band was observed after SDS-PAGE as compared with uninduced bacterial extracts (data not shown). The 14-kDa recombinant polypeptide was further purified as described under “Experimental Procedures,” and a polyclonal antibody was raised against the purified protein in rabbit. The serum was collected and purified in a protein G column. The eluted fraction served as anti-MRP2 polyclonal antibody.
Western blot analysis showed that there was an 8-fold overexpression of LdMRP2-like transporter in the pB25R cell type compared with wild type parasites (Fig. 4A). To our surprise, the antibody produced no signal when analyzed by flow cytometry. When the cell membrane was permeabilized with 0.1% Triton X-100, the resistant parasites showed 6.2 times greater MRP2 expression compared with the wild type counterparts. To rule out the possibility of cross-reactivity of anti-mouse MRP2 antibody, we repeated the same experiment but with the anti-mouse MRP2 antibody. We did not observe any change in the mean fluorescence intensity in our flow cytometric experiment, and this antibody was unresponsive in Western blot (data not shown).Upon prolonged incubation in drug-free medium, the expression of MRP2 protein fell appreciably, and the percent population was decreased to 3.5-fold compared with BLN-treated promastigotes (Fig. 4B). This suggests that MRP2 transporters are not localized on the cell surface but are intracellularly localized.
FIGURE 4.
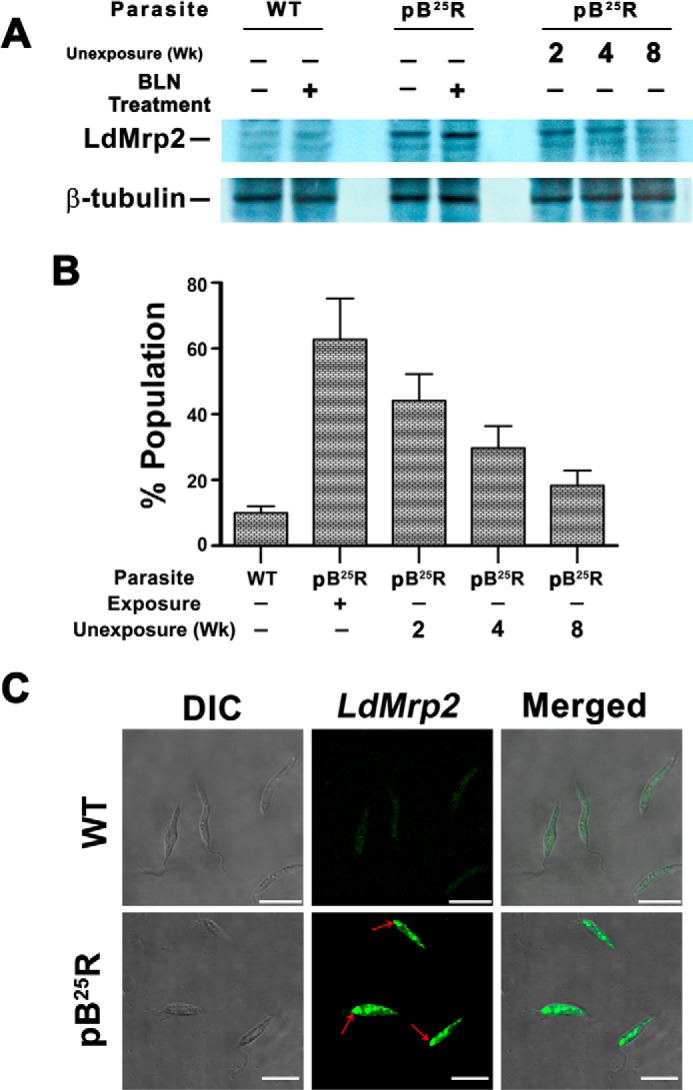
A, up-regulated expression of LdMRP2 in pB25R parasites. Shown is Western blot analysis of total proteins from sensitive Leishmania parasites (WT) and L. donovani parasites resistant to 25 μm BLN (pB25R) either incubated with BLN (lane 2 for WT and lane 4 for pB25R parasites) or in the absence of BLN (lane 1 for WT and lane 3 for pB25R parasites) using an anti-LdMRP2 antibody raised in rabbit using the first 130 amino acids of LdMRP2 protein. Molecular mass standards (kDa) are from Roche Applied Science. The reversal of phenotypic expression was also checked after long term exposure to BLN as indicated. B, flow cytometry analysis of a population of MRP2 protein-expressing pB25R parasites. WT or pB25R parasites were fixed followed by permeabilization with 0.1% Triton X-100. The parasites were incubated with polyclonal rabbit LdMRP2 antiserum (1:200) and finally visualized by fluorescein-conjugated secondary antibody (Atto 488), and the population of positive cells was counted using a BD Biosciences flow cytometer. C, intracellular localization of LdMRP2 inside parasites. WT or pB25R parasites were fixed and permeabilized with 0.1% Triton X-100. The parasites were stained with polyclonal rabbit antiserum followed by fluorescein-conjugated secondary antibody (Atto 488). The images shown are differential interference contrast images (DIC) (left), fluorescence only images (Atto 488) (middle), and a merge of the differential interference contrast and GFP images (right). Arrows indicate the region of the flagellar pocket. Scale bars, 10 μm. Error bars represent S.D. wk, weeks.
To gain insight into the intracellular localization of L. donovani MRP2 protein, the MRP2 antibody raised in rabbit was used to probe fixed permeabilized parasites. When antiserum against recombinant LdMRP2 was used, fluorescence signals to a network of intracellular vesicle-like organelles oriented along the longitudinal axis of the parasite and to the posterior end of the parasites (Fig. 4C) were observed, whereas no fluorescence was observed when preimmune serum was used as the first antibody.
Infected Resistant Intracellular Parasites Stimulate Mf-MRP2 and Are Incurable with Baicalein Treatment
One clonal population of pB25R parasites showing high resistance to BLN was subjected to tests for intracellular amastigote susceptibility to BLN. Briefly, peritoneally lavaged macrophages were infected either with wild type AG83 or pB25R at a ratio of 10:1. After subsequent washing, infected macrophages were incubated with different concentrations (0.5, 1, 2.5, 5, 10, 20, and 25 μm) of BLN for 24 h and fixed, and intracellular amastigotes were counted following Giemsa staining (Fig. 5A). The EC50 of intracellular parasite clearance was determined using nonlinear curve fitting in GraphPad Prism 5.0 (Table 3). Infected Mf ranged from 82 to 94%, and the number of amastigotes/100 Mf ranged from 590 to 820. The parasite burden was reduced by almost 91% at 10 μm and no visible parasites were seen at 25 μm concentration in wild type parasites, whereas in pB25R-infected macrophages, BLN up to 25 μm could only eliminate up to 15% of intracellular amastigotes.
FIGURE 5.

