

Background: GLI1 is required for pancreatic tumor initiation; however, its role at later stages of carcinogenesis remains elusive.
Results: Genetic inactivation of GLI1 accelerates pancreatic cancer progression.
Conclusion: GLI1 can act as both a promoter and suppressor of pancreatic carcinogenesis depending on the tumor stage.
Significance: This knowledge increases our understanding of pancreatic carcinogenesis and may help the design of therapies targeting GLI1-related pathways.
Keywords: Cancer Biology, Gene Expression, Pancreatic Cancer, Transcription Factors, Tumor Microenvironment, GLI1, Tumor Progression
Abstract
The role of GLI1 in pancreatic tumor initiation promoting the progression of preneoplastic lesions into tumors is well established. However, its function at later stages of pancreatic carcinogenesis remains poorly understood. To address this issue, we crossed the gli1 knock-out (GKO) animal with cre-dependent pancreatic activation of oncogenic kras concomitant with loss of the tumor suppressor tp53 (KPC). Interestingly, in this model, GLI1 played a tumor-protective function, where survival of GKO/KPC mice was reduced compared with KPC littermates. Both cohorts developed pancreatic cancer without significant histopathological differences in survival studies. However, analysis of mice using ultrasound-based imaging at earlier time points showed increased tumor burden in GKO/KPC mice. These animals have larger tumors, decreased body weight, increased lactate dehydrogenase production, and severe leukopenia. In vivo and in vitro expression studies identified FAS and FAS ligand (FASL) as potential mediators of this phenomenon. The FAS/FASL axis, an apoptotic inducer, plays a role in the progression of pancreatic cancer, where its expression is usually lost or significantly reduced in advanced stages of the disease. Chromatin immunoprecipitation and reporter assays identified FAS and FASL as direct targets of GLI1, whereas GKO/KPC mice showed lower levels of this ligand compared with KPC animals. Finally, decreased levels of apoptosis were detected in tumor tissue in the absence of GLI1 by TUNEL staining. Together, these findings define a novel pathway regulated by GLI1 controlling pancreatic tumor progression and provide a new theoretical framework to help with the design and analysis of trials targeting GLI1-related pathways.
Introduction
Pancreatic cancer is a devastating disease with a poor prognosis due in part to the complex network of pathways driving aberrant gene expression and tumor progression (1–9). Thus, an understanding of the mechanisms controlling pancreatic cancer growth and maintenance are essential for the development of future therapeutic approaches for this disease.
Our group is particularly interested in understanding the role of transcription factor GLI1,2 a member of the GLI family of transcription factors, in pancreatic carcinogenesis. GLI1 has been shown to exert an oncogenic function working as an effector of the Hedgehog pathway in multiple malignancies (8, 10–13). This transcription factor is expressed at low levels in normal adult tissue, but it is often re-expressed and de-regulated in many human cancers carrying signature mutations in the Kras gene (10–13). Recently, we demonstrated that the loss of GLI1 in a genetically engineered mouse model (GEMM) blocks the progression of pancreatic intraepithelial neoplasms (PanIN) induced by oncogenic KRAS (10). In agreement with these findings, using a transgenic model overexpressing a repressor form of the transcription factor GLI3 to antagonize GLI1 activity, Rajurkar et al. (11) showed a requirement for this transcription factor in pancreatic cancer initiation. Furthermore, several studies demonstrate that blocking Sonic Hedgehog (SHH), the most well established upstream regulator of GLI1, down-regulates GLI1 expression and inhibits tumor growth in established xenograft models, suggesting a role for GLI1 in pancreatic tumor progression (13).
To define the biological significance and molecular mechanisms associated with expression of GLI1 in the advanced stages of tumor progression, and the mechanisms driving this phenomenon, we generated a quadruple cross GEMM resembling these late stages of the disease. This mouse model contained p48 cre-dependent activation of oncogenic Kras and loss of tumor suppressor tp53 (KPC) and gli1 resulting in the GKO/KPC mouse. We hypothesized that the loss of Gli1 in this model would result in prolonged survival and lower frequency of pancreas tumors. Interestingly, both cohorts retained the common biological and molecular features of advanced pancreatic cancer, but GKO/KPC mice developed early signs of tumor burden and perished within the 1st month of life. Loss of GLI1 accelerated disease progression as shown by decreased survival, body weight, fatigue, increased tumor volume, increased serum lactate dehydrogenase (LDH), and leukopenia. Analysis of the mechanism showed that both FAS and FAS ligand (FASL) expression were significantly decreased in the GKO/KPC cohort. Down-regulation of these pro-apoptotic proteins is often associated with the progression of many cancers in conjunction with loss of tumor suppressor p53 (14, 15), but more recent evidence suggests other factors may contribute to this phenomenon (16). Using ChIP assays, we demonstrate endogenous GLI1 binding to the FAS and FASL promoters. Furthermore, overexpression and knockdown studies confirm a role for GLI1 in the regulation of the expression of these genes. Finally, TUNEL staining of pancreas sections resulted in decreased levels of apoptosis in the GLI1-null group demonstrating the existence of a novel GLI1-FASL/FAS axis in pancreatic cancer. We hope this knowledge will provide a new theoretical framework to help with the analysis of existing studies targeting GLI1-related pathways as well as the design of future clinical trials.
EXPERIMENTAL PROCEDURES
Breeding
Mice were housed in pathogenic free conditions and maintained in facilities approved by the American Association for Accreditation of Laboratory Animal Care in accordance with current regulations and standards of the United States Department of Agriculture, Department of Health and Human Services, and National Institutes of Health/IACUC. LSL-kras mouse strain 1XJ6 B6.129-Ktm4Tyj (LSL-krasG12D), p48-cre, WT, and gli1lacZ (GKO) were described previously (10), and FVB.129-tp53tm1Brn strain 01xC2 (TRPF) were obtained from the mouse repository at National Institutes of Health, NCI Frederick. Two simultaneous crosses were used to produce gli1±; krasG12D; p48-cre; tp53± breeding pairs as follows: 1) GLI1LacZ mice were crossed with LSL-krasG12D and p48-cre mice producing GLI1±; krasG12D; p48-cre mice; and 2) gli11lacZ was crossed with p48-cre; tp53F2–10 mice producing gli1±; p48-cre; tp53±. Progeny from these groups were crossed to produce gli1±; krasG12D; p48-cre; tp53±. Finally, this group of mice was crossed to yield three groups as follows: 1) krasG12D; p48-cre-tp53F (KPC); 2) gli1±; krasG12D; p48-cre; tp53F (GHet/KPC); and 3) gli1lacZ; krasG12D; p48-cre; tp53F (GKO/KPC). p48-cre;krasG12D (KC) animals were used as controls in experiments (10).
Genotyping
Allele-specific PCR verified the presence of mutant gene arrangements in the DNA as described previously (16) with the addition of tp53F primers as follows: tp53tm1Brn, forward 5′-aaggggtatgagggacaagg-3′ and reverse 5′-gaagacagaaaaggggaggg-3′.
Immunohistochemistry
Paraffin-embedded tissue sections were processed using methods described previously (10). Primary antibodies used are as follows: CK19 (TROMA III, Developmental Studies Hybridoma Bank, Iowa City, IA); PCNA (Abcam, Cambridge, MA); F4/80 (Abcam), and CD31 (Santa Cruz Biotechnology). Masson's trichrome staining was performed using standard methods described previously (13). Images were taken with an Olympus BX-51 microscope, Olympus DP71 digital camera, and DP Controller (Axioplan, II-Zeiss, Thornwood, NY).
