

Background: The haptoglobin genotype is a determinant of atherosclerotic plaque oxidation and instability.
Results: The amount of lysosomal iron and membrane injury is haptoglobin genotype-dependent.
Conclusion: Hemoglobin-mediated lysosomal oxidative injury of the macrophage is haptoglobin genotype-dependent.
Significance: The haptoglobin genotype may modulate plaque stability by determining the macrophage response to free hemoglobin within the atherosclerotic plaque.
Keywords: α-Tocopherol, Apoptosis, Atherosclerosis, Genetic Polymorphism, Hemoglobin, Lysosome, Macrophage, Oxidative Stress
Abstract
The major function of the Haptoglobin (Hp) protein is to control trafficking of extracorpuscular hemoglobin (Hb) thru the macrophage CD163 receptor with degradation of the Hb in the lysosome. There is a common copy number polymorphism in the Hp gene (Hp 2 allele) that has been associated with a severalfold increased incidence of atherothrombosis in multiple longitudinal studies. Increased plaque oxidation and apoptotic markers have been observed in Hp 2-2 atherosclerotic plaques, but the mechanism responsible for this finding has not been determined. We proposed that the increased oxidative injury in Hp 2-2 plaques is due to an impaired processing of Hp 2-2-Hb complexes within macrophage lysosomes, thereby resulting in redox active iron accumulation, lysosomal membrane oxidative injury, and macrophage apoptosis. We sought to test this hypothesis in vitro using purified Hp-Hb complex and cells genetically manipulated to express CD163. CD163-mediated endocytosis and lysosomal degradation of Hp-Hb were decreased for Hp 2-2-Hb complexes. Confocal microscopy using lysotropic pH indicator dyes demonstrated that uptake of Hp 2-2-Hb complexes disrupted the lysosomal pH gradient. Cellular fractionation studies of lysosomes isolated from macrophages incubated with Hp 2-2-Hb complexes demonstrated increased lysosomal membrane oxidation and a loss of lysosomal membrane integrity leading to lysosomal enzyme leakage into the cytoplasm. Additionally, markers of apoptosis, DNA fragmentation, and active caspase 3 were increased in macrophages that had endocytosed Hp 2-2-Hb complexes. These data provide novel mechanistic insights into how the Hp genotype regulates lysosomal oxidative stress within macrophages after receptor-mediated endocytosis of Hb.
Introduction
Growth of atherosclerotic lesions is accompanied by plaque neovascularization (1, 2)., Microvessels within advanced atherosclerotic lesions are involved in intra-plaque hemorrhage (IPH)2 and may play a role in plaque rupture and atherothrombosis (3, 4). Neovascularization is augmented in ruptured atherosclerotic lesions of human aorta as well as in coronary lesions from patients with acute myocardial infarction (5, 6). In plaques from individuals with diabetes mellitus (DM), microvessel content, macrophage infiltration, and IPH are substantially increased (7). However, the mechanistic association between IPH and plaque instability is still unclear.
During IPH, free extracorpuscular hemoglobin (Hb) is rapidly captured by haptoglobin (Hp) and endocytosed as a complex via the macrophage CD163 scavenger receptor (8, 9). After Hp-Hb complex endocytosis, Hb is degraded within macrophage lysosomes resulting in intralysosomal iron accumulation and potential increase in iron-mediated oxidative stress (10). Therefore, IPH may be a potent oxidative stimulus for macrophage activation and cell injury. Although it is not fully understood, macrophage apoptosis has been shown to be an important response to intracellular oxidative stress and cholesterol accumulation within the advanced atherosclerotic lesions and is ultimately responsible for plaque destabilization and rupture (11–13).
There exists a common copy number variant polymorphism in the Hp gene defined by the presence (Hp 2 allele with allele frequency of 60%) or absence (Hp 1 allele with allele frequency of 40%) of a 1.7-kb in-frame duplication of exons 3 and 4 of the Hp 1 allele (14). The Hp 2 protein has been shown to be inferior to the Hp 1 protein in protecting against Hb-driven oxidative injury in a variety of cell free systems and has been associated with increased oxidative injury in vivo (including in the atherosclerotic plaque in man) (15–20). Individuals with the Hp 2-2 genotype have been shown to have a 2–3-fold greater risk of atherothrombosis as compared with non-Hp 2-2 individuals, specifically in the setting of DM (21–25). The mechanism explaining why increased hemoglobin-driven oxidative injury exists in Hp 2-2 and how this leads to increased atherothrombosis in this population is not known.
In this study we sought to test our hypothesis that the increased oxidative injury and atherothrombotic events that have been observed in Hp 2-2 and DM are due to an impaired processing and injury induced by the Hp 2-2-Hb complex within macrophage lysosomes.
EXPERIMENTAL PROCEDURES
Reagents
Hp was purified from human plasma using a polyclonal goat anti-haptoglobin antibody affinity column. Hb was isolated from human red blood cells as previously described (15). Met-Hb was prepared by incubation of 1.1 mm potassium ferrocyanide with 1 mm oxy-Hb for 30 min at room temperature and then purification of met-Hb using PD-10 columns as previously described (17). Hb was glycosylated in vitro using glycolaldehyde (Fluka AG) as previously described (26). Hp-Hb complex was formed by incubating the complex at different concentrations at room temperature for 15 min before it was used. Radiochemicals were from Amersham Biosciences. A LysoSensor yellow/blue DND-160 probe was purchased from Molecular Probes. The caspase 3 fluorometric assay kit was purchased from Biovision, and caspase 3 inhibitor was purchased from Mercury. All other chemicals were purchased from Sigma. Hp was labeled as previously described (16).
Low Density Lipoprotein (LDL) Isolation and Modification
All LDL preparations were isolated from pooled plasma of healthy volunteers. Vitamin E-enriched LDL was prepared as described previously with some modifications (27). Briefly, water-miscible α-tocopherol was added to plasma to reach a final concentration of 460 mm. The mixture was vortexed then incubated at 37 °C in the dark overnight with continuous shaking. Native LDL was prepared as described above for with the addition of phosphate-buffered saline (PBS) to plasma instead of α-tocopherol. LDL (native or vitamin E-enriched LDL) were then isolated by sequential ultracentrifugation as previously described (28). LDL was acetylated using acetic anhydride as previously described (29). After dialysis against PBS for 48 h, LDL concentration was determined by Lowry (55).
Labeling of Hp
Purified Hp was radio-iodinated using the chloramine-T method as previously described (16). Briefly, 5 μg (Hp 1-1 or Hp 2-2) was mixed with 50 μl of NaH2PO4, 0.5 mCi of 125I, and 5 μl of chloramine T (8.8 mm) and stirred for 45 s at room temperature. Then 12.5 μl of Na2S2O5 (10.5 mm) was added to terminate the reaction. The labeled protein was purified using a PD-10 column (Amersham Biosciences) with PBS running buffer containing 1% bovine serum albumin (BSA). The specific activity of 125I-Hp was about 50,000 cpm/ng of protein for both Hp 1-1 and Hp 2-2 proteins.
Cell Culture
CHO cells were stably transfected with the human CD163 receptor, and its expression was confirmed by Western blot using a CD163 monoclonal antibody. CHO/CD163 cells were grown in large plates in F-12 medium supplemented with 10% FCS (v/v), glutamine (2 mm), penicillin (50 IU/ml), and streptomycin (86 nm) for 48 h. Cells were washed 3 times with PBS and incubated in F-12 medium containing glutamine, penicillin/streptomycin, and 1% BSA and with 1% FCS before they were used for lysosomes assay. THP-1 cells were grown in suspension with RPMI medium with 10% FCS. The THP-1 cells were differentiated to macrophages by treatment with 16.2 nm phorbol 12-myristate 13-acetate for 3 days. LDL-loaded cells were prepared by incubating THP-1-differentiated macrophages for an additional day with 300 μg/ml native LDL or vitamin E-enriched LDL and then with 30 μg/ml acetylated LDL for another 6 h to get foam cell-like macrophages. Control (non-loaded) cells were incubated with medium only without any addition of LDL.
Hp-Hb Complex Endocytosis Assay
Hb alone or complexed to Hp 1-1 or Hp 2-2 was added to CHO/CD163 or differentiated THP-1 cells (control or LDL-loaded cells) grown in medium containing 1% BSA at a final concentration of 1 μm Hb or 1 μm Hb-Hp complex (1:1 molar ratio of Hp and Hb). Cells were incubated at 37 °C in a 5% CO2 incubator overnight, after which complexes were removed, and the cells were washed 3 times with PBS containing 1% BSA. For the hydrogen peroxide assay, cells were incubated with medium containing 50 μm of H2O2 with or without the Hp-Hb complexes for overnight before the cells were washed 3 times as described above and prepared for the lysosomes-rich fraction (LRF) purification. For conditions designed to mimic DM, glycosylated Hb was used instead of native Hb or cells were grown in high glucose medium (22.2 mm).
Intralysosomal Degradation of the Hp-Hb Complex
THP-1 cells were grown and differentiated to macrophages in 12-well tissue culture plates as described above. The cells were then washed 3 times with PBS containing 1% BSA and incubated with 1 million cpm of 125I-Hp (with or without 1:1 molar ratio of Hb) in RPMI tissue culture medium with 0.5% serum at 37 °C in a CO2 incubator for 2 h. Subsequently, the cells were washed 3 times (removing all label and prohibiting further association and uptake of radiolabeled Hp-Hb complex), and the amount of radioactivity (counts) remaining with the cells at that time was taken as 100% (T0). The amount of cell associated counts was then monitored over the next 24 h. Cell-associated radioactivity was assessed at the defined time points after first washing the cells by lysing them with 1 ml of 1% SDS and then subjecting the solubilized cell extract to γ-scintillation counting. Degradation was measured as a function of time by calculating the percentage of remaining cell-associated counts at different time points relative to the counts measured at T0.