A, effectiveness of clearance of internalized wild type L. donovani (AG83) and resistant L. donovani pB25R infection by baicalein from in vitro infected mouse Mf. Macrophages from peritoneal exudate of a Balb/c mouse were infected either with wild type AG83 or BLN-resistant pB25R parasites. Twenty-four hours postinfection, cultures were treated with BLN as indicated under “Experimental Procedures.” Incubations were carried out for 24 h. Cells were fixed, stained with Giemsa, and counted under a bright field microscope. For pB25R parasitic infection, probenecid and MK571 pretreatment were performed followed by baicalein treatment. Results shown are the means of three independent experiments and are plotted as mean ± S.D. ***, p < 0.001 compared with 1 μm inhibitor treatment. B, accumulation of BLN in macrophages infected either with wild type or BLN-resistant parasites. Intracellular total BLN content was measured by fluorescence spectroscopy. Baicalein at a concentration of 25 μm was added to macrophages after infection for 24 h with wild type AG83 or pB25R parasites. Baicalein accumulation was measured up to 10 h of incubation. To confirm the role of ABC transporters, pretreatment with probenecid, verapamil, or MK571 was followed by incubation with BLN. The fitted lines (sigmoidal) from these data points (n = 3) have R2 values of 0.9904, 0.8243, 0. 9126, 0.8947, and 0.8843, respectively. C, flow cytometry analysis of Mf-MRP2 in infected cells. Macrophages from peritoneal exudates were cultured, and then the cells were left uninfected or infected with either WT AG83 parasites or pB25R parasites for the indicated time (hours). Cells were then isolated, stained with anti-mouse MRP2 antibody, and counterstained with FITC. The cells were analyzed in a BD FACSAria II cytometer. MFI, mean fluorescence intensity. D, Western blot analysis of MRP2 level in infected macrophages. Equal numbers of cells were left uninfected or infected with either WT AG83 or pB25R parasites for the indicated time (hours). Cells were isolated and lysed as described under “Experimental Procedures.” Total cell proteins (25 μg) were separated on a 7.5% polyacrylamide gel, and immunochemical analysis was performed using anti-MRP2 antibody. As loading controls, cytoplasmic extracts were probed for GAPDH. E, confocal images of macrophage MRP2 expression after pB25R infection. Macrophages from peritoneal exudate of Balb/c mice were infected either with wild type AG83 or BLN-resistant pB25R parasites. After 6 or 12 h of infection, the excess parasites were washed off, and anti-mouse MRP2 antibody was used to probe the status of MRP2 expression on the cell surface. pB25R-Mf show up-regulated MRP2 as early as 6 h postinfection, and MRP2 increases up to 12 h of infection. Scale bars, 10 μm. Error bars represent S.D. mu, murine; DIC, differential interference contrast.
TABLE 3.
Sensitivity of WT and resistant parasites inside macrophages
| Parasite | EC50 |
|---|---|
| μm | |
| AG83 | 2.32 ± 0.51 |
| pB25R | >60 |
Because the efflux of drug from the cell is well known to be an important mechanism of drug resistance, especially in cancer and leishmaniasis (28), we measured the total intracellular BLN content in Mf infected with wild type and BLN-resistant L. donovani parasites. The BLN content in AG83-Mf increased sharply after 30 min following drug treatment, reaching a steady state level at 8 h post-treatment. On the contrary, BLN content in Mf infected with pB25R increased slightly until 3 h post-BLN treatment and rapidly declined thereafter, reaching almost the basal level (Fig. 5B). This suggested that, in contrast to AG83-Mf, pB25R-Mf were unable to retain BLN intracellularly upon BLN treatment.
Overexpression of ABC transporters causes multiple drug resistance in different diseases like cancers, HIV infection, mycobacterium-HIV coinfection, and resistant Leishmania isolates. To determine whether higher expression levels of permeability glycoprotein and MRP are indeed involved in the removal of BLN, pretreatment with inhibitors of ABC transporters, viz. permeability glycoprotein-specific inhibitor verapamil, MRP-specific inhibitor probenecid, MRP1-specific inhibitor lovastatin, and MRP2-specific inhibitor MK571, were tested for BLN retention in pB25R-infected macrophages (Fig. 5B). BLN was retained in the presence of the MRP inhibitor probenecid but not in the presence of verapamil, suggesting that MRP transporters are active during resistant parasite infection. To identify the specific MRP transporter, we determined BLN retention activity after pretreating the infected cells with MRP1-specific inhibitor lovastatin or MRP2-specific inhibitor MK571. Pretreatment with MK571 only led to retention of BLN inside pB25R-Mf. The combination of probenecid or MK571 with BLN was also effective in intracellular pB25R killing, suggesting that MRP2 transporter is indeed necessary for BLN resistivity. This combinational treatment allowed clearance of more than 90% of the parasite by 24 h of BLN treatment (Fig. 5A). But the cell line WT+G-Abcc2 upon infection failed to overexpress MRP2 transporter on host macrophages (data not shown).
We further analyzed the expression status of MRP2 on the host cells, i.e. in pB25R-Mf and AG83-Mf. We observed an increased MRP2 mean fluorescence intensity in pB25R-Mf (Fig. 5C) compared with AG83-Mf. This flow cytometric observation was further confirmed by Western blot analysis (Fig. 5D) and confocal microscopy (Fig. 5E) of pB25R-Mf where higher degrees of MRP2 overexpression were observed compared with AG83-infected macrophages. A time-dependent increment of surface MRP2 of pB25R infection was observed up to 12 h. In contrast to this result, when we infected cells with WT+G and WT+G-Abcc2 parasite cultures individually, the macrophage transporter levels were found to be unaltered at the same time period.
pB25R-driven Mf-MRP2 Activation Is Associated with the Hyperactivation of 5′-Flanking Region of the Mouse Mrp2 Gene and Is Mediated by ARE1 Promoter Region
It is already known that specific stimulation with butylated hydroxyanisole or β-naphthoflavone lead to hyperactivation of 5′-flanking region of mouse Mrp2 gene. The isolated region of mouse Mrp2 promoter (Fig. 6A) contains transcription factor-binding sites for CCAAT/enhancer-binding protein, hepatocyte nuclear factor 3β (HNF-3β), HNF-4, pregnane X receptor/constitutively active receptor/farnesoid X receptor, and everted repeat 8 abundantly expressed in liver. In addition, the flanking region contains binding sites for ubiquitous factors including activator protein 1, nuclear factor Y, nuclear factor κB, and specificity protein 1 expressed in multiple tissues. Interestingly, two ARE-like sequences were found in the isolated region: one at positions −1391 to −1381 (ARE2; cTGACatgGCa with lowercase letters indicating nonconserved bases) and the other at positions −95 to −85 (ARE1; aTGACataGCa) from the transcription start site (17). For the reporter assay, the Mrp2 promoter at −1895/+99 (1995 bp) spanning all the potential transcription sites was cloned in pGL3 Basic vector (data not shown). The murine macrophage cell line RAW264.7 was transfected with this promoter construct and infected either with wild type L. donovani (AG83) or pB25R. There was a basal level of luciferase gene expression in the vector control (Fig. 6B). The enhancement was almost nil in AG83 infection, whereas in pB25R infection, there was a 5-fold luciferase activity (6 h postinfection) that increased up to 8-fold after 12 h of infection (Fig. 6B).
FIGURE 6.

