

Background: Low-grade endotoxemia is a risk factor for chronic non-resolving inflammatory diseases.
Results: A super-low dose of LPS induces cellular stress, mitochondrial fission, and necroptosis.
Conclusion: IRAK-1 is required for cellular and molecular events leading to mitochondrial fission and cellular necroptosis.
Significance: This study reveals novel mechanisms responsible for cellular stress, necroptosis, and low-grade inflammation associated with low-grade endotoxemia.
Keywords: Cellular Immune Response, Chronic Stress, Inflammation, Innate Immunity, Lipopolysaccharide (LPS)
Abstract
Super-low-dose endotoxemia in experimental animals and humans is linked to low-grade chronic inflammatory diseases. However, the underlying molecular and cellular mechanisms are not well understood. In this study, we examined the effects of a super-low dose of LPS on low-grade inflammation in macrophages as well as underlying mechanisms. We observed that a super-low dose of LPS induces mitochondrial fission and cell necroptosis in primary murine macrophages, dependent upon interleukin 1 receptor-associated kinase (IRAK-1). Mechanistically, our study reveals that a super-low dose of LPS causes protein ubiquitination and degradation of mitofusin 1 (Mfn1), a molecule required for maintaining proper mitochondrial fusion. A super-low dose of LPS also leads to dephosphorylation and activation of Drp1, a molecule responsible for mitochondrial fission and cell necroptosis. Furthermore, we demonstrated that a super-low dose of LPS activates receptor interacting protein 3 kinase (RIP3), a key molecule critical for the assembly of the necrosome complex, the initiation of Drp1 dephosphorylation, and necroptosis. The effects of a super-low dose of LPS are abolished in macrophages harvested from IRAK-1-deficient mice. Taken together, our study identified a novel molecular pathway that leads to cellular stress and necroptosis in macrophages challenged with a super-low dose of endotoxin. This may reconcile low-grade inflammation often associated with low-grade endotoxemia.
Introduction
Individuals with adverse health conditions and lifestyles tend to maintain mildly elevated levels of the circulating endotoxin LPS, a phenomenon referred to as subclinical low-grade endotoxemia (1). This may contribute to chronic low-grade inflammation as manifested in chronic diseases such as diabetes, Parkinson disease, and atherosclerosis (2–6). Mechanistically, a super-low dose of endotoxin results in a mildly sustained activation of proinflammatory mediators without the activation of anti-inflammatory mediators, allowing low-grade, non-resolving inflammation to persist (7). In contrast, medium to higher dosages of LPS induce a robust yet transient immune response where both pro- and anti-inflammatory mediators are activated, providing the host with a compensatory mechanism to resolve inflammation and maintain homeostasis (8).
Recent progress suggests unique molecular and cellular mechanisms that underlie acute resolving inflammation versus chronic non-resolving inflammation (9, 10). With particular interest to this study, cell apoptosis may contribute to the resolution of acute inflammation (11). In contrast, the propagation of non-resolving, low-grade inflammation can be potentially achieved by cell necroptosis instead of apoptosis (12, 13). Necroptosis may contribute to prolonged inflammation through the release of inflammation-propagating damage-associated molecular patterns (14–16). The processes of apoptosis and necroptosis tend to be mutually inhibitory, as indicated by the positive role of caspase during the initiation of apoptosis and suppressive role during necroptosis (17, 18). Although apoptosis may be facilitated by caspase activation, necroptosis is initiated through the activation of receptor-interacting protein 3 kinase (RIP3) and the assembly of a complex “necrosome” near mitochondrion membranes (14). The RIP3 necrosome leads to the activation of a critical phosphatase, PGAM5, that subsequently dephosphorylates and activates dynamin-related protein 1 (Drp1) through its dephosphorylation (19). Drp1 dephosphorylation triggers mitochondrial fission and the generation of reactive oxygen species and other unidentified events that ultimately lead to necroptosis and chronic inflammation (19).
In contrast to Drp1, mitofusins (Mfn1/2) act to prevent mitochondrial fission and facilitate mitochondrial fusion (20). Both Mfn1 and Mfn2 are believed to be regulated via posttranslational modifications such as ubiquitination. A previous study has demonstrated that proteasome inhibition can stabilize Mfn1 and prevent mitochondrial fission (21).
Higher dosages of LPS are known to trigger compensatory tolerance in innate immune cells, as reflected in the reduced expression of proinflammatory cytokines as well as increased mitochondrial bioenergetics and function (8). However, the mechanisms responsible for the non-resolving, low-grade inflammation initiated by a super-low dose of LPS are not well understood. To address this critical question, we examined the effects of a super-low dose of LPS on cellular necroptosis as well as key upstream signaling pathways. We demonstrate that a super-low dose of LPS potently induces necroptosis through an interleukin 1 receptor-associated kinase 1 (IRAK-1)-dependent pathway that leads to selective activation of Drp1 and degradation of Mfn1.