Purification of LRF from CHO/CD163 and THP-1 Cells
LRF was purified from CHO/CD163 or THP-1 cells as previously described with some modifications (30). Cells were detached from the plates using trypsin, washed once with homogenization medium (HM) (containing 0.25 m sucrose, 1 mm EDTA, 10 mm HEPES, adjusted to PH 7.0), and pelleted by centrifugation at 500 × g for 10 min. The cells were then suspended in HM and homogenized in a ball-bearing cell homogenizer using 5 passes. The cell homogenate was washed with HM and centrifuged at 800 × g for 10 min to pellet the nuclei, cell debris, and any unbroken cells. This step was repeated twice after which the LRF was used in the lysosomal fragility assay. Protein concentrations were determined using the Bradford reagent (Sigma B6916) according to the manufacturer's instructions. The specific activity of the lysosomal preparations was assessed by determining the activity of acid phosphatase (lysosomal marker, Sigma kit #CS0740) and cathepsin D (lysosomal marker, Sigma kit #CS0800), all according to the manufacturer's instructions.
Lysosomal Membrane Loss of Integrity Assay
Lysosomal membrane integrity was determined by incubating the suspended LRF at 37 °C for 30 min, and then the activity of the lysosomal enzyme β-hexosaminidase was measured using the fluorimetric substrate 4-methylumbelliferyl-2-acetamido-2-deoxy-β-d-Glucopyranoside (30, 31). Because intact organelle membranes prevent substrate access to the luminal enzyme, no substrate turnover indicates integrity of the organelle, whereas in the presence of the detergent Triton X-100 total enzyme activity was assessed. Briefly, 50 μl of portion of the LRF suspension pool was incubated for 1 min at 37 °C with 250 μl of 100 mm citrate buffer, pH 4.5, containing 0.25 m sucrose, 20 mm sodium phosphate, pH 4.5, and 1.2 mm 4-methylumbelliferyl-2-acetamido-2-deoxy-β-d-glucopyranoside with and without 0.2% Triton X-100. The reaction was terminated after 1 min by the addition of 1 ml of 0.5 m glycine, sodium carbonate buffer, pH 10. The liberated 4-methylumbelliferone was determined by measuring the fluorescence in a BMG GalaxyFlouroStar microplate reader with a 365/450-nm excitation/emission filter pair. The degree of lysosomal loss of integrity (leakage) was expressed as % fragility (activity in the absence of detergent/activity in the presence of detergent) × 100.
LDL Oxidation Assay
Hb-driven oxidation of LDL was assessed at pH 7 using PBS as a reaction buffer and pH 4.5 using citric acid as a reaction buffer. LDL (300 μg/ml) was incubated for 2 h at 37 °C with free oxy-Hb, Met-Hb or glycosylated Hb alone (10 μm) in the presence of H2O2 (20 μm). The change in LDL oxidation using Hp was assessed using the same method but in the presence of increasing concentrations of Hp 1-1 or Hp 2-2 (2.5 μm up to 20 μm for each type) complexed to Hb at different pH. Oxidation of LDL lipids was determined using the thiobarbituric reactive substances assay as previously described (32). The data are presented as the percentage of reduction in LDL oxidation in the presence of Hp compared with LDL oxidation in the absence of Hp.
Quantitative Assessment of Heme Transfer from Hb to LDL
For preparation of radiolabeled Hb, MEL cells (a kind gift of Professor Ben-Zion Levi) were maintained in DMEM medium supplemented with 10% fetal calf serum, 100 units/ml penicillin, and 0.1 mg/ml streptomycin and grown at 37 °C, 5% CO2. For preparation of radiolabeled Hb, cells were seeded at 2 × 105 cells/ml in DMEM/F-12 1:1 medium (Beit HaEmek) supplemented with 10 mg/ml BSA, 5 μg/ml insulin, 12 μg/ml cholesterol, 5 ng/ml sodium selenite, 5 μg/ml Fe55 loaded (see protocol below for loading transferring with iron) transferrin, and 2% DMSO. Cells were harvested on day 4, washed twice with PBS, and lysed in 0.5% Nonidet P-40 in PBS for 30 min at 37 °C. Lysed cells were spun at 5000 rpm (4696 × g) in a tabletop centrifuge for 15 min. The supernatant was passed over a 0.5-ml nickel-nitrilotriacetic acid-agarose column (Qiagen) and washed with 10 ml of 10 mm imidazole, and the bound fraction was eluted with 1 ml of 250 mm imidazole. The eluate was spun over a PD-10 column (GE Healthcare) to remove the imidazole. Further purification was achieved by adding 0.25 ml of Hemoglobind resin (Biotech Support Group) and rotating for 30 min at room temperature. The resin was spun down at 1000 × g for 2 min in a microcentrifuge. Purified Hb was eluted using 2 × 0.5 ml of 0.1 m sodium carbonate, and the buffer was exchanged for PBS using a PD-10 column. A single band at 16,000 kDa was observed by Coomassie staining on a polyacrylamide reducing gel. The specific activity of the radiolabeled Hb was 10–50 cpm/ng. For preparation of transferrin loaded with 55Fe, nitrilotriacetate (NTA), pH 3, was mixed at a 2:1 molar ratio at room temperature for 30 min with Fe55Cl3 (18.5 mCi/mg, catalog no. NEZ043, PerkinElmer Life Sciences) to form ferric nitrilotriacetate (Fe(NTA)2). Apotransferrin (Kamada, Ltd) was prepared in Hepes-buffered saline (50 mm, pH 7.4) and mixed with Fe(NTA)2 at a molar ratio of 4:1 in the presence of sodium bicarbonate (0.05 m final) and incubated for 1 h at room temperature (this mixture should be reddish; 1 n NaOH can be added to the Fe(NTA)2 until the mixture becomes reddish before mixing with apotransferrin. The pH should be 7.4) Radiolabeled transferrin was stored in aliquots and kept at −20 °C. For heme transfer assay, human LDL was isolated by density gradient centrifugation according to established protocols and kept covered at 4 °C. LDL was then conjugated to Sepharose 4B beads (GE Healthcare) according to the manufacturer's recommendations. Fe55 Hb was converted to met-Hb by incubation with a 1.1 molar excess of potassium ferricyanide at room temperature for 30 min followed by purification of over a PD-10 column. Fe55 met-Hb (4.5 μg) was mixed with 20 μl of LDL Sepharose beads (26 μg of LDL) and various components in a total of 50 μl of PBS, pH 7.4. Mixtures at low pH contained 5 μl of 1 m sodium acetate, pH 4.3. The mixture was placed in a microtiter dish and mixed at room temperature for 3 h. Beads were washed 3 times with 1 ml of PBS containing 1% BSA and 0.05% Tween 20 and spun down at 1000 × g for 1 min. Beads were resuspended in 100 μl of 0.1% SDS and counted in 4 ml of scintillation fluid. Background was determined by incubating Fe55 met-Hb and LDL beads separately at room temperature for 3 h followed by mixing the two together and washing immediately. Percent heme transferred was calculated as the number of counts recovered in the beads minus the number of counts obtained in the background samples divided by the number of counts added to the reaction multiplied by 100.
Assessment of Iron-dependent and Iron-independent Oxidative Stress in Lysosomal Extracts with Dihydrorhodamine (DHR)
Total and iron-dependent oxidative stress of LRF was assessed using DHR as a sensitive fluorescent indicator of oxidative activity and carried out as described previously with some modifications (17). Briefly, quadruplicates of 40 μl of LRF purified from cells treated with different combinations of Hp and Hb complexes were incubated with DHR (50uM) in 180 μl of iron-free Hepes-buffered saline containing 40 μm ascorbic acid in the presence and absence of 50 μm iron chelator (deferiprone). The kinetics of fluorescence increase was assessed in a BMG GalaxyFluoroStar microplate reader with a 485/538-nm excitation/emission filter pair for 40 min. The rate of total oxidation was measured by calculating the slope of DHR fluorescence intensity over time in the absence of iron chelator. To calculate the iron-dependent oxidation associated with LRF, the difference in the rate of oxidation of DHR in the presence and absence of the chelator was then assessed, representing the component of iron that is catalytically redox-active. Redox-active iron concentrations were then determined in each preparation from calibration curves using Hepes-buffered saline with increasing concentrations of Fe-NTA.
Confocal Microscopy for Assessing Lysosomal Injury
A LysoSensor yellow/blue DND-160 probe (Molecular Probes) was used to assess lysosomal injury by confocal microscopy. A LysoSensor yellow/blue DND-160 probe is a fluorescent pH indicator used to detect intact lysosomes in live cells. Intact lysosomes with pH around 4.5 elicit yellow fluorescence when labeled with the LysoSensor probe, whereas destabilized lysosomes with higher pH values elicit blue fluorescence. For these experiments THP-1 cells were resuspended to 0.8 × 106 cells/ml in RPMI medium (containing 10% FCS) and grown in Fluorodish plates (World Precision Instruments, Inc.). After differentiation to macrophages, cells were washed twice with PBS and incubated overnight with 1 μm Hb (native Hb or glycosylated Hb) alone or as a complex with 1 μm Hp (Hp 1-1 or Hp 2-2). For using LDL-loaded foam cells, the cells were washed after differentiation and loaded with 300 μg/ml LDL (native or vitamin E-enriched LDL) and then with 30 μg/ml acetylated LDL as described above. Cells were then washed again with PBS and incubated with the Hp-Hb complex. Chloroquine (50 μm), a lysosome inhibitor that causes a dramatic increase in lysosomal pH, was used as a positive control for lysosomal inactivation. The cells were then washed with PBS and incubated with 5 μm LysoSensor yellow/blue DND-160 probe for 5 min at 37 °C, washed again, and then incubated with LysoSensor-free medium for an additional 15 min before analysis. Fluorescent images were collected using a Zeiss LSM 510 confocal laser-scanning microscope (Carl Zeiss Inc.) with excitation at 405 nm and emission measured at 420–480 nm (blue) and 505–550 nm (yellow). Lysosomal injury was assessed qualitatively by overlapping of blue and yellow emission images and quantitatively by calculating the fluorescence intensity ratio of the blue and the yellow emission for each stimulus after subtracting the nonspecific fluorescence from each image using the LSM 510 Image Browser software. More than 15 fields per condition from 5 independent experiments were measured.