A, promoter analysis of the 5′-flanking region of mouse Mrp2 gene. The full nucleotide sequence comprising all the probable promoter sites is available at GenBank under accession number AY905402. The 5′-flanking region of mouse Mrp2 (from nucleotides −1895 to +99) was cloned in pGL3 Basic vector from genomic DNA of the RAW264.7 murine macrophage cell line as described under “Experimental Procedures.” The transcription start site (+1; arrow) is indicated in the sequence; and the ATG start codon is shown in boldface. The consensus binding sites for putative regulatory elements are underlined. B, transcriptional analysis of the 5′-flanking region of the Mrp2 gene. The 5′-flanking region constructs were transiently transfected into RAW264.7 cells, and infection was established either with AG83 or pB25R parasites. After 24 h, the LUC activities were determined. The luciferase activity was normalized to respective protein load as determined by Bradford reagent. The results are expressed as the relative luciferase activity where the RAW264.7/pGL3 empty vector is the baseline of measurement. Transfections were performed in triplicate, and results are means ± S.D. C, deletion analysis of the 5′-flanking region of the Mrp2 gene. The deletion constructs p−60/+99-LUC (comprising HNF-4), p−120/+99-LUC (comprising HNF-4 and ARE1), and p−854/+99-LUC (comprising all the promoter sites) and the full-length construct p−1895/+99-LUC were transiently transfected individually to RAW264.7 cells. In another set, the p−60/+99-LUC construct underwent site-directed mutagenesis to ARE1M as described under “Experimental Procedures.” The transfected cells were further infected with pB25R parasites, and 24 h postinfection, luciferase activity was measured. The results are expressed as the percent activity of the entire isolated 5′-flanking region (with p−1895/+99-LUC normalized to 100%) after subtraction of the activity of the empty plasmid reporter. C/EBP, CCAAT/enhancer-binding protein; PXR/CAR/FXR, pregnane X receptor/constitutively active receptor/farnesoid X receptor; AP-1, activator protein 1; NF-Y, nuclear factor Y; NF-κB, nuclear factor κB; SP-1, specificity protein 1. Error bars represent S.D.
To identify the sequences in the 5′-flanking region of the Mrp2 gene that mediate constitutive gene expression, LUC reporter gene assays with constructs of 5′-unidirectionally deleted inserts (data not shown) were performed in RAW264.7 transfected cells. The deletion construct p−60/+99-LUC comprising the HNF-4 transcription activator site but excluding all other promoters was cloned in pGL3 vector, and another construct comprising only ARE1 and the HNF-4 region was cloned (p−120/+99-LUC). The constructs were transfected individually in the RAW264.7 cell line. A significant reduction in luciferase activity was noticed in p−60/+99-LUC when infected with pB25R parasites compared with that of the full-length promoter (p−1895/+99-LUC) construct (Fig. 6C). In the case of the p−120/+99-LUC construct, the luciferase activity was restored and was comparable with that of full-length promoter construct (p−1895/+99-LUC). To further ascertain the activation of the ARE1 region by pB25R parasites, we mutated ARE1 sequences in the p−120/+99-LUC construct by site-directed mutagenesis as described under “Experimental Procedures” (p−120/+99(ARE1M)-LUC). This construct was transfected into the RAW264.7 cell line, and after infection with pB25R, the luciferase activity was significantly lowered to 25% of wild type activity. Mutation of the in vivo occupied ARE1 nearly abolished luciferase activity, indicating that it is absolutely critical for transcription. These results suggested that the hyperactivation of mouse Mrp2 gene is associated with the transcription activator site ARE1 on the 5′-flanking region of Mrp2 gene.
Resistant Parasite Infection Utilizes Translocation of Nrf2 Transcription Factor into Nucleus of Macrophage via F-actin Depolymerization to Form Active ARE1-Nrf2 Complex
The activation of ARE promoter led us to investigate whether resistant parasite infection triggers translocation of Nrf2 transcription factor, which is otherwise bound with Keap1 (38) and Kelch protein. Kelch protein is highly homologous to Keap1 and is known to bind with actin (39). Confocal images of pB25R-Mf clearly showed that Nrf2 was translocated into the nucleus of the macrophages of the parasitized cells, whereas AG83 infection failed to show any signal of Nrf2 in the nucleus (Fig. 7A). Immunoblot analysis revealed that the level of cytosolic Nrf2 was gradually decreased from 3 to 24 h of infection. This was followed by a concomitant increase in the level of protein in the nucleus (Fig. 7B). Previously it was observed that the PI 3-kinase cascade is involved in the translocation of Nrf2. To probe the involvement of PI 3-kinase in the nuclear translocation of Nrf2 in the case of resistant parasite infection, the cells were pretreated with wortmannin or LY294002 followed by pB25R infection and BLN (25 μm) treatment. Western blot analysis of nuclear fractions supports the involvement of the PI3K pathway during the infection process as evidenced by a drastic decrease in band intensity of nuclear Nrf2 (Fig. 7C).
FIGURE 7.

A, activation and translocation of Nrf2 into the nuclei of macrophages infected with BLN-resistant parasites. Mouse macrophage cells were infected with either WT AG83 or BLN-resistant pB25R parasites for 6 or 12 h. Excess parasites were washed off thoroughly and fixed followed by permeabilization. Nrf2 localization was immunochemically detected using anti-Nrf2 antibody overnight followed by incubation with an Atto 594-conjugated secondary antibody. For each panel, an image of the cell nuclei stained with DNA-specific DAPI, a phase-contrast image of the cells (differential interference contrast (DIC)), and merged images are also presented. B, fractionation of mouse peritoneal macrophage cells into nuclear and cytoplasmic fractions. An equal number of cells was left uninfected or infected with either WT AG83 parasites or pB25R parasites for the indicated time (hours). Cells were isolated and fractionated into nuclear and cytoplasmic extracts (see “Experimental Procedures”). Equal amounts of protein extracts were loaded in each lane, resolved by SDS-polyacrylamide gel electrophoresis, and probed for nuclear (lower panel) and cytosolic proteins (upper panel) by immunoblot analysis. As loading controls, cytoplasmic extracts were probed for β-tubulin, and nuclear fractions were probed for H2B. C, the effects of PI 3-kinase inhibitors on the subcellular localization of Nrf2. Nrf2 localization was immunochemically assessed in cells infected either with WT AG83 parasites or pB25R parasites or BLN-treated pB25R-infected macrophages for 12 h in combination with PI 3-kinase inhibitors. The effects of PI 3-kinase inhibitors (wortmannin (WO), 0.5 μm; LY294002 (LY), 25 μm) on pB25R-inducible Nrf2 induction were determined by Western blot analysis, which confirmed decreased nuclear translocation of Nrf2. Each lane was loaded with 25 μg of nuclear proteins. Nuclear H2B served as a loading control. Scale bars, 10 μm.
