

Background: The RNase H activity of HIV-1 reverse transcriptase (RT) is an under-explored target.
Results: GSK5750 is a novel RNase H active site inhibitor that displays slow dissociation kinetics.
Conclusion: Tight binding may compensate for the inability of active site inhibitors to access the RT-substrate complex.
Significance: The GSK5750 scaffold may lead to the development of clinically relevant RNase H inhibitors.
Keywords: Drug Development, HIV-1, Retrovirus, Reverse Transcription, Ribonuclease
Abstract
Compounds that efficiently inhibit the ribonuclease (RNase) H activity of the human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) have yet to be developed. Here, we demonstrate that GSK5750, a 1-hydroxy-pyridopyrimidinone analog, binds to the enzyme with an equilibrium dissociation constant (Kd) of ∼400 nm. Inhibition of HIV-1 RNase H is specific, as DNA synthesis is not affected. Moreover, GSK5750 does not inhibit the activity of Escherichia coli RNase H. Order-of-addition experiments show that GSK5750 binds to the free enzyme in an Mg2+-dependent fashion. However, as reported for other active site inhibitors, binding of GSK5750 to a preformed enzyme-substrate complex is severely compromised. The bound nucleic acid prevents access to the RNase H active site, which represents a possible biochemical hurdle in the development of potent RNase H inhibitors. Previous studies suggested that formation of a complex with the prototypic RNase H inhibitor β-thujaplicinol is slow, and, once formed, it dissociates rapidly. This unfavorable kinetic behavior can limit the potency of RNase H active site inhibitors. Although the association kinetics of GSK5750 remains slow, our data show that this compound forms a long lasting complex with HIV-1 RT. We conclude that slow dissociation of the inhibitor and HIV-1 RT improves RNase H active site inhibitors and may circumvent the obstacle posed by the inability of these compounds to bind to a preformed enzyme-substrate complex.
Introduction
Human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) is a heterodimeric (p66/p51) multifunctional enzyme that catalyzes the conversion of the single-stranded, viral RNA genome into double-stranded DNA (1). Both polymerase and ribonuclease H (RNase H)4 active sites reside on the larger, catalytic p66 subunit. The N-terminal polymerase active site catalyzes nucleotidyl transfer reactions, whereas the C-terminal RNase H active site hydrolyzes the RNA strand of DNA/RNA hybrids that are generated during reverse transcription (2–5). Both active sites are necessary for successful reverse transcription; however, presently, all clinically available drugs that target HIV-1 RT inhibit DNA synthesis, whereas specific RNase H inhibitors that show antiviral effects are yet to be developed (6).
The distance between the polymerase and RNase H active sites is ∼60 Å (3, 4, 7). A ternary complex with a chain-terminated DNA primer and a bound deoxynucleoside triphosphate yields a specific cut at position −18 on the RNA template, with the scissile bond located between residues −18 and −19 (5, 8, 9). Here, the complex exists in its post-translocated conformation, in which the nucleotide binding site is accessible (10). In contrast, a pretranslocated complex that exists immediately following the nucleotidyl transfer yields a cut at template position −19. Such a complex can be trapped in the presence of the pyrophosphate analog phosphonoformic acid (PFA) (11). In general, these cuts are referred to as polymerase-dependent RNase H activity to reflect the reliance on specific interactions between the polymerase active site and the 3′-end of the primer (12). Conversely, polymerase-independent cuts collectively refer to RNase H cleavage events that occur, whereas the polymerase active site is not positioned at the 3′-primer terminus. Variations of this mechanism have been proposed (13). This activity is required for non-specifically degrading the transcribed viral RNA genome during (−)-strand DNA synthesis, as well as for creating and removing the (+)-strand primer or polypurine tract, and for removing the (−)-strand or tRNALys-3 primer (14–19).
The RNase H active site is comprised of a DEDD catalytic motif (Asp-443, Glu-478, Asp-498, Asp-549), which coordinates two divalent metal ions as essential cofactors (2). This architecture has driven the design of small molecule inhibitors of RNase H activity. Several distinct chemical compounds with a three-oxygen pharmacophore, or equivalent structures that are capable of chelating the two catalytic metal ions have been described (20, 21). As such, these compounds are generally referred to as active site RNase H inhibitors. Crystal structures of HIV-1 RT RNase H with a bound active site inhibitor confirmed that these compounds are anchored at the RNase H active site and interact with the bound divalent metal ions (22–25). Allosteric RNase H inhibitors can bind in the vicinity of the polymerase active site as shown for N-acyl hydrazones (26, 27), or in the vicinity of the p51 thumb subdomain as shown for vinylogous ureas (28, 29).
The development of small molecule effectors of RNase H that compete with substrate poses a biochemical obstacle. Indeed, the nucleic acid substrates bind to HIV-1 RT with equilibrium dissociation constants (Kd) in the low nanomolar range (30), and most contacts are seen in close proximity to the polymerase active site (3, 4). Consistent with this notion, order-of-addition experiments revealed that the tropolone-based, prototypic RNase H active site inhibitor β-thujaplicinol does not bind to a preformed RT-nucleic acid complex (10). Inhibition of the primary, endonucleolytic RNase H cut is only seen if the inhibitor is preincubated with RT and Mg2+ ions. The added DNA/RNA primer/template is, however, rapidly cleaved, after an initial delay on the time scale of seconds. These findings suggested that β-thujaplicinol rapidly dissociates from the complex and rebinding of the inhibitor is blocked by the nucleic acid substrate. The structure of RT in complex with β-thujaplicinol, along with a modeled RNA/DNA hybrid indeed point to a steric conflict between the inhibitor and the intact RNA/DNA substrate (22). Notably, β-thujaplicinol and derivatives appear to be able to bind to the nicked product of the primary cleavage reaction as subsequent secondary, polymerase-independent cuts are effectively inhibited (31).
Here, we show that the 1-hydroxy-pyridopyrimidinone derivative, GSK5750, represents a novel, potent inhibitor of the HIV-1 RT-associated RNase H activity, which, in contrast to β-thujaplicinol, can form long lasting complexes with the enzyme. This is likely due to a greater intrinsic affinity for the active site and may ultimately translate into a compound class with promise in delivering a potent, mechanism-specific, antiviral effect.
EXPERIMENTAL PROCEDURES
Enzymes and Nucleic Acids
Heterodimeric, HIV-1 RT (p66/p51) was expressed and purified as described previously (32). Escherichia coli RNase H was purchased from Invitrogen. All nucleic acids used in this study were synthesized by IDT DNA Technologies. The following sequences were used: PBS-22dpol, 5′-AGGTCCCTGTTCGGGCGCCACT-3′; PBS-52r, 5′-aaaucucuagcaguggcgcccgaacagggaccugaaagcgaaagggaaac-3′; PBS-42r, 5′-ucucuagcaguggcgcccgaacagggaccugaaagcuccucc-3′; and PBS-14r8d, 5′-cuguucgggcgccaCTGCTAGA-3′. Nucleotides were purchased from Fermentas Life Sciences, and PFA was purchased from Sigma. 5′-Radiolabeling was performed essentially as described previously (12). Briefly, 5′-radiolabeling was performed with [γ-32P]ATP (PerkinElmer Life Sciences) and T4 polynucleotide kinase (Fermentas). Reactions were allowed to proceed for 1 h at 37 °C. Labeled DNA or RNA was subjected to phenol-chloroform purification and further purified using P-30 size exclusion columns (Bio-Rad). 3′-Radiolabeling of RNA was performed with [5′-32P]pCp and T4 RNA ligase, allowed to proceed overnight at 4 °C, and purified as described above.
DNA Synthesis Assay
5′-Radiolabeled PBS-22dpol was heat-annealed to a 3-fold excess of PBS-42r and allowed to cool to room temperature for 45 min. 500 nm HIV-1 RT was then preincubated for 10 min with 100 nm of the preformed DNA/RNA hybrid, and inhibitor in a buffer containing 50 mm Tris-HCl (pH 7.8), 50 mm NaCl, and 0.2 mm EDTA. Reactions were initiated with 6 mm MgCl2 and allowed to proceed for 0.13, 0.33, 0.5, 1, 1.5, 2, 3, 4, 5, 8, and 10 min. Reactions were stopped with the addition of formamide and 20 mm EDTA. Samples were resolved on a 12% denaturing polyacrylamide gel and visualized by PhosphorImaging. Bands were quantified by QuantityOne software (Bio-Rad). Results were graphed using GraphPad Prism (version 5.0).
IC50 Determination for Primary and Secondary RNase H Cleavages of HIV-1 RT and E. coli RNase H
To compare activity and inhibition of E. coli RNase H with HIV-1 RNase H, the concentration of the E. coli was adjusted to the efficiency of 500 nm HIV-1 RT in the absence of inhibitor. For IC50 determination, 100 nm of 5′-radiolabeled PBS-14r8d was heat annealed to a 3-fold molar excess as described above. 100 nm preformed DNA-RNA/DNA hybrid was incubated with 500 nm HIV-1 RT or 10 nm E. coli RNase Hin a buffer of 50 mm Tris-HCl (pH 7.8), 50 mm NaCl, 0.2 mm EDTA, and varying concentrations of GSK5750 and β-thujaplicinol. Reactions were initiated by the addition of 6 mm MgCl2 and allowed to proceed at 37 °C for 6 min. Samples were analyzed as described above.
Order-of-Addition Experiments
DNA/RNA hybrids were prepared as described above with PBS-22dpol primer and 3′-radiolabeled PBS-52r template. 100 nm DNA/RNA hybrid was added to 1 μm HIV-1 RT in a buffer containing 50 mm Tris-HCl (pH 7.8), 50 mm NaCl, 1 mm EDTA, and 50 μm GSK5750, as well as 6 mm MgCl2. Components in the preincubation mixes were incubated at 37 °C for 10 min. Reactions were allowed to proceed for 0.05, 0.1, 0.25, 0.5, 0.75, 1, 2, 4, 8, 16, 24, and 30 s and were rapidly quenched with 100 μl of 0.5 m EDTA. These experiments were conducted with a RQF-3 rapid quench-flow instrument (Kintek; Austin, TX). The order of component addition was varied as described in the results section. Samples were analyzed as described above.
Determination of the Equilibrium Dissociation Constant (Kd) for GSK5750
Multiple time course experiments were performed with a quench-flow apparatus as described for order-of-addition experiments. In this case, 1 μm HIV-1 RT was added to 50 mm Tris-HCl (pH 7.8), 50 mm NaCl, 6 mm MgCl2, and varying concentrations of GSK5750 to form enzyme-inhibitor (E-I) complexes. E-I complexes were rapidly mixed with 100 nm DNA/RNA hybrid (PBS-22dpol/PBS-50r) and 1 mm EDTA. Concentrations of GSK5750 were 0, 250, 500, 1000, 2000, and 5000 nm. The resulting curves were fit to the first order exponential equation (Y = Ymax × (1 − exp(−K × X))). The burst amplitudes were then plotted against the GSK5750 concentration, and the resulting curve was fit to a quadratic equation (Y = E − 0.5{(K + E + X) − [(K + E + X)2 − 4(E)X]0.5}) to give the equilibrium dissociation constant Kd for the E-I complex. The entire experiment was performed in triplicate, and the reported Kd values represent the mean ± S.D.
Complex Stabilization Time Course
DNA/RNA hybrids were prepared as described above with PBS-22dpol primer and 3′-radiolabeled PBS-52r template. 2 μm GSK5750 or 1% dimethyl sulfoxide was added to 500 nm HIV-1 RT in a buffer containing 50 mm Tris-HCl (pH 7.8), 50 mm NaCl, 1 mm EDTA, and 6 mm MgCl2. Components in the preincubation mixes were incubated at 37 °C for 10 min. Reactions were initiated with 50 nm DNA/RNA hybrid and/or 0.5 μm GSK5750 and were allowed to proceed for 0, 1, 5, 10, 30, 60, 120, 240, and 360 min.
Preincubation Time Course
Experiments were performed essentially as described above under “Complex Stabilization Time Course,” except GSK5750 and β-thujaplicinol concentrations were 0.5 μm and 50 μm, respectively. Time = 0 was defined as the addition of MgCl2 to the reaction. Samples were taken at preincubation times of 1, 2, 4, 8, 16, 24, 32, 60, and 90 min. Reactions were started by the addition of DNA/RNA hybrid and allowed to proceed for 30 s. Samples were resolved and quantified as described above.
Computational Methods
The three-dimensional structures of β-thujaplicinol and 4-amino-1-hydroxypyridopyrimidin-2-one, the minimal pharmacophore of GSK5750, were generated using Concord software (version 4.0.7, Tripos, Intl.; St. Louis, MO). Similarly, two anionic structures of β-thujaplicinol were generated: one with a negatively ionized, 2-hydroxyl group and the second with a negatively ionized, 7-hydroxyl group. An anionic structure of 4-amino-1-hydroxypyridopyrimidin-2-one was also generated with its 1-hydroxyl group negatively ionized. The three-dimensional structures of all molecules were geometry optimized at the HF/6-311++G** level of theory. Single point energies, total electron densities, and electrostatic potentials were then calculated at the MP2 level with the same basis set. For each anion, the electrostatic potential was mapped onto the 0.002 electron density surface with the resultant maps using the same scale, −0.28 to 0.08 eV, for equivalent color coding. All quantum mechanics calculations were performed with Gaussian 03 (revision C.02, Gaussian, Inc.; Wallingford, CT), and all figures were generated using GaussView (Semichem, Inc.; Shawnee Mission, KS).
RESULTS
Effects of GSK5750 on the Polymerase and RNase H Activities of HIV-1 RT
GSK5750 was designed as an active site inhibitor of the HIV-1 RT-associated RNase H. Similar to β-thujaplicinol and other active site inhibitors, GSK5750 features structural motifs capable of chelating divalent metal ions (Fig. 1). GSK5750 shows two exo-cyclic oxygen atoms and one aromatic nitrogen atom. The goal of this study was to characterize the biochemical properties of GSK5750. We focused on its ability to compete with the nucleic acid substrate at the RNase H active site to determine whether the development of tight binding RNase H inhibitors is conceivable. Initially, we employed a gel-based assay to study potential effects of GSK5750 on the polymerase activity of HIV-1 RT (Fig. 2). We devised a model DNA/RNA primer/template substrate to monitor DNA synthesis over a short stretch of eight nucleotides (Fig. 2A). We performed time course experiments with a high concentration of 20 μm of the polymerase inhibitor PFA, GSK5750, and β-thujaplicinol, respectively (Fig. 2B). Although the addition of PFA shows inhibition of DNA synthesis, neither GSK5750 nor β-thujaplicinol affect the polymerase activity under these conditions (Fig. 2C).
FIGURE 1.
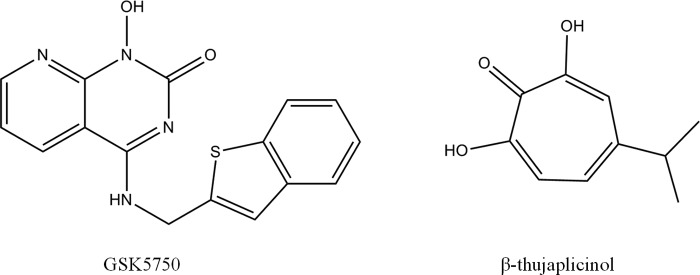
Structures of active site RNase H inhibitors GSK5750 and β-thujaplicinol.
FIGURE 2.

Effect of RNase H inhibitors on DNA synthesis by HIV-1 RT. A, sequence of polymerase-dependent substrate used in this assay. * indicates 5′- radiolabeled primer. B, time course of DNA synthesis (0–10 min) in the absence and presence of PFA, GSK5750, and β-thujaplicinol. C, results from B shown graphically. Polymerase activity in the absence of inhibitor (○), with GSK5750 (×), with β-thujaplicinol (♦), and with PFA (□) was plotted and fitted to a single exponential function.
Previous studies have shown that HIV-1 RNase H inhibitors weakly inhibit primary cleavage events on intact substrates, as free access to the active site is likely impeded. However, secondary cleavage events are more efficiently inhibited with β-thujaplicinol and derivatives. To assess the extent with which GSK5750 interferes with RNase H-induced primary and secondary cleavage events, we used a chimeric DNA-RNA/DNA primer/template substrate that mimics the tRNA (−)-strand primer removal reaction (Fig. 3A). At the same time, we tested the inhibitory effects of GSK5750 and β-thujaplicinol against E. coli RNase H to assess specificity for inhibition. In this experiment, we observed primary RNase H cleavage one base pair downstream of the DNA-RNA junction, followed by downstream secondary cleavages, as described previously (10). Similar to β-thujaplicinol, GSK5750 inhibits secondary cleavages, but only weakly inhibits the primary cleavage (Fig. 3B). The lack of appreciable primary cleavage inhibition points to similar obstacles that limit the access of small molecule inhibitors to the RNase H active site. However, inhibition of secondary cuts is specific to the HIV-1 enzyme. Weak inhibition of E. coli RNase H is seen only at high concentrations of 50 μm β-thujaplicinol. GSK5750 shows no significant inhibitory effect against the E. coli enzyme. The potency of inhibition of the secondary cuts was also stronger than that seen with β-thujaplicinol (GSK5750, IC50 = 0.33 ± 0.11 μm; β-thujaplicinol, IC50 = 3.8 ± 0.65 μm) (Fig. 3C).
FIGURE 3.

Inhibition of RNase H activity of HIV-1 RT and E. coli RNase H by GSK5750 and β-thujaplicinol. A, a chimeric DNA-RNA/DNA substrate was used in the assay, which mimics the (−)-strand primer removal reaction. The primary RNase H cleavage occurs at the DNA/RNA junction +1, as indicated. * indicates 5′- radiolabeled primer. B, comparative effects of GSK5750 and β-thujaplicinol on the RNase H activity of HIV-1 RT and E. coli RNase H on the chimeric substrate shown in A. C, results from B for the HIV-1 RT primary (○) and secondary (■) cuts are shown graphically. IC50 values for HIV-1 RT secondary cleavages are 0.33 ± 0.11 μm and 3.8 ± 0.65 μm for GSK5750 and β-thujaplicinol, respectively.
Dissociation Kinetics of GSK5750
We next performed order-of-addition experiments in attempts to determine whether GSK5750 binds preferentially to the free enzyme in the presence of divalent metal ions, similar to that shown previously with β-thujaplicinol, We used the model DNA/RNA substrate shown in Fig. 4A. There are no significant effects on RNase H activity due to the order-of-addition in the absence of inhibitor: preincubation of HIV-1 RT with or without MgCl2, or with DNA/RNA substrate (Fig. 4B). The experimental setup in the presence of GSK5750 was as follows: (i) preincubation of RT with DNA/RNA and GSK5750, starting the reaction with MgCl2; (ii) preincubation of RT with GSK5750 alone, starting the reaction with DNA/RNA and MgCl2; and (iii) preincubation of RT with GSK5750 and MgCl2 and starting the reaction with DNA/RNA. Inhibition was observed solely when GSK5750 was pre-incubated with enzyme and catalytic metal ions, which provides strong evidence for active site binding (Fig. 4C). Intriguingly, inhibition of RNase H activity is sustained for 30 s at a point when substrate cleavage has completely rebounded in the presence of β-thujaplicinol (10). These findings suggest that dissociation of GSK5750 from the RNase H active site is significantly reduced when compared with β-thujaplicinol. Hence, we next determined the apparent rate of dissociation of inhibitor from the E-I complex. We preformed the RT-GSK5750 complex in the presence of MgCl2, added RNA/DNA substrate, and followed the RNase H cleavage reaction over longer periods of time (Fig. 4D). In the absence of GSK5750, the substrate was completely cleaved within the first minute of the reaction. Remarkably, in the presence of GSK5750, 50% of the substrate remained intact after 60 min, and complete cleavage was observed after 400 min (Fig. 4B). By comparison, 100% of the substrate was cleaved after 20 s in the presence of β-thujaplicinol under the same conditions (10).
FIGURE 4.

Effects of the order-of-addition on inhibition of RNase H activity. A, sequence of the polymerase-dependent substrate used in this assay. Primary RNase H cleavages are indicated. * indicates 3′-radiolabeled primer. B, graphic representation of the effects of the order-of-addition of reaction components in the absence of inhibitor. Percent activity refers to the percentage of RNA template converted into the −18 and −19 major products. C, effects of the order-of-addition of reaction components during inhibition by GSK5750 (0.5 μm) under pre-steady-state conditions. D, results of extended time course experiment using the inhibited pre-formed E-I-S complex from C.
Association Kinetics of GSK5750
To determine the rate of inhibitor association, we investigated the effect of the pre-incubation period in the formation of an E-I complex on RNase H inhibition using the same model DNA/RNA substrate. In this assay, we varied the time between the addition of inhibitor to free enzyme and the initiation of the reaction with substrate (Fig. 5). We observed that maximum inhibition by GSK5750 occurred only after ∼8 min of pre-incubation. This demonstrates that binding of GSK5750 to HIV-1 RT is slow. The rapid dissociation of β-thujaplicinol prevents accurate measurements of rates of association under these conditions (Fig. 5).
FIGURE 5.
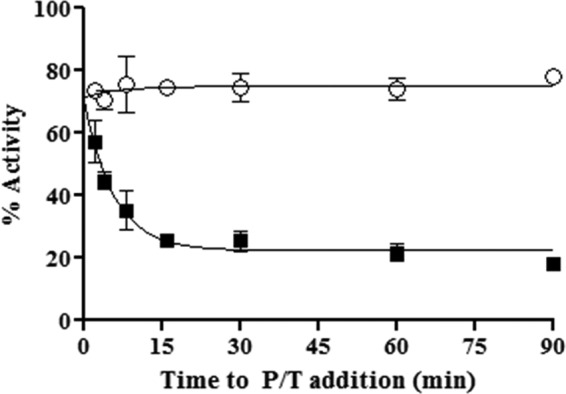
Time course assay showing association of GSK5750 with free enzyme. x axis represents time from the creation of the E-I complex to the addition of primer/template (P/T) substrate for both 50 μm β-thujaplicinol (○) and 0.5 μm GSK5750 (■).
Affinity of GSK5750 to HIV-1 RT RNase H
To further assess binding of GSK5750 to HIV-1 RT, we measured the equilibrium dissociation constant (Kd) of GSK5750 (Fig. 6, A and B). RNase H activity was determined on a DNA/RNA primer/template with a recessed 3′-primer end in the presence of varying concentrations of GSK5750. The resulting curves show a change in the maximum product (Ymax) but not in the observed rate (kobs). Plotting the values for Ymax against inhibitor concentration gives a Kd value for GSK5750 of 386.5 ± 177.5 nm.
FIGURE 6.
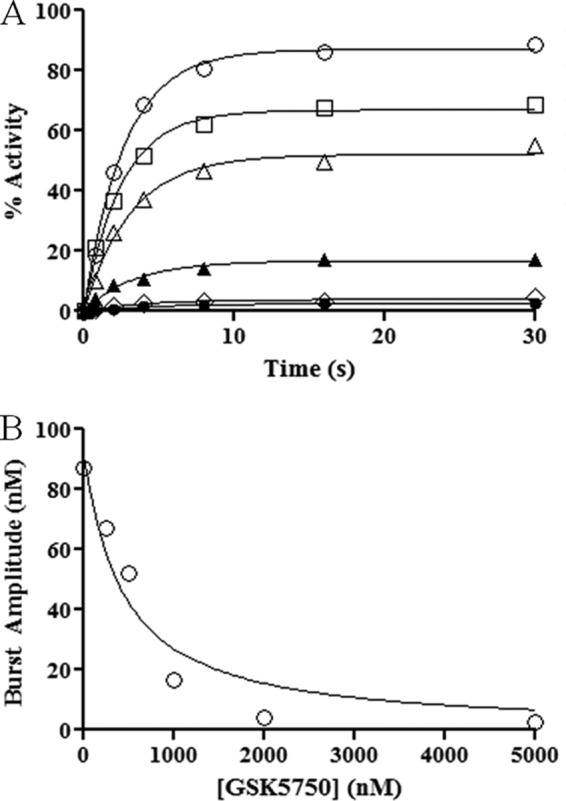
Pre-steady-state RNase H activity in the presence of GSK5750. A, various concentrations of GSK5750 were pre-incubated with MgCl2 and RT. Reactions were started with DNA/RNA. Concentrations of GSK5750 are 0 nm (●), 100 nm (□), 250 nm (▴), 500 nm (▾), 1250 nm (♦), and 2500 nm (○). B, burst amplitudes from A (○) plotted against the concentration of GSK5750 and fitted to a quadratic equation. A Kd (inhibitor) value of 386.5 ± 177.5 nm was calculated from the experiment done in triplicate.
DISCUSSION
In this study, we evaluated the properties of a novel HIV-1 RT RNase H inhibitor, referred to as GSK5750. Despite the development of compounds that show RNase H inhibition in cell-free assays, antiviral activity of compounds that unequivocally target the RNase H active site is yet to be demonstrated. The rather shallow active site is generally viewed as a problem in the development of HIV-1 RT RNase H inhibitors (2, 33). However, the recent approval of boceprevir and telaprevir for the hepatitis C virus NS3-4A serine protease demonstrates that effective small molecules can be designed for shallow binding sites (34–36). We and others have demonstrated that competition with the nucleic acid substrate is a related potential obstacle in this context (10, 23, 31). Order-of-addition experiments revealed that β-thujaplicinol does not bind to a pre-formed complex composed of HIV-1 RT and substrate (10). GSK5750 shows the same behavior in this regard. These experiments also provide strong evidence that GSK5750 is bound at the RNase H active site through a metal ion chelation mechanism. When substrate binding is allowed prior to the start of the reaction, no RNase H inhibition is observed over the time course of a single turnover event. Therefore, similar to β-thujaplicinol, GSK5750 is denied access to the Mg2+ ions in the RNase H active site by the bound substrate. However, RNase H inhibition with GSK5750 is seen over protracted periods of time in the range of hours, provided that the E-I complex is preformed. In contrast, β-thujaplicinol appears to dissociate from the complex within seconds.
The slower dissociation of GSK5750 from the RNase H active site is likely due to its larger metal-chelating scaffold compared with that of β-thujaplicinol and, thus, the amount of electron density available for coordinate bond formation. This is illustrated by the electrostatic potential maps, in which the metal-ligating atoms of GSK5750 are more electron-rich compared with those of β-thujaplicinol (Fig. 7). For the β-thujaplicinol anion, we speculate that its negative charge may not be as readily available for coordinate bond formation due to its delocalization across the tropolone ring, carbonyl, and hydroxyl groups. This is generally consistent with the perspective of Himmel et al. (22), with the exception that they suggest that β-thujaplicinol binds as a neutral molecule.
FIGURE 7.
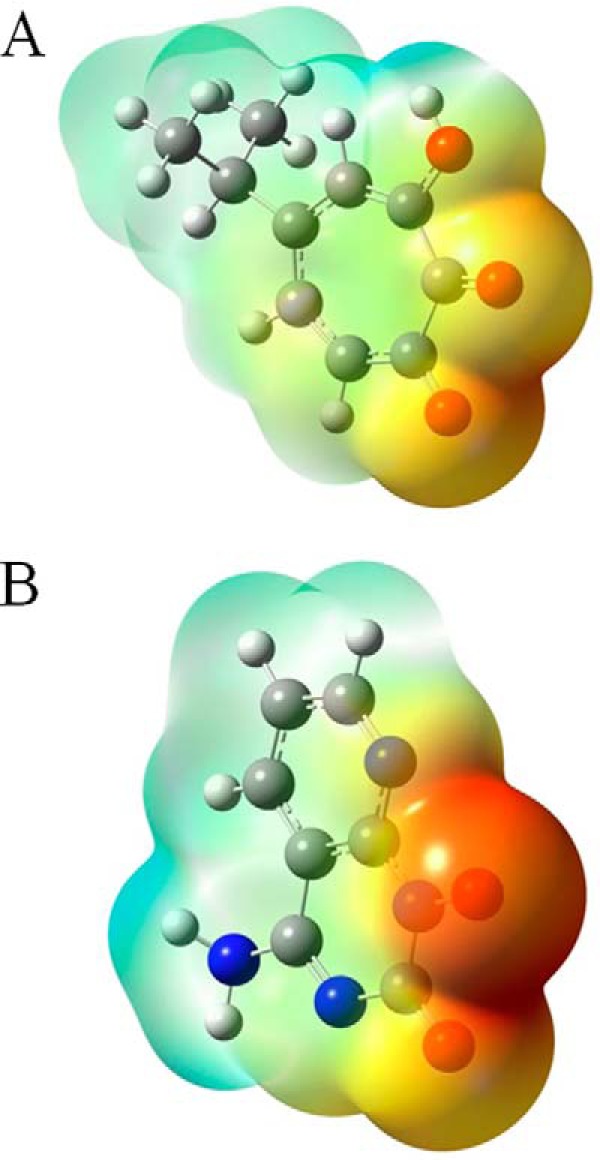
The geometry-optimized molecular structures of β-thujaplicinol (A) and GSK5750 (B). The 2-hydroxyl group of β-thujaplicinol and the 1-hydroxyl group of GSK5750 are negatively ionized. Their electrostatic potential maps are color-coded with shades of red representing electron-rich regions, whereas shades of blue represent electron-poor regions.
In addition to the slow dissociation of GSK5750 from the E-I complex, the formation of a stable E-I-S complex, in which the inhibitor is trapped by the nucleic acid substrate, may also contribute to the increased duration of inhibition and potency of GSK5750. The existence of such a complex has been postulated in the context of β-thujaplicinol (22). It is conceivable that the benzothiophene moiety of GSK5750 contributes to this interaction by directly binding to the substrate, perhaps by intercalation.
It is also of interest that GSK5750 requires an unexpectedly long pre-incubation with enzyme to achieve maximal inhibition. We propose that this apparent slow rate of formation of an active, inhibitory E-I complex is in part due to the rate of deprotonation of the hydroxyl groupof GSK5750; a prerequisite for the formation of coordinate bonds with the active site metals. The metal cations themselves should facilitate the deprotonation process by lowering the free energy associated with proton abstraction. Although the slow rate of dissociation of GSK5750 allows this observation to be made by the methods employed here, the fast dissociation rate of β-thujaplicinol precludes its ability to demonstrate similar kinetics.
Collectively, this report illustrates methods for the biochemical characterization and comparison of metal-chelating RNase H inhibitors. By these methods, GSK5750 was found to be superior to β-thujaplicinol, most notably in its ability to form a long-lasting complex with HIV-1 RT, which may in part neutralize the obstacle presented by the competing nucleic acid substrate. Thus, inhibitors based on this scaffold or other scaffolds with similar or better binding profiles that also include faster rates of association, may help to advance the development of antiviral drugs that target the RNase H activity of HIV-1 RT.
Acknowledgments
We thank Suzanne McCormick and Anick Auger for invaluable technical help.
This work was supported in part by the Canadian Foundation for AIDS Research (CANFAR) and by GlaxoSmithKline (GSK).
- RNase H
- ribonuclease H
- PFA
- phosphonoformic acid
- E-I
- enzyme-inhibitor.
REFERENCES
- 1. Telesnitsky A., Goff S. (eds) (1997) Retroviruses, pp. 121–160, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 2. Davies J. F., 2nd, Hostomska Z., Hostomsky Z., Jordan S. R., Matthews D. A. (1991) Crystal structure of the ribonuclease H domain of HIV-1 reverse transcriptase. Science 252, 88–95 [DOI] [PubMed] [Google Scholar]
- 3. Huang H., Chopra R., Verdine G. L., Harrison S. C. (1998) Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: implications for drug resistance. Science 282, 1669–1675 [DOI] [PubMed] [Google Scholar]
- 4. Jacobo-Molina A., Ding J., Nanni R. G., Clark A. D., Jr., Lu X., Tantillo C., Williams R. L., Kamer G., Ferris A. L., Clark P. (1993) Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 A resolution shows bent DNA. Proc. Natl. Acad. Sci. U.S.A. 90, 6320–6324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sarafianos S. G., Das K., Tantillo C., Clark A. D., Jr., Ding J., Whitcomb J. M., Boyer P. L., Hughes S. H., Arnold E. (2001) Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 20, 1449–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. De Clercq E. (2007) The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 6, 1001–1018 [DOI] [PubMed] [Google Scholar]
- 7. Kohlstaedt L. A., Wang J., Friedman J. M., Rice P. A., Steitz T. A. (1992) Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science 256, 1783–1790 [DOI] [PubMed] [Google Scholar]
- 8. Gopalakrishnan V., Peliska J. A., Benkovic S. J. (1992) Human immunodeficiency virus type 1 reverse transcriptase: spatial and temporal relationship between the polymerase and RNase H activities. Proc. Natl. Acad. Sci. U.S.A. 89, 10763–10767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Götte M., Fackler S., Hermann T., Perola E., Cellai L., Gross H. J., Le Grice S. F., Heumann H. (1995) HIV-1 reverse transcriptase-associated RNase H cleaves RNA/RNA in arrested complexes: implications for the mechanism by which RNase H discriminates between RNA/RNA and RNA/DNA. EMBO J. 14, 833–841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Beilhartz G. L., Wendeler M., Baichoo N., Rausch J., Le Grice S., Götte M. (2009) HIV-1 reverse transcriptase can simultaneously engage its DNA/RNA substrate at both DNA polymerase and RNase H active sites: implications for RNase H inhibition. J. Mol. Biol. 388, 462–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marchand B., Götte M. (2003) Site-specific footprinting reveals differences in the translocation status of HIV-1 reverse transcriptase. Implications for polymerase translocation and drug resistance. J. Biol. Chem. 278, 35362–35372 [DOI] [PubMed] [Google Scholar]
- 12. Furfine E. S., Reardon J. E. (1991) Reverse transcriptase.RNase H from the human immunodeficiency virus. Relationship of the DNA polymerase and RNA hydrolysis activities. J. Biol. Chem. 266, 406–412 [PubMed] [Google Scholar]
- 13. Schultz S. J., Champoux J. J. (2008) RNase H activity: structure, specificity, and function in reverse transcription. Virus Res. 134, 86–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fuentes G. M., Rodríguez-Rodríguez L., Fay P. J., Bambara R. A. (1995) Use of an oligoribonucleotide containing the polypurine tract sequence as a primer by HIV reverse transcriptase. J. Biol. Chem. 270, 28169–28176 [DOI] [PubMed] [Google Scholar]
- 15. Götte M., Maier G., Onori A. M., Cellai L., Wainberg M. A., Heumann H. (1999) Temporal coordination between initiation of HIV (+)-strand DNA synthesis and primer removal. J. Biol. Chem. 274, 11159–11169 [DOI] [PubMed] [Google Scholar]
- 16. Huber H. E., Richardson C. C. (1990) Processing of the primer for plus strand DNA synthesis by human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 265, 10565–10573 [PubMed] [Google Scholar]
- 17. Palaniappan C., Fay P. J., Bambara R. A. (1995) Nevirapine alters the cleavage specificity of ribonuclease H of human immunodeficiency virus 1 reverse transcriptase. J. Biol. Chem. 270, 4861–4869 [DOI] [PubMed] [Google Scholar]
- 18. Pullen K. A., Champoux J. J. (1990) Plus-strand origin for human immunodeficiency virus type 1: implications for integration. J. Virol. 64, 6274–6277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith J. S., Roth M. J. (1992) Specificity of human immunodeficiency virus-1 reverse transcriptase-associated ribonuclease H in removal of the minus-strand primer, tRNA(Lys3). J. Biol. Chem. 267, 15071–15079 [PubMed] [Google Scholar]
- 20. Beilhartz G. L., Götte M. (2010) HIV-1 ribonuclease H: structure, catalytic mechanism and inhibitors. Viruses 2, 900–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tramontano E., Di Santo R. (2010) HIV-1 RT-associated RNase H function inhibitors: Recent advances in drug development. Curr. Med. Chem. 17, 2837–2853 [DOI] [PubMed] [Google Scholar]
- 22. Himmel D. M., Maegley K. A., Pauly T. A., Bauman J. D., Das K., Dharia C., Clark A. D., Jr., Ryan K., Hickey M. J., Love R. A., Hughes S. H., Bergqvist S., Arnold E. (2009) Structure of HIV-1 reverse transcriptase with the inhibitor β-thujaplicinol bound at the RNase H active site. Structure 17, 1625–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kirschberg T. A., Balakrishnan M., Squires N. H., Barnes T., Brendza K. M., Chen X., Eisenberg E. J., Jin W., Kutty N., Leavitt S., Liclican A., Liu Q., Liu X., Mak J., Perry J. K., Wang M., Watkins W. J., Lansdon E. B. (2009) RNase H active site inhibitors of human immunodeficiency virus type 1 reverse transcriptase: design, biochemical activity, and structural information. J. Med. Chem. 52, 5781–5784 [DOI] [PubMed] [Google Scholar]
- 24. Klumpp K., Mirzadegan T. (2006) Recent progress in the design of small molecule inhibitors of HIV RNase H. Curr. Pharm. Des. 12, 1909–1922 [DOI] [PubMed] [Google Scholar]
- 25. Su H. P., Yan Y., Prasad G. S., Smith R. F., Daniels C. L., Abeywickrema P. D., Reid J. C., Loughran H. M., Kornienko M., Sharma S., Grobler J. A., Xu B., Sardana V., Allison T. J., Williams P. D., Darke P. L., Hazuda D. J., Munshi S. (2010) Structural basis for the inhibition of RNase H activity of HIV-1 reverse transcriptase by RNase H active site-directed inhibitors. J. Virol. 84, 7625–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Arion D., Sluis-Cremer N., Min K. L., Abram M. E., Fletcher R. S., Parniak M. A. (2002) Mutational analysis of Tyr-501 of HIV-1 reverse transcriptase: effects on ribonuclease H activity and inhibition of this activity by N-acylhydrazones. J. Biol. Chem. 277, 1370–1374 [DOI] [PubMed] [Google Scholar]
- 27. Himmel D. M., Sarafianos S. G., Dharmasena S., Hossain M. M., McCoy-Simandle K., Ilina T., Clark A. D., Jr., Knight J. L., Julias J. G., Clark P. K., Krogh-Jespersen K., Levy R. M., Hughes S. H., Parniak M. A., Arnold E. (2006) HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl naphthyl hydrazone bound at a novel site. ACS Chem. Biol. 1, 702–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chung S., Wendeler M., Rausch J. W., Beilhartz G., Gotte M., O'Keefe B. R., Bermingham A., Beutler J. A., Liu S., Zhuang X., Le Grice S. F. (2010) Structure-activity analysis of vinylogous urea inhibitors of human immunodeficiency virus-encoded ribonuclease H. Antimicrob. Agents Chemother. 54, 3913–3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wendeler M., Lee H. F., Bermingham A., Miller J. T., Chertov O., Bona M. K., Baichoo N. S., Ehteshami M., Beutler J., O'Keefe B. R., Götte M., Kvaratskhelia M., Le Grice S. (2008) Vinylogous ureas as a novel class of inhibitors of reverse transcriptase-associated ribonuclease H activity. ACS Chem. Biol. 3, 635–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kati W. M., Johnson K. A., Jerva L. F., Anderson K. S. (1992) Mechanism and fidelity of HIV reverse transcriptase. J. Biol. Chem. 267, 25988–25997 [PubMed] [Google Scholar]
- 31. Herman B. D., Sluis-Cremer N. (2013) Transient kinetic analyses of the ribonuclease H cleavage activity of HIV-1 reverse transcriptase in complex with efavirenz and/or a β-thujaplicinol analogue. Biochem. J. 455, 179–184 [DOI] [PubMed] [Google Scholar]
- 32. Le Grice S. F., Grüninger-Leitch F. (1990) Rapid purification of homodimer and heterodimer HIV-1 reverse transcriptase by metal chelate affinity chromatography. Eur. J. Biochem. 187, 307–314 [DOI] [PubMed] [Google Scholar]
- 33. Garvey E. P., Romines K. R., Boone L. R. (2003) Discovery and Development of New HIV Medicines in AIDS and Other Manifestations of HIV Infection (Wormser G. P., ed.), 4th Ed., pp. 855–866, Academic Press, San Diego [Google Scholar]
- 34. Burney T., Dusheiko G. (2011) Overview of the PROVE studies evaluating the use of telaprevir in chronic hepatitis C genotype 1 patients. Expert Rev. Anti Infect. Ther. 9, 151–160 [DOI] [PubMed] [Google Scholar]
- 35. Foote B. S., Spooner L. M., Belliveau P. P. (2011) Boceprevir: a protease inhibitor for the treatment of chronic hepatitis C. Ann. Pharmacother. 45, 1085–1093 [DOI] [PubMed] [Google Scholar]
- 36. Lin C. (2006) HCV NS3–4A serine protease in Hepatitis C Viruses: Genomes and Molecular Biology (Tan S.-L., ed) pp. 163–206, Horizon Bioscience, Norfolk, UK [Google Scholar]