Lysosomal Cathepsin D and Cathepsin L Assays
After THP-1-differentiated cells were washed 3 times with cold PBS containing 1% BSA, cells were detached from the plates, washed twice with HM, and then homogenized in a ball-bearing cell homogenizer using five passes with minimum lysosomal damage as assayed by lysosomal enzyme activity of control (untreated) cells. The cell homogenate was washed with HM and centrifuged at 14,000 × g for 15 min at 4 °C to sediment intact lysosomes. Cathepsin D and cathepsin L activity was then measured in the suspension (non-sedimented fraction not containing the cytosol) as well as in the pellet (sediment fraction containing the intact lysosomes) using a fluorimetric assay kit (BioVision). The fluorescence of the reaction products was measured by the FluoroStar instrument at excitation/emission 328/460 nm and 400/505 nm for cathepsin D and cathepsin L assays, respectively. Lysosomal destabilization in the different cell groups was assessed by measuring the percentage of cytosolic cathepsin activity out of the total (both cytosolic and lysosomal) activity.
Active Caspase 3 Assay
Caspase 3 activity was measured in THP-1 differentiated cells using a fluorometric assay kit (Biovision). Briefly, after differentiation to macrophages, THP-1 cells were directly incubated or preloaded with LDL or vitamin E-loaded LDL before being incubated with the different Hp-Hb complexes for 24 h. For caspase 3 inhibitor experiments, cell-permeable Ac-DMQD-CHO caspase 3 inhibitor (50 μm) was added to the cells 2 h before the addition of the Hp-Hb complexes. Then, 5 × 106 cells were washed twice with cold PBS and lysed with lysis buffer. After incubation with DEVD-aminofluoromethylcoumarin substrate (50 μm final concentration) for 3 h at 37 °C, caspase 3 activity was assessed in cell lysates by measuring the fluorescence in a 96-well plate reader with a 400/505-nm excitation/emission filter after subtracting the aminofluoromethylcoumarin fluorescence measured in control cells (untreated with Hp or Hb).
DNA Fragmentation Assay
DNA fragmentation was determined as described previously (33). After incubation with Hp-Hb for 24 h, cells were detached from the plate, and 1 × 106 cells were washed twice with PBS, suspended in lysis buffer (100 mm Tris-HCl, pH 8, 5 mm EDT, 200 mm NaCl, 0.5% w/v SDS, and 200 μg/ml protease K), and incubated at 37 °C overnight. The cell lysate was resuspended with NaCl to reach a final concentration of 1.5 m then centrifuged at 10,000 × g for 30 min at room temperature. After discarding the pellet, the DNA was precipitated by mixing the suspension with 2:1 (v/v) of 100% chilled ethanol, incubated at −20 °C for 1 h, and then centrifuged at 14,000 × g for 10 min. The DNA pellet was washed once with 70% ethanol and dissolved at 37 °C for 30 min in Tris-EDTA containing 100 μg/ml RNase A. For DNA fragmentation analysis, 1 μg of DNA was loaded onto a 1.2% agarose gel, subjected to electrophoresis for ∼6 h, and then visualized under UV illumination.
Statistical Analysis
All results are expressed as the means ± S.E. Comparisons between groups were analyzed for statistical significance by two-way analysis of variance using SPSS software (Version 20.0) or the unpaired Student's t test. p values less than 0.05 were considered significant in all experiments.
RESULTS
Intracellular Degradation Rate of Hp-Hb Complex in THP-1 Differentiated Cells
We sought to investigate whether there might be Hp type-dependent differences in the complex degradation rate that may result in differences in the amount of intralysosomal iron available for oxidation. To assess the rate of degradation, we incubated 125I-labeled Hp-Hb complexes with THP-1 differentiated macrophages, which endogenously express the CD163 scavenger receptor and then followed the degradation of the complex. After the complex had been endocytosed we found that the rate of degradation for Hp 2-2-Hb complexes was up to 3-fold less as compared with the Hp 1-1-Hb complexes (Fig. 1).
FIGURE 1.
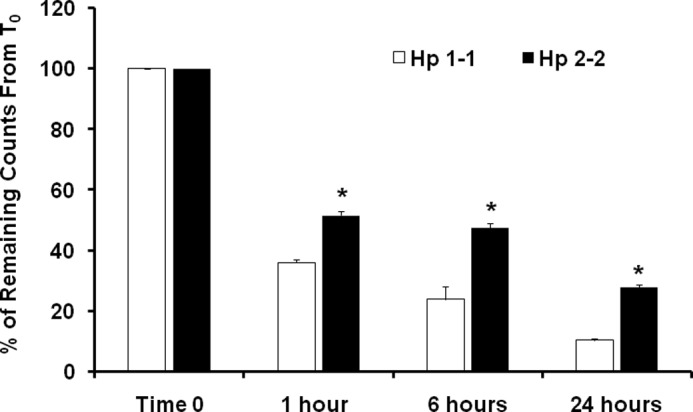
Intralysosomal Hp 2-2-Hb complex degradation is impaired in THP-1 differentiated cells. The degradation rate is presented as the percentage of remaining counts, taking the zero time point (T0) as 100% remaining counts immediately after cell loading. There was a significant reduction in Hp 2-2-Hb complex degradation as compared with Hp 1-1-Hb complex degradation. 24 h after loading the cells, 28% of the Hp 2-2-Hb complexes were accumulated within the lysosomes as compared with only 10% of the Hp 1-1-Hb complexes accumulated within the lysosomes (n = 3, p < 0.001). Total counts at T0 for Hp 1-1-Hb complexes were 1620 ± 145 cpm and for Hp 2-2-Hb were 1858 ± 237 cpm.
Lysosomal Membrane Loss of Integrity after Hp-Hb Complex Uptake by CHO/CD163 and THP-1 Cells
We sought to investigate lysosomal oxidative injury after uptake and intralysosomal degradation of the different Hp-Hb complexes using CHO cells stably transfected with human CD163 receptor as well as human differentiated THP-1 macrophages endogenously expressing high levels of the CD163 receptor. CHO/CD163 cells were incubated with Hp 1-1-Hb or Hp 2-2-Hb complex overnight, and lysosomal membrane loss of integrity was then assessed in the LRF by measuring the specific activity of a lysosomal enzyme, β-hexosaminidase, as described under “Experimental Procedures.”
We found a modest increase in lysosomal membrane destabilization in the LRF purified from cells exposed to Hp 2-2-Hb complex as compared with LRF purified from cells exposed to Hp 1-1-Hb complex (28.7 ± 3.3% for Hp 2-2-Hb versus 14.1 ± 3.6% for Hp 1-1-Hb complexes, n = 5, p = 0.02). These differences were dramatically increased when the cells were exposed to an additional oxidative stimulus using hydrogen peroxide (39.7 ± 4.6% for Hp 2-2-Hb versus 18.3 ± 2.9% for Hp 1-1-Hb complexes, n = 5, p = 0.004) (Fig. 2A). Consistent with the results found in CD163-transfected CHO cells, we found a significant increase in lysosomal loss of integrity associated with the Hp 2-2-Hb complexes and hydrogen peroxide incubation using THP-1 differentiated macrophages (Fig. 2B).
FIGURE 2.
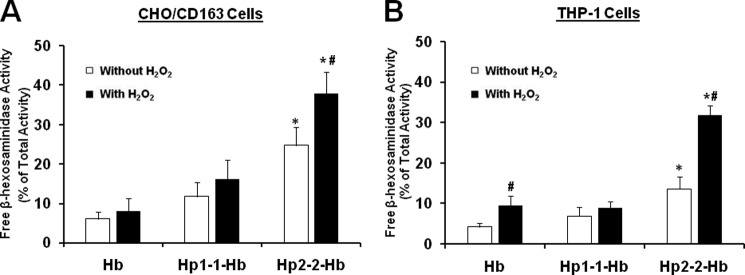
Lysosomal membrane leakage is increased in CHO/CD163 and THP-1 differentiated cells after incubation with Hp 2-2-Hb complex as well as hydrogen peroxide. Lysosomal membrane leakage is presented as the percentage of extralysosomal β-hexosaminidase activity out of the total enzyme activity for each lysosomal preparation. Data shown represent the mean ± S.E. of five independent experiments. A, CHO/CD163 cells. There was an increase in lysosomal injury when the cells were incubated with Hp 2-2-Hb complexes as compared with cells incubated with free Hb (28.7 ± 3.3 versus 8.2 ± 2.1, n = 5, p < 0.001) or Hp 1-1-Hb complexes (28.7 ± 3.3 versus 14.1 ± 3.6, n = 5, p = 0.02). These differences in lysosomal injury were further increased when the cells were preincubated with hydrogen peroxide, and there was significantly higher lysosomal injury associated with Hp 2-2-Hb as compared with Hp 1-1-Hb complexes (39.7 ± 4.6 versus 18.3 ± 2.9, n = 5, p = 0.004). B, there was significantly increased lysosomal leakage in hydrogen peroxide-treated THP-1 cells associated with Hp 2-2-Hb as compared with Hp 1-1-Hb complexes or Hb alone (p < 0.01). * indicates significant differences compared with Hp 1-1 or Hb treatment, and # indicates significant differences between hydrogen peroxides-treated and non-treated conditions.
Hp Type-dependent Reduction in Hb-driven Oxidation of LDL at Normal and Acidic pH
We previously demonstrated that Hp 2-2 is an inferior antioxidant through its reduced ability to block Hb-mediated peroxidation of LDL and linolenic acid (15). Moreover, we demonstrated a marked impairment in the ability of Hp to block the Hb-driven oxidation of LDL in vitro (16). Leake and co-workers (34, 35) show that LDL oxidation catalyzed by iron is much faster at acidic pH, presumably because iron is more redox-active and the protonation of superoxide anions to hydroxyl radicals is increased at acidic pH.
We hypothesized that LDL oxidation by Hb is also pH-dependent and that this oxidation is blocked differently by the different Hp types. To test this hypothesis, we incubated LDL with native Hb, glycosylated Hb, or met-Hb and measured LDL oxidation without and with Hp 1-1 or Hp 2-2 at pH 7.4 and pH 4.5.