Actin cytoskeleton dynamics is the major target for many intracellular events, and several transcription factors important for hypertrophy are likely controlled by free G-actin. Given the possible interaction of Nrf2 with actin-related proteins, we monitored whether pB25R infection changed the cellular filamentous structure of actin. Confocal images confirmed that in pB25R-Mf the actin cytoskeletal was partially depolymerized 6 h postinfection, whereas a substantial amount of depolymerization was observed 12 h postinfection (Fig. 8A). To our surprise, some of the phalloidin molecule localized in the nucleus of the pB25R-Mf cells (Fig. 8A, 3D Section) was absent in uninfected and AG83-Mf nuclei. To validate our confocal microscopy observation, fractionated cell extracts containing nonpolymerized G-actin and F-actin were prepared and analyzed for G- and F-actin contents to quantitate the depolymerization of the actin cytoskeleton (Fig. 8B). Uninfected control macrophages exhibited 85% of filamentous actin in the pelleted fraction, and this remained almost unaltered in wild type AG83-infected macrophages (82%). A gradual loss of F-actin content, i.e. 66 and 41%, was observed after 6 and 12 h of infection with pB25R promastigotes, respectively, with a concomitant increase in G-actin in the supernatant fraction (32 and 58%, respectively). Hence, we analyzed the interaction of actin with Nrf2 in the nuclear extracts (Fig. 8C). Like Nrf2, nuclear actin increased in a time-dependent manner. The level was saturated at the 12-h time point and was maintained at a high level in the nucleus. Previous study proved that the Nrf2 pathway is PI3K-dependent. Incubation with a PI3K inhibitor such as wortmannin or LY294002 drastically decreased the level of Nrf2-precipitable actin in the nucleus (Fig. 8D). Thus, pB25R infection increased the level of nuclear actin coimmunoprecipitated with anti-Nrf2 antibody in the cells. The level of Nrf2-bound actin was not changed in the samples of total cell lysates (Fig. 8D). The samples precipitated after reaction with rabbit IgG in the absence of anti-Nrf2 antibody did not produce any signal. The results were further confirmed by confocal microscopy where both actin and Nrf2 colocalized in the nucleus (Fig. 8E). These results demonstrated that the PI 3-kinase pathway indeed controlled the cytoskeletal rearrangements (i.e. actin depolymerization and repolymerization) in pB25R-Mf cells and might be responsible for the nuclear migration of Nrf2.
FIGURE 8.

A, effect of pB25R infection on actin filament rearrangement in mouse macrophage cells. Mouse macrophage cells were infected with either WT AG83 or BLN-resistant pB25R parasites for 6 or 12 h. The cells were fixed with paraformaldehyde followed by staining for actin with Atto 594-conjugated phalloidin and optically visualized using a confocal laser-scanning microscope (Nikon A1R). For each panel, an image of the cell nuclei stained with DNA-specific DAPI, a phase-contrast image of the cells (differential interference contrast (DIC)), and merged images are also presented. The boxed panel is the three-dimensional reconstruction (3D Section) of images of pB25R-Mf. The total field (panel I) is rotated 90° counterclockwise (panel II). x and y axis sections were cut keeping only the DAPI panel on and the red channel off (panel III). Panel IV represents another angle view of panel III. z axis sections were cut as for both DAPI and red channels to visualize violet patches inside the nucleus (blue) (panel V). Scale bars, 10 μm. B, analysis of filamentous actin depolymerization by Western blotting (WB). An equal number of cells was left uninfected or infected with either WT AG83 parasites (for 12 h) or pB25R parasites (for 6 and 12 h). Cells were isolated, and G- and F-actin were separated by ultracentrifugation followed by Western blotting of supernatants (G-actin) and pellets (F-actin). Representative Western blots (upper panel) were analyzed for F/G-actin ratios by Bio-Rad Quantity One software. Total actin served as a loading control. Lower panel, stacked bar graph of the G/F-actin ratios in the cytosol of the above stated samples (quantified from three Western blots as described under “Experimental Procedures”). C, determination of actin in the nuclei by immunoblot analysis. Immunoblot analyses of actin in the immunoprecipitates of nuclear extracts or cell lysates with anti-Nrf2 antibody are shown. The nuclear extracts or lysates prepared from the cells infected with either WT AG83 or BLN-resistant pB25R parasites for the indicated time points were subjected to immunoprecipitation (IP) with anti-Nrf2 antibody and immunoblotted for actin. D, the effects of PI 3-kinase inhibitors on nuclear actin in the immunoprecipitate with Nrf2. The cells were infected with either WT AG83 or BLN-resistant pB25R parasites for 12h in the presence or absence of wortmannin (WO; 0.5 μm) or LY294002 (LY; 25 μm) as indicated. Immunoprecipitation and immunoblot analyses were carried out as in B. E, colocalization of actin and Nrf2 in pB25R-Mf. To assess colocalization of Nrf2 with actin microfilaments, macrophage cells infected with pB25R for 24 h were stained for actin with anti-actin antibody (Ab) and for Nrf2 with anti-Nrf2 antibody and optically visualized using confocal laser-scanning microscopy. Corresponding images were superimposed to determine the degrees of overlap. Superposition of actin with Nrf2 showed a complete overlap of two staining patterns with DAPI-stained nucleus. Scale bars, 10 μm. F, actin depolymerization-mediated Nrf2 translocation in nucleus. The levels of nuclear Nrf2 in the immunoprecipitate with actin was analyzed from the isolated nuclei of the peritoneal macrophage cells infected with either WT AG83 or BLN-resistant pB25R parasites for 12 h in the presence or absence of 2 μm phalloidin (Pha). Immunoprecipitation was carried out with anti-actin antibody, and the immunoblot analyses were carried out with anti-Nrf2 antibody. pB25R parasite-infected macrophages were treated with BLN (20 μm) in another set. G, BLN-resistant pB25R parasites increased the binding of Nrf2-protein complexes to the ARE1 sequence. Lane 1, double-stranded ARE1 probe. EMSA was performed using nuclear extracts (5 μg) obtained from control mouse peritoneal macrophage cells (lane 2) or those infected with either WT AG83 parasites (lane 3) or pB25R resistant parasites (lane 4) for 12 h and the radiolabeled probe containing the ARE1 or ARE1M sequence. A supershift assay (lane 5) was performed using nuclear extracts (15 μg) from mouse peritoneal macrophage cells infected with pB25R resistant parasites for the above stated periods containing the ARE1 sequence in the presence of anti-Nrf2 antibody. The shifted bands are denoted by a single arrow, and the supershifted bands are denoted by a bold arrow. EMSA also was performed with ARE1M probe in the presence of nuclear extracts (5 μg) obtained from nuclei of pB25R-Mf (lane 6).
Studies were extended to confirm the interaction between Nrf2 and actin in the cells treated with phalloidin, an agent that prevents depolymerization of actin filaments (40, 41). Phalloidin at a concentration of 5 μm actively inhibited actin depolymerization in pB25R-Mf cells. As a result, the increase in the level of nuclear actin coimmunoprecipitated with Nrf2 in pB25R-Mf was also abolished in the cells treated with phalloidin (Fig. 8F), which ultimately decreased the MRP2-level on pB25R-Mf (data not shown). These results further supported the conclusion that MRP2 induction by pB25R infection was dependent on actin rearrangements.
To assess whether pB25R infection induced ARE1-activated nuclear transcription factor, nuclear extracts from pB25R-Mf cells were analyzed for binding capabilities on the MRP2 ARE1 sequences. Remarkably, as shown in Fig. 8G, the presence of nuclear extracts from pB25R-Mf increased the binding of protein complexes to the ARE1 sequence compared with AG83-Mf nuclear extracts. The increase of activity closely paralleled the loss of activity observed in the luciferase reporter assay. The presence of Nrf2 in ARE1-protein complexes was determined by a supershift assay using anti-Nrf2 antibody (sc-722x). The addition of anti-Nrf2 antibody resulted in the appearance of a strongly supershifted band, indicating that Nrf2 is present in the ARE1-protein complex. When ARE1M mutant probe was used, the shifted signal decreased appreciably, confirming the binding of Nrf2 to the ARE1 sequence.