Ultrasound in Vivo Imaging
Mice pancreata were imaged via ultrasound using the VEVO2100 small imaging system. Briefly, mice were anesthetized using 1.5 liters/min oxygen/isofluorane mixture and then gently taped to the imaging stage. One-two ml of normal saline was injected into the right flank via intraperitoneal injection. Visual signs of distress as well as vital signs were monitored on the VEVO2100 (VisualSonics Inc, Ontario, Canada). Mice were imaged in both supine and oblique positions for better imaging. Still images were captured using the abdominal package in M-mode, and three images per pancreas (10 fields/analysis) were captured via three-dimensional Doppler for volume analysis following manufacturer's (VisualSonics) and published literature procedures (17, 18). KPC tumors have a highly fibrotic nature and appeared extremely bright in the ultrasound field; thus, we used WT or KC tumors as controls. These tumors appeared seedy/fluid and dark/dense as described for WT animals (Fig. 3A) (18).
FIGURE 3.

Loss of GLI1 increases tumor burden. A, representative ultrasound images of WT, KPC, and GKO/KPC mice captured at day 25 after birth. Developing pancreatic tumors (T), spleen (S), kidney (K), and normal pancreas (P). B, tumors measured by three-dimensional mode ultrasound show a lower tumor volume in KPC mice compared with the GKO/KPC animals at 25 days. C, similarly, GKO/KPC average body weight was lower compared with the KPC mice at 30 days after birth. D, GKO/KPC average pancreas weight was significantly higher. E, GKO/KPC mice have higher levels of LDH compared with KPC. F, CD4+/CD8+ T-cell ratio in GKO/KPC is significantly decreased compared with KPC. G, WT and GKO (no mutated kras) CD4+/CD8+ subpopulations in the spleen, mesenteric lymph nodes (MLN), or thymus.
Lactate Dehydrogenase (LDH) Assay
Serum LDH was measured using a colorimetric kit (Abcam) following the manufacturer's recommended protocol. Briefly, 2.5 μl of serum was mixed with assay buffer, at time 0 and 30 min after incubation at 37 °C, and activity was measured by spectrophotometry at 450 λ. LDH activity was analyzed utilizing a linear standard curve equation (nmol/min/ml (in milliunits/ml)) by using the following equation: B × sample dilution/(t1 − t0) × V; where B is NADH (Mr = 763 g/mol) generated between t0 and t1 in nanomoles, and t0 and t1 indicate the absorbance at time 0 and after 30 min, respectively. V is the pretreated sample volume added into the reaction well (in milliliters).
Lymphocyte Isolation and Analysis
Mice were autopsied at 1 month of age. CD4+ and CD8+ T cells were isolated from KPC and GKO/KPC spleens, and lymphocytes from spleen, mesenteric lymph node, thymus, and blood (WT and GKO not expressing oncogenic mutant kras). T-cells were isolated using the standard MACs isolation kit (Miltenyi, Cambridge, MA) and biotin/antibody mixture/microbeads. The FACS analysis was performed using a FACSCanto system (BD Biosciences) and FACSDiva software. Data were analyzed using FloJo software.
Quantitative RT-PCR (qPCR)
Tissues were homogenized in TRIzol reagent (Invitrogen), and RNA was extracted according to the manufacturer's instructions. Reverse transcription reactions were conducted using High Capacity cDNA reverse transcription kit (Applied Biosystems, Carlsbad, CA). To detect the expression of krasG12D mutant, pancreatic tissue from KPC and GKO/KPC mice was minced, and RNA was extracted. To amplify the 243-bp product from both the wild-type and mutant transcripts, the following primers were used: KRAS forward 5′-aggcctgctgaaaatgactg-3′ and reverse 5′-ccctcccgagttctcatgta-3′. Verification of the krasG12D allele was performed using HindIII digestion of the PCR product, which yields a 213- and 30-bp product present only in the mutant but not in the wild-type form of kras. Products were verified by sequencing to verify the G→A mutation in the cDNA. qPCRs in pancreas tumors were prepared with 1× SYBR Green PCR Master Mix (Bio-Rad) and various primers. All primers were optimized for amplification under reaction conditions as follows: 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 15 s, and 72, 76, and 80 °C for 10 s (with capture). GAPDH and 18 S were used as housekeeping genes. The sequence of the primers were as follows: 18 S forward 5′-aacccgttgaaccccattcgtgat-3′ and reverse 5′-caggttcacctacggaaaccttgt-3′ NC_000083.6 (19), GAPDH forward 5′-gcattgtggaagggctca and reverse 5′-gggtaggaacacggaagg-3′ NM_008084.2; IL-12p40 forward 5′-ccactcacatctgctgctgctccacaag-3′ and reverse 5′-acttctcatagtcccttggtccag-3′ NM_008352.2; IFNγ forward 5′-aacgctacacactgcatcttg-3′ and reverse 5′-gacttcaaagagtctgagg-3′ NM_008337.3; IL-10a forward 5′-cgggaagacaataactg-3′ and reverse 5′-catttccgataaggcttgg-3′ NM_010548; IL-4 forward 5′-acaggagaagggacgccat-3′ and reverse 5′-gaagccctacagacgagctca-3′ NM_021283.2; IL-1b forward 5′-caaccaacaagtgatattctccatg-3′ and reverse 5′-gatccacactctccagctgca-3′ AK156449.1; IL-2 forward 5′-cctgagcaggatggagaattaca-3′ and reverse 5′-tccagaacatgccgcafa-3′ NM_008366.3; FASL forward 5′-gaaggaaaccctttcctg-3′ and reverse 5′-ccccggagtatactttg-3′ NM_001205243.1; FAS forward 5′-cacagttaagagttcatactcaaggtactaat-3′ and reverse 5′-caacaaccataggcgatttctgggacttt-3′ NM_007987.2; TSLP1 forward 5′-ctgagagaaatgacggtactcagg-3′ and reverse 5′-gtggtattccttcatgcaatctcc-3′ NM_021367.2; TSLP2 forward 5′-ggcgacagcatggttcttct-3′ and reverse 5′-ttttgtcggggagtgaaggg-3′ NM_021367.2; MCP-1 forward 5′-gtgctgaccccaagaaggaa-3′ and reverse 5′-gtgctgaagaccttagggca-3′ NM_011333.3; TNFα forward 5′-ggcaggtctactttggagtcattgc-3′ and reverse 5′-acattcgaggctccagtgaattcgg-3′ NM_013693.2; and IL-8/KC forward 5′-cttgaaggtgttgccctcag-3′ and reverse 5′-tggggacaccttttagcatc-3′ NM_008176.3. Experiments were performed in triplicate. The results were calculated following the method 2ΔCp.
Tissue Culture and Transfections
Immortalized mouse embryonic fibroblasts were transfected with GLI1 overexpression and shRNA plasmids using XtremeGENE transfection reagent (Roche Applied Science). Overexpression studies were analyzed at 48 h and knockdown studies at 72 h.
Chromatin Immunoprecipitation Assay (ChIP)
ChIP was performed in mouse fibroblasts with the EZ-Magna ChIPTM G kit (Millipore, Billerica, MA) using 1 μg of rabbit IgG or GLI1 antibody (Novus Biologicals, Littleton, CO) as described previously (10) using the following primer sets for fas and fasl upstream of the first exon in each of the promoters as follows: fas forward 5′-gagttgtgtgggtctcagttt-3′ and reverse 5′-catgaaggtgatctatagccaaataatg-3′ ENSMUST00000025691; and fasl forward 5′-tggttaaggcacacttgctat-3′ and reverse 5′-gaagtcccttccctgaacatc-3′ ENSMUST00000000834. Alternative primers sets to confirm the specificity of the GLI1 antibody were designed to amplify regions upstream of the first exon in both fas and fasl at least 200 bp from the canonical GLI1-binding sites. The primers used were fas forward 5′-gcctattatctctacctctgctg-3′ and reverse 5′-ggcctctgaatggtaattaccac-3′; fasl forward 5′-ggctgattccgtggaatcatgag-3′ and reverse 5′-gtagaacttagctctgtagctgc-3′. Results were represented as percentage of input, where each antibody signal was relative to its respective input.