EXPERIMENTAL PROCEDURES
Reagents
LPS (Escherichia coli 0111:B4) and z-VAD-FMK (V116) were purchased from Sigma-Aldrich. Anti-Mfn1(H-65) antibody was obtained from Santa Cruz Biotechnology. Anti-Drp1(catalog no. 5391S), anti-phospho-Drp1 (Ser-637, catalog no. 4867), anti-ubiquitin (catalog no. 3933), anti-phospho-JNK (catalog no. 9251S), anti-JNK (catalog no. 9252S), and anti-β-actin (catalog no. 4967) antibodies were obtained from Cell Signaling Technology. Anti-phospho-RIP3K antibody was provided by Dr. Jiahuai Han (22). Anti-mouse IgG and anti-rabbit IgG HRP-linked antibodies were purchased from Cell Signaling Technology. MitoTracker Red (catalog no. M7512) was purchased from Invitrogen.
Mice and Cell Culture
WT C57BL/6 mice were purchased from the Charles River Laboratories. IRAK-1−/− mice from the C57BL/6 background were provided by Dr. James Thomas (University of Texas Southwestern Medical School). All mice were housed and bred at the Virginia Tech animal facility in compliance with approved Animal Care and Use Committee protocols of Virginia Tech. BMDMs3 were isolated from the tibias and femurs of WT and IRAK-1−/− mice by flushing the bone marrow with DMEM. The cells were cultured in untreated tissue culture dishes with 50 ml of DMEM containing 30% L929 cell supernatant. On the third day of culture, the cells were fed with an additional 20 ml of fresh medium and cultured for another 3 days. Cells were harvested with PBS, resuspended in DMEM supplemented with 1% fetal bovine serum, and allowed to rest overnight before further treatment. As shown by our previous studies, BMDMs harvested through this approach possess relevant activities of macrophages (23–25).
Confocal and Electron Microscopy
WT and IRAK-1−/− BMDMs were plated in 35-mm glass-bottom Petri dishes (MatTek). For staining of mitochondria, the cells were incubated with 75 nm MitoTracker Red (Invitrogen) for 20 min at 37 °C in darkness. After washing three times with PBS, the cells were fixed with paraformaldehyde (4%) in PBS for 15 min at room temperature and then washed three times with PBS. The nuclei were stained using DAPI. Fluorescence images were obtained with a laser-scanning confocal microscope (Zeiss LSM510). MitoTracker Red was excited with a 543-nm laser line, and its emission was collected between 590–640 nm. The percentages of cells undergoing mitochondrial fission were counted and presented. For the purpose of electron microscopy, WT and IRAK-1−/− BMDMs were harvested after specified treatments and washed with PBS. The cells were pelleted in a microcentrifuge tube, and 1 ml of 2% glutaraldehyde with 0.1 m cacodylate buffer (]pH 7.4) fixative was placed on top of the cells. Samples were sliced and prepared on grids for visualization on a JEOL JEM 1400 transmission electron microscope. Cells with mitochondrial fission were counted under at least three different viewing fields. The average numbers of cells with mitochondrial fission in untreated controls were adjusted to 1. The fold increases in cells with mitochondrial fission after LPS treatment were represented from three experiments.
Assays for Cell Viability, Proliferation, and Death
Cell viability was determined using the Vybrant® MTT cell proliferation assay kit (Invitrogen). Briefly, cells were cultured in a 96-well plate in phenol red-free medium with at least three empty wells that served as the background. The cells were then treated with or without a super-low dose of LPS for the specified time periods, and the medium was replaced with 100 μl of fresh medium containing 10 μl of 12 mm stock MTT. The plates were covered in aluminum foil and incubated for 4 h at 37 °C. 50 μl of dimethyl sulfoxide was added to each well, mixed thoroughly with a pipette, and incubated at 37 °C for 10 min, and then the absorbance was measured at 540 nm. For the quantitative analyses of necroptosis, treated cells were stained with 50 nm propidium iodide and subsequently analyzed by FACSCanto flow cytometer. The mean fluorescence intensities of propidium iodide staining were collected.
Assays for Caspase 3 Activity
The caspase 3 activity assay was performed using the caspase 3 colorimetric assay kit from Biovision as recommended by the manufacturer.
Cellular Protein Extraction, Immunoprecipitation, and Immunoblot Analysis
Cells were harvested after specified treatments and washed with cold PBS. The cells were resuspended in SDS lysis buffer containing protease inhibitor mixture (Sigma) and subjected to SDS-PAGE. The protein bands were transferred to an immunoblot PVDF membrane (Bio-Rad) and subjected to immunoblot analysis with the indicated antibodies. For immunoprecipitation analysis, cells were lysed in TBS buffer (50 mm Tris-Cl (pH 7.4) and 150 mm NaCl) supplemented with 1% Triton X-100, 1 mm PMSF, and a protease inhibitor mixture (Roche). After preclearing for 1 h, lysates were incubated with the appropriate antibody for 4 h to overnight at 4 °C. Two hours after adding protein A/G-agarose, the immunoprecipitates were washed extensively with lysis buffer and eluted with SDS loading buffer by boiling for 3 min. For immunoblot analysis, the samples were resolved by SDS-PAGE and transferred to a PVDF membrane (Bio-Rad). Immunoblotting was performed with the indicated antibodies. The proteins were visualized by using a chemiluminescence ECL kit (Pierce).
Statistical Analysis
Results are expressed as mean ± S.D. Statistical significances between groups were determined using two-tailed Student's t test. p < 0.05 was considered statistically significant.
RESULTS
Super-low-dose LPS Induces Low-grade Stress and Necroptosis in Macrophages
To test whether a super-low dose of LPS can indeed lead to low-grade cellular stress, we tested the phosphorylation status of a key stress kinase, JNK. WT BMDMs were treated with either a super-low dose (50 pg/ml) or a higher dose (100 ng/ml) of LPS. As shown in Fig. 1A, 100 ng/ml LPS induced a robust yet transient activation of JNK phosphorylation that peaked at 30 min post-stimulation and diminished quickly to a resting state by 1 h post-LPS stimulation. In contrast, a super-low dose of LPS induced a mild and sustained phosphorylation of JNK that lasted throughout the 2-h treatment period.
FIGURE 1.

A super-low dose of LPS induces low-grade stress and necroptotic cell death. A, WT macrophages were treated with either 50 pg/ml or 100 ng/ml LPS for the indicated times. The phosphorylation levels of JNK (p-JNK) were determined by Western blot analysis. The same blots were probed for total levels of JNK as a loading control. B, WT macrophages were treated with the specified doses of LPS for 18 h. The effect of LPS on cell death was measured by MTT assay. C, WT macrophages were left untreated or treated with 50 pg/ml LPS for 18 h. The activities of caspase 3 were measured, and the relative activities of caspase 3 in control cells were adjusted to 1. D, WT macrophages were treated with 50 pg/ml LPS, 20 μm Z-VAD-fmk, or LPS plus Z-VAD-fmk overnight, stained with propidium iodide (PI), and analyzed by flow cytometry. All data are representative of three experiments. MFI, mean fluorescence intensity. *, p < 0.05.
Low-grade inflammation induces cellular stress and death, which, in turn, may sustain non-resolving inflammation (26). Next, we tested the effect of a super-low dose of LPS on macrophage viability and death. As shown in Fig. 1B, a super-low dose of LPS (50 pg/ml) significantly reduced cell viability and proliferation, as measured by MTT assay. To examine the potential mechanisms, we determined the selective contribution of apoptosis and necroptosis. WT BMDMs were treated with 50 pg/ml LPS for 18 h, followed by a caspase activation assay. As shown in Fig. 1C, a super-low dose of LPS failed to cause a significant induction in the caspase 3 activity. This suggests that a super-low dose of LPS would likely not cause programmed cell apoptosis. Because caspase activity generally suppresses necroptosis (17, 18), we subsequently studied the effect of the caspase inhibitor Z-VAD-fmk. As shown in Fig. 1D, application of Z-VAD-fmk exacerbated cell death induced by a super-low dose of LPS, as measured by propidium iodide staining and flow cytometry. Collectively, these data indicate that a super-low dose of LPS leads to cellular stress and necroptosis in macrophages.
Induction of Mitochondrial Fission by a Super-low Dose of LPS in WT Macrophages
Mitochondrial dynamics play a key role in the modulation of cellular stress, with mitochondrial fission leading to necroptosis and chronic low-grade inflammation (27, 28). Thus, we analyzed the effect of a super-low dose of LPS on mitochondrial fission and fusion. WT BMDMs were either left untreated or treated with 50 pg/ml LPS and then labeled with the mitochondrion-specific MitoTracker dye. Cell nuclei were counterstained by DAPI, and the mitochondrial morphology was observed by confocal fluorescence microscope. As shown in Fig. 2A, a super-low dose of LPS induced the conversion of an elongated mitochondrial morphology to a fragmented one, as indicated in the magnified panel, indicative of mitochondrial fission. To further confirm mitochondrial fragmentation in response to super-low dose LPS at the ultra-structural level, we performed electron microscopy analyses using WT BMDMs. As shown in Fig. 2B, consistent with our observation using confocal microscopy, WT BMDMs treated with a super-low dose of LPS exhibited fractionated mitochondria, potentially because of increased fission, compared with the untreated cells.
FIGURE 2.
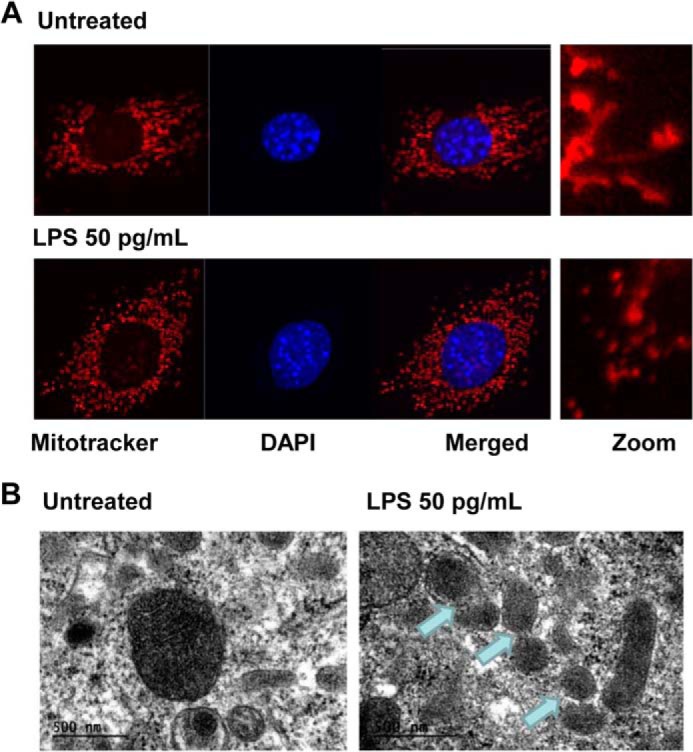
A super-low dose of LPS induces mitochondrial fission in murine macrophages. A, WT BMDMs were treated with a super-low dose of LPS (50 pg/ml) for 1 h and then labeled with MitoTracker Red to stain the mitochondria. The nuclei were stained using DAPI (blue). The cells were visualized under a Zeiss LSM510 laser-scanning confocal microscope (original magnification ×400). The merged images were magnified and are shown at the right. B, WT BMDMs were treated with 50 pg/ml LPS for 1 h. Cells were prepared and visualized under a JEOL JEM 1400 transmission electron microscope. The arrows denote fragmented mitochondria.
Cellular Stress Mediated by a Super-low Dose of LPS Is Dependent upon IRAK-1
On the basis of previous studies showing that IRAK-1 is preferentially responsible for mediating the cellular responses to a super-low dose of LPS (7, 29, 30), we tested the effect of a super-low dose of LPS on cell stress in IRAK-1-deficient macrophages. As shown in Fig. 3A, JNK1 phosphorylation induced by a super-low dose of LPS was reduced in IRAK-1-deficient cells. Furthermore, the effect of a super-low dose of LPS on cell death was alleviated in IRAK-1-deficient cells. (Fig. 3B).
FIGURE 3.

A super-low dose of LPS promotes cellular stress and mitochondrial fission through an IRAK1-dependent mechanism. A, macrophages derived from WT and IRAK-1-deficient mice were treated with a super-low dose of LPS (50 pg/ml) for the indicated times. The levels of JNK phosphorylation (p-JNK) were determined via Western blot analysis, and total JNK levels were detected as a loading control. B, macrophages derived from WT and IRAK-1-deficient mice were treated with a super-low dose of LPS (50 pg/ml) for 18 h. The effect of LPS on cell death was measured by MTT assay. C, WT and IRAK1-deficient BMDMs were treated with a super-low dose of LPS (50pg/ml) for 1 h. Cells were stained with MitoTracker Red to stain the mitochondria. The nuclei were counterstained using DAPI (blue). The cells were visualized under a laser-scanning confocal microscope (original magnification ×400). D, cells with mitochondrial fission were counted under at least three different viewing fields. The average numbers of cells with mitochondria fission in untreated controls were adjusted to 1. The fold increases in cells with mitochondrial fission after LPS treatment were represented from three experiments. All data are representative of three experiments and are represented as the mean ± S.D. *, p < 0.05.
We further tested the effect of IRAK-1 on mitochondria dynamics. In contrast to WT cells, a super-low dose of LPS failed to induce mitochondrial fragmentation in the absence of IRAK1 (Fig. 3, C and D).
A Super-low Dose of LPS Activates Drp1 and Degrades Mfn1 in an IRAK-1-dependent Manner
Cellular necroptosis and mitochondrial fission are regulated by the phosphorylation status of dynamin-related protein 1 (Drp1) (18, 31). Phosphorylation of Drp1 at Ser-637 inhibits both cell necroptosis as well as mitochondrial fission (32, 33). On the other hand, dephosphorylation of Drp1 is critical for the initiation of necroptosis and mitochondrial fission. Thus, we tested the phosphorylation status of Drp1 in BMDMs harvested from WT and IRAK-1-deficient mice treated with a super-low dose of LPS. As shown in Fig. 4A, a super-low dose of LPS significantly reduced the phosphorylation of Drp1 at Ser-637 in WT BMDMs, corresponding to elevated cell necroptosis and mitochondrial fission. This effect was ablated in IRAK1-deficient cells. Besides Drp1, members of the dynamin family of proteins, mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), are involved in fusion between mitochondria by tethering adjacent mitochondria. We determined the effect of a super-low dose of LPS on the levels of Mfn1 in macrophages. We observed that Mfn1 levels were reduced drastically in response to a super-low dose of LPS in WT but not in IRAK-1-deficient BMDMs (Fig. 4B). There was no change in the levels of Mfn2 upon treatment with a super-low dose of LPS (data not shown). These results suggest that a super-low dose of LPS may induce necroptosis and cellular stress by modulating key regulatory molecules, such as Drp1 and Mfn1, through an IRAK-1-dependent mechanism.
FIGURE 4.

A super-low dose of LPS promotes mitochondrial fission while simultaneously decreasing mitochondrial fusion. A, WT and IRAK-1-deficient BMDMs were treated with a super-low dose of LPS (50 pg/ml) for the indicated times. The levels of phosphorylated Drp1 (p-Drp1, Ser-637) were determined by Western blot analysis. The blots were probed with β-actin as loading controls. B, WT and IRAK1-deficient BMDMs were treated with a super-low dose of LPS (50 pg/ml) for the indicated times. The levels of Mfn1 were determined by Western blot analysis. The blots were probed with β-actin as loading controls. Band intensities of phospho-Drp1 (A) and Mfn1 (B) were quantified and normalized relative to the β-actin levels. All data are representative of three experiments and are represented as mean ± S.D. *, p < 0.05.
A Super-low Dose of LPS Activates RIP3 Responsible for the Dephosphorylation of Drp1 through the IRAK-1-dependent Pathway
The RIP3 pathway has been closely associated with the initiation of cellular necroptosis (14, 31). RIP3 activation triggers the activation of the phosphatase PGAM5, which subsequently dephosphorylates Drp1 (19). To test whether a super-low dose of LPS may induce cellular stress and necroptosis through the RIP3 pathway, we examined the phosphorylation status of RIP3. As shown in Fig. 5, 50 pg/ml LPS induced rapid phosphorylation of RIP3, indicative of RIP3 activation. In contrast, this effect was abolished in IRAK-1-deficient cells.
FIGURE 5.

A super-low dose of LPS activates RIP3 kinase dependent upon IRAK-1. WT and IRAK-1-deficient BMDMs were treated with 50 pg/ml LPS for the indicated times. The phosphorylation status of RIP3 (p-RIP3) was determined by Western blot analysis using a specific antibody against phosphorylated RIP3. The blots were probed with total RIP3 and β-actin antibodies as loading controls. All data are representative of three experiments, *, p < 0.05.
A Super-low Dose of LPS Causes the Ubiquitination and Degradation of Mfn1 Dependent upon IRAK-1
With regard to the regulation of Mfn1, protein stability and degradation play a key role (21). We further tested whether protein degradation is responsible for the decreased Mfn1 levels in cells challenged with a super-low dose of LPS. As shown in Fig. 6A, the proteasomal inhibitor MG132 blocked the reduction of Mfn1 in response to a super-low dose of LPS (Fig. 6A). To further test whether Mfn-1 undergoes ubiquitination, immunoprecipitation experiments were performed to detect the ubiquitination status of Mfn1 in the presence of a super-low dose of LPS. We detected ubiquitinated Mfn1 in the presence of a super-low dose of LPS (Fig. 6B). In contrast, the ubiquitination of Mfn1 was completely abolished in IRAK-1-deficient cells, indicating that IRAK-1 is responsible for Mfn1 ubiquitination and degradation mediated by a super-low dose of LPS. Taken together, this study reveals a novel molecular pathway in innate macrophages that is preferentially induced by a super-low dose of LPS and is responsible for triggering cellular stress and necroptosis (Fig. 7).
FIGURE 6.

A super-low dose of LPS promotes Mfn1 degradation. A, WT macrophages were treated with 50 pg/ml LPS in the absence or presence of the proteasome inhibitor MG132, followed by immunoblot analysis of Mfn1 protein levels. The blots were probed with β-actin as loading controls. DMSO, dimethyl sulfoxide. B, immunoprecipitation (IP) analysis was performed using cell lysates derived from WT and IRAK1-deficient macrophages treated with or without 50 pg/ml LPS with either an isotype control or Mfn1-specific antibodies, as indicated on the blots. The gels were immunoblotted (IB) with either ubiquitin-specific (Ub, top row) or Mfn1-specific (bottom row) antibodies. All data are representative of three experiments.
FIGURE 7.

Schematic depicting the mechanism responsible for low-grade inflammation triggered by a super-low dose of LPS. A super-low dose of LPS selectively induces the activation of RIP3, activation of downstream kinase mixed lineage kinase domain-like (MLKL), dephosphorylation of Drp1 (P Drp1 to the active Drp1), and degradation of Mfn1 in an IRAK-1-dependent fashion. Collectively, these molecular and cellular events may lead to cellular necroptosis and low-grade inflammation.
DISCUSSION
This study reveals novel mechanisms that underlie cell necroptosis and low-grade inflammation preferentially induced by a super-low dose of LPS. Our findings indicate that a super-low dose of LPS selectively induces cell stress and necroptosis. This effect may be facilitated by enhanced mitochondrial fission, reduced fusion mediated by reduced Drp1 phosphorylation, and Mfn1 degradation. All of these processes are IRAK-1-dependent. This conclusion is corroborated by various molecular and cellular lines of evidence. First, a super-low dose of LPS triggers low-grade cellular stress and necroptosis in wild-type but not in IRAK-1-deficient macrophages. Second, a super-low dose of LPS leads to mitochondrial fission, the dephosphorylation and activation of Drp1 in macrophages dependent upon IRAK-1. Third, a super-low dose of LPS degrades Mfn1, a protein involved in maintaining proper mitochondrial fusion and antagonizing the function of Drp1, in an IRAK-1-dependent manner.
Our data complement and extend recent studies that support an intriguing link between low-grade endotoxemia, cell necroptosis, and chronic inflammatory diseases (4, 18, 34, 35). Subclinical dosages of circulating endotoxin in experimental mice and humans (1–100 pg/ml) are associated with chronic inflammatory conditions (36, 37). Despite its significance, most mechanistic studies regarding low-dose endotoxin used dosages in the nanogram per milliliter range, 10- to 100-fold higher than the pathologically relevant super-low dose (38). Low to higher dosages of endotoxin (>100 ng/ml) can trigger both robust inflammatory responses as well as compensatory anti-inflammatory responses in innate immune cells and tissues (39, 40). To our knowledge, this is the first report that reveals a novel role of a super-low dose of endotoxin. Our data indicate that a super-low dose of endotoxin is highly potent in inducing macrophage necroptosis instead of apoptosis. Unlike programmed apoptotic cell death, which resolves inflammation, necroptosis is associated with non-resolving inflammation and persistent activation of stress kinases such as JNK (41, 42). As a consequence, cell necroptosis is associated with a multitude of inflammatory complications such as atherosclerosis, reduced wound repair, inflammatory bowel diseases, and ischemic injury (13, 42–46). Our finding that a super-low dose of endotoxin elicits cell necroptosis potentially explains the detrimental effect of super-low-grade endotoxemia in humans.
Biochemically, our study provides a new perspective regarding the novel connection between IRAK-1 and the necroptosis pathway. Conventionally, IRAK-1 has been examined primarily in the context of NFκB signaling and transcriptional regulation of inflammatory gene expression (47). Instead, these data reveal that IRAK-1 is also critically involved in mediating the activation of RIP3, a key component of the necrosome complex, in cells challenged with a super-low dose of endotoxin. The RIP3 pathway eventually leads to the dephosphorylation and activation of Drp1, causing mitochondrial fission and necroptosis (18, 19).
In addition to Drp1 activation and mitochondrial fission, a reduction in the compensatory mitochondrial fusion process may further perpetuate non-resolving inflammation and necroptosis. Our data indicate that a super-low dose of LPS not only activates Drp1 but also reduces the protein levels of Mfn1 through an IRAK-1-dependent pathway. We documented that IRAK-1 is required for Mfn1 ubiquitination and degradation induced by a super-low dose of LPS. Future studies are needed to better define the biochemical process responsible for Mfn1 degradation mediated by IRAK-1.
Although this study reveals a novel connection between IRAK-1 and cell necroptosis, we realize that this is only the beginning of a full understanding of underlying complex mechanisms. Recent studies reveal that cellular stress may be modulated by a multitude of cellular death pathways that include inflammasome-mediated pyroptosis (48, 49). Higher doses of LPS have been shown to activate the inflammasome and subsequent pyroptosis through an IRAK-1-dependent mechanism (49). Our preliminary study suggests that a super-low dose of LPS fails to activate caspase 1. Future studies are warranted to tease out the relative contribution of necroptosis, pytoptosis, and other cellular forms of cell death during the progression of non-resolving inflammation. Another missing link is the detailed mechanism responsible for the dephosphorylation of Drp1 initiated by a super-low dose of LPS. So far, studies on protein phosphatases in the area of innate immunity are scarce. Additional studies are needed to determine the dynamics of phosphatase regulation by varying dosages of LPS.
In terms of pathophysiological relevance, it is likely that a super-low dose of LPS may exert similar effects on other cells in addition to macrophages. Indeed, our preliminary data with primary embryonic fibroblasts demonstrated similar regulatory patterns of cell death, Drp1 dephosphorylation, and Mfn1 degradation by a super-low dose of LPS (data not shown). Given the prevalent role of mitochondria in vital tissues, such effects may have a general implication in various forms of low-grade, non-resolving inflammatory conditions such as atherosclerosis, diabetes, and aging. Our data should guide future studies with animal models of low-grade inflammation, and human clinical studies should be conducted.
Taken together, our study reveals a novel IRAK-1-mediated pathway that is responsible for low-grade inflammation and necroptosis induced by a super-low dose of LPS. Given recent findings showing that selective inhibition of necroptosis may hold promise in treating chronic inflammatory diseases (46, 50), this study suggests that IRAK-1 may be a viable target in the potential intervention of chronic inflammation mediated by low-grade endotoxemia.
Acknowledgments
We thank the members of our laboratory for discussions and technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant R01HL115835.
- BMDM
- bone marrow-derived macrophage
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone.
REFERENCES
- 1. Chang S. L., L. (2011) Metabolic endotoxemia: a novel concept in chronic disease pathology. J. Med. Sci. 31, 191–209 [Google Scholar]
- 2. Laugerette F., Vors C., Géloën A., Chauvin M. A., Soulage C., Lambert-Porcheron S., Peretti N., Alligier M., Burcelin R., Laville M., Vidal H., Michalski M. C. (2011) Emulsified lipids increase endotoxemia: possible role in early postprandial low-grade inflammation. J. Nutr. Biochem. 22, 53–59 [DOI] [PubMed] [Google Scholar]
- 3. Lassenius M. I., Pietiläinen K. H., Kaartinen K., Pussinen P. J., Syrjänen J., Forsblom C., Pörsti I., Rissanen A., Kaprio J., Mustonen J., Groop P. H., Lehto M. (2011) Bacterial endotoxin activity in human serum is associated with dyslipidemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 34, 1809–1815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiesner P., Choi S. H., Almazan F., Benner C., Huang W., Diehl C. J., Gonen A., Butler S., Witztum J. L., Glass C. K., Miller Y. I. (2010) Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor κb and activator protein-1: possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ. Res. 107, 56–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sun L., Yu Z., Ye X., Zou S., Li H., Yu D., Wu H., Chen Y., Dore J., Clément K., Hu F. B., Lin X. (2010) A marker of endotoxemia is associated with obesity and related metabolic disorders in apparently healthy Chinese. Diabetes Care 33, 1925–1932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Manco M., Putignani L., Bottazzo G. F. (2010) Gut microbiota, lipopolysaccharides, and innate immunity in the pathogenesis of obesity and cardiovascular risk. Endocr. Rev. 31, 817–844 [DOI] [PubMed] [Google Scholar]
- 7. Maitra U., Deng H., Glaros T., Baker B., Capelluto D. G., Li Z., Li L. (2012) Molecular mechanisms responsible for the selective and low-grade induction of proinflammatory mediators in murine macrophages by lipopolysaccharide. J. Immunol. 189, 1014–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu T. F., Brown C. M., El Gazzar M., McPhail L., Millet P., Rao A., Vachharajani V. T., Yoza B. K., McCall C. E. (2012) Fueling the flame: bioenergy couples metabolism and inflammation. J. Leukocyte Biol. 92, 499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ingersoll M. A., Platt A. M., Potteaux S., Randolph G. J. (2011) Monocyte trafficking in acute and chronic inflammation. Trends Immunol. 32, 470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nathan C., Ding A. (2010) Nonresolving inflammation. Cell 140, 871–882 [DOI] [PubMed] [Google Scholar]
- 11. Gautier E. L., Ivanov S., Lesnik P., Randolph G. J. (2013) Local apoptosis mediates clearance of macrophages from resolving inflammation in mice. Blood 122, 2714–2722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roychowdhury S., McMullen M. R., Pisano S. G., Liu X., Nagy L. E. (2013) Absence of receptor interacting protein kinase 3 prevents ethanol-induced liver injury. Hepatology 57, 1773–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Günther C., Neumann H., Neurath M. F., Becker C. (2013) Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium. Gut 62, 1062–1071 [DOI] [PubMed] [Google Scholar]
- 14. Moriwaki K., Chan F. K. (2013) RIP3: a molecular switch for necrosis and inflammation. Genes Dev. 27, 1640–1649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Vanlangenakker N., Vanden Berghe T., Vandenabeele P. (2012) Many stimuli pull the necrotic trigger, an overview. Cell Death Differ. 19, 75–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lau A., Wang S., Jiang J., Haig A., Pavlosky A., Linkermann A., Zhang Z. X., Jevnikar A. M. (2013) RIPK3-mediated necroptosis promotes donor kidney inflammatory injury and reduces allograft survival. Am. J. Transplant. 13, 2805–2818 [DOI] [PubMed] [Google Scholar]
- 17. Kim S. J., Li J. (2013) Caspase blockade induces RIP3-mediated programmed necrosis in Toll-like receptor-activated microglia. Cell Death Dis. 4, e716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Remijsen Q., Goossens V., Grootjans S., Van den Haute C., Vanlangenakker N., Dondelinger Y., Roelandt R., Bruggeman I., Goncalves A., Bertrand M. J., Baekelandt V., Takahashi N., Berghe T. V., Vandenabeele P. (2014) Depletion of RIPK3 or MLKL blocks TNF-driven necroptosis and switches towards a delayed RIPK1 kinase-dependent apoptosis. Cell Death Dis. 5, e1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Z., Jiang H., Chen S., Du F., Wang X. (2012) The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell 148, 228–243 [DOI] [PubMed] [Google Scholar]
- 20. Chen Y., Liu Y., Dorn G. W., 2nd. (2011) Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ. Res. 109, 1327–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tanaka A., Cleland M. M., Xu S., Narendra D. P., Suen D. F., Karbowski M., Youle R. J. (2010) Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J. Cell Biol. 191, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen W., Zhou Z., Li L., Zhong C. Q., Zheng X., Wu X., Zhang Y., Ma H., Huang D., Li W., Xia Z., Han J. (2013) Diverse sequence determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288, 16247–16261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Xie Q., Gan L., Wang J., Wilson I., Li L. (2007) Loss of the innate immunity negative regulator IRAK-M leads to enhanced host immune defense against tumor growth. Mol. Immunol. 44, 3453–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Maitra U., Parks J. S., Li L. (2009) An innate immunity signaling process suppresses macrophage ABCA1 expression through IRAK-1-mediated downregulation of retinoic acid receptor α and NFATc2. Mol. Cell. Biol. 29, 5989–5997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maitra U., Singh N., Gan L., Ringwood L., Li L. (2009) IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of antioxidative enzymes. J. Biol. Chem. 284, 35403–35411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ishikawa J., Tamura Y., Hoshide S., Eguchi K., Ishikawa S., Shimada K., Kario K. (2007) Low-grade inflammation is a risk factor for clinical stroke events in addition to silent cerebral infarcts in Japanese older hypertensives: the Jichi Medical School ABPM Study, wave 1. Stroke 38, 911–917 [DOI] [PubMed] [Google Scholar]
- 27. Ferrari L. F., Chum A., Bogen O., Reichling D. B., Levine J. D. (2011) Role of Drp1, a key mitochondrial fission protein, in neuropathic pain. J. Neurosci. 31, 11404–11410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dai D. F., Rabinovitch P. S., Ungvari Z. (2012) Mitochondria and cardiovascular aging. Circ. Res. 110, 1109–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Deng H., Maitra U., Morris M., Li L. (2013) Molecular mechanism responsible for the priming of macrophage activation. J. Biol. Chem. 288, 3897–3906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maitra U., Li L. (2013) Molecular mechanisms responsible for the reduced expression of cholesterol transporters from macrophages by low-dose endotoxin. Arterioscler. Thromb. Vasc. Biol. 33, 24–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhou Z., Han V., Han J. (2012) New components of the necroptotic pathway. Protein Cell 3, 811–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cho S. G., Du Q., Huang S., Dong Z. (2010) Drp1 dephosphorylation in ATP depletion-induced mitochondrial injury and tubular cell apoptosis. Am. J. Physiol. Renal Physiol. 299, F199–F206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cereghetti G. M., Stangherlin A., Martins de Brito O., Chang C. R., Blackstone C., Bernardi P., Scorrano L. (2008) Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A. 105, 15803–15808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moreno-Navarrete J. M., Manco M., Ibanez J., Garcia-Fuentes E., Ortega F., Gorostiaga E., Vendrell J., Izquierdo M., Martinez C., Nolfe G., Ricart W., Mingrone G., Tinahones F., Fernandez-Real J. M. (2010) Metabolic endotoxemia and saturated fat contribute to circulating NGAL concentrations in subjects with insulin resistance. Int. J. Obes. 34, 240–249 [DOI] [PubMed] [Google Scholar]
- 35. Szeto C. C., Kwan B. C., Chow K. M., Lai K. B., Chung K. Y., Leung C. B., Li P. K. (2008) Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin. J. Am. Soc. Nephrol. 3, 431–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lira F. S., Rosa J. C., Pimentel G. D., Souza H. A., Caperuto E. C., Carnevali L. C., Jr., Seelaender M., Damaso A. R., Oyama L. M., de Mello M. T., Santos R. V. (2010) Endotoxin levels correlate positively with a sedentary lifestyle and negatively with highly trained subjects. Lipids Health Dis. 9, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Erridge C., Attina T., Spickett C. M., Webb D. J. (2007) A high-fat meal induces low-grade endotoxemia: evidence of a novel mechanism of postprandial inflammation. Am. J. Clin. Nutr. 86, 1286–1292 [DOI] [PubMed] [Google Scholar]
- 38. Morris M., Li L. (2012) Molecular mechanisms and pathological consequences of endotoxin tolerance and priming. Arch. Immunol. Ther. Exp. 60, 13–18 [DOI] [PubMed] [Google Scholar]
- 39. Shnyra A., Brewington R., Alipio A., Amura C., Morrison D. C. (1998) Reprogramming of lipopolysaccharide-primed macrophages is controlled by a counterbalanced production of IL-10 and IL-12. J. Immunol. 160, 3729–3736 [PubMed] [Google Scholar]
- 40. Li L., Jacinto R., Yoza B., McCall C. E. (2003) Distinct post-receptor alterations generate gene- and signal-selective adaptation and cross-adaptation of TLR4 and TLR2 in human leukocytes. J. Endotoxin Res. 9, 39–44 [DOI] [PubMed] [Google Scholar]
- 41. Kaczmarek A., Vandenabeele P., Krysko D. V. (2013) Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 38, 209–223 [DOI] [PubMed] [Google Scholar]
- 42. Linkermann A., Hackl M. J., Kunzendorf U., Walczak H., Krautwald S., Jevnikar A. M. (2013) Necroptosis in immunity and ischemia-reperfusion injury. Am. J. Transplant. 13, 2797–2804 [DOI] [PubMed] [Google Scholar]
- 43. Pierdomenico M., Negroni A., Stronati L., Vitali R., Prete E., Bertin J., Gough P. J., Aloi M., Cucchiara S. (2014) Necroptosis is active in children with inflammatory bowel disease and contributes to heighten intestinal inflammation. Am. J. Gastroenterol. 109, 279–287 [DOI] [PubMed] [Google Scholar]
- 44. Bao L., Li Y., Deng S. X., Landry D., Tabas I. (2006) Sitosterol-containing lipoproteins trigger free sterol-induced caspase-independent death in ACAT-competent macrophages. J. Biol. Chem. 281, 33635–33649 [DOI] [PubMed] [Google Scholar]
- 45. Wang Y. Q., Wang L., Zhang M. Y., Wang T., Bao H. J., Liu W. L., Dai D. K., Zhang L., Chang P., Dong W. W., Chen X. P., Tao L. Y. (2012) Necrostatin-1 suppresses autophagy and apoptosis in mice traumatic brain injury model. Neurochem. Res. 37, 1849–1858 [DOI] [PubMed] [Google Scholar]
- 46. You Z., Savitz S. I., Yang J., Degterev A., Yuan J., Cuny G. D., Moskowitz M. A., Whalen M. J. (2008) Necrostatin-1 reduces histopathology and improves functional outcome after controlled cortical impact in mice. J. Cereb. Blood Flow Metab. 28, 1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liu G., Tsuruta Y., Gao Z., Park Y. J., Abraham E. (2007) Variant IL-1 receptor-associated kinase-1 mediates increased NF-κ B activity. J. Immunol. 179, 4125–4134 [DOI] [PubMed] [Google Scholar]
- 48. Fink S. L., Cookson B. T. (2005) Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect. Immun. 73, 1907–1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin K. M., Hu W., Troutman T. D., Jennings M., Brewer T., Li X., Nanda S., Cohen P., Thomas J. A., Pasare C. (2014) IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc. Natl. Acad. Sci. U.S.A. 111, 775–780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koshinuma S., Miyamae M., Kaneda K., Kotani J., Figueredo V. M. (2014) Combination of necroptosis and apoptosis inhibition enhances cardioprotection against myocardial ischemia-reperfusion injury. J. Anesth. 28, 235–241 [DOI] [PubMed] [Google Scholar]