DISCUSSION
A rational approach to chemotherapy is always the mainstay in treatment of leishmaniasis. A better understanding of chemoresistance with molecular insights and its relation to parasite protection inside the host underlies the need for new treatments. The unusual bisubunit topoisomerase IB of Leishmania is known to be an attractive target for chemotherapy (42). Among the topoisomerase inhibitors, flavones such as BLN, luteolin, and quercetin are potent antileishmanial compounds (23, 43). Although the most potent antileishmanial compound, SAG, has been used for almost 10 decades, recent clinical isolates of Leishmania show SAG unresponsiveness. This resistance has been found to be due to the interplay among defective drug uptake, enhanced efflux from the host, and sequestration of active molecules in the parasite (4, 44). Resistant clinical isolates not only overexpress P-glycoprotein A and down-regulate aquaglyceroporin for active antimony accumulation but also enhance host MDR1 and MRP1 on the Mf surfaces for active efflux of SAG (32). Therefore, a detailed study on the active pump associated with BLN transport and altered signaling cascade inside host macrophages is needed for understanding the drug-mediated resistance.
In the present study, we identified an MRP2 transporter that is different from the eukaryotic counterpart. Unlike clinical isolates of Leishmania resistant to SAG, BLN resistance is due to enhanced efflux of the drug from the parasite from its flagellar pocket region, which is a major site for exocytosis-mediated processes (27). BLN is cytotoxic to Leishmania cells as it stabilizes the topoisomerase I-DNA complex (5) and activates LdEndoG to form the LdFEN1-TatD-EndoG complex for DNA degradation (23). Hence, the development of BLN resistance in Leishmania is a gradual process that includes different cascades in parasites to circumvent the cell death. This study will help in understanding the changes in altered drug transport that these parasites develop to manifest a chemoresistant phenotype before generation of spontaneous mutations to its target gene. The steep increase in ED50 value for pB25R indicates that the parasites are about 32-fold resistant to the drug over WT promastigotes. The stability of the drug resistance is transient as the removal of drug pressure reverts parasites back to wild type conditions. Analysis of resistance mechanisms revealed that there is an up-regulation of the efflux pump. Removal of drug pressure gradually reverts resistant parasites to WT phenotype, and thus, this unstable resistance cannot be corroborated with involvement of target gene mutation or altered metabolic adaptation.
Previous studies show that baicalein is an apoptotic agent. Altered efflux of drug leads to less baicalein accumulation and as a result escape from programmed cell death. Depletion of cellular ATP levels and destabilization of cellular protein of pB25R parasites confirm that the efflux is an active transport process, and thus, a lower accumulation of drug is observed within 3 h of treatment. An increase in calcein fluorescence indicates higher MRP-like activity in resistant promastigotes, whereas pretreatment with probenecid showed no significant accumulation of BLN, confirming that MDR activity is not associated with drug efflux. Overexpression of different types of transporters is associated with chemoresistance. Among them, ABCG6 has been linked to CPT resistance (17). In clinical SAG-resistant isolates, an up-regulation of MRPA has been characterized (4), and ABCG4 is associated with miltefosine-mediated resistance (45). Among the known ABCC transporters, pB25R parasites have an increased number of ABCC2 transporters. By reducing its target gene expression (LdTOP1L), the parasite gains a survival advantage. A previous study on mammalian cells shows that MRP2 is primarily responsible for the baicalein-mediated resistance phenotype (46). A striking difference from mammalian MRP2 localization is that the transporter is located inside the parasites. This overexpression with localization in flagellar pockets results in enhanced efflux of drug.
The induction of phase II genes in response to reactive chemical stress is regulated primarily at the transcriptional level by a cis-acting element, ARE (47). Activation of gene transcription in response to cellular defense through the ARE is mediated primarily by Nrf2. The transcription factor Nrf2 complexes with Maf family proteins to transactivate ARE1 promoter. Under the basal condition, the cytosolic regulator protein Keap1 binds tightly to Nrf2 at its N-terminal Neh2 domain (38). In addition to being a requirement for Nrf2, increased Akt activity also elevates Nrf2 activity (48). Earlier it was found that in cancer cell lines gain of function of the PI3K/Akt pathway regulates Nrf2 (49). PI3K is activated by membrane receptor tyrosine kinase(s) and forms a complex with phosphotyrosine residues in the activator receptor. Relocation and rearrangement of cytoskeletal actin are dependent on the activities of these kinases (50, 51). The subcellular localization of Nrf2 is completely dependent on the actin microfilament network. A time course study revealed that in BLN-resistant pB25R infection actin was translocated to the nucleus as early as 3 h. Immunocytochemistry revealed that pB25R infection stimulates Nrf2 to be completely colocalized with the nucleus in the cells. Colocalization of Nrf2 with actin and dependence on the PI3K pathway for actin rearrangements strongly support the possibility that nuclear translocation of Nrf2 and subsequent ARE activation are mediated by actin binding and rearrangement (52). Time course analysis revealed that actin-bound Nrf2 translocated into the nucleus, and this was supported by coimmunoprecipitation analysis.
Nrf2 activity is regulated in part by the actin-associated Keap1 protein, which was initially proposed to act by binding and tethering the transcription factor in the cytoplasm. Because of the large number of cysteine residues within its primary structure, Keap1 is an attractive target for ubiquitin-mediated proteolysis of Nrf2 (53). Upon infection, PI3K-mediated Akt is activated, suggesting that Akt-mediated phosphorylation of Nrf2 may be the reason for the reduced affinity of Keap1 for Nrf2, ultimately allowing Nrf2 to enter the nucleus (54). The action of Keap1 is analogous to that of IKκB, which prevents activation and translocation of transcription factor NFκB (55, 56).
The regulation of the Nrf2-ARE pathway may also extend to the regulation of the human MRP1 gene, which is the homologue of MRP2 in the basolateral domain of polarized cells (57). In fact, two ARE-like sequences (located at positions −1098 to −1087 and −327 to −335) and the NF-E2-like elements have been identified in the 5′-flanking region of human MRP1 gene. Apart from this element, there is an additional NF-E2-like consensus binding site located at positions −235 to −227 in the investigated part of the MRP2 promoter region (35). Analysis of sequences of the 5′-flanking region of human MRP2 gene (GenBankTM accession number AF144630) revealed an additional ARE site (at positions −2865 to −2856), indicating that distribution of ARE sequences in the promoter region is similar in human MRP2 and mouse Mrp2 genes (58).
Previous studies have demonstrated that phosphorylation of Nrf2 and subsequent nuclear translocation are critical for cell survival (59, 60). It can be postulated that cross-talk between PI3K and the Nrf2 pathway leads to activation of Akt and Bad through Nrf2 activation in pB25R-infected cells, suggesting a role for Nrf2 in the induction of cell survival, a situation favorable for Leishmania pathogenicity and colonization. The molecular mechanisms underlying Nrf2-mediated cell survival are still not clearly understood. Based on previous studies and our results, it is likely that Nrf2-dependent induction of phase II detoxifying enzyme expression will contribute to cell survival by Leishmania infection.