Plasmids
The mouse fas and fasl promoters were cloned in the pGL3 vector (Promega, Madison, WI) using standard recombinant DNA methodology. The following primers were used to clone these promoters: fas forward 5′-agctcaggtaccgtctgcagcaagggaaaacagcgggc-3′ and reverse 5′-agctcactcgagcctaggtccctgggctatctagtctcagg-3′; and fasl forward 5′-agctcagctagcctgagctgctaaacgagcatcctggaggtacc-3′ and reverse 5′-agctcactcgagctcctgttctcttctctttgcttctctgtgagac-3′. The human GLI1 and shRNA vectors were previously reported (8, 10).
Luciferase Reporter Assay
For the assays, mouse embryonic fibroblasts were plated in triplicate into six-well plates in medium containing 10% FBS. Twenty four hours later, cells were transfected as indicated above. Samples were collected and processed in accordance with the manufacturer's protocol (Promega, Madison, WI). Intersample variations were controlled by measuring the total protein of samples in each well (Bio-Rad), and luciferase readouts were normalized to protein content. Relative luciferase activity represents luciferase readouts/protein concentrations normalized to control cells within each experiment.
Patient Expression Study
Patient samples analyzed for desmoplasia and GLI1 expression were cases from the Mayo Clinic that were recruited from October 1, 2000 to July 1, 2010. Recruitment, consent, and specimen collection were IRB-approved (IRB protocol number 06-002459) by our institution. Patient records were reviewed by a gastrointestinal specialist and confirmed as pancreatic adenocarcinoma. This study was done without the bias of underlying pathologies, prior treatment or demographics (gender, age, etc). Staging was performed using American Joint Committee on Cancer (AJCC) 6th Edition criteria. Patients were identified as resected, locally advanced, and/or metastatic. Resected patients were further categorized by stage IA, IIB, IIA, and IIB. Blinded histopathology analysis was performed by two expert pathologists at the Mayo Clinic, and desmoplasia was scored using the following criteria: 1) none; 2) mild; 3) moderate; and 4) severe. Median desmoplasia expression level was categorized into three groups as follows: (a) none/mild versus moderate versus severe; (b) severe versus none/mild/moderate; (c) moderate/severe versus none/mild. Analyses were performed using SAS software, version 9.3 (SAS Institute, Cary, NC). RNA was reverse-transcribed into cDNA (as described above). qPCR was performed using TaqMan primer/probe sets for human GLI1 and 18 S using ABI7900 technology and software (Applied Biosystems, Foster City, CA). Relative target gene expression was normalized to the reference gene using the 2ΔCp method, and all reactions were performed in triplicate. Wilcoxon/Kruskal Wallis test (p value < 0.05) was used to test for statistically significant differences in expression across desmoplasia groups in both histology scoring and expression studies.
Western Blot
Pancreas total protein lysates were extracted and run on 10% acrylamide gels. Blots were probed with total ERK (Cell Signaling, Danvers, MA), phosphorylated ERK (pERK) (Cell Signaling), and γ-tubulin (Sigma) as described previously (10).
Terminal Deoxynucleotidyltransferase dUTP Nick End Labeling (TUNEL)
The DeadEnd fluorometric TUNEL system (Promega) was used to detect fragmented DNA of apoptotic cells in mouse pancreas tissues. PCNA (Abcam, Cambridge, MA) double staining was used to detect areas of proliferation. Seven mouse pancreas tissue sections per group, and four fields at ×10 were visualized, analyzed, and captured using the LSM 510 confocal microscopy (Zeiss, Thornwood, NY) and ImageJ software (National Institutes of Health).
Statistical Analysis
All data except the results from the human samples expression studies are expressed as the mean ± S.E. Differences between groups were compared using a two-tailed Student's t test. One-way analysis of variance was used to compare samples from the different time points in the same group.
RESULTS
Loss of GLI1 Accelerates Pancreatic Tumorigenesis
To define the biological significance of GLI1 at later stages of pancreatic carcinogenesis, we established cohorts of mice containing common features of advanced pancreatic cancer. This model includes the well established cre-dependent, pancreas-specific activation of oncogenic mutant kras and loss of the tumor suppressor tp53 (2), coupled with the loss of gli1 (GKO/KPC) (10). Mice from the GKO/KPC cohort succumbed to death at 1 month of age on average compared with 2.5 months in the KPC cohort containing normal expression levels of GLI1 (p = 0.0001) (Fig. 1A). Furthermore, gli1 heterozygous mouse (GHet)/KPC survival was similar to KPC (p = 0.2210) mice suggesting that one wild-type gli1 allele is sufficient to maintain functional GLI1 activity (Fig. 1B). All three cohorts (KPC, GKO/KPC, and GHet/KPC) developed fibrotic pancreas tumors that were similar in size (Fig. 1C) and showed similar oncogenic KRAS activity, as determined by the activation of the downstream effectors ERK (Fig. 1D and data not shown for GHet/KPC). Two independent expert pathologists completed blinded histopathological analysis of the three cohorts of experimental mice. Surprisingly, pancreas tumors were phenotypically indistinguishable between the varying degrees of GLI1 expression. Both KPC and GKO/KPC pancreata contained high grade PanIN II and III lesions, and more than 50% of pancreata in the cohorts presented with moderate fibrosis, inflammation, and pancreatic cancer (Fig. 2A). Furthermore, no significant or very minor differences were observed between the cohorts with respect to epithelial cell content (CK19), inflammatory macrophages (F4/80), microvessel density (CD31), and fibrosis/collagen content or deposition (Trichrome staining) in areas of pancreas tumors in this end point survival study (Fig. 2B). Finally, we engaged in a detailed study of the mice lacking oncogenic kras re-arranged alleles to assess the transgenic colony for anomalies associated with cre-p48 or cre-tp53-null mediated effects. Histopathology of the pancreas, liver, spleen, stomach, heart, and lungs was completed over an extended period of time, and it was determined that no developmental defects or cre-mediated effects were present at 2, 4, 6 and 8 months of age, and the tissues appeared normal at all time points considered. No differences in survival were noted between cohorts over an 18-month period (data not shown). Together, these results suggest that the loss of GLI1 hastens mortality in the KPC GEMM with similar pancreas pathology in this end point study.
FIGURE 1.

GKO/KPC mice develop pancreatic cancer but have lower survival. A, Kaplan-Meir survival curve shows a statistically significant difference in survival between the GKO/KPC (1 month) compared with 2.5 months in KC mice. B, no differences in survival were detected in GHet/KPC mice compared with the KPC animals. C, representative pancreatic tumors of KPC, GHet/KPC, and GKO/KPC on autopsy/death. D, representative Western blot demonstrating activation of the KRAS downstream effector ERK by looking at the levels of phosphoylated-ERK (pERK) in KPC and GKO/KPC. Densitometry of total ERK (tERK), pERK, and tubulin was used to quantify levels of the proteins in both cohorts.
FIGURE 2.