Consistent with our previous findings, we found that the antioxidant activity of Hp against Hb-mediated oxidation is significantly impaired with glycosylated Hb and that Hp 1-1 is superior to Hp 2-2 in blocking Hb-mediated oxidation of LDL at pH 7.4. Hp antioxidant activity was further impaired when incubating these complexes at pH 4.5. We found a pro-oxidant effect of Hp 2-2 on LDL-oxidation at pH 4.5. Although Hp 1-1 still conferred antioxidant activity against Hb (2.2 ± 4.7% reduction for native Hb, 7.0 ± 1.4% reduction for met-Hb, and 7.6 ± 4.1% reduction for glycosylated Hb, n = 3, p < 0.01 for the effect of Hp on oxidation, p = not significant for comparisons between the different Hb groups), Hp 2-2 was paradoxically associated with a significant increase in Hb-mediated oxidation at pH 4.5, with more dramatic oxidation seen against met- and glycosylated Hb (4.2 ± 1.2% increase for native Hb, 13 ± 1.1% increase for met-Hb, and 7.0 ± 2.0% increase for glycosylated Hb, n = 3, p < 0.01 for the Hp effect on oxidation as well as for glycosylated Hb and met-Hb versus native Hb) (Fig. 3).
FIGURE 3.
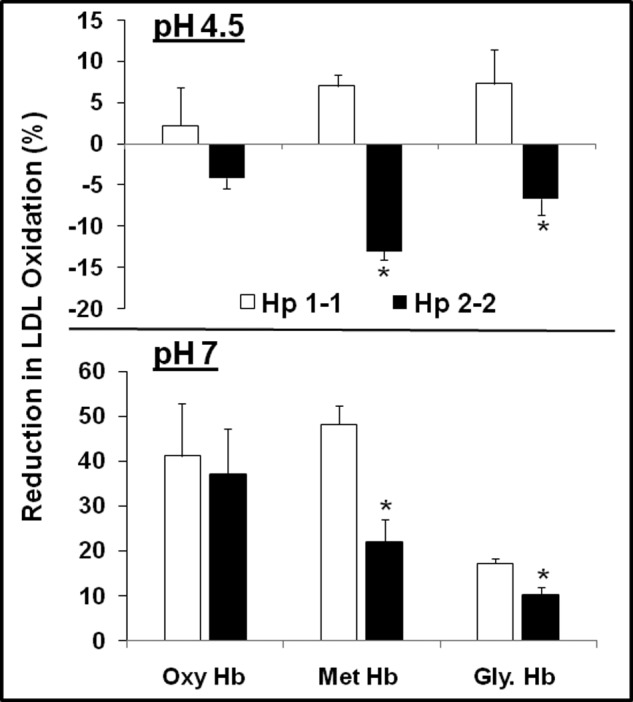
Hp 2-2 confers a paradoxical pro-oxidant activity against Hb-mediated oxidation of LDL at acidic pH in vitro. Data represent the mean ± S.E. of three independent experiments. Data are presented as a percentage inhibition of Hb-induced LDL oxidation conferred by Hp. There was a severe impairment in the antioxidant activity of Hp 2-2 against LDL oxidation and even a paradoxical increase in LDL oxidation specifically at acidic pH with a more dramatic impairment associated with glycosylated Hb (Gly. Hb) and oxidized Hb (met-Hb) as compared with native Hb (oxy-Hb).
Heme Transfer from Hp 2-2-Hb Complexes Is Accelerated, Is Greater Than in Hp 1-1-Hb Complexes, and Is Accelerated by the Acidic pH Found in Lysosomes
The ability of Hp to prevent Hb-induced oxidation has been shown to be the result of increased stabilization of the heme group within Hb (36). Bamm et al. (37) show that the prosthetic heme group within the heme pocket of Hb in the Hp 2-2-Hb complex is more readily lost or transferred to other proteins (like LDL) than the heme group within the Hp 1-1-Hb complex. The heme group, once outside of the heme pocket of Hb, is an extremely potent oxidant. We developed a novel method to quantitatively examine the role of an acidic environment in the transfer of heme from Hb-Hp complexes. Hb containing Fe55 heme was produced in erytholeukemia cells, and transfer of the heme from Hb to LDL was monitored as described under “Experimental Procedures.” We found significantly more heme transfer to LDL from Hp 2-2-Hb than Hp 1-1-Hb complexes, and this transfer was significantly increased by performing the transfer reaction at the physiological acidic pH of the lysosome (Fig. 4).
FIGURE 4.
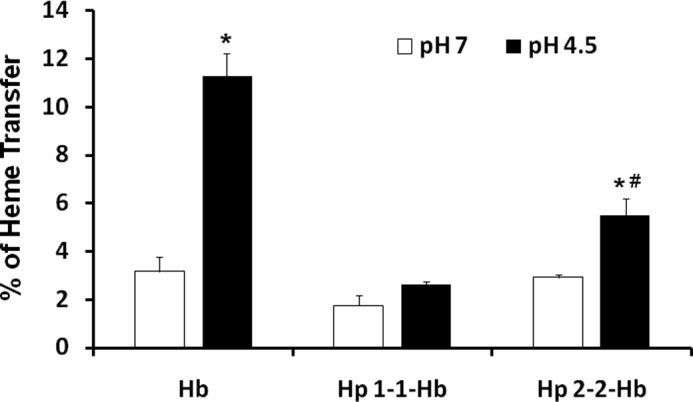
Heme transfer from Hp 2-2-Hb complexes is increased relative to Hp 1-1-Hb and is enhanced in an acidic environment. Data represent the mean ± S.E. of three independent experiments. Data are presented as the percentage of heme transfer from Hb to LDL. There was a significant increase in heme transfer from Hp 2-2-Hb complexes as compared with Hp 1-1-Hb complexes, and this transfer was accelerated at acidic pH.
Increased Lysosomal Loss of Integrity with Hp 2-2 Glycosylated Hb Complexes and LDL-loaded Macrophages
The interaction between the Hp genotype and cardiovascular disease is specific for DM. Hb undergoes increased glycosylation in the setting of DM. In vitro we have shown that the antioxidant properties of Hp 2-2 against Hb are significantly reduced when Hb is glycosylated, with a more dramatic impairment as shown above occurring at acidic pH. Therefore, we sought to investigate lysosomal membrane integrity in macrophages incubated with different Hp types complexed to glycosylated Hb. We found a further increase in lysosomal destabilization associated with Hp 2-2-glycosylated Hb as compared with Hp 2-2-native Hb complexes (21.8 ± 2.4 versus 10.6 ± 1.1%, n = 5, p = 0.008). There was no difference in lysosomal destabilization associated with Hp 1-1-glycosylated Hb compared with Hp 1-1-native Hb complexes (11.4 ± 2.1 versus 7.7 ± 1.6%, n = 5, p = 0.2).
LDL has been shown to undergo oxidation within lysosomes after its uptake, and this oxidation may be mediated by intralysosomal iron (35). We hypothesized that there may be a synergy between LDL- and Hp 2-2-glycosylated Hb within lysosomes leading to more lysosomal oxidative injury. To test our hypothesis, we generated foam cell-like cells (mimicking the foam cells that exist within the atherosclerotic plaques) by loading THP-1 differentiated macrophages with native and acetylated LDL before exposure to Hp-Hb complexes and measured lysosomal membrane destabilization using the same method described above. Loading cells with LDL had no significant deleterious effect on lysosomal membrane integrity. We found that there was a 2-fold further increase in lysosomal membrane destabilization associated with LDL-loaded macrophages as compared with non-LDL-loaded macrophages when these cells were incubated with Hp 2-2-glycosylated Hb (40.8 ± 4.2% for LDL-loaded versus 21.8 ± 2.4% for non-LDL-loaded cells, n = 5, p = 0.004) (Fig. 5). The deleterious effect of LDL and glycosylated Hb on lysosomal membrane integrity seen with Hp 2-2 was not observed for Hp 1-1 (p = 0.3 for the interaction between LDL and non-LDL-loaded cells incubated with Hp 1-1-glycosylated Hb), indicating a synergistic effect exists only between Hp 2-2-glycosylated Hb and LDL. This is consistent with the paradoxical pro-oxidant properties of Hp 2-2 against LDL oxidation promoted by glycosylated Hb at acidic pH (Fig. 3).
FIGURE 5.
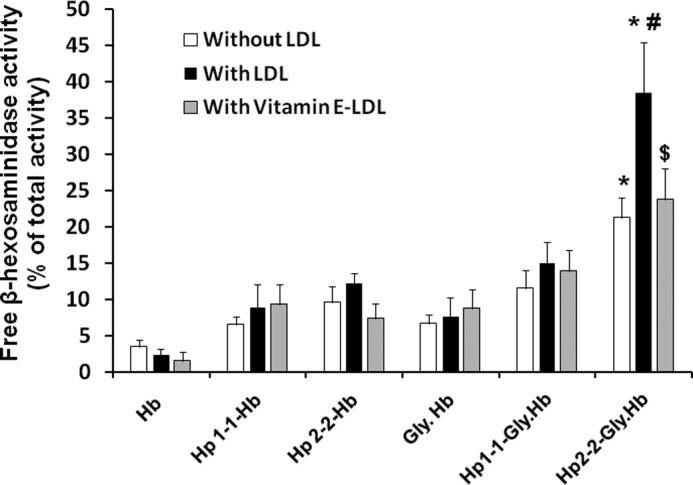
Lysosomal injury was increased with Hp 2-2-glycosylated Hb complexes incubated with THP-1 differentiated macrophages after loading the cells with LDL, and vitamin E conferred significant protection. Data shown represent the mean ± S.E. of five independent experiments. Three THP-1 differentiated cell groups were used in this experiment before the addition of the different Hp-Hb complexes; THP-1 cells without any treatment, THP-1 cells loaded with native LDL (300 μg/ml) for 24 h and then with acetylated LDL (30 μg/ml) for 6 h, and THP-1 cells prepared the same as the second group except for using vitamin E-enriched LDL (300 μg/ml) instead of native LDL. Lysosomal injury was increased 3-fold in cells loaded with LDL and incubated with Hp 2-2-glycosylated Hb as compared with Hp 1-1-glycosylated Hb (p < 0.001). Vitamin E treatment resulted in ∼40% reduction in lysosomal injury associated with Hp 2-2-glycosylated Hb (p = 0.01). * indicates significant differences between Hp 1-1 and Hp 2-2 within the same cell group. # indicates significant differences between non-LDL- and LDL-loaded cells for each condition. $ indicates significant differences between Vitamin E-LDL- and LDL-loaded cells for each condition. Gly, glycosylated.