In agreement with earlier reports that emphasize the notion that drug resistance is multifactorial (61, 62), our study shows that drug resistance in parasites is not only due to reprogramming of expression of host cell ABC transporters but also due to overexpression of ABC transporters on the parasites to evade the adverse action of the drug. The mechanisms of ABC transporter overexpression on parasites and parasite-mediated overexpression of ABC transporters may be entirely unrelated and different. It was shown earlier (63) that both a laboratory-generated antimony-resistant promastigote (GE1) and a clinical field isolate (K29) induce overexpression of host cell MDR1 (32). Mukhopadhyay et al. (4) reported that a recent antimony-resistant clinical field isolate shows overexpression of promastigote MRPA, and this isolate induces macrophage MDR1 overexpression by transcriptional regulation. They also showed that a unique surface glycan specific for drug-resistant parasites is responsible for inducing MDR1 overexpression on the surface of the host cell (64). Similarly, there may be several other factors specific for baicalein-resistant parasites that are responsible for inducing MRP2 overexpression in infected macrophages that have yet to be determined.
In conclusion, the ARE-Nrf2 transcriptional pathway plays an important role in the regulation of genes that control the expression of protein as well as transporters critical in the extrusion of drugs that may be associated with other clinical resistant phenotypes. In this regard, Nrf2, a master regulator of the induction of a battery of transporters as well as phase II detoxifying genes in the xenobiotic detoxification process, may be a promising target to counteract multidrug resistance phenomenon and its consequences to dreaded leishmaniasis.
Acknowledgments
We thank Prof. S. Roy, the director of the Indian Institute of Chemical Biology, for interest in this work. We also thank Dr. Anupam Banerjee for confocal imaging analysis.
This research work was supported by Department of Biotechnology, Government of India Grant BT/PR4456/MED/29/355/2012.
- BLN
- baicalein
- ARE
- antioxidant response element
- Nrf2
- NFE2-related factor 2
- Mf
- macrophage(s)
- MRP
- multidrug-resistant protein
- LdTOP1L
- L. donovani topoisomerase 1 large subunit
- ABC
- ATP-binding cassette
- M199
- Medium 199
- ED
- effective dose
- SAG
- sodium antimony gluconate
- DIM
- 3,3′-diindolylmethane
- HBS
- HEPES-buffered saline
- Rh123
- Rhodamine 123
- NEM
- N-ethylmaleimide
- SAP
- secreted acid phosphatase
- LdTOP1LS
- bisubunit L. donovani topoisomerase I
- LUC
- luciferase
- CPT
- camptothecin
- MDR
- multidrug resistance protein
- HNF
- hepatocyte nuclear factor
- EndoG
- endonuclease G
- Ld
- L. donovani
- PI
- phosphoinositol
- aMRP2
- antisense MRP2.
REFERENCES
- 1. Kobets T., Grekov I., Lipoldova M. (2012) Leishmaniasis: prevention, parasite detection and treatment. Curr. Med. Chem. 19, 1443–1474 [DOI] [PubMed] [Google Scholar]
- 2. Kedzierski L. (2011) Leishmaniasis. Hum. Vaccin. 7, 1204–1214 [DOI] [PubMed] [Google Scholar]
- 3. Kaye P., Scott P. (2011) Leishmaniasis: complexity at the host-pathogen interface. Nat. Rev. Microbiol. 9, 604–615 [DOI] [PubMed] [Google Scholar]
- 4. Mukhopadhyay R., Mukherjee S., Mukherjee B., Naskar K., Mondal D., Decuypere S., Ostyn B., Prajapati V. K., Sundar S., Dujardin J. C., Roy S. (2011) Characterisation of antimony-resistant Leishmania donovani isolates: biochemical and biophysical studies and interaction with host cells. Int. J. Parasitol. 41, 1311–1321 [DOI] [PubMed] [Google Scholar]
- 5. Das B. B., Sen N., Roy A., Dasgupta S. B., Ganguly A., Mohanta B. C., Dinda B., Majumder H. K. (2006) Differential induction of Leishmania donovani bi-subunit topoisomerase I-DNA cleavage complex by selected flavones and camptothecin: activity of flavones against camptothecin-resistant topoisomerase I. Nucleic Acids Res. 34, 1121–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Marquis J. F., Hardy I., Olivier M. (2005) Topoisomerase I amino acid substitutions, Gly185Arg and Asp325Glu, confer camptothecin resistance in Leishmania donovani. Antimicrob. Agents Chemother. 49, 1441–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sauvage V., Aubert D., Escotte-Binet S., Villena I. (2009) The role of ATP-binding cassette (ABC) proteins in protozoan parasites. Mol. Biochem. Parasitol. 167, 81–94 [DOI] [PubMed] [Google Scholar]
- 8. Dean M., Annilo T. (2005) Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genomics Hum. Genet. 6, 123–142 [DOI] [PubMed] [Google Scholar]
- 9. Leprohon P., Légaré D., Girard I., Papadopoulou B., Ouellette M. (2006) Modulation of Leishmania ABC protein gene expression through life stages and among drug resistant parasites. Eukaryot. Cell 5, 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fairlamb A. H., Cerami A. (1992) Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 46, 695–729 [DOI] [PubMed] [Google Scholar]
- 11. Légaré D., Richard D., Mukhopadhyay R., Stierhof Y. D., Rosen B. P., Haimeur A., Papadopoulou B., Ouellette M. (2001) The Leishmania ATP binding cassette protein PGPA is an intracellular metal-thiol transporter ATPase. J. Biol. Chem. 276, 26301–26307 [DOI] [PubMed] [Google Scholar]
- 12. Dey S., Ouellette M., Lightbody J., Papadopoulou B., Rosen B. P. (1996) An ATP-dependent As(III)-glutathione transport system in membrane vesicles of Leishmania tarentolae. Proc. Natl. Acad. Sci. U.S.A. 93, 2192–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jedlitschky G., Leier I., Buchholz U., Barnouin K., Kurz G., Keppler D. (1996) Transport of glutathione, glucuronate, and sulfate conjugates by the MRP gene-encoded conjugate export pump. Cancer Res. 56, 988–994 [PubMed] [Google Scholar]
- 14. Gueiros-Filho F. J., Viola J. P., Gomes F. C., Farina M., Lins U., Bertho A. L., Wirth D. F., Lopes U. G. (1995) Leishmania amazonensis: multidrug resistance in vinblastine-resistant promastigotes is associated with rhodamine 123 efflux, DNA amplification, and RNA overexpression of a Leishmania mdr1 gene. Exp. Parasitol. 81, 480–490 [DOI] [PubMed] [Google Scholar]
- 15. Araújo-Santos J. M., Parodi-Talice A., Castanys S., Gamarro F. (2005) The overexpression of an intracellular ABCA-like transporter alters phospholipid trafficking in Leishmania. Biochem. Biophys. Res. Commun. 330, 349–355 [DOI] [PubMed] [Google Scholar]
- 16. Castanys-Muñoz E., Alder-Baerens N., Pomorski T., Gamarro F., Castanys S. (2007) A novel ATP-binding cassette transporter from Leishmania is involved in transport of phosphatidylcholine analogues and resistance to alkylphospholipids. Mol. Microbiol. 64, 1141–1153 [DOI] [PubMed] [Google Scholar]
- 17. BoseDasgupta S., Ganguly A., Roy A., Mukherjee T., Majumder H. K. (2008) A novel ATP-binding cassette transporter, ABCG6 is involved in chemoresistance of Leishmania. Mol. Biochem. Parasitol. 158, 176–188 [DOI] [PubMed] [Google Scholar]
- 18. Leprohon P., Légaré D., Ouellette M. (2009) Intracellular localization of the ABCC proteins of Leishmania and their role in resistance to antimonials. Antimicrob. Agents Chemother. 53, 2646–2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Doyle P. S., Engel J. C., Pimenta P. F., da Silva P. P., Dwyer D. M. (1991) Leishmania donovani: long-term culture of axenic amastigotes at 37 °C. Exp. Parasitol. 73, 326–334 [DOI] [PubMed] [Google Scholar]
- 20. Kaur J., Dey C. S. (2000) Putative P-glycoprotein expression in arsenite-resistant Leishmania donovani down-regulated by verapamil. Biochem. Biophys. Res. Commun. 271, 615–619 [DOI] [PubMed] [Google Scholar]
- 21. Mandal G., Sarkar A., Saha P., Singh N., Sundar S., Chatterjee M. (2009) Functionality of drug efflux pumps in antimonial resistant Leishmania donovani field isolates. Indian J. Biochem. Biophys. 46, 86–92 [PubMed] [Google Scholar]
- 22. Pérez-Victoria F. J., Castanys S., Gamarro F. (2003) Leishmania donovani resistance to miltefosine involves a defective inward translocation of the drug. Antimicrob. Agents Chemother. 47, 2397–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. BoseDasgupta S., Das B. B., Sengupta S., Ganguly A., Roy A., Dey S., Tripathi G., Dinda B., Majumder H. K. (2008) The caspase-independent algorithm of programmed cell death in Leishmania induced by baicalein: the role of LdEndoG, LdFEN-1 and LdTatD as a DNA ‘degradesome’. Cell Death Differ. 15, 1629–1640 [DOI] [PubMed] [Google Scholar]
- 24. Dodge M. A., Waller R. F., Chow L. M., Zaman M. M., Cotton L. M., McConville M. J., Wirth D. F. (2004) Localization and activity of multidrug resistance protein 1 in the secretory pathway of Leishmania parasites. Mol. Microbiol. 51, 1563–1575 [DOI] [PubMed] [Google Scholar]
- 25. Vince J. E., Tull D. L., Spurck T., Derby M. C., McFadden G. I., Gleeson P. A., Gokool S., McConville M. J. (2008) Leishmania adaptor protein-1 subunits are required for normal lysosome traffic, flagellum biogenesis, lipid homeostasis, and adaptation to temperatures encountered in the mammalian host. Eukaryot. Cell 7, 1256–1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cuvillier A., Redon F., Antoine J. C., Chardin P., DeVos T., Merlin G. (2000) LdARL-3A, a Leishmania promastigote-specific ADP-ribosylation factor-like protein, is essential for flagellum integrity. J. Cell Sci. 113, 2065–2074 [DOI] [PubMed] [Google Scholar]
- 27. Mullin K. A., Foth B. J., Ilgoutz S. C., Callaghan J. M., Zawadzki J. L., McFadden G. I., McConville M. J. (2001) Regulated degradation of an endoplasmic reticulum membrane protein in a tubular lysosome in Leishmania mexicana. Mol. Biol. Cell 12, 2364–2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mukherjee A., Padmanabhan P. K., Singh S., Roy G., Girard I., Chatterjee M., Ouellette M., Madhubala R. (2007) Role of ABC transporter MRPA, γ-glutamylcysteine synthetase and ornithine decarboxylase in natural antimony-resistant isolates of Leishmania donovani. J. Antimicrob. Chemother. 59, 204–211 [DOI] [PubMed] [Google Scholar]
- 29. Das B. B., Sen N., Ganguly A., Majumder H. K. (2004) Reconstitution and functional characterization of the unusual bi-subunit type I DNA topoisomerase from Leishmania donovani. FEBS Lett. 565, 81–88 [DOI] [PubMed] [Google Scholar]
- 30. Chowdhury S., Mukherjee T., Mukhopadhyay R., Mukherjee B., Sengupta S., Chattopadhyay S., Jaisankar P., Roy S., Majumder H. K. (2012) Niranthin, a lignan, intercepts Leishmania donovani replication by poisoning topoisomerase IB of parasite and ameliorates Th1 response to cure infected-BALB/c mice. EMBO Mol. Med. 4, 1126–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chowdhury S., Mukherjee T., Sengupta S., Chowdhury S. R., Mukhopadhyay S., Majumder H. K. (2011) Novel betulin derivatives as antileishmanial agents with mode of action targeting type IB DNA topoisomerase. Mol. Pharmacol. 80, 694–703 [DOI] [PubMed] [Google Scholar]
- 32. Mookerjee Basu J., Mookerjee A., Banerjee R., Saha M., Singh S., Naskar K., Tripathy G., Sinha P. K., Pandey K., Sundar S., Bimal S., Das P. K., Choudhuri S. K., Roy S. (2008) Inhibition of ABC transporters abolishes antimony resistance in Leishmania infection. Antimicrob. Agents Chemother. 52, 1080–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Holling T. M., van der Stoep N., Quinten E., van den Elsen P. J. (2002) Activated human T cells accomplish MHC class II expression through T cell-specific occupation of class II transactivator promoter III. J. Immunol. 168, 763–770 [DOI] [PubMed] [Google Scholar]
- 34. Lin Y. Z., Yao S. Y., Veach R. A., Torgerson T. R., Hawiger J. (1995) Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 270, 14255–14258 [DOI] [PubMed] [Google Scholar]
- 35. Stöckel B., König J., Nies A. T., Cui Y., Brom M., Keppler D. (2000) Characterization of the 5′-flanking region of the human multidrug resistance protein 2 (MRP2) gene and its regulation in comparison with the multidrug resistance protein 3 (MRP3) gene. Eur. J. Biochem. 267, 1347–1358 [DOI] [PubMed] [Google Scholar]
- 36. Kang K. W., Lee S. J., Park J. W., Kim S. G. (2002) Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2-related factor 2 through actin rearrangement in response to oxidative stress. Mol. Pharmacol. 62, 1001–1010 [DOI] [PubMed] [Google Scholar]
- 37. Smith A. J., Humphries S. E. (2009) Characterization of DNA-binding proteins using multiplexed competitor EMSA. J. Mol. Biol. 385, 714–717 [DOI] [PubMed] [Google Scholar]
- 38. Itoh K., Wakabayashi N., Katoh Y., Ishii T., Igarashi K., Engel J. D., Yamamoto M. (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 13, 76–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Way M., Sanders M., Garcia C., Sakai J., Matsudaira P. (1995) Sequence and domain organization of scruin, an actin-cross-linking protein in the acrosomal process of Limulus sperm. J. Cell Biol. 128, 51–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lader A. S., Kwiatkowski D. J., Cantiello H. F. (1999) Role of gelsolin in the actin filament regulation of cardiac L-type calcium channels. Am. J. Physiol. Cell Physiol. 277, C1277–C1283 [DOI] [PubMed] [Google Scholar]
- 41. Sullivan R., Price L. S., Koffer A. (1999) Rho controls cortical F-actin disassembly in addition to, but independently of, secretion in mast cells. J. Biol. Chem. 274, 38140–38146 [DOI] [PubMed] [Google Scholar]
- 42. Reguera R. M., Díaz-González R., Pérez-Pertejo Y., Balaña-Fouce R. (2008) Characterizing the bi-subunit type IB DNA topoisomerase of Leishmania parasites: a novel scenario for drug intervention in trypanosomatids. Curr. Drug Targets 9, 966–978 [DOI] [PubMed] [Google Scholar]
- 43. Mittra B., Saha A., Chowdhury A. R., Pal C., Mandal S., Mukhopadhyay S., Bandyopadhyay S., Majumder H. K. (2000) Luteolin, an abundant dietary component is a potent anti-leishmanial agent that acts by inducing topoisomerase II-mediated kinetoplast DNA cleavage leading to apoptosis. Mol. Med. 6, 527–541 [PMC free article] [PubMed] [Google Scholar]
- 44. Singh N., Kumar M., Singh R. K. (2012) Leishmaniasis: current status of available drugs and new potential drug targets. Asian Pac. J. Trop. Med. 5, 485–497 [DOI] [PubMed] [Google Scholar]
- 45. Seifert K., Matu S., Javier Pérez-Victoria F., Castanys S., Gamarro F., Croft S. L. (2003) Characterisation of Leishmania donovani promastigotes resistant to hexadecylphosphocholine (miltefosine). Int. J. Antimicrob. Agents 22, 380–387 [DOI] [PubMed] [Google Scholar]
- 46. Akao T., Sato K., Hanada M. (2009) Hepatic contribution to a marked increase in the plasma concentration of baicalein after oral administration of its aglycone, baicalein, in multidrug resistance-associated protein 2-deficient rat. Biol. Pharm. Bull. 32, 2079–2082 [DOI] [PubMed] [Google Scholar]
- 47. Nguyen T., Nioi P., Pickett C. B. (2009) The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 284, 13291–13295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Martin D., Rojo A. I., Salinas M., Diaz R., Gallardo G., Alam J., De Galarreta C. M., Cuadrado A. (2004) Regulation of heme oxygenase-1 expression through the phosphatidylinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in response to the antioxidant phytochemical carnosol. J. Biol. Chem. 279, 8919–8929 [DOI] [PubMed] [Google Scholar]
- 49. Min K. J., Lee J. T., Joe E. H., Kwon T. K. (2011) An IκBα phosphorylation inhibitor induces heme oxygenase-1(HO-1) expression through the activation of reactive oxygen species (ROS)-Nrf2-ARE signaling and ROS-PI3K/Akt signaling in an NF-κB-independent mechanism. Cell. Signal. 23, 1505–1513 [DOI] [PubMed] [Google Scholar]
- 50. Fukami K., Endo T., Imamura M. (1994) α-Actinin and vinculin are PIP2-binding proteins involved in signaling by tyrosine kinase. J. Biol. Chem. 269, 1518–1522 [PubMed] [Google Scholar]
- 51. Grassmé H. U., Ireland R. M., van Putten J. P. (1996) Gonococcal opacity protein promotes bacterial entry-associated rearrangements of the epithelial cell actin cytoskeleton. Infect. Immun. 64, 1621–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Beverley S. M. (1996) Hijacking the cell: parasites in the driver's seat. Cell 87, 787–789 [DOI] [PubMed] [Google Scholar]
- 53. McMahon M., Itoh K., Yamamoto M., Hayes J. D. (2003) Keap1-dependent proteasomal degradation of transcription factor Nrf2 contributes to the negative regulation of antioxidant response element-driven gene expression. J. Biol. Chem. 278, 21592–21600 [DOI] [PubMed] [Google Scholar]
- 54. Huang H. C., Nguyen T., Pickett C. B. (2002) Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J. Biol. Chem. 277, 42769–42774 [DOI] [PubMed] [Google Scholar]
- 55. Itoh K., Mochizuki M., Ishii Y., Ishii T., Shibata T., Kawamoto Y., Kelly V., Sekizawa K., Uchida K., Yamamoto M. (2004) Transcription factor Nrf2 regulates inflammation by mediating the effect of 15-deoxy-Δ12,14-prostaglandin J2. Mol. Cell. Biol. 24, 36–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gong P., Stewart D., Hu B., Li N., Cook J., Nel A., Alam J. (2002) Activation of the mouse heme oxygenase-1 gene by 15-deoxy-Δ12,14-prostaglandin J2 is mediated by the stress response elements and transcription factor Nrf2. Antioxid. Redox Signal. 4, 249–257 [DOI] [PubMed] [Google Scholar]
- 57. Hayashi A., Suzuki H., Itoh K., Yamamoto M., Sugiyama Y. (2003) Transcription factor Nrf2 is required for the constitutive and inducible expression of multidrug resistance-associated protein 1 in mouse embryo fibroblasts. Biochem. Biophys. Res. Commun. 310, 824–829 [DOI] [PubMed] [Google Scholar]
- 58. Vollrath V., Wielandt A. M., Iruretagoyena M., Chianale J. (2006) Role of Nrf2 in the regulation of the Mrp2 (ABCC2) gene. Biochem. J. 395, 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cullinan S. B., Zhang D., Hannink M., Arvisais E., Kaufman R. J., Diehl J. A. (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23, 7198–7209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cullinan S. B., Diehl J. A. (2004) PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 279, 20108–20117 [DOI] [PubMed] [Google Scholar]
- 61. Haldar A. K., Yadav V., Singhal E., Bisht K. K., Singh A., Bhaumik S., Basu R., Sen P., Roy S. (2010) Leishmania donovani isolates with antimony-resistant but not -sensitive phenotype inhibit sodium antimony gluconate-induced dendritic cell activation. PLoS Pathog. 6, e1000907. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62. Decuypere S., Rijal S, Yardley V., De Doncker S., Laurent T., Khanal B., Chappuis F., Dujardin J. C. (2005) Gene expression analysis of the mechanism of natural Sb(V) resistance in Leishmania donovani isolates from Nepal. Antimicrob. Agents Chemother. 49, 4616–4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Basu R., Bhaumik S., Basu J. M., Naskar K., De T., Roy S. (2005) Kinetoplastid membrane protein-11 DNA vaccination induces complete protection against both pentavalent antimonial-sensitive and -resistant strains of Leishmania donovani that correlates with inducible nitric oxide synthase activity and IL-4 generation: evidence for mixed Th1- and Th2-like responses in visceral leishmaniasis. J. Immunol. 174, 7160–7171 [DOI] [PubMed] [Google Scholar]
- 64. Mukherjee B., Mukhopadhyay R., Bannerjee B., Chowdhury S., Mukherjee S., Naskar K., Allam U. S., Chakravortty D., Sundar S., Dujardin J. C., Roy S. (2013) Antimony-resistant but not antimony-sensitive Leishmania donovani up-regulates host IL-10 to overexpress multidrug-resistant protein 1. Proc. Natl. Acad. Sci. U.S.A. 110, E575–E582 [DOI] [PMC free article] [PubMed] [Google Scholar]