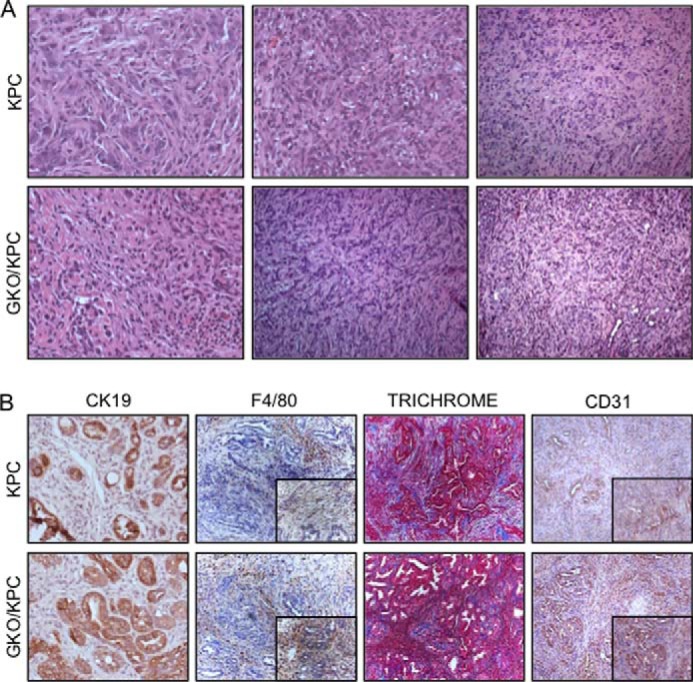
Pancreas histopathology in KPC and GKO/KPC mice is similar post-mortem. A, H&E stain shows that the KPC and GKO/KPC develop high grade PanIN III lesions and poorly differentiated invasive adenocarcinoma on death. B, immunohistochemistry shows similar epithelial component (CK19) in representative areas of PanIN lesions and adenocarcinoma and inflammation/monocyte infiltration (F4/80), fibrosis/collagen (Trichrome), and CD31 (vasculature compartment) in areas of tumor development.
GLI1 Loss Accelerates Cancer Progression
To determine the cause of accelerated morbidity in the GLI1-null cohorts, we evaluated mice at earlier time points by ultrasound imaging at 25 days of age, and biological specimens were collected on autopsy at day 30. Both supine and oblique images were captured and analyzed by ultrasound enabling a detailed capture of the pancreas and tumors (Fig. 3A). Using three-dimensional mode imaging, 10 fields per tumor region were captured and analyzed. The average tumor volume was smaller in KPC pancreas (56.14 ± 14.27 mm3, n = 14) compared with GKO/KPC pancreas (162.1 ± 15.8 mm3, n = 12) (p < 0.0001) (Fig. 3B). On autopsy, GKO/KPC average body weight was decreased (15.734 ± 0.3725 g, n = 15) compared with KPC (21.46 ± 0.6697 g, n = 15) (p = 0.0001) (Fig. 3C), and average pancreas weight was augmented in GKO/KPC (0.590 ± 0.0208 g, n = 15) compared with the KPC cohort (0.290 ± 0.0195 g, n = 15) (p < 0.0001) (Fig. 3D). At 30 days, the GKO/KPC mice present with the physical attributes of advanced disease or cancer progression compared with KPC littermates. Increased LDH activity is associated with tissue damage and is often used as a prognostic tool to measure progression of several diseases, including cancer (20–24). Next, we analyzed the serum of mice at 30 days post-natal. A direct correlation between increased tumor burden and elevated LDH activity was noted in the GKO/KPC cohort (0.397 ± 0.005 nmol/min/ml, n = 9) compared with KPC (0.363 ± 0.009 nmol/min/ml, n = 9) (p = 0.0015) (Fig. 3E). Together, these results reflect animals that are experiencing accelerated cachexia due to accelerated tumor growth that is reminiscent of human pancreatic ductal adenocarcinoma progression. Because advanced stage cancer is often associated with progressive lymphopenia, particularly within the CD4+ compartment (25–27), we evaluated CD4+/CD8+ ratios from splenocyte populations as a clinically relevant marker of advanced disease. Lymphocytes isolated from the spleen were analyzed by FACS and showed a reduction in CD4+/CD8+ T-cell ratio in the GKO/KPC cohort (1.745 ± 0.1179) compared with KPC (2.440 ± 0.2367) (p = 0.0545) (Fig. 3F). Overall, these results resemble the dismal features of clinical disease that many patients present at the time of diagnosis. T-cell ratios in wild-type and GKO mice in the absence of kras or tp53 mutations were analyzed to determine whether the loss of GLI1 alone caused lymphopenia in the experimental mice. No significant alterations within the lymphocyte CD4+/CD8+ subpopulations were noted in the spleen (WT 2.379 ± 0.3629 and GKO 3.038 ± 0.197, p = 0.4145), mesenteric lymph (WT 2.41 ± 0.2631 and GKO 2.822 ± 0.553, p = 0.5782), or thymus (WT 86.17 ± 0.4333 and GKO 86.30 ± 0.7506, p = 0.8875) (Fig. 3G).
Not surprisingly, at day 30 GKO/KPC histopathology showed a predominance of late stage PanIN III lesions and carcinoma. Alternatively, PanIN II and few PanIN III lesions were noted in the majority of the pancreata in the KPC cohort (Fig. 4A). Immunohistochemical analysis of pancreas markers in time point studies failed to reveal statistically significant changes in other pathology between the cohorts, but the analysis was limited due to the rapid development of tumor and extensive fibrosis in GKO/KPC and a more normal architecture in the KPC pancreas. Hence, analysis and comparisons were confined to similar areas between the cohorts (Fig. 4B).
FIGURE 4.
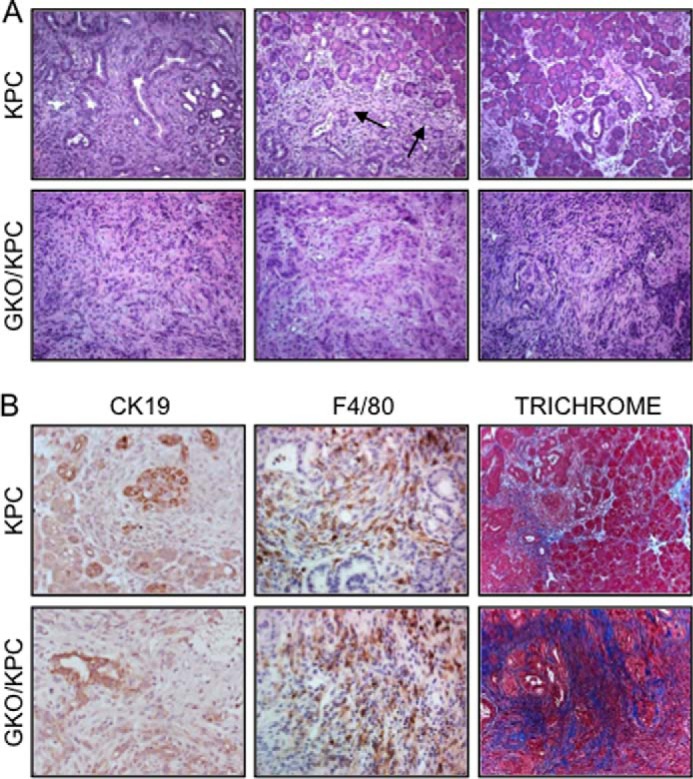
Inactivation of GLI1 accelerates carcinogenesis in GKO/KPC mice. A, representative H&E; KPC mice develop entire spectrum of PanIN lesions (left upper panel), small areas of invasive adenocarcinoma (arrows), and preserved normal pancreas (right upper panel) at 30 days. Lower panel, GKO/KPC pancreas develops extensive high grade PanIN lesions and invasive adenocarcinoma within 30 days. B, epithelial differentiation (CK19) is not significantly different between cohorts. The numbers of infiltrating monocytes (F4/80) are extensive around PanIN lesions in KPC and throughout the tumor and stroma in GKO/KPC. Fibrosis/collagen (Trichrome) content is higher in GKO/KPC tumor compared with (KPC), where several areas contain normal pancreas architecture at 30 days.