Proof of Concept That the Lysosomal Injury Associated with Hp 2-2-glycosylated Complexes Incubated with LDL Was Due to an Oxidative Mechanism Provided by Assessing the Ability of Vitamin E to Block This Injury
For this purpose we enriched plasma with vitamin E overnight before LDL was isolated resulting in a 4-fold increase in vitamin E concentration in LDL particles. THP-1 differentiated cells were loaded with vitamin E-enriched LDL before Hp-Hb complexes were added. We found a significant reduction in lysosomal membrane destabilization when Hp 2-2-glycosylated Hb complexes were incubated with cells loaded with vitamin E-enriched LDL as compared with incubation of these complexes with cells loaded with LDL without vitamin E enrichment (23.2 ± 3.2% for vitamin E-LDL-loaded versus 40.8 ± 4.2% for LDL-loaded cells, n = 5, p = 0.01). We found no significant change in lysosomal membrane integrity using vitamin E enriched LDL with cells incubated with the Hp 1-1.
Demonstration That the Increased Oxidative Stress in Lysosomes of Hp 2-2-Hb-treated Cells Is Due to Iron
We have assessed oxidative stress in lysosomes using DHR, which becomes fluorescent when it is oxidized. We have differentiated the amount of oxidative stress that is derived from iron and iron-dependent mechanisms by assessing oxidation in the presence and absence of an iron chelator. We found a significant increase in redox-active iron associated with lysosomes purified from cells treated with Hp 2-2-Hb complexes as compared with those treated with Hp 1-1-Hb complexes, specifically when using glycosylated Hb. Differences in the total amount of lysosomal oxidative stress between Hp 1-1 and Hp 2-2-Hb complex-treated cells could be completely accounted for by iron-mediated oxidative stress (Fig. 6).
FIGURE 6.
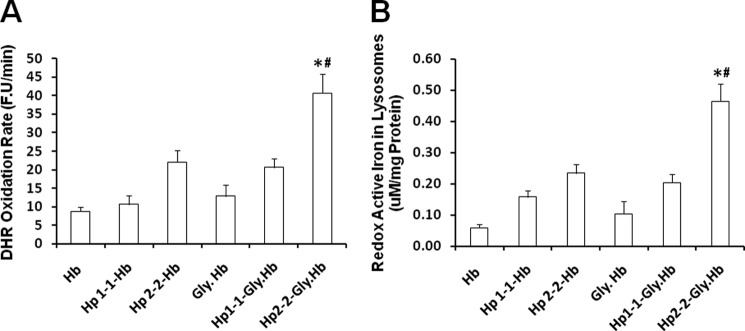
Differences in lysosomal oxidative stress between Hp 1-1 and Hp 2-2-Hb complexes is iron-dependent. Total lysosomal oxidative stress (A) and the amount of lysosomal redox-active iron (B) were assessed by measuring DHR oxidation rate in the presence and absence of iron chelator as described under “Experimental Procedures.” Using an iron calibration curve (17), we found that each 90 fluorescence units/min (F.U/min) accounts for 1 μm redox-active iron. Data represent the mean ± S.E. of three independent experiments. There was significantly more oxidative stress associated with lysosomal fractions from cells incubated with Hp 2-2-glycosylated Hb complexes as compared with cells incubated with Hp 1-1-glycosylated Hb complexes (A). The Hp-dependent differences in lysosomal oxidative stress were mainly explained by iron redox activity (B). * indicates significant differences between Hp 2-2 versus Hp 1-1. # indicates significant differences between Hp complexed to native versus glycosylated Hb. Gly, glycosylated.
Lysosomal Inactivation Assessed by Confocal Microscopy
We sought to further investigate the effect of Hp-Hb complex uptake on lysosomal function and integrity by determining the change in pH of lysosomes in THP-1 cells using the LysoSensor yellow/blue DND-160 probe and confocal microscopy. The LysoSensor probe exhibits a pH-dependent fluorescent pattern of yellow fluorescence in acidic environments (the physiological pH of lysosomes is about 4.5) and a significant blue fluorescent shift as lysosomal pH increases (when the lysosomal cytoplasmic pH gradient is lost and the pH of the lysosome approaches 7.0). Loss of lysosomal acidity was evaluated by overlapping the yellow and blue fluorescent channels (as presented in the fluorescent images) (Fig. 7, A–D) and by calculating the specific ratio of blue and yellow fluorescence in the different conditions (as presented in the histogram) (Fig. 7E).
FIGURE 7.

Lysosomal pH is significantly increased in LDL-loaded THP-1 macrophages after incubation with Hp 2-2-glycosylated Hb complexes. The change in lysosomal pH was monitored using the LysoSensor DND-160 probe and confocal microscopy as described under “Experimental Procedures.” Fluorescent images are presented as overlap of the blue and yellow emissions for each condition (A–D). A, four channels are presented for clarification (upper right square presents the yellow fluorescent channel, upper left square presents the blue channel, lower right square presents the overlap between the blue and yellow channels, and lower left square presents cell contrast). Chloroquine was used as positive control to increase lysosomal pH, whereas medium alone was used as negative control to show normal lysosomal pH. B–D, blue and yellow overlap channel for each condition are presented. We used three different cell conditions: THP-1 differentiated cells without LDL (B), LDL-loaded THP-1 cells (C), and vitamin E-enriched LDL-loaded cells (D). E, lysosomal injury was assessed quantitatively by calculating the ratio of the blue and yellow fluorescent intensity of 15 fields in 5 independent experiments for each condition after subtracting the nonspecific fluorescence from each field. There was a significant increase in the lysosomal pH with Hp 2-2 complexed with glycosylated Hb and Hp 2-2-Hb complex incubated with cells grown at high glucose medium (22.2 mmol/liter). After loading the cells with LDL, there was no change in lysosomal pH without additional treatment, whereas there was a remarkable increase in the pH after incubating the cells with Hp 2-2-glycosylated Hb complexes (p < 0.001). Vitamin E-enriched LDL was able to significantly reduce lysosomal pH associated with LDL and Hp 2-2-glycosylated Hb complexes (p < 0.001). Data shown represent the mean ± S.E. of five independent experiments. * indicates significant differences between Hp 1-1 and Hp 2-2 within the same cell group. # indicates significant differences between non-LDL- and LDL-loaded cells for each condition. $ indicates significant differences between LDL- and vitamin E-LDL-loaded cells for each condition. Gly, glycosylated.
We found no lysosomal injury in THP-1 cells unexposed to the Hp-Hb complex (negative control) and dramatic lysosomal dysfunction (loss of acidity) when these cells were treated for 3 h with chloroquine (positive control) (Fig. 7A). After incubation of these cells with Hp-Hb complexes overnight, we found no significant change in lysosomal pH in cells incubated with Hp 1-1-Hb complex or Hb alone, whereas there was a slight increase in lysosomal pH when the cells were incubated with Hp 2-2-Hb complex, indicating a modest lysosomal dysfunction. When using glycosylated Hb instead of native Hb, we found a remarkable increase in lysosomal pH with Hp 2-2-glycosylated Hb complex, whereas there was a modest and non-significant increase in lysosomal pH with the Hp 1-1-glycosylated Hb. Moreover, incubating the cells with high glucose medium (about 22.2 mm) resulted in a similar though less significant effect than this seen by glycosylated Hb.
Jerome et al. (38) have demonstrated that oxidized LDL accumulation within lysosomes inhibits subsequent lipoprotein hydrolysis and that lipid-engorged lysosomes failed to maintain an acidic pH. We proposed that the Hp genotype-dependent oxidative effect on LDL accumulated within lysosomes may further increase its pH leading to more lysosomal dysfunction. As shown in Fig. 7C, lysosomes of THP-1 cells that were loaded with LDL were enlarged but without significant change in pH, indicating no lysosomal inactivation. However, when LDL-loaded cells were incubated with Hp-Hb complex, there was a significant increase in lysosomal pH with a greater increase in pH seen when the cells were incubated with Hp 2-2-Hb complex as compared with those incubated with Hp 1-1-Hb complex. Again, when using glycosylated Hb there was a striking increase in lysosomal pH when complexed to Hp 2-2, indicating a more deleterious effect in the diabetic setting. No significant change in pH was found when these cells were incubated with Hp 1-1-glycosylated Hb or glycosylated Hb alone.
Furthermore, we assessed the ability of vitamin E to reduce the deleterious increase in lysosomal pH by loading macrophages with LDL enriched with vitamin E and found a significant reduction in lysosomal pH in cells treated with Hp 2-2-glycosylated Hb compared with cells loaded with LDL without vitamin E (Fig. 7, D and E). No significant change in lysosomal pH was seen with vitamin E in the Hp 1-1-treated groups.
Increased Lysosomal Markers of Apoptosis in LDL-loaded Macrophages Incubated with Hp 2-2 but Not with Hp 1-1 Complexed with Glycosylated Hb
The oxidative lysosomal injury demonstrated in Hp 2-2- and LDL-loaded macrophages, specifically in the setting of DM, may be associated with a release into the cytoplasm of lysosomal enzymes, which are involved in induction of apoptosis. Thus, we assessed the increase in two of the most potent lysosomal enzymes associated with apoptosis in the cytosol of LDL and non-LDL-loaded macrophages incubated with the different Hp-Hb complexes. First, we measured cathepsin D activity, as described under “Experimental Procedures” and found an ∼2-fold increase in cathepsin D activity in THP-1 differentiated macrophages incubated with Hp 2-2-glycosylated Hb complexes as compared with those incubated with Hp 1-1-glycosylated Hb complexes (18 ± 1.7 versus 9.7 ± 0.9%, n = 5, p = 0.004) (Fig. 8A). Moreover, we found that the differences between Hp 1-1 and Hp 2-2 were exaggerated when the cells were preloaded with LDL with an up to 3-fold increase in cathepsin D activity associated with Hp 2-2-glycosylated Hb as compared with Hp 1-1-glycosalyted Hb complexes (40.7 ± 4.8 versus 14.7 ± 2.7%, n = 5, p = 0.002). Second, consistent with the cathepsin D results, we found a marked increase in cathepsin L activity associated with Hp 2-2-glycosylated Hb incubated with LDL-loaded cells, but no significant increase was seen with Hp 1-1 (p < 0.001) (Fig. 8B). We found no significant increase in these lysosomal markers in cells incubated with Hb alone or with Hp complexed with native Hb.
FIGURE 8.
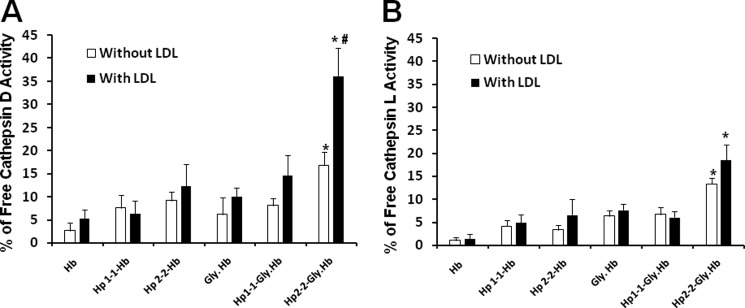
Increased lysosomal markers of apoptosis in LDL-loaded macrophages after incubation with Hp 2-2-glycosylated Hb complexes. Data represent the mean ± S.E. of five independent experiments. There was significantly more activity of both cathepsin D and cathepsin L in the cytosol of cells incubated with Hp 2-2-glycosylated Hb as compared with cells incubated with Hp 1-1-glycosylated Hb complexes. These differences were more exaggerated when using LDL-loaded THP-1 cells. * indicates significant differences between Hp 1-1 and Hp 2-2 within the same cell group. # indicates significant differences between non-LDL- and LDL-loaded cells for each condition. Gly, glycosylated.
Hp 2-2-Glycosylated Hb Endocytosis Induces Apoptosis in LDL-loaded Macrophages, and Apoptosis Can Be Blocked by Vitamin E
We sought to further examine whether lysosomal oxidative injury associated with the accumulation of Hp 2-2-glycosylated Hb and LDL could result in induction of apoptosis. We assessed two specific apoptotic markers: caspase 3 activity and DNA fragmentation in non-loaded and LDL-loaded macrophages treated with different Hp-Hb complexes. We found that caspase 3 activity was significantly increased in macrophages incubated only with Hp 2-2 complexed with glycosylated Hb (Fig. 9A). No significant increase in caspase 3 activity was seen in cells incubated with Hb alone or with Hp 1-1 complexed with native or glycosylated Hb. When macrophages were loaded with LDL before incubation with the different Hp-Hb complexes, we found a marked increase in caspase 3 activity associated with Hp 2-2-glycosylated Hb, indicating a further induction of apoptosis, whereas there was no change in caspase 3 activity when these cells were incubated with Hp 1-1-glycosyltaed Hb. Growing the cells in a high glucose medium resulted in no significant effect on caspase 3 activation with either incubation with Hp 1-1 or Hp 2-2 complexed with native Hb. Moreover, we found that the marked increase in caspase 3 activation associated with Hp 2-2-glycosylated Hb uptake by LDL-loaded macrophages was blocked when these cells were loaded with vitamin E-enriched LDL instead of native LDL (about 40% reduction in caspase 3 activity, n = 5, p = 0.01) (Fig. 9B). No significant change in caspase 3 activity was seen with Hp 1-1-glycosylated Hb with vitamin E treatment. The increased caspase 3 activity found in Hp 2-2-glycosylated Hb and LDL-loaded macrophages was dramatically reduced when these cells were pretreated with a specific cell-permeable caspase 3 inhibitor (>80% reduction in caspase 3 activity, n = 5, p < 0.001).
FIGURE 9.

Increased caspase 3 activation in LDL-loaded macrophages after endocytosis of Hp 2-2-glycosylated Hb complexes and the protective effect of vitamin E. Data are presented as the mean ± S.E. of fluorescent units relative to control cells for each cell group of five independent experiments. A, there was significant increase in caspase 3 activity associated with Hp 2-2-glycosylated Hb complexes as compared with Hp 2-2-Hb (p = 0.02) and as compared with Hp 1-1-glycosylated Hb complexes (p = 0.03). Caspase activity was dramatically increased when the cells were preloaded with LDL, and the differences associated with Hp 2-2 were exaggerated. B, caspase 3 activity associated with Hp 2-2-glycosylated Hb complexes was significantly reduced when the cells were preloaded with vitamin E-enriched LDL ($ indicates significant differences between these groups) and the addition of caspase 3 inhibitor to LDL-loaded cells blocked, the caspase 3 activity seen with Hp-Hb complexes (& indicates significant differences between LDL-loaded cells treated and non-treated with caspase 3 inhibitor).Gly, glycosylated.
DNA fragmentation was also induced by Hp 2-2-glycosylated Hb endocytosis, suggesting increased macrophage apoptotic signaling (Fig. 10). When THP-1 differentiated macrophages were preloaded with LDL, DNA fragmentation was substantially induced after Hp 2-2-glycosylated Hb uptake as compared with Hp 1-1-glycosylated Hb uptake. There was also a mild induction of DNA fragmentation after uptake of Hp 2-2-Hb complexes by LDL-loaded cells grown in a high glucose medium, whereas no DNA fragmentation was seen with Hp 1-1-Hb complexes in the same conditions. Consistent with caspase 3 activation data, we found that DNA fragmentation associated with the uptake of Hp 2-2-Hb complexes by LDL-loaded cells was markedly attenuated when the cells were loaded with vitamin E-enriched LDL, indicating a protective effect of vitamin E against apoptosis induced by Hp 2-2-Hb complexes.
FIGURE 10.

Induction of DNA fragmentation in LDL-loaded macrophages after endocytosis of Hp 2-2-glycosylated Hb complexes and the protective effect of vitamin E. DNA fragmentation was examined in control (non-LDL-loaded) (I), LDL-loaded (II), and vitamin E-enriched LDL-loaded (III) THP-1 differentiated cells after different treatments: 1, control (no Hp or Hb treatment); 2, Hb alone; 3, glycosylated Hb alone; 4, Hp 1-1-Hb complex; 5, Hp 2-2-Hb complex; 6, Hp 1-1-glycosylated Hb complex; 7, Hp 2-2-glycosylated Hb complex; 8, Hp 1-1-Hb complex in high glucose medium; 9, Hp 1-1-Hb complex in high glucose medium. M, 1-kb DNA marker. DNA fragmentation was markedly induced in LDL-loaded cells treated with Hp 2-2-glycosylated Hb complexes and significantly attenuated when using cells loaded with vitamin E-enriched-LDL.
DISCUSSION
In this study we have demonstrated that the Hp genotype modulates the response to free Hb by modulating Hb degradation and iron-driven oxidation within macrophage lysosomes. We propose that the Hp 2-2-Hb complex acts like a Trojan horse. After it is endocytosed and directed to the lysosome, the toxicity of Hb iron derived from the Hp 2-2-Hb complex is not properly contained and results in macrophage lysosomal injury and apoptosis. These data provide a mechanism for the recent demonstration of increased oxidation-specific epitopes and apoptosis in human Hp 2-2 atheroslcerotic plaques as compared with Hp 2-1 and Hp 1-1 plaques (39). Macrophage apoptosis has been demonstrated to promote inflammation, and in advanced atherosclerotic lesions it is a key event in producing the cell debris and lipid-rich core of the vulnerable plaques (11–13). Consequently, these findings provide a possible explanation for the association of the Hp 2-2 genotype with increased atherothrombotic cardiovascular disease events in DM (Fig. 11).
FIGURE 11.

Proposed model of accelerated atherosclerotic cardiovascular disease in Hp 2-2 DM individuals. The Hp 1 and Hp 2 proteins are produced from the Hp 1 or Hp 2 alleles at the Hp locus as a single polypeptide that is subsequently cleaved into an α and β chain. The Hp 1 and Hp 2 monomeric proteins differ only in their α chain (exons 1–4) but their identical β chains (exon 5) are what is principally responsible for binding Hb. Hp 1 and Hp 2 monomers oligomerize with a stoichiometry that is Hp genotype dependent. In individuals with the Hp 1-1 genotype (who have Hp 1 protein) there are only dimers of Hp. In individuals with the Hp 2-2 genotype large cyclic polymers of Hp are found. Hb is released from erythrocytes extravasated during intraplaque hemorrhage and then complexed to Hp. The Hp-Hb complex is rapidly endocytosed by the CD163 scavenger receptor expressed on atherosclerotic plaque macrophages and degraded within the lysosome. The Hp 2-2-Hb complex has a higher affinity for CD163 as compared with Hp 1-1-Hb. This is probably a direct result of the multimeric nature of Hp 2-2. However, CD163-mediated endocytosis is more rapid with Hp 1-1-Hb complexes (16). The increased redox activity of the Hp 2-2-Hb complex particularly in the acidic environment of the lysosome results in the formation of large cross-linked aggregates that are degraded more slowly by lysosomal hydrolases. Glycosylation of Hb as occurs in DM increases the amount of redox injury produced by Hp 2-2-Hb complexes most likely by accelerating heme loss from the complex. The increased redox activity of the Hp 2-2-Hb complex results in lysosomal injury leading to intracellular cascades that result in induction of macrophage apoptosis leading to atherosclerotic plaque rupture and myocardial infarction. Gly, Hb, glycosylated Hb.
In this study non-enzymatic glycosylation of Hb was achieved with glycoaldehyde (26). This results in the covalent attachment of glucose to any of the N-terminal α amino groups on the α or β chains of Hb or on any of the lysine ϵ amino groups from these chains. This same interaction between glucose and Hb is markedly increased in the diabetic state due to hyperglycemia. Why should glycosylation of Hb interfere with the ability of Hp to inhibit the oxidative activity of Hb? One of the lysine residues in Hb that is preferentially glycosylated in DM and by glycoaldehyde has been directly implicated in binding to the heme group (where the lysine ϵ amino group of Lys66 of the β chain of Hb is thought to make contact with a propionic acid carboxyl of the heme of Hb) (40). Interference with the interaction between Lys66 and heme by the covalent attachment of a glucose molecule to the Lys66 ϵ amino group would be anticipated to decrease the stability of heme in the heme pocket. Once the heme leaves the heme pocket and is transferred to a different hydrophobic environment (such as the lysosomal membrane or LDL) it may act as a potent oxidant (16).
We have shown using a lysotropic pH-sensitive fluorescent reporter that the normal acidic pH within the lysosome is lost upon treatment of cells with glycosylated Hb-Hp 2-2 complexes. We can hypothesize that this is due either to an impaired function (perhaps due to oxidative damage by Hb-Hp 2-2) of the lysosomal integral proteins, which maintain the pH gradient across the lysosomal membrane, or due to loss of the barrier function of a lysosomal membrane that has lost its integrity.
Degradation of the Hp 2-2 complex may be slowed due to the redox activity of the complex and its ability to produce large heavily cross-linked complexes that are resistant to degradation by lysosomal hydrolases. The presence of iron-enriched complexes that are heavily oxidized and cross-linked and that are resistant to digestion is well documented in aged and diseased lysosomes and is often referred to as lipofuscin (41).
Lysosomes play a critical role in the breakdown of a variety of ferruginous materials through the operations of professional scavengers by endocytosis (heterophagy) as well as through autophagocytosis of iron-containing macromolecules such as mitochondrial electron-transport complexes and ferritin (42). Therefore, lysosomes that have been recently engaged in degradation of iron-rich compounds contain a large amount of iron. Several studies have demonstrated that intralysosomal iron increases lysosomal susceptibility for oxidative stress leading to lysosomal instability and cell death (42, 43). However, lysosomal injury needs large amounts of redox iron or additional oxidative trigger, and as long as the oxidative stress is not overwhelmingly strong, lysosomes are typically resistant to oxidative damage (44). The finding that the membranes of rat liver lysosomes contain a 37-fold higher content of α-tocopherol compared with other cellular membranes supports the notion that lysosomes are under substantial oxidative stress and anti-oxidants are needed to protect lysosomes from oxidative damage (45).
Morgan and Leake (34) demonstrated that iron-mediated oxidation of LDL in vivo is substantially greater in the acidic environment of the lysosome. One explanation for increased iron toxicity at acidic pH is that the ferrous iron (Fe2+), the favored iron redox state at acidic pH, is catalytically more active by the Fenton reaction and has a higher affinity for lipoproteins, thus enabling it to oxidize LDL faster than in the ferric iron (Fe3+) state. Intralysosomal LDL oxidation has been shown to be iron-dependent by demonstrating that iron chelation could block LDL oxidation (35). Lysosomal oxidative damage generally leads to lysosomal membrane permeabilization and was recently found to be increasingly involved in subsequent triggering a cascade of intracellular degradative events that eventually lead to apoptosis (46, 47). Cathepsin D microinjection has been shown to induce caspase-dependent apoptosis (48), and cathepsin L was found to be significantly associated with macrophage apoptosis and plaque destabilization in human carotid atherosclerotic lesions (49). DNA fragmentation is generally associated with caspase-dependent apoptotic signaling, but the mechanisms by which caspases promote DNA fragmentation remain unclear (50). The combined increase in caspase 3 activity and DNA fragmentation in our experiments supports a caspase-dependent pathway for apoptosis.
Several studies have previously shown that vitamin E increases LDL resistance to oxidation induced by iron (27, 51). Furthermore, vitamin E was found to inhibit oxidized LDL-induced apoptosis in human monocytic U937 cells (52) as well as to protect T cells from apoptosis by inhibiting CD95 apoptotic ligand expression (53). We found that enrichment of LDL with vitamin E (up to 4-fold increase) before its uptake by macrophages was enough to cause significant reduction in lysosomal oxidative injury and to substantially reduce apoptosis in macrophages. This finding indicates that lysosomal injury was mediated by increased oxidative stress and LDL oxidative modification associated with Hp 2-2-Hb uptake. This protective effect of vitamin E on lysosomal injury and macrophage apoptosis may explain in part the ability of vitamin E to decrease atherothrombotic events in Hp 2-2 DM individuals (54).
This work was supported, in whole or in part, by National Institutes of Health Grant RO1DK085226. This work was also supported by the Israel Science Foundation and the Rappaport Fund for Medical Research (to A. P. L.) and by a grant from the Slava Smolakovski Fund to the Rambam-Atidim Academic Excellence Program (to R. A.). Dr. Levy is an employee of the Rappaport Faculty of Medicine at the Technion-Israel Institute of Technology, which owns patents related to the use of haptoglobin typing to predict the risk of diabetic vascular complications.
- IPH
- intraplaque hemorrhage
- DM
- diabetes mellitus
- HM
- homogenization medium
- Hp
- haptoglobin
- LRF
- lysosomes-rich fraction
- DHR
- dihydrorhodamine
- NTA
- nitrilotriacetate.
REFERENCES
- 1. Moreno P. R., Purushothaman K. R., Sirol M., Levy A. P., Fuster V. (2006) Neovascularization in human atherosclerosis. Circulation 113, 2245–2252 [DOI] [PubMed] [Google Scholar]
- 2. Doyle B., Caplice N. (2007) Plaque neovascularization and antiangiogenic therapy for atherosclerosis. J. Am. Coll. Cardiol. 49, 2073–2080 [DOI] [PubMed] [Google Scholar]
- 3. Virmani R., Kolodgie F. D., Burke A. P., Finn A. V., Gold H. K., Tulenko T. N., Wrenn S. P., Narula J. (2005) Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 25, 2054–2061 [DOI] [PubMed] [Google Scholar]
- 4. Kolodgie F. D., Gold H. K., Burke A. P., Fowler D. R., Kruth H. S., Weber D. K., Farb A., Guerrero L. J., Hayase M., Kutys R., Narula J., Finn A. V., Virmani R. (2003) Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 349, 2316–2325 [DOI] [PubMed] [Google Scholar]
- 5. Moreno P. R., Purushothaman K. R., Fuster V., Echeverri D., Truszczynska H., Sharma S. K., Badimon J. J., O'Connor W. N. (2004) Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: implications for plaque vulnerability. Circulation 110, 2032–2038 [DOI] [PubMed] [Google Scholar]
- 6. Barger A. C., Beeuwkes R., 3rd (1990) Rupture of coronary vasa vasorum as a trigger of acute myocardial infarction. Am. J. Cardiol. 66, 41G–43G [DOI] [PubMed] [Google Scholar]
- 7. Moreno P. R., Fuster V. (2004) New aspects in the pathogenesis of diabetic atherothrombosis. J. Am. Coll. Cardiol. 44, 2293–2300 [DOI] [PubMed] [Google Scholar]
- 8. Kristiansen M., Graversen J. H., Jacobsen C., Sonne O., Hoffman H. J., Law S. K., Moestrup S. K. (2001) Identification of the haemoglobin scavenger receptor. Nature 409, 198–201 [DOI] [PubMed] [Google Scholar]
- 9. Nielsen M. J., Moestrup S. K. (2009) Receptor targeting of hemoglobin mediated by the haptoglobins: roles beyond heme scavenging. Blood 114, 764–771 [DOI] [PubMed] [Google Scholar]
- 10. Madsen M., Graversen J. H., Moestrup S. K. (2001) Haptoglobin and CD163: captor and receptor gating hemoglobin to macrophage lysosomes. Redox. Rep. 6, 386–388 [DOI] [PubMed] [Google Scholar]
- 11. Kockx M. M., De Meyer G. R., Muhring J., Jacob W., Bult H., Herman A. G. (1998) Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 97, 2307–2315 [DOI] [PubMed] [Google Scholar]
- 12. Tabas I. (2009) Macrophage apoptosis in atherosclerosis: consequences on plaque progression and the role of endoplasmic reticulum stress. Antioxid. Redox. Signal. 11, 2333–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Littlewood T. D., Bennett M. R. (2003) Apoptotic cell death in atherosclerosis. Curr. Opin. Lipidol. 14, 469–475 [DOI] [PubMed] [Google Scholar]
- 14. Bowman B. H., Kurosky A. (1982) Haptoglobin: the evolutionary product of duplication, unequal crossing over, and point mutation. Adv. Hum. Genet. 12, 189–261 [DOI] [PubMed] [Google Scholar]
- 15. Melamed-Frank M., Lache O., Enav B. I., Szafranek T., Levy N. S., Ricklis R. M., Levy A. P. (2001) Structure-function analysis of the antioxidant properties of haptoglobin. Blood 98, 3693–3698 [DOI] [PubMed] [Google Scholar]
- 16. Asleh R., Marsh S., Shilkrut M., Binah O., Guetta J., Lejbkowicz F., Enav B., Shehadeh N., Kanter Y., Lache O., Cohen O., Levy N. S., Levy A. P. (2003) Genetically determined heterogeneity in hemoglobin scavenging and susceptibility to diabetic cardiovascular disease. Circ. Res. 92, 1193–1200 [DOI] [PubMed] [Google Scholar]
- 17. Asleh R., Guetta J., Kalet-Litman S., Miller-Lotan R., Levy A. P. (2005) Haptoglobin genotype- and diabetes-dependent differences in iron-mediated oxidative stress in vitro and in vivo. Circ. Res. 96, 435–441 [DOI] [PubMed] [Google Scholar]
- 18. Moreno P. R., Purushothaman K. R., Purushothaman M., Muntner P., Levy N. S., Fuster V., Fallon J. T., Lento P. A., Winterstern A., Levy A. P. (2008) Haptoglobin genotype is a major determinant of the amount of iron in the human atherosclerotic plaque. J. Am. Coll. Cardiol. 52, 1049–1051 [DOI] [PubMed] [Google Scholar]
- 19. Kalet-Litman S., Moreno P. R., Levy A. P. (2010) The haptoglobin 2-2 genotype is associated with increased redox active hemoglobin derived iron in the atherosclerotic plaque. Atherosclerosis 209, 28–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Levy A. P., Levy J. E., Kalet-Litman S., Miller-Lotan R., Levy N. S., Asaf R., Guetta J., Yang C., Purushothaman K. R., Fuster V., Moreno P. R. (2007) Haptoglobin genotype is a determinant of iron, lipid peroxidation, and macrophage accumulation in the atherosclerotic plaque. Arterioscler. Thromb. Vasc. Biol. 27, 134–140 [DOI] [PubMed] [Google Scholar]
- 21. Levy A. P., Roguin A., Hochberg I., Herer P., Marsh S., Nakhoul F. M., Skorecki K. (2000) Haptoglobin phenotype and vascular complications in patients with diabetes. N. Engl. J. Med. 343, 969–970 [DOI] [PubMed] [Google Scholar]
- 22. Levy A. P., Hochberg I., Jablonski K., Resnick H. E., Lee E. T., Best L., Howard B. V. (2002) Haptoglobin phenotype is an independent risk factor for cardiovascular disease in individuals with diabetes: The Strong Heart Study. J. Am. Coll. Cardiol. 40, 1984-1990 [DOI] [PubMed] [Google Scholar]
- 23. Suleiman M., Kapeliovich M. R., Roguin A., Aronson D., Meisel S. R., Shochat M., Reisner S. A., Hammerman H., Lotan R., Levy N. S., Levy A. P. (2003) Haptoglobin phenotype and mortality in the 30 day period following acute myocardial infarction. Diabetes Care 26, 2699–2700 [DOI] [PubMed] [Google Scholar]
- 24. Roguin A., Koch W., Kastrati A., Aronson D., Schomig A., Levy A. P. (2003) Haptoglobin genotype is predictive of major adverse cardiac events in the one year period after PTCA in individuals with diabetes. Diabetes Care 26, 2628–2631 [DOI] [PubMed] [Google Scholar]
- 25. Levy A. P., Larson M. G., Corey D., Lotan R., Vita J. A., Benjamin E. J. (2004) Haptoglobin phenotype and prevalent coronary heart disease in the Framingham Cohort. Atherosclerosis 172, 361–365 [DOI] [PubMed] [Google Scholar]
- 26. Twigg S. M., Chen M. M., Joly A. H., Chakrapani S. D., Tsubaki J., Kim H. S., Oh Y., Rosenfeld R. G. (2001) Advanced glycosylation end products up-regulate connective tissue growth factor (insulin-like growth factor binding protein related protein 2) in human fibroblasts; a potential mechanism for expansion of extracellular matrix in diabetes mellitus. Endocrinology 142, 1760–1769 [DOI] [PubMed] [Google Scholar]
- 27. Bowen H. T., Omaye S. T. (1998) Oxidative changes associated with β-carotene and α-tocopherol enrichment of human low-density lipoproteins. J. Am. Coll. Nutr. 17, 171–179 [DOI] [PubMed] [Google Scholar]
- 28. Schumaker V. N., Puppione D. L. (1986) Sequential flotation ultracentrifugation. Methods Enzymol. 128, 155–170 [DOI] [PubMed] [Google Scholar]
- 29. Maor I., Aviram M. (1994) Oxidized low density lipoprotein leads to macrophage accumulation of unesterified cholesterol as a result of lysosomal trapping of the lipoprotein hydrolyzed cholesterol ester. J. Lipid Res. 35, 803–819 [PubMed] [Google Scholar]
- 30. Graham J. M. (2000) Isolation of Lysosomes from tissues and cells by differential and density gradient centrifugation. Curr. Protoc. Cell Biol., 10.1002/0471143030.cb0306s07 [DOI] [PubMed] [Google Scholar]
- 31. Schütt F., Bergmann M., Holz F. G., Kopitz J. (2002) Isolation of intact lysosomes from human RPE cells and effects of A2-E on the integrity of the lysosomal and other cellular membranes. Graefes Arch. Clin. Exp. Ophthalmol. 240, 983–988 [DOI] [PubMed] [Google Scholar]
- 32. el-Saadani M., Esterbauer H., el-Sayed M., Goher M., Nassar A. Y., Jürgens G. (1989) Spectrophotometric assay for lipid peroxides in serum lipoproteins using a commercially available reagent. J. Lipid Res. 30, 627–630 [PubMed] [Google Scholar]
- 33. Lu Z. G., Zhang C. M., Zhai Z. H. (2004) LDFF, the large molecular weight DNA fragmentation factor, is responsible for the large molecular weight DNA degradation during apoptosis in Xenopus egg extracts. Cell Res. 14, 134–140 [DOI] [PubMed] [Google Scholar]
- 34. Morgan J., Leake D. S. (1995) Oxidation of low density lipoprotein by iron or copper at acidic pH. J. Lipid Res. 36, 2504–2512 [PubMed] [Google Scholar]
- 35. Wen Y., Leake D. S. (2007) Low density lipoprotein undergoes oxidation within lysosomes in cells. Circ. Res. 100, 1337–1343 [DOI] [PubMed] [Google Scholar]
- 36. Bunn H. F., Jandl J. H. (1968) Exchange of heme among hemoglobins and between hemoglobin and albumin. J. Biol. Chem. 243, 465–475 [PubMed] [Google Scholar]
- 37. Bamm V. V., Tsemakhovich V. A., Shaklai M., Shaklai N. (2004) Haptoglobin phenotypes differ in their ability to inhibit heme transfer from hemoglobin to LDL. Biochemistry 43, 3899–3906 [DOI] [PubMed] [Google Scholar]
- 38. Jerome W. G., Cox B. E., Griffin E. E., Ullery J. C. (2008) Lysosomal cholesterol accumulation inhibits subsequent hydrolysis of lipoprotein cholesteryl ester. Microsc. Microanal. 14, 138–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Purushothaman K. R., Purushothaman M., Levy A. P., Lento P. A., Evrard S., Kovacic J. C., Briley-Saebo K. C., Tsimikas S., Witztum J. L., Krishnan P., Kini A., Fayad Z. A., Fuster V., Sharma S. K., Moreno P. R. (2012) Increased expression of oxidation-specific epitopes and apoptosis are associated with haptoglobin genotype: possible implications for plaque progression in human atherosclerosis. J. Am. Coll. Cardiol. 60, 112–119 [DOI] [PubMed] [Google Scholar]
- 40. Shapiro R., McManus M. J., Zalut C., Bunn H. F. (1980) Sites of non enzymatic glycosylation of human hemoglobin. J. Biol. Chem. 255, 3120–3127 [PubMed] [Google Scholar]
- 41. Höhn A., Jung T., Grimm S., Grune T. (2010) Lipofuscin bound iron is a major intracellular source of oxidants: role in senescent cells. Free Rad. Biol. Med. 48, 1100–1108 [DOI] [PubMed] [Google Scholar]
- 42. Kurz T., Eaton J. W., Brunk U. T. (2010) Redox activity within the lysosomal compartment: implications for aging and apoptosis. Antioxid. Redox. Signal. 13, 511–523 [DOI] [PubMed] [Google Scholar]
- 43. Yu Z., Persson H. L., Eaton J. W., Brunk U. T. (2003) Intralysosomal iron: a major determinant of oxidant-induced cell death. Free Radic. Biol. Med. 34, 1243–1252 [DOI] [PubMed] [Google Scholar]
- 44. Nilsson E., Ghassemifar R., Brunk U. T. (1997) Lysosomal heterogeneity between and within cells with respect to resistance against oxidative stress. Histochem. J. 29, 857–865 [DOI] [PubMed] [Google Scholar]
- 45. Rupar C. A., Albo S., Whitehall J. D. (1992) Rat liver lysosome membranes are enriched in α-tocopherol. Biochem. Cell Biol. 70, 486–488 [DOI] [PubMed] [Google Scholar]
- 46. Turk B., Turk V. (2009) Lysosomes as “suicide bags” in cell death: myth or reality? J. Biol. Chem. 284, 21783–21787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yuan X. M., Li W., Brunk U. T., Dalen H., Chang Y. H., Sevanian A. (2000) Lysosomal destabilization during macrophage damage induced by cholesterol oxidation products. Free Radic. Biol. Med. 28, 208–218 [DOI] [PubMed] [Google Scholar]
- 48. Roberg K., Kågedal K., Ollinger K. (2002) Microinjection of cathepsin d induces caspase-dependent apoptosis in fibroblasts. Am. J. Pathol. 161, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li W., Kornmark L., Jonasson L., Forssell C., Yuan X. M. (2009) Cathepsin L is significantly associated with apoptosis and plaque destabilization in human atherosclerosis. Atherosclerosis 202, 92–102 [DOI] [PubMed] [Google Scholar]
- 50. Wolf B. B., Schuler M., Echeverri F., Green D. R. (1999) Caspase-3 is the primary activator of apoptotic DNA fragmentation via DNA fragmentation factor-45/inhibitor of caspase-activated DNase inactivation. J. Biol. Chem. 274, 30651–30656 [DOI] [PubMed] [Google Scholar]
- 51. Esterbauer H., Dieber-Rotheneder M., Striegl G., Waeg G. (1991) Role of vitamin E in preventing the oxidation of low-density lipoprotein. Am. J. Clin. Nutr. 53, 314S–321S [DOI] [PubMed] [Google Scholar]
- 52. Lyons N. M., Woods J. A., O'Brien N. M. (2001) α-Tocopherol, but not γ-tocopherol inhibits 7 β-hydroxycholesterol-induced apoptosis in human U937 cells. Free Radic. Res. 35, 329–339 [DOI] [PubMed] [Google Scholar]
- 53. Li-Weber M., Weigand M. A., Giaisi M., Süss D., Treiber M. K., Baumann S., Ritsou E., Breitkreutz R., Krammer P. H. (2002) Vitamin E inhibits CD95 ligand expression and protects T cells from activation-induced cell death. J. Clin. Invest. 110, 681–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Blum S., Vardi M., Brown J. B., Russell A., Milman U., Shapira C., Levy N. S., Miller-Lotan R., Asleh R., Levy A. P. (2010) Vitamin E reduces cardiovascular disease in individuals with diabetes mellitus and the haptoglobin 2-2 genotype. Pharmacogenomics 11, 675–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lowry O. H., Rosebrough N. J., Farr A. L., Randall R. J. (1951) Protein measurement with the Folin phenol reagent. J. Biol. Chem. 193, 265–275 [PubMed] [Google Scholar]