

Background: Naturally occurring anticoagulant proteins provide models for new medications with highly desirable properties.
Results: The crystal structure of the active region of mosquito protein AAPP has been solved.
Conclusion: The mosquito protein AAPP uses a small turn region to block coagulation extremely effectively by binding collagen.
Significance: New small molecule anti-coagulants may be developed with completely new mechanisms and none of the drawbacks of current treatments.
Keywords: Antibody Engineering, Blood Coagulation Factors, Collagen, Crystal Structure, Malaria, Anti-coagulant
Abstract
Blood clotting is a vitally important process that must be carefully regulated to prevent blood loss on one hand and thrombosis on the other. Severe injury and hemophilia may be treated with pro-coagulants, whereas risk of obstructive clotting or embolism may be reduced with anti-coagulants. Anti-coagulants are an extremely important class of drug, one of the most widely used types of medication, but there remains a pressing need for novel treatments, however, as present drugs such as warfarin have significant drawbacks. Nature provides a number of examples of anti-coagulant proteins produced by blood-sucking animals, which may provide templates for the development of new small molecules with similar physiological effects. We have, therefore, studied an Anopheles anti-platelet protein from a malaria vector mosquito and report its crystal structure in complex with an antibody. Overall the protein is extremely sensitive to proteolysis, but the crystal structure reveals a stable domain built from two helices and a turn, which corresponds to the functional region. The antibody raised against Anopheles anti-platelet protein prevents it from binding collagen. Our work, therefore, opens new avenues to the development of both novel small molecule anti-clotting agents and anti-malarials.
Introduction
For blood-sucking insects, leeches, or other animals, it is important to prevent the blood of the host clotting after puncture of the skin (1, 2). These animals produce a number of factors in their saliva to block the action of host defense mechanisms and ensure blood flow (1, 2). Anti-clotting factors from these parasitic animals are of medical interest, and leeches have been used historically for cleaning wounds (1, 2). Because blood coagulation depends on numerous proteins, it is not surprising that different parasites have evolved anti-clotting factors with different targets (1). Bivalirudin, an oligopeptide analog of the leech anti-clotting factor hirudin, is a direct thrombin inhibitor (3), and desmoteplase, from the saliva of bats, is a plasminogen activator (4–6). Both have undergone clinical trials. We have identified an abundant protein in the saliva of the female mosquito Anopheles stephensi, a known malaria vector. This protein, anopheline anti-platelet protein (AAPP),5 is found to bind directly to type-I and type-III collagen and blocks platelet adhesion with an IC50 of ∼30 nm (7). Collagen is only exposed to circulating plasma proteins at sites of damage to blood vessels (8–10). It causes platelets to aggregate through the action of the plasma protein von Willebrand factor, which initiates platelet tethering and blood clotting at sites of vascular injury. Activated platelets subsequently adhere directly to subendothelial collagen through glycoprotein 1b so that blocking exposed collagen prevents the initiation and progression of platelet aggregation. AAPP shows similarity to salivary gland proteins from other mosquito species, including aegyptin from Anopheles aegypti, which also binds collagen (11–13). These proteins are the first anti-coagulants found to have this mode of action. Earlier work by us has shown AAPP is able to inhibit blood aggregation without prolonging bleeding time, which holds out the promise of safer treatments for thrombotic disease (14). Other types of anti-platelet medication such as cyclooxygenase inhibitors have a risk of leading to excessive bleeding (15). Among present anticoagulants, warfarin in particular is known to be problematic, having a narrow dose window, complications due to interactions with food-derived molecules, and a very different pharmacological profile in different patients (16).
A molecular model of AAPP is highly desirable in order to understand the ability of the protein to block collagen and platelets from interacting. In particular it would be helpful for efforts to design small molecule drugs with the same mode of action as AAPP. Despite the ability to express and purify AAPP in significant amounts, crystallization has proved extremely challenging. Here we describe the use of an 8H7 Fab monoclonal antibody (mAb) fragment to stabilize AAPP sufficiently to allow well ordered crystals to be grown. The structure of the complex is described.
EXPERIMENTAL PROCEDURES
Cloning, Expression, and Purification of AAPP
Cloning and purification were essentially carried out as for the AAPP protein described previously (7, 14, 17). The gene sequence from A. stephensi mosquitoes was cloned into pET22 with a hexahistidine tag and tobacco etch virus cleavage site at the C terminus. The resulting expression plasmid was transformed into Escherichia coli BL21(DE3) strain, and cells were cultured at 15 °C overnight after induction with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside. The AAPP was purified by chromatography using nickel-nitrilotriacetic acid-agarose (Qiagen) followed by Q Sepharose (GE Healthcare). The histidine tag was removed by tobacco etch virus protease digestion after nickel-nitrilotriacetic acid chromatography, and the purified complex was then concentrated to 10 mg/ml by Centricon YM-3 (Millipore) for crystallization.
Mutagenesis
The cDNAs of AAPP mutants were amplified by polymerase chain reaction (PCR) using Pfu Ultra (Stratagene). Primers are listed in Table 1. A primer pair of pGEX6P2-F2/p8H7-pep3-R1 was used for the PCR of AAPP225–244, whereas a primer pair pGEX6P2-F2/pAnSG-R17 was used for C4. Primer pairs pAnSG-F8/pAnSG-R20 and pAnSG-F20/pAnSG-R1 were used for the overlapping PCR of C3, whereas primer pairs pAnSG-F8/pAnSG-R21 and pAnSG-F21/pAnSG-R1 were used for 4A. The two PCR products were gel-purified using NucleoSpin® Gel and PCR Clean-up (Takara, Otsu, Japan) followed by a second PCR using pAnSG-F8 and pAnSG-R1. All PCR products were cloned into pENTR-TOPO vector (Invitrogen) and then digested with NcoI/NotI for C3 and 4A and NdeI/XhoI for C4 and AAPP225–244 for the cloning into the pET22-GEX6P2 vector (18). cDNA of C3/C4 was amplified from pET22-GEX6P2-AAPPex3–4 C4 using primer pairs pAnSG-F8/pAnSG-R20 and pAnSG-F20/pAnSG-R17, and a second PCR was carried out using pAnSG-F8 and pAnSG-R17. After cloning into the pENTR vector, a DNA fragment encoding C3/C4 was excised by digested with NcoI/XhoI and cloned into the pET22-GEX6P2 vector. The mutants were purified by chromatography using nickel-nitrilotriacetic acid agarose and eluted with imidazole. Slide-A-Lyzer dialysis cassettes with a Mr cutoff of 10,000 (Pierce) were used to dialyze samples into PBS (pH 7.4).
TABLE 1.
Primers used in AAPP mutants
| Primer | Sequence |
|---|---|
| pGEX6P2-F2 | CACCATATGTCCCCTATACTAGGTTATT |
| p8H7-pep3-R1 | CTCGAGTGTACCTTCACTGCCGCAGTGAGGGTTATTCTTGTCCTTGCTGCACTCTTCGTACGATGTGAATTCCGGGGATCCCATGGGCCCCTGGAA |
| pAnSG-F8 | CACCATGGGTGAAGAGTCCCCCGTTAATAC |
| pAnSG-F20 | AACCCTCACGCAGGCAGTGAA |
| pAnSG-F21 | GAAGAGTGCAGCGCAGCTGCTGCTAACCCTCACTGC |
| pAnSG-R1 | GGCGGCCGCCTCTGAATCACGCTTTTCGACGATGC |
| pAnSG-R17 | CTCGAGCTCTGAATCACGCTTTTCGACGATCGCATCCGATAGCTT |
| pAnSG-R20 | TTCACTGCCTGCGTGAGGGTT |
| pAnSG-R21 | GCAGTGAGGGTTAGCAGCAGCTGCGCTGCACTCTTC |
Production of Anti-AAPP Antibodies
A DNA fragment encoding AAPP exon 3–4 was excised from pET32-AAPPex3–4 (17) by digested with NcoI and XhoI and cloned into NcoI/XhoI sites of the E. coli expression vector pET22-GEX6P2. The resulting expression plasmid, pET22-GEX6P2-AAPPex3–4, was transformed into E. coli BL21(DE3) strain, and cells were cultured at 37 °C for 2 h after induction with 1 mm isopropyl 1-thio-β-d-galactopyranoside. The AAPPex3–4 was purified by chromatography using glutathione-Sepharose 4B (GE Healthcare). The GST tag was removed by PreScission Protease (GE Healthcare) digestion after GST chromatography. After immunization of BALB/c mice with the AAPPex3–4, the spleen cells were fused with P3X63Ag8.U1 myeloma cells (American Type Culture Collection, Manassas, VA) using an established procedure (19). Hybridoma lines were screened by enzyme-linked immunosorbent assay (ELISA) using the AAPPex3–4. Moreover, the ELISA-positive hybridoma lines were rescreened to obtain inhibitory monoclonal antibodies for AAPP-collagen interaction by AAPP binding assay described previously (7). Briefly, the AAPPex3–4 was preincubated with each mAb, and the mixture was added to 96-well collagen-coated microtiter plates (Nunc, Rochester, NY). Binding of the AAPPex3–4 to collagen was detected using the ExpressDetector nickel-HRP (KPL, Gaithersburg, MD), which can bind to the His tag at the C terminus of the AAPPex3–4. One of the inhibitory monoclonal antibodies, 8H7, was maintained in RPMI 1640 supplemented with 10% fetal calf serum. The 8H7 mAb was purified from ascites fluid using Protein G affinity column (GE Healthcare).
Preparation of 8H7 IgG and Fab
The 8H7 IgG mAb was purified using the Protein G affinity column (GE Healthcare) from the supernatant of cultured hybridoma cells expressing the murine mAb 8H7 IgG. After the filtration of the supernatant, the sample was loaded onto the column equilibrated with 20 mm potassium phosphate (pH 7.0) buffer. The mAb fraction was eluted with 100 mm glycine (pH 2.7). The eluate was neutralized immediately after elution 1 m Tris-HCl (pH 9.0) and dialyzed overnight against 20 mm potassium phosphate (pH 7.0). The 8H7 Fab fragment was prepared through limited digestion with immobilized papain (Thermo Scientific). The reaction was carried out in 20 mm potassium phosphate (pH 7.0) and 20 mm l-cysteine. 10 mg of IgG was added per 0.5 ml of immobilized papain and incubated for 6 h at 37 °C. After the reaction the sample was separated with immobilized papain using the spin column by centrifugation at 4000 rpm for 15 min and dialyzed overnight against 20 mm Tris-HCl (pH 8.0). The protein was then loaded onto Q-Sepharose (GE Healthcare) to remove undigested IgG and Fc. The 8H7 Fab was passed through Q-Sepharose (GE Healthcare) to remove minor proteins before loading onto Superdex 200 (GE Healthcare) equilibrated with the same buffer. Finally, 8H7 Fab was concentrated to 10 mg/ml by ultrafiltration using a Centriprep YM-30 (Millopore) for crystallization.
Cloning and Sequencing of the Variable Heavy and Light Chain Genes of 8H7 mAb
mRNA was extracted from 1 × 107 hybridoma cell line 8H7 using the FastTrack 2.0 mRNA Isolation kit (Invitrogen), and first-strand cDNA was synthesized from the mRNA with reverse transcriptase using the First-strand cDNA Synthesis kit (Novagen). The cDNA was used as a template for PCR amplification of the variable heavy and light chain genes of 8H7 mAb using Taq 2000 DNA polymerase (Stratagene) and the primer sets in the Mouse Ig-Prime kit (Novagen). The PCR fragments were cloned into pCR2.1 (Invitrogen), the variable heavy and light chain genes were sequenced, and the nucleotide sequence data have been deposited in GenBankTM database under the accession numbers AB903029 and AB903030.
Crystallization and Structure Determination
The AAPP-8H7 Fab complex was crystallized by vapor diffusion using the sitting drop method. Protein (20 mm Tris-HCl (pH 8.0), 150 mm NaCl, 5 mm CaCl2) and reservoir solution (0.1 m HEPES (pH 7.0), 15% PEG 20,000) were mixed in a 1:1 ratio then equilibrated against 1 ml of reservoir solution at 20 °C. Crystals grew in space-group P212121, with a = 93.8 Å, b = 99.4 Å, c = 166.0 Å and contained two molecules in the asymmetric unit. Diffraction data were collected at −180 °C using crystal flash-frozen in crystallization buffer containing 18% (v/v) glycerol. Diffraction data were collected at 1.0 Å on beam line BL17A stations at the Photon Factory, Tsukuba, Japan using an ADSC Quantum 315 CCD detector. All data were processed and scaled using HKL2000 (20). The AAPP-8H7 Fab complex structure was solved by molecular replacement using Phaser (21) and the previously reported Fab structure (22) as a starting model. Two solutions of 8H7-Fab dimer were obtained with final TFZ (translation function Z-score) of 19.0. The model of 8H7-Fab was subjected to rigid-body refinement using PHENIX (23) giving an R-factor of 0.35. The resultant |2Fo − Fc| and |Fo − Fc| electron density maps clearly showed two AAPP molecules. The electron density was interpreted and traced using COOT (24), and the model was refined with PHENIX (23). Solvent molecules were placed at positions where spherical electron density peaks were found above 1.3σ in the |2Fo − Fc| map and above 3.0σ in the |Fo − Fc| map and where stereochemically reasonable hydrogen bonds could form. Structural evaluation of the final models of the AAPP-8H7 Fab complex using MolProbity (25) indicated that 98.9% of the residues are in the most favorable regions of the Ramachandran plot. A summary of the data collection and refinement statistics is given in Table 2. Figures were prepared with PyMOL (26) and LIGPLOT (27). Atomic coordinates and structure factors of the complex have been deposited in the Protein Data Bank under accession code 4OKV.
TABLE 2.
Crystal parameters, data collection, and structure refinement
| Data set | AAPP-8H7 Fab |
|---|---|
| Data collection | |
| Resolution range (Å) | 50.0–1.80 |
| Space group | P212121 |
| Unit cell dimensions (Å) | a = 93.8, b = 99.4, c = 166.0 |
| Reflections (measured/unique) | 508,041/135,319 |
| Completeness (overall/outer shell,a %) | 94.5/80.8 |
| Rmergeb (overall/outer shell,a %) | 9.3/43.3 |
| Mean 〈I〉/〈σ(I)〉 (overall) | 9.6 |
| Redundancy (overall) | 3.9 |
| Wilson B-factor (Å2) | 14 |
| Refinement statistics | |
| R-factor (%)c/free R-factor (%) | 21.8/25.0 |
| Root mean square deviation bond lengths (Å)/bond angles (°) | 0.007/1.134 |
| Number of water molecules | 704 |
| Average B-factor (Å2) | |
| AAPP (131 amino acid residues) | 31 |
| Fab (865 amino acid residues) | 24 |
| Water molecules | 29 |
| Ramachandran plot | |
| Residues in most favorable regions (%) | 98.9 |
| Residues in allowed regions (%) | 1.0 |
| Residues in outlier regions (%) | 0.1 |
a Values in outer shell are for the highest shell with a resolution of 1.83-1.80 Å.
b Rmerge = Σ |Ii − 〈I〉|/Σ|Ii|, where Ii is the intensity of an observation, 〈I〉 in the mean value for that reflection, and the summations are over all reflections. Free R-factor was calculated with 5% of the data.
c R-factor = Σh‖Fo(h)| − |Fc(h)‖/ΣhFo(h), where Fo and Fc are the observed and calculated structure factor amplitudes, respectively.
Collagen Binding Assay
The collagen binding assay was carried out as described previously (7, 17). In brief, soluble collagen type-I was immobilized in 96-well enzyme immunoassay plates followed by blocking with blocking buffer (PBS containing 1% BSA). AAPP was serially diluted and incubated for 1 h with indicated doses of the 8H7 Fab and whole IgG. The proteins were incubated for 1 h at room temperature on the plates, and binding of AAPP to collagen was detected using anti-His Abs conjugated with HRP (Qiagen).
Pulldown Assay
GST (5.7 μm), GST-fused AAPPex3–4 (4.1 μm), C3 (9.6 μm), and 4A (4.6 μm) were incubated with the 8H7 Fab (5.7 μm) in a total volume of 50 μl of PBS at room temperature for 30 min. The volume of PBS was raised to 500 μl, and 30 μl of glutathione Sepharose resin (GE Healthcare) was applied to the mixture and then incubated at room temperature for 1 h with rotating. Glutathione resin carrying the proteins was pelleted and washed 3 times with PBS. Proteins were eluted by boiling in 25 μl of buffer with 2% 2-mercaptoethanol and loaded onto 12% SDS-PAGE gels.
Mass Spectrometry
Full-length and selenomethionine AAPP samples were analyzed by MALDI-TOF mass spectrometry (Bruker Daltonics). 2 μl of the protein solution (30 μm) was mixed with 0.5 μl of the matrix solution (10 mg/ml 3,5-dimethoxy-4-hydroxycinnamic acid [sinapinic acid] in a 0.1% trifluoroacetic acid, 70% acetonitrile aqueous solution) on the MALDI sample target. The mixture was allowed to dry at room temperature before analysis. Each MALDI-TOF mass spectrum was acquired by 200–250 laser shots. FlexAnalysis Version 2.0 (Bruker Daltonics) was used for data processing.
RESULTS AND DISCUSSION
Molecular Characteristics of AAPP
The cloning and expression of AAPP in E. coli with an N-terminal histidine tag has been described. Full-length AAPP can be readily expressed in a soluble form to a level of ∼3 mg/liter of culture by overnight expression at 15 °C. A simple procedure to remove the affinity tag followed by gel filtration yields samples that appear pure by gel electrophoresis. After purification the protein mass was measured by MALDI-TOF spectrometry. The experimental mass of 27302.7 Da agrees well with the predicted mass of 27292.1 Da (Fig. 1A). AAPP has a low complexity region of ∼90 residues, rich in glycine and glutamic acid, from residue 60 to 150.
FIGURE 1.

Mass spectra and amino sequence of the AAPP protein. MALDI-TOF spectrum of full-length (A) and SeMet-AAPP (B) is shown. Degradation from the C terminus removes residues one at a time so that adjacent peaks are separated by the mass of the corresponding amino acid. The N terminus of the fragments shown is Ser-203, and their C termini are close to that of the full-length protein. C, protein sequence of AAPP showing the exon structure of the gene. The secondary structure of the crystal structure is shown over residues 202–269. a.u., absorbance units.
Thousands of crystallization trials with freshly purified protein proved fruitless. A mAb was, therefore, raised against full-length AAPP, and the AAPP-8H7 Fab complex was subjected to crystallization trials. Thin, needle-shaped crystals were obtained that diffracted to 1.8 Å resolution. Phases were obtained by molecular replacement of the previously reported Fab structure (22) as a starting model. This allowed a model of AAPP to be built from Tyr-202 to Glu-269, but no further ordered residues appear in the structure. To highlight other parts of the structure in the electron density map, a number of mutants were made changing individual leucine residues to methionine in order to place selenium atoms at selected points in the structure. Overall four leucine residues (162, 181, 184, 251) were mutated, and the selenomethionine protein was purified. In each case the mutant crystallized in space group P212121 with similar cell parameters to the native crystal (Table 2). Diffraction data from these crystals, however, failed to reveal any selenium atoms outside the core region previously identified, indicating these residues are either absent or simply highly disordered.
After storage of the selenomethionine mutant samples at 4 °C for 1 month, a repeat MALDI-TOF experiment showed complete loss of the full-length protein (Fig. 1B). The major peak was found to have a mass of 8128.7 Da, suggesting significant degradation. Testing samples at intermediate stages after storage of a few weeks yielded a greater range of sharply-defined peaks. Comparing the masses of these fragments revealed a succession of cleavage events removing single amino acid residues (Table 3). Reference to the sequence of AAPP showed the polypeptide fragments present in the sample (Fig. 1B). The low complexity region is found roughly 50 residues to the N terminus of the stable domain.
TABLE 3.
Observed and theoretical masses of degradation in selenomethionine AAPP
| Residues number | Sequence | Theoretical mass | Observed mass |
|---|---|---|---|
| Da | Da | ||
| 182–249 | SKIKE---A | 7748.4 | 7743.5 |
| 182–250 | SKIKE---AA | 7819.5 | 7812.7 |
| 182–251 | SKIKE---AAA | 7890.6 | 7886.6 |
| 182–252 | SKIKE---AAAE | 8019.7 | 8014.4 |
| 182–253 | SKIKE---AAAEN | 8133.8 | 8128.7 |
| 182–254 | SKIKE---AAAENL | 8246.9 | 8241.6 |
| 182–255 | SKIKE---AAAENLY | 8410.1 | 8405.0 |
Overall Structure of AAPP-8H7 Fab Complex
There are two independent copies of the AAPP-8H7 Fab complex in the model (Fig. 2A) that overlay closely (root mean square deviation = 0.678). Residues from Tyr-202 to Arg-266 of both copies of AAPP are visible in the electron density map, and residue 201 is also modeled in one copy. The ordered residues form two equi-length helices, one severely kinked near one end, connected by a turn that brings the helices into close contact nearly anti-parallel to each other (Fig. 2A). The two copies of AAPP in the asymmetric unit make a major contact through a classic knobs-in-holes interaction, as one helix pair lies against another. Although the Matthews coefficient (VM) is 2.32 A3 Da−1, suggesting a solvent content of ∼50%, large open cavities appear in the crystal lattice due to the highly helical AAPP holding apart the mAb molecules. The AAPP domain is stabilized by two disulfide bonds, one near each end of the helices (Fig. 2B). There are also salt-bridge interactions across the helices between Asp-215 and Arg-254 and between Glu-207 and Lys-265 (Fig. 2A). Such a two-dimensional structure has little hydrophobic core in any meaningful sense, but there are also hydrophobic contacts such as between residues Leu-219, Leu-247, and Leu-251 (Fig. 2B). Without the disulfide bridges the structure shows little evidence of being stable. The turn region consists of residues Asp-233, Lys-234, Asn-235, Asn-236, and Pro-237 (Fig. 2C). Both asparagine residues form hydrogen bonds to main-chain atoms through their side chains. Asn-236 also bonds to the imidazole of His 238 (Fig. 3A). These interactions suggest the turn region is generally stable, which is supported by the temperature factors of atoms in these residues. The average temperature factors of all atoms in the two AAPP subunits are 29.7 and 32.7 Å2 and are 16.1 and 18.6 Å2 in the turn regions.
FIGURE 2.
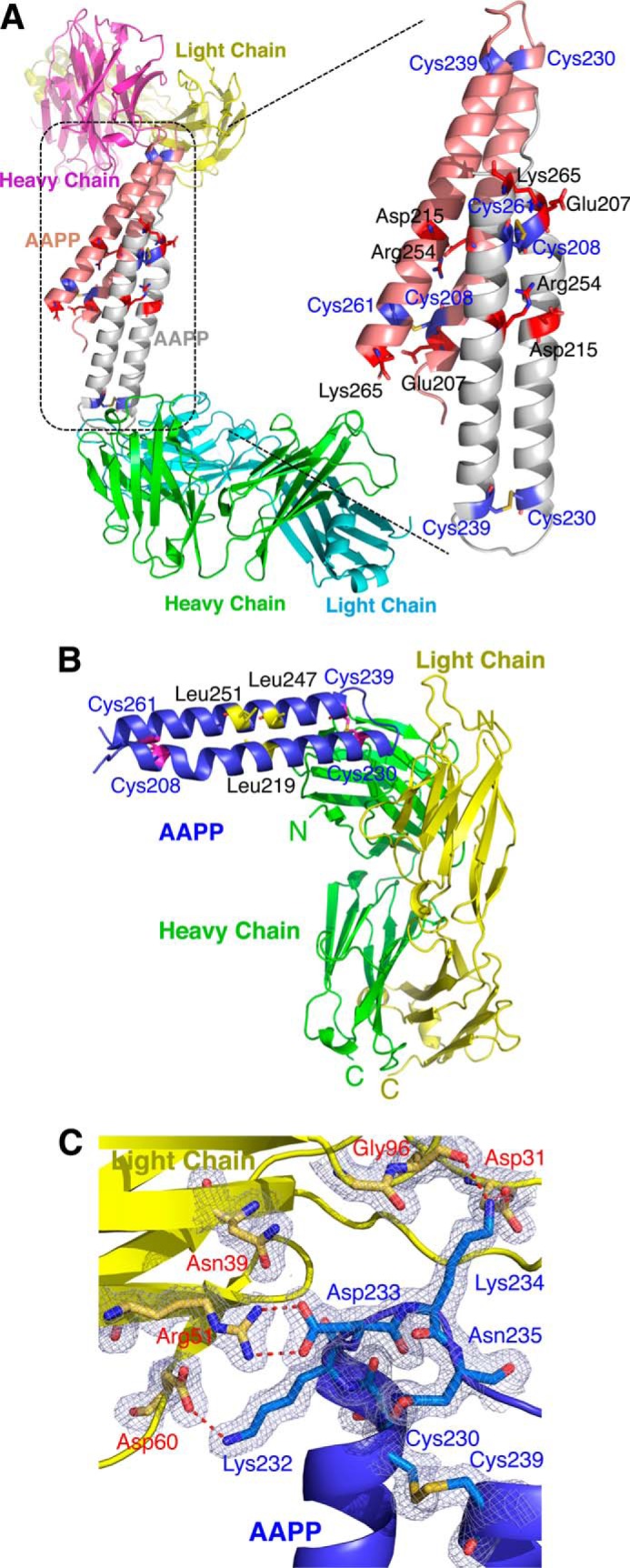
Overall schematic diagram of AAPP complex with 8H7 Fab. A, a ribbon diagram showing the two copies of the AAPP-8H7 Fab complex found in the asymmetric unit. Secondary structure is indicated with arrows and coils to show β strands and α helices. The AAPP chains are shown in gray or pink, with the cysteine residues marked in blue. B, Cα trace of AAPP bound to the 8H7 Fab, with α-helices shown as coils and β-strands shown as arrows. The AAPP is shown in purple, with the cysteine residues are marked. The disulfide bonds are shown in red. Light chain (yellow) consists of residues 1–218, and the heavy chain (green) consists of residues 1–215 of 8H7 Fab. C, the 2mFo − DFc electron density map (contoured at 1σ) showing the interaction between AAPP and the 8H7 Fab. Residues involved in binding are shown as sticks, with carbon atoms colored blue for AAPP and yellow for the 8H7 Fab light chain. Hydrogen bonds are shown as red dotted lines.
FIGURE 3.

A stick diagram and space-filling model of the AAPP with 8H7 Fab. A, stereo view of the interactions between AAPP and the heavy and light chains of the 8H7 Fab. For Tyr-32 of the heavy chain and Tyr 37 of the light chain the van der Waals volume of the side chain is indicated by a dotted surface. B, two-dimensional plot of the interactions between AAPP and the 8H7 Fab. Hydrogen bonds are shown as red dotted lines, and hydrophobic interactions are shown as arcs. C, molecular surface of the 8H7 Fab bound to AAPP, colored by charge with red negative and blue positive. The AAPP itself is shown as a green ribbon trace with the turn between helices fitting into pockets on the antibody surface.
The 8H7 Fab structure shows the classical IgG domain structure of anti-parallel β-sheet sandwiches with an antigen binding pocket formed from loops on both the heavy and light chains (Fig. 2B). It interacts solely with the turn region of AAPP so that the helical region points away from the antibody (Fig. 2C and Fig. 3A). Only AAPP residues from Glu-228 to Cys-239 make contact with the 8H7 Fab, but these turn residues make very close contact, including several salt bridges and hydrogen bonds (Fig. 3A). The SS bond formed between Cys-230 and Cys-239 also comes within van der Waals distance of Tyr-32 of the heavy chain (Fig. 3A). As well as the charge-charge interactions, such as between Asp-233 of AAPP and Arg 51 of the light chain, there are substantial apolar interactions such as Trp-33 of the heavy chain lying against the peptide bond formed by Asn-235 and Asn-236 (Fig. 3B). The surface area of AAPP buried by the 8H7 Fab is roughly 800 Å2, shared roughly equally between the heavy and light chains. This small interfacial area suggests that rigidity of the AAPP binding site contributes strongly to tight, specific binding (Fig. 3C).
Inhibition of Collagen Binding of AAPP by the 8H7 Fab
AAPP inhibits platelet aggregation via direct binding to collagen (7, 14). Experiments were carried out to determine whether the 8H7 Fab can block this interaction. To this end, AAPP was preincubated with the 8H7 Fab or whole IgG, and the binding ability of AAPP to immobilized soluble collagen type I was assessed by a plate assay. Free AAPP effectively bound to soluble collagen in a concentration-dependent manner, whereas both 8H7 Fab and whole IgG significantly inhibited the interaction in a dose-dependent manner (Fig. 4, A and B). The EC50 of collagen binding of free AAPP was 4.6 nm (95% CI, 4.3–5.0 nm). The 8H7 Fab at 1.0 μm significantly reduced the effective Kd value to 24.0 nm (95% CI, 22.9–25.2 nm), and whole IgG at 1.0 μm reduced it to 22.7 nm (95% CI, 21.2–24.3 nm) (Fig. 4B). IC50 values of the 8H7 Fab for blocking with 5 and 2.5 nm AAPP were 32.4 nm (95% CI, 25.2–41.8 nm) and 19.4 nm (95% CI, 15.4–24.5 nm), respectively (Fig. 4C). The nonspecific mouse IgG did not significantly inhibit the interaction (Fig. 4D).
FIGURE 4.

Inhibition of collagen binding of AAPP by antibody. AAPPex1–4 was serially diluted and incubated for 1 h with indicated doses of the 8H7 Fab (A) and whole IgG (B) and then applied into the collagen-coated plates. Plate-bound AAPP was detected by anti-His antibody conjugated with horseradish peroxidase. C, 5 or 2.5 nm AAPPex1–4 was incubated with serially diluted 8H7 Fab and then applied into the collagen-coated plates. Plate-bound AAPP was detected by anti-His antibody conjugated with HRP. D, AAPPex1–4 was serially diluted and incubated for 1 h with 1000 nm control mouse IgG and then applied into the collagen-coated plates. Plate-bound AAPP was detected by anti-His antibody conjugated with HRP. The error bars show the S.D. of three independent experiments.
Involvement of a Stable Structure of AAPP with Collagen Binding
The aapp gene of A. stephensi includes four exons separated by three introns (17). The region of AAPP involved in collagen binding has been tested previously by expression of recombinant truncated genes carrying different combinations of the four exons making up the coding sequence (17). Exons 3 and 4, which encode residues 148–204 and 205–269, respectively (Fig. 1B), were found to be absolutely required for collagen binding, and the full-length protein binds soluble collagen with similar affinity to AAPPex3–4 (17). To locate the site of collagen interaction more accurately, several mutants of GST-fused AAPPex3–4 were produced including a multiple mutant with Lys 232, Asp-233, Lys-234, and Asn-235 changed to alanines (termed “4A”), Cys-239 to alanine (termed “C3”), and both Cys-239 and Cys 261 to alanines (termed “C3/C4”) (Fig. 5A). The pulldown assay demonstrated that the 8H7 Fab bound to wild-type AAPPex3–4 but not to 4A or C3 (Fig. 5B), further confirming that the 8H7 Fab binds to the turn region between helices. The loss of interaction if one cysteine residue is mutated to alanine suggests that the tertiary structure of the loop region is also required for tight binding of AAPP to the 8H7 Fab (Fig. 5B).
FIGURE 5.
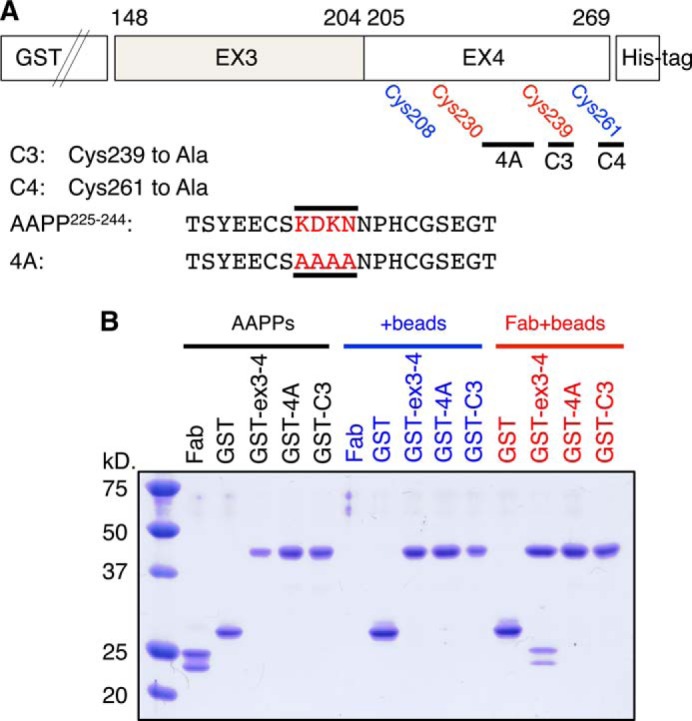
GST pulldown assay using AAPPex3–4 mutants. A, schematic diagram illustrating the AAPPex3–4 mutants. The several mutants of GST-fused AAPPex3–4 were produced, including a multiple mutant with Lys-232, Asp-233, Lys-234, and Asn-235 changed to alanines (termed 4A), Cys-239 to alanine (termed C3), and Cys-261 to alanine (termed C4). B, GST, GST-AAPPex3–4 (GST-ex3-4; wild type), GST-AAPPex3–4 4A (GST-4A), and GST-AAPPex3–4 C3 (GST-C3) were incubated with or without anti-AAPP 8H7 Fab before purification with glutathione resin (beads). The pelleted resin was washed 3 times, and then proteins were eluted and loaded in 12% SDS-PAGE.
The 4A mutant, missing key residues making close contact with the 8H7 Fab, was found to bind to collagen with very similar affinity to wild-type AAPP (Fig. 6A). The binding of C3 was markedly reduced (EC50 = 164.1 nm; 95% CI, 118.6–227.1 nm) compared with the wild type (EC50 = 5.9 nm; 95% CI, 5.7–6.2 nm), and C3/C4 did not bind to collagen detectibly at 100 nm (Fig. 6A). The loss of collagen binding on mutating the cysteine residues seems likely to be due to destabilization of the protein fold. This is supported by the fact that the GST fusion peptide of AAPP225–244 (corresponding to the helix-turn-helix region) does not bind collagen (Fig. 6B), indicating a stable structure is apparently required for this interaction.
FIGURE 6.

Collagen binding assay of AAPPex3–4 mutants. A, GST, GST-AAPPex3–4 (GST-ex3-4; wild type), GST-AAPPex3–4 4A (GST-4A), GST-AAPPex3–4 C3 (GST-C3), and GST-AAPPex3–4 C3/C4 (GST-C3/C4) were serially diluted and applied into the collagen-coated plates. Plate-bound AAPPs were detected by anti-His antibody conjugated with HRP. B, GST-AAPPex3–4 (GST-ex3-4; wild type) and GST-AAPP225–244 (GST-225–244) were serially diluted and applied into the collagen-coated plates. Plate-bound AAPPs were detected by anti-His Abs conjugated with HRP. The error bars show the S.D. of three independent experiments.
Conclusions
Nearly all biological processes involve some sort of interaction between one protein and another, and protein-protein interactions have recently come strongly into focus as a target for drug design. Although it was previously believed that protein-protein interactions would prove extremely difficult to block with small molecule inhibitors, recent trends suggest that in fact this strategy for drug design can prove a very fruitful one (28). For example the binding of the von Willebrand factor to the glycoprotein Ibα receptor on blood platelets is a protein-protein interaction of major importance in the control of blood clotting. von Willebrand factor is a multimeric glycoprotein whose A3 domain binds to subendothelial collagen (29) and whose A1 domain binds to glycoprotein Ibα (30). The clinical importance of the latter interaction is underlined by multiple efforts to produce effective inhibitors. The nanobody ALX-0081 and the aptamer ARC1779 have been described that are active in vitro (31–34) but unsuitable for oral administration as they are peptides. This has prompted work using fragment-based drug design and the discovery of several lead molecules. The work described here takes a different approach of using natural inhibitor proteins and mimicking their action through structure-based design of small molecule drugs. To this end we have solved the structure of the C-terminal region of AAPP, an effective anti-coagulant protein that functions by masking subendothelial collagen at sites of vascular injury, the principal trigger in platelet aggregation. AAPP prevents collagen types I and III from interacting with the platelet receptors and so prevents release of internal calcium, the second messenger that activates platelets (7). The mass spectrometry results show full-length AAPP to be highly unstable to proteolysis, even in highly purified form, but reveal a structural domain at the C terminus, which corresponds to the active region of the protein. This more stable region, although small and consisting essentially of only two α-helices, is tightly bound by a cognate antibody, which proved essential in obtaining crystals suitable for x-ray analysis.
Acknowledgment
We thank the staff at the beam-line BL17A of the Photon Factory for assistance with data collection.
This work was supported in part by Grant-in-aid for Scientific Research (B) 25305007 from the Japan Society for the Promotion of Science (to S. Y.).
The atomic coordinates and structure factors (code 4OKV) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AAPP
- anopheline anti-platelet protein.
REFERENCES
- 1. Francischetti I. M. (2010) Platelet aggregation inhibitors from hematophagous animals. Toxicon 56, 1130–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Francischetti I. M., Sa-Nunes A., Mans B. J., Santos I. M., Ribeiro J. M. (2009) The role of saliva in tick feeding. Front. Biosci. 14, 2051–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Warkentin T. E., Greinacher A., Koster A. (2008) Bivalirudin. Thromb. Haemost. 99, 830–839 [DOI] [PubMed] [Google Scholar]
- 4. Paciaroni M., Medeiros E., Bogousslavsky J. (2009) Desmoteplase. Expert. Opin. Biol. Ther. 9, 773–778 [DOI] [PubMed] [Google Scholar]
- 5. Furlan A. J., Eyding D., Albers G. W., Al-Rawi Y., Lees K. R., Rowley H. A., Sachara C., Soehngen M., Warach S., Hacke W. (2006) Dose escalation of desmoteplase for acute ischemic stroke (DEDAS): evidence of safety and efficacy 3 to 9 hours after stroke onset. Stroke 37, 1227–1231 [DOI] [PubMed] [Google Scholar]
- 6. Gardell S. J., Duong L. T., Diehl R. E., York J. D., Hare T. R., Register R. B., Jacobs J. W., Dixon R. A., Friedman P. A. (1989) Isolation, characterization, and cDNA cloning of a vampire bat salivary plasminogen activator. J. Biol. Chem. 264, 17947–17952 [PubMed] [Google Scholar]
- 7. Yoshida S., Sudo T., Niimi M., Tao L., Sun B., Kambayashi J., Watanabe H., Luo E., Matsuoka H. (2008) Inhibition of collagen-induced platelet aggregation by anopheline antiplatelet protein, a saliva protein from a malaria vector mosquito. Blood 111, 2007–2014 [DOI] [PubMed] [Google Scholar]
- 8. Penz S., Reininger A. J., Brandl R., Goyal P., Rabie T., Bernlochner I., Rother E., Goetz C., Engelmann B., Smethurst P. A., Ouwehand W. H., Farndale R., Nieswandt B., Siess W. (2005) Human atheromatous plaques stimulate thrombus formation by activating platelet glycoprotein VI. FASEB J. 19, 898–909 [DOI] [PubMed] [Google Scholar]
- 9. Ruggeri Z. M. (2002) Platelets in atherothrombosis. Nat. Med. 8, 1227–1234 [DOI] [PubMed] [Google Scholar]
- 10. Fuster V., Badimon L., Badimon J. J., Chesebro J. H. (1992) The pathogenesis of coronary artery disease and the acute coronary syndromes (2). N. Engl. J. Med. 326, 310–318 [DOI] [PubMed] [Google Scholar]
- 11. Calvo E., Tokumasu F., Marinotti O., Villeval J. L., Ribeiro J. M., Francischetti I. M. (2007) Aegyptin, a novel mosquito salivary gland protein, specifically binds to collagen and prevents its interaction with platelet glycoprotein VI, integrin α2β1, and von Willebrand factor. J. Biol. Chem. 282, 26928–26938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Calvo E., Tokumasu F., Mizurini D. M., McPhie P., Narum D. L., Ribeiro J. M., Monteiro R. Q., Francischetti I. M. (2010) Aegyptin displays high-affinity for the von Willebrand factor binding site (RGQOGVMGF) in collagen and inhibits carotid thrombus formation in vivo. FEBS J. 277, 413–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mizurini D. M., Francischetti I. M., Monteiro R. Q. (2013) Aegyptin inhibits collagen-induced coagulation activation in vitro and thromboembolism in vivo. Biochem. Biophys. Res. Commun. 436, 235–239 [DOI] [PubMed] [Google Scholar]
- 14. Hayashi H., Kyushiki H., Nagano K., Sudo T., Matsuoka H., Yoshida S. (2012) Anopheline anti-platelet protein from a malaria vector mosquito has anti-thrombotic effects in vivo without compromising hemostasis. Thromb. Res. 129, 169–175 [DOI] [PubMed] [Google Scholar]
- 15. Van de Werf F. (2010) Balancing benefit and bleeding risk of antithrombotic agents in the individual patient with an acute coronary syndrome. Circulation 121, 5–7 [DOI] [PubMed] [Google Scholar]
- 16. Kuruvilla M., Gurk-Turner C. (2001) A review of warfarin dosing and monitoring. Proc. Bayl. Univ. Med. Cent. 14, 305–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hayashi H., Kyushiki H., Nagano K., Sudo T., Iyori M., Matsuoka H., Yoshida S. (2013) Identification of the active region responsible for the anti-thrombotic activity of anopheline anti-platelet protein from a malaria vector mosquito. Platelets 24, 324–332 [DOI] [PubMed] [Google Scholar]
- 18. Yamamoto D. S., Nagumo H., Yoshida S. (2010) Flying vaccinator; a transgenic mosquito delivers a Leishmania vaccine via blood feeding. Insect Mol. Biol. 19, 391–398 [DOI] [PubMed] [Google Scholar]
- 19. Carter P. B., Beegle K. H., Gebhard D. H. (1986) Monoclonal antibodies: clinical uses and potential. Vet. Clin. North Am. Small. Anim. Pract. 16, 1171–1179 [DOI] [PubMed] [Google Scholar]
- 20. Otwinowski Z., Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 21. McCoy A. J. (2007) Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr. D Biol. Crystallogr. 63, 32–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Uysal H., Sehnert B., Nandakumar K. S., Böiers U., Burkhardt H., Holmdahl R., Thunnissen M. M. (2008) The crystal structure of the pathogenic collagen type II-specific mouse monoclonal antibody CIIC1 Fab: structure to function analysis. Mol. Immunol. 45, 2196–2204 [DOI] [PubMed] [Google Scholar]
- 23. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Emsley P., Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 25. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. DeLano W. L. (2010) The PyMOL Molecular Graphics System, Version 1.3r1, Schrodinger, LLC, New York [Google Scholar]
- 27. Wallace A. C., Laskowski R. A., Thornton J. M. (1995) LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 8, 127–134 [DOI] [PubMed] [Google Scholar]
- 28. Mullard A. (2012) Protein-protein interaction inhibitors get into the groove. Nat. Rev. Drug. Discov. 11, 173–175 [DOI] [PubMed] [Google Scholar]
- 29. Huizinga E. G., Martijn van der Plas R., Kroon J., Sixma J. J., Gros P. (1997) Crystal structure of the A3 domain of human von Willebrand factor: implications for collagen binding. Structure 5, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 30. Reininger A. J. (2008) VWF attributes: impact on thrombus formation. Thromb. Res. 122, S9–S13 [DOI] [PubMed] [Google Scholar]
- 31. Ulrichts H., Silence K., Schoolmeester A., de Jaegere P., Rossenu S., Roodt J., Priem S., Lauwereys M., Casteels P., Van Bockstaele F., Verschueren K., Stanssens P., Baumeister J., Holz J. B. (2011) Antithrombotic drug candidate ALX-0081 shows superior preclinical efficacy and safety compared with currently marketed antiplatelet drugs. Blood 118, 757–765 [DOI] [PubMed] [Google Scholar]
- 32. Mayr F. B., Knöbl P., Jilma B., Siller-Matula J. M., Wagner P. G., Schaub R. G., Gilbert J. C., Jilma-Stohlawetz P. (2010) The aptamer ARC1779 blocks von Willebrand factor-dependent platelet function in patients with thrombotic thrombocytopenic purpura ex vivo. Transfusion 50, 1079–1087 [DOI] [PubMed] [Google Scholar]
- 33. Knöbl P., Jilma B., Gilbert J. C., Hutabarat R. M., Wagner P. G., Jilma-Stohlawetz P. (2009) Anti-von Willebrand factor aptamer ARC1779 for refractory thrombotic thrombocytopenic purpura. Transfusion 49, 2181–2185 [DOI] [PubMed] [Google Scholar]
- 34. Gilbert J. C., DeFeo-Fraulini T., Hutabarat R. M., Horvath C. J., Merlino P. G., Marsh H. N., Healy J. M., Boufakhreddine S., Holohan T. V., Schaub R. G. (2007) First-in-human evaluation of anti von Willebrand factor therapeutic aptamer ARC1779 in healthy volunteers. Circulation 116, 2678–2686 [DOI] [PubMed] [Google Scholar]