To ascertain whether this study recapitulates the clinical features of this disease, samples from patients with varying stages of the disease were analyzed by qPCR and histopathology. GLI1 expression levels were grouped as containing either high or low levels of GLI1. Matched H&E slides were reviewed and scored by a trained pancreas pathologist. Interestingly, patients with severe desmoplasia, a histopathological feature associated with poor prognosis, appeared to have a significantly lower median level of GLI1 expression when compared with those patients with none, mild, or moderate desmoplasia (0.48 versus 2.08 median expression, Kruskal Wallis (p = 0.0238)). These results, coupled with recent reports (28, 29), suggest that lower levels of GLI1 at later stages of pancreas tumorigenesis promote tumor progression. Together, these findings demonstrate that the loss of GLI1 results in increased tumor burden ultimately leading to accelerated morbidity and poor prognosis.
Regulation of FASL Underlies the Role of GLI1 during Tumor Progression
Cancer progression is often marked by aberrant expression of cytokines (30–32). These molecules modulate growth, proliferative, migration, and apoptotic pathways, often resulting in metastasis and advanced stage disease (33–40). Next, we evaluated expression of a panel of cytokines associated with disease progression using pancreas tumor tissues from the same cohort in the previous section by qPCR. Several cytokines showed trends in differential expression patterns, but only FASL (18-fold, p = 0.010) was significantly decreased in GKO/KPC pancreata (Table 1). The FASL receptor, FAS, was also decreased (14-fold, p = 0.008) in GKO/KPC samples (Table 1). We confirmed FASL and FAS expression results in a separate larger cohort of animals (n = 7/group) with the same results (Fig. 5A). Interestingly, the changes in cytokine expression were accompanied by changes in the SHH pathway activation, the most well characterized regulator of GLI1 function. Expression studies demonstrate that the levels of receptor Patched, a marker of the activity of SHH, were down-regulated in GKO/KPC mice compared with the KPC animals. Similarly, Smoothened, GLI2, and GLI3 were significantly diminished in the GKO/KPC, but the levels of SHH remained unchanged (Fig. 5, B–G).
TABLE 1.
Cytokine expression in GKO/KPC and KPC pancreatic tumor
Pancreatic tumors/tissues were analyzed by qPCR. FAS and FASL expression levels are significantly decreased in GKO/KPC mice
| Cytokine (2ΔCp) | GKO/KPC n = 4 |
KPC n = 4 |
p value | ||
|---|---|---|---|---|---|
| Mean | S.E. | Mean | S.E. | ||
| FAS (CD95) | 1.54 | 0.5 | 21.17 | 3.2 | *0.008 |
| FASL (CD95L) | 0.99 | 0.7 | 18.55 | 3.9 | *0.010 |
| IL-1b | 151.50 | 90.8 | 114.30 | 50.2 | 0.736 |
| IL-2 | 8.280 | 6.187 | 302.74 | 174.8 | 0.191 |
| IL-6 | 32.01 | 19.1 | 6.11 | 2.6 | 0.343 |
| IL-8 | 19.61 | 30.8 | 24.46 | 9.7 | 0.829 |
| IL-12p40 | 3.78 | 3.6 | 2.98 | 1.6 | 0.657 |
| IL-17 | 1.25 | 1.1 | 3.87 | 1.9 | 0.171 |
| IL-22 | 3.79 | 2.3 | 8.11 | 5.3 | 0.914 |
| INFγ | 3.14 | 2.1 | 6.17 | 2.3 | 0.343 |
| MCP-1 | 756.5 | 160.0 | 1008.00 | 374.8 | 0.570 |
| TGFβ | 13.54 | 9.7 | 7.52 | 2.8 | 0.587 |
| TNFα | 92.36 | 38.0 | 79.66 | 15.0 | 0.772 |
| TSLP | 15.11 | 6.2 | 14.57 | 2.29 | 0.939 |
FIGURE 5.

GLI1 regulates FAS and FASL expression in pancreatic tumors. A, mRNA expression analysis by qPCR in mouse pancreas 30 days post-partum showed that FAS and FASL levels are reduced in GKO/KPC group compared with the KPC group. B–G, qPCR analysis of the components of the Sonic Hedgehog pathway in mouse pancreas. B, as expected, GLI1 expression was reduced in the GKO/KPC. C–G, GLI2 (C), GLI3 (D), PATCHED1 (F), and SMOOTHENED (G) showed as well a reduced expression in the GKO/KPC compared with KPC. SHH levels did not change in both experimental groups.
Bioinformatics analysis identified six candidate GLI1-binding sites in the promoter sequences of both FASL and FAS proteins (data not shown). ChIP assays confirmed the binding of endogenous GLI1 to the FAS promoter at −1147 to −943 bp upstream of the first exon. Alternative FAS primers amplifying a region without GLI1 consensus sequences were used to control for antibody specificity at −613 to −486 bp on the FAS promoter (Fig. 6A, left panel). GLI1 also bound to the FASL promoter at a region from −539 to −293 bp upstream of the first exon. Similar to FAS promoter, alternative FASL primers without GLI1 consensus sites in the promoter region −1646 to −1517 bp were also negative (Fig. 6A, right panel).
FIGURE 6.

FAS and FASL are direct targets of GLI1 in pancreatic cancer. A, endogenous GLI1 binds to the fas promoter at regions spanning from −1147 to −943 bp upstream of the first exon. No binding was detected outside this region in an area lacking canonical GLI1-binding site (negative control region/−613 to −486, left panel). Binding of endogenous GLI1 to the fasl promoter at −539 to −293 bp upstream of the first exon. No binding of GLI1 was seen outside this region (negative control region/−1646 to −1517 bp) in this promoter (right panel). B, FAS and FASL mRNA expression in cells overexpressing GLI1 (left panel) or knockdown for GLI1 (right panel). C, reporter assays using the mouse fas and fasl promoters. Cells were cotransfected with the indicated reporter vectors and control or GLI1 expression vectors (left panel) or scramble or shGLI1 vectors (right panel). D, PCNA (proliferation), TUNEL (apoptosis), and DAPI (nuclei). TUNEL staining is positive in KPC pancreas, and fewer areas of apoptosis are noted in GKO/KPC. Proliferation is equal between both cohorts indicating that lack of expression in GKO/KPC is not due to tissue necrosis.
Overexpression of GLI1 in fibroblasts increased expression of FAS expression by 2-fold (p = 0.0198) and FASL by 4.5-fold (p = 0.0211) (Fig. 6B, left panel). Conversely, knockdown of GLI1 resulted in a decrease in FAS expression by 1.8-fold (p = 0.003), and a decrease in FASL expression by >8-fold (p = 0.0285) (Fig. 6B, right panel). Finally, we demonstrate that GLI1 overexpression increased both mouse FAS (control 100 ± 3.986 versus GLI1 191 ± 18.66, n = 3) and FASL (control 100 ± 8.768 versus GLI1 203.9 ± 15.23, n = 3) promoter activity (Fig. 6C, left panel). Conversely, knockdown of GLI1 using shRNA constructs decreased activity of these regulatory sequences in mouse fibroblasts (FAS Scramble 100 ± 6.137 versus shGLI1 65.89 ± 2.671, n = 3) and (FASL Scramble 100 ± 9.191 versus shGLI1 32.76 ± 1.356, n = 3) (Fig. 6C, right panel). Together, these studies identify FAS and FASL as direct targets of GLI1 and define a novel transcriptional mechanism for these pro-apoptotic molecules.
FASL and FAS are members of the tumor necrosis ligand and receptor superfamily, and they play a critical role in apoptosis in multiple cell types (41–43). Thus, we analyzed proliferation (PCNA) and apoptosis (TUNEL) using triple fluorescence staining of pancreatic tumor tissues from both cohorts. GKO/KPC showed a decrease in the number of apoptotic cells (0.78 ± 0.466, n = 7) compared with KPC (4.75 ± 1.48, n = 7) (p = 0.002) (Fig. 6D). Cell proliferation (PCNA stain) in both cohorts confirmed that the cellular tumor bed of the tissue analyzed is not necrotic. DAPI staining provided reference for cell nuclei. Together, these results provide evidence of a novel GLI1-FAS/FASL interaction that may play a role in the progression of pancreatic tumorigenesis.
DISCUSSION
GLI1, like other molecules (e.g. TGFβ), can act as both a positive and negative regulator of tumor development (8, 10, 11, 29, 44–47). Examination of human and mouse tumor samples by immunohistochemistry or global arrays (49, 52–55) demonstrates that the expression of GLI1 in many cancers often correlates with poor prognosis. Therefore, GLI1 expression signatures are often associated with cancer progression and metastasis (50, 51). However, several in vitro and in vivo studies either inhibiting effectors upstream of GLI1 or GLI1 directly with genetic or pharmacological tools show that loss of this transcription factor decreases processes like epithelial-to-mesenchymal transition and cellular de-differentiation due to a less aggressive tumorigenic phenotype (56). Although GLI1 expression in pancreatic cancer clinical samples and pancreatic cancer GEMMs is well documented, the biological relevance of this result in pancreatic cancer progression is not completely understood. This is important because several clinical trials inhibiting pathways known to induce GLI1 activity (e.g. Hedgehog) are underway (12, 48, 49, 57–59). The success of these trials has been mixed, possibly due to the complexity of the signaling processes involved in pancreatic cancer progression. Recently, our group and others have identified GLI1 as a mediator in the process of cellular transformation in vivo where loss of GLI1 in the presence of oncogenic KRAS inhibited late stage PanIN formation and pancreas cancer initiation (10, 11). However, the significance of GLI1 expression and the molecular mechanisms regulating expression in pancreatic cancer progression are conflicting. Thus, we generated a GLI1-null GEMM retaining the molecular and biological features of advanced pancreatic cancer. We hypothesized that the reduction of GLI1 in this model of tumor progression would slow tumorigenesis and cancer progression thereby extending survival. Surprisingly, the GKO/KPC died at a rate that was 50% faster than the KPC littermates. Histologically, the pancreas was similar in both cohorts. There appeared to be minor increases in fibrosis in the GKO/KPC cohort, but these alterations could not be scored by the pathologist as significantly different between the two cohorts. Marker analysis failed to provide insight to the relevant pathways that might be involved in progression in part due to a fairly normal phenotype in the KPC mice at 30 days. Thus, accurate comparisons could not be made and were primarily focused on comparing fibrotic areas of the pancreas that were present on both cohorts. Although the vasculature was diagnosed as normal in appearance, we saw an increase in microvessel density in GKO/KPC in the survival cohort, but the result was not significant. Only one section was stained and analyzed, and a more detailed study, including blood flow analysis, is warranted to conclude whether or not the findings are significant. Finally, to define the mechanism underlying these biological findings, we explored the role of cytokines known to modulate tumor progression, especially in the tumor microenvironment that might participate in escalating this phenotype. The panels of cytokines evaluated showed trends, but significant results were not achieved for most cytokines. However, expression of both FASL and FAS was significantly reduced in the GKO/KPC mice with a tight fit compared with KPC littermates. Both proteins are critical modulators in tumorigenesis and intimately involved in homeostasis inducing apoptosis on target cells (60, 61). We demonstrated endogenous GLI1 binding to both FASL and FAS promoters and define these pro-apoptotic molecules as novel direct transcriptional targets of GLI1 in fibroblasts. Similar results were seen in other cell types that are known to express GLI1 in pancreatic cancer, such as cancer cells and lymphocytes (data not shown). Furthermore, we define a novel regulatory role for GLI1 in the FAS/FASL axis suggesting a possible explanation for the transformed cellular phenotype to propagate. To confirm this hypothesis, we examined the functional output of FAS/FASL axis and noted lower levels of apoptosis in the GKO/KPC cohort. Thus, we defined a GLI1-FAS/FASL axis as a novel regulator of cell survival in the advanced stages of pancreatic tumorigenesis.
Together, these results define GLI1 as a novel regulator of tumor progression and expand the list of pathways playing a dual role at different stages of tumor development. We provide evidence that the loss of GLI1 in a cancer progression model hastens morbidity over animals with wild-type levels of GLI1 suggesting that inhibition of GLI1 may be detrimental in some contexts in patients diagnosed with pancreatic cancer. Future studies will include the identification of genetic and epigenetic modulators regulating GLI1, FAS, and FASL transcription and expression with the hope that novel therapies to treat this dismal disease and other types of cancer will finally come to fruition.
Acknowledgments
We are grateful to Drs. Jordan Miller and Grace Verzosa for use of the VEVO2100. We express our gratitude to Drs. Isaiah J. Fidler and Eugenie S. Kleinerman for their insightful discussions and mentorship. We also thank Dr. Luciana Almada for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant CA136526, Grant P50 CA102701 (Mayo Clinic Pancreatic SPORE), and P30 DK84567 (Mayo Clinic Center for Cell Signaling in Gastroenterology) (to M. E. F.-Z.) from NCI.
- GLI1
- glioma-associated protein
- LDH
- lactate dehydrogenase
- GKO
- gli1 knock-out animal
- FASL
- FAS ligand
- qPCR
- quantitative PCR
- PanIN
- pancreatic intraepithelial neoplasm
- SHH
- Sonic Hedgehog
- GEMM
- genetically engineered mouse model
- PCNA
- proliferating cell nuclear antigen
- KPC
- krasG12D; tp53F; p48-cre.
REFERENCES
- 1. Izeradjene K., Combs C., Best M., Gopinathan A., Wagner A., Grady W. M., Deng C. X., Hruban R. H., Adsay N. V., Tuveson D. A., Hingorani S. R. (2007) Kras(G12D) and Smad4/Dpc4 haploinsufficiency cooperate to induce mucinous cystic neoplasms and invasive adenocarcinoma of the pancreas. Cancer Cell 11, 229–243 [DOI] [PubMed] [Google Scholar]
- 2. Hingorani S. R., Wang L., Multani A. S., Combs C., Deramaudt T. B., Hruban R. H., Rustgi A. K., Chang S., Tuveson D. A. (2005) Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell 7, 469–483 [DOI] [PubMed] [Google Scholar]
- 3. Plentz R., Park J. S., Rhim A. D., Abravanel D., Hezel A. F., Sharma S. V., Gurumurthy S., Deshpande V., Kenific C., Settleman J., Majumder P. K., Stanger B. Z., Bardeesy N. (2009) Inhibition of γ-secretase activity inhibits tumor progression in a mouse model of pancreatic ductal adenocarcinoma. Gastroenterology 136, 1741–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bardeesy N., Aguirre A. J., Chu G. C., Cheng K. H., Lopez L. V., Hezel A. F., Feng B., Brennan C., Weissleder R., Mahmood U., Hanahan D., Redston M. S., Chin L., Depinho R. A. (2006) Both p16(Ink4a) and the p19(Arf)-p53 pathways constrain progression of pancreatic adenocarcinoma in the mouse. Proc. Natl. Acad. Sci. U.S.A. 103, 5947–5952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hezel A. F., Kimmelman A. C., Stanger B. Z., Bardeesy N., Depinho R. A. (2006) Genetics and biology of pancreatic ductal adenocarcinoma. Genes Dev. 20, 1218–1249 [DOI] [PubMed] [Google Scholar]
- 6. Kornmann M., Danenberg K. D., Arber N., Beger H. G., Danenberg P. V., Korc M. (1999) Inhibition of cyclin D1 expression in human pancreatic cancer cells is associated with increased chemosensitivity and decreased expression of multiple chemoresistance genes. Cancer Res. 59, 3505–3511 [PubMed] [Google Scholar]
- 7. Prasad N. B., Biankin A. V., Fukushima N., Maitra A., Dhara S., Elkahloun A. G., Hruban R. H., Goggins M., Leach S. D. (2005) Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 65, 1619–1626 [DOI] [PubMed] [Google Scholar]
- 8. Nolan-Stevaux O., Lau J., Truitt M. L., Chu G. C., Hebrok M., Fernández-Zapico M. E., Hanahan D. (2009) GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 23, 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pasca di Magliano M., Sekine S., Ermilov A., Ferris J., Dlugosz A. A., Hebrok M. (2006) Hedgehog/Ras interactions regulate early stages of pancreatic cancer. Genes Dev. 20, 3161–3173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mills L. D., Zhang Y., Marler R. J., Herreros-Villanueva M., Zhang L., Almada L. L., Couch F., Wetmore C., Pasca di Magliano M., Fernandez-Zapico M. E. (2013) Loss of the transcription factor GLI1 identifies a signaling network in the tumor microenvironment mediating KRAS oncogene-induced transformation. J. Biol. Chem. 288, 11786–11794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rajurkar M., De Jesus-Monge W. E., Driscoll D. R., Appleman V. A., Huang H., Cotton J. L., Klimstra D. S., Zhu L. J., Simin K., Xu L., McMahon A. P., Lewis B. C., Mao J. (2012) The activity of Gli transcription factors is essential for Kras-induced pancreatic tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 109, E1038–E1047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang S. H., Hsu C. H., Lee J. C., Tien Y. W., Kuo S. H., Cheng A. L. (2013) Nuclear expression of glioma-associated oncogene homolog 1 and nuclear factor-κB is associated with a poor prognosis of pancreatic cancer. Oncology 85, 86–94 [DOI] [PubMed] [Google Scholar]
- 13. Thayer S. P., di Magliano M. P., Heiser P. W., Nielsen C. M., Roberts D. J., Lauwers G. Y., Qi Y. P., Gysin S., Fernández-del Castillo C., Yajnik V., Antoniu B., McMahon M., Warshaw A. L., Hebrok M. (2003) Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature 425, 851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boldrini L., Faviana P., Pistolesi F., Gisfredi S., Di Quirico D., Lucchi M., Mussi A., Angeletti C. A., Baldinotti F., Fogli A., Simi P., Basolo F., Fontanini G. (2001) Alterations of Fas (APO-1/CD 95) gene and its relationship with p53 in non small cell lung cancer. Oncogene 20, 6632–6637 [DOI] [PubMed] [Google Scholar]
- 15. Volkmann M., Schiff J. H., Hajjar Y., Otto G., Stilgenbauer F., Fiehn W., Galle P. R., Hofmann W. J. (2001) Loss of CD95 expression is linked to most but not all p53 mutants in European hepatocellular carcinoma. J. Mol. Med. 79, 594–600 [DOI] [PubMed] [Google Scholar]
- 16. Maas S., Warskulat U., Steinhoff C., Mueller W., Grimm M. O., Schulz W. A., Seifert H. H. (2004) Decreased Fas expression in advanced-stage bladder cancer is not related to p53 status. Urology 63, 392–397 [DOI] [PubMed] [Google Scholar]
- 17. Olive K. P., Jacobetz M. A., Davidson C. J., Gopinathan A., McIntyre D., Honess D., Madhu B., Goldgraben M. A., Caldwell M. E., Allard D., Frese K. K., Denicola G., Feig C., Combs C., Winter S. P., Ireland-Zecchini H., Reichelt S., Howat W. J., Chang A., Dhara M., Wang L., Rückert F., Grützmann R., Pilarsky C., Izeradjene K., Hingorani S. R., Huang P., Davies S. E., Plunkett W., Egorin M., Hruban R. H., Whitebread N., McGovern K., Adams J., Iacobuzio-Donahue C., Griffiths J., Tuveson D. A. (2009) Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324, 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sastra S. A., Olive K. P. (2013) Quantification of murine pancreatic tumors by high-resolution ultrasound. Methods Mol. Biol. 980, 249–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harmsma M., Schutte B., Ramaekers F. C. (2013) Serum markers in small cell lung cancer: opportunities for improvement. Biochim. Biophys. Acta 1836, 255–272 [DOI] [PubMed] [Google Scholar]
- 20. Steyerberg E. W., Pencina M. J., Lingsma H. F., Kattan M. W., Vickers A. J., Van Calster B. (2012) Assessing the incremental value of diagnostic and prognostic markers: a review and illustration. Eur. J. Clin. Invest. 42, 216–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rattigan Y. I., Patel B. B., Ackerstaff E., Sukenick G., Koutcher J. A., Glod J. W., Banerjee D. (2012) Lactate is a mediator of metabolic cooperation between stromal carcinoma associated fibroblasts and glycolytic tumor cells in the tumor microenvironment. Exp. Cell Res. 318, 326–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Butt A. A., Michaels S., Kissinger P. (2002) The association of serum lactate dehydrogenase level with selected opportunistic infections and HIV progression. Int. J. Infect. Dis. 6, 178–181 [DOI] [PubMed] [Google Scholar]
- 23. Ferraris A. M., Giuntini P., Gaetani G. F. (1979) Serum lactic dehydrogenase as a prognostic tool for non-Hodgkin lymphomas. Blood 54, 928–932 [PubMed] [Google Scholar]
- 24. Maftouh M., Avan A., Sciarrillo R., Granchi C., Leon L. G., Rani R., Funel N., Smid K., Honeywell R., Boggi U., Minutolo F., Peters G. J., Giovannetti E. (2014) Synergistic interaction of novel lactate dehydrogenase inhibitors with gemcitabine against pancreatic cancer cells in hypoxia. Br. J. Cancer 110, 172–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fogar P., Basso D., Fadi E., Greco E., Pantano G., Padoan A., Bozzato D., Facco M., Sanzari M. C., Teolato S., Zambon C. F., Navaglia F., Semenzato G., Pedrazzoli S., Plebani M. (2011) Pancreatic cancer alters human CD4+ T lymphocyte function: a piece in the immune evasion puzzle. Pancreas 40, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 26. Trédan O., Manuel M., Clapisson G., Bachelot T., Chabaud S., Bardin-dit-Courageot C., Rigal C., Biota C., Bajard A., Pasqual N., Blay J. Y., Caux C., Ménétrier-Caux C. (2013) Patients with metastatic breast cancer leading to CD4+ T cell lymphopaenia have poor outcome. Eur. J. Cancer 49, 1673–1682 [DOI] [PubMed] [Google Scholar]
- 27. Balmanoukian A., Ye X., Herman J., Laheru D., Grossman S. A. (2012) The association between treatment-related lymphopenia and survival in newly diagnosed patients with resected adenocarcinoma of the pancreas. Cancer Invest. 30, 571–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moutasim K. A., Mellows T., Mellone M., Lopez M. A., Tod J., Kiely P. C., Sapienza K., Greco A., Neill G. W., Violette S., Weinreb P. H., Marshall J. F., Ottensmeier C. H., Sayan A. E., Jenei V., Thomas G. J. (2014) Suppression of hedgehog signalling promotes pro-tumourigenic integrin expression and function. J. Pathol., in press 10.1002/path.4342 [DOI] [PubMed] [Google Scholar]
- 29. Joost S., Almada L. L., Rohnalter V., Holz P. S., Vrabel A. M., Fernandez-Barrena M. G., McWilliams R. R., Krause M., Fernandez-Zapico M. E., Lauth M. (2012) GLI1 inhibition promotes epithelial-to-mesenchymal transition in pancreatic cancer cells. Cancer Res. 72, 88–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mantovani A., Allavena P., Sica A., Balkwill F. (2008) Cancer-related inflammation. Nature 454, 436–444 [DOI] [PubMed] [Google Scholar]
- 31. Mantovani A., Pierotti M. A. (2008) Cancer and inflammation: a complex relationship. Cancer Lett. 267, 180–181 [DOI] [PubMed] [Google Scholar]
- 32. Terzić J., Grivennikov S., Karin E., Karin M. (2010) Inflammation and colon cancer. Gastroenterology 138, 2101–2114.e5 [DOI] [PubMed] [Google Scholar]
- 33. Chen M. F., Lin P. Y., Wu C. F., Chen W. C., Wu C. T. (2013) IL-6 expression regulates tumorigenicity and correlates with prognosis in bladder cancer. PLoS One 8, e61901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carlsson A., Wingren C., Ingvarsson J., Ellmark P., Baldertorp B., Fernö M., Olsson H., Borrebaeck C. A. (2008) Serum proteome profiling of metastatic breast cancer using recombinant antibody microarrays. Eur. J. Cancer 44, 472–480 [DOI] [PubMed] [Google Scholar]
- 35. Orchekowski R., Hamelinck D., Li L., Gliwa E., vanBrocklin M., Marrero J. A., Vande Woude G. F., Feng Z., Brand R., Haab B. B. (2005) Antibody microarray profiling reveals individual and combined serum proteins associated with pancreatic cancer. Cancer Res. 65, 11193–11202 [DOI] [PubMed] [Google Scholar]
- 36. Huang F. T., Zhuan-Sun Y. X., Zhuang Y. Y., Wei S. L., Tang J., Chen W. B., Zhang S. N. (2012) Inhibition of hedgehog signaling depresses self-renewal of pancreatic cancer stem cells and reverses chemoresistance. Int. J. Oncol. 41, 1707–1714 [DOI] [PubMed] [Google Scholar]
- 37. Inoue K., Slaton J. W., Kim S. J., Perrotte P., Eve B. Y., Bar-Eli M., Radinsky R., Dinney C. P. (2000) Interleukin 8 expression regulates tumorigenicity and metastasis in human bladder cancer. Cancer Res. 60, 2290–2299 [PubMed] [Google Scholar]
- 38. Yadav A., Kumar B., Datta J., Teknos T. N., Kumar P. (2011) IL-6 promotes head and neck tumor metastasis by inducing epithelial-mesenchymal transition via the JAK-STAT3-SNAIL signaling pathway. Mol. Cancer Res. 9, 1658–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ravishankaran P., Karunanithi R. (2011) Clinical significance of preoperative serum interleukin-6 and C-reactive protein level in breast cancer patients. World J. Surg. Oncol. 9, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mohamed M. M., El-Ghonaimy E. A., Nouh M. A., Schneider R. J., Sloane B. F., El-Shinawi M. (2014) Cytokines secreted by macrophages isolated from tumor microenvironment of inflammatory breast cancer patients possess chemotactic properties. Int. J. Biochem. Cell Biol. 46, 138–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Strand S., Hofmann W. J., Hug H., Müller M., Otto G., Strand D., Mariani S. M., Stremmel W., Krammer P. H., Galle P. R. (1996) Lymphocyte apoptosis induced by CD95 (APO-1/Fas) ligand-expressing tumor cells–a mechanism of immune evasion? Nat. Med. 2, 1361–1366 [DOI] [PubMed] [Google Scholar]
- 42. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 43. Itoh N., Yonehara S., Ishii A., Yonehara M., Mizushima S., Sameshima M., Hase A., Seto Y., Nagata S. (1991) The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 66, 233–243 [DOI] [PubMed] [Google Scholar]
- 44. Dahmane N., Lee J., Robins P., Heller P., Ruiz i Altaba A. (1997) Activation of the transcription factor Gli1 and the Sonic hedgehog signalling pathway in skin tumours. Nature 389, 876–881 [DOI] [PubMed] [Google Scholar]
- 45. Dahmane N., Sánchez P., Gitton Y., Palma V., Sun T., Beyna M., Weiner H., Ruiz i Altaba A. (2001) The Sonic Hedgehog-Gli pathway regulates dorsal brain growth and tumorigenesis. Development 128, 5201–5212 [DOI] [PubMed] [Google Scholar]
- 46. Stecca B., Mas C., Clement V., Zbinden M., Correa R., Piguet V., Beermann F., Ruiz I Altaba A. (2007) Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. U.S.A. 104, 5895–5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Clement V., Sanchez P., de Tribolet N., Radovanovic I., Ruiz i Altaba A. (2007) HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Curr. Biol. 17, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li Y., Yang W., Yang Q., Zhou S. (2012) Nuclear localization of GLI1 and elevated expression of FOXC2 in breast cancer is associated with the basal-like phenotype. Histol. Histopathol. 27, 475–484 [DOI] [PubMed] [Google Scholar]
- 49. Ciucci A., De Stefano I., Vellone V. G., Lisi L., Bottoni C., Scambia G., Zannoni G. F., Gallo D. (2013) Expression of the glioma-associated oncogene homolog 1 (gli1) in advanced serous ovarian cancer is associated with unfavorable overall survival. PLoS One 8, e60145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kwon Y. J., Hurst D. R., Steg A. D., Yuan K., Vaidya K. S., Welch D. R., Frost A. R. (2011) Gli1 enhances migration and invasion via up-regulation of MMP-11 and promotes metastasis in ERα negative breast cancer cell lines. Clin. Exp. Metastasis 28, 437–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu J. T., Zhao W. D., He W., Wei W. (2012) Hedgehog signaling pathway mediates invasion and metastasis of hepatocellular carcinoma via ERK pathway. Acta Pharmacol. Sin. 33, 691–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cui D., Xu Q., Wang K., Che X. (2010) Gli1 is a potential target for alleviating multidrug resistance of gliomas. J. Neurol. Sci. 288, 156–166 [DOI] [PubMed] [Google Scholar]
- 53. Katoh Y., Katoh M. (2009) Integrative genomic analyses on GLI1: positive regulation of GLI1 by Hedgehog-GLI, TGFbeta-Smads, and RTK-PI3K-AKT signals, and negative regulation of GLI1 by Notch-CSL-HES/HEY, and GPCR-Gs-PKA signals. Int. J. Oncol. 35, 187–192 [DOI] [PubMed] [Google Scholar]
- 54. Sheng W., Dong M., Zhou J., Li X., Liu Q., Dong Q., Li F. (2014) The clinicopathological significance and relationship of Gli1, MDM2, and p53 expression in resectable pancreatic cancer. Histopathology 64, 523–535 [DOI] [PubMed] [Google Scholar]
- 55. Husain K., Centeno B. A., Chen D. T., Fulp W. J., Perez M., Zhang Lee G., Luetteke N., Hingorani S. R., Sebti S. M., Malafa M. P. (2013) Prolonged survival and delayed progression of pancreatic intraepithelial neoplasia in LSL-KrasG12D/+;Pdx-1-Cre mice by vitamin E δ-tocotrienol. Carcinogenesis 34, 858–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Feig C., Gopinathan A., Neesse A., Chan D. S., Cook N., Tuveson D. A. (2012) The pancreas cancer microenvironment. Clin. Cancer Res. 18, 4266–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Dijkgraaf G. J., Alicke B., Weinmann L., Januario T., West K., Modrusan Z., Burdick D., Goldsmith R., Robarge K., Sutherlin D., Scales S. J., Gould S. E., Yauch R. L., de Sauvage F. J. (2011) Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 71, 435–444 [DOI] [PubMed] [Google Scholar]
- 58. LoRusso P. M., Rudin C. M., Reddy J. C., Tibes R., Weiss G. J., Borad M. J., Hann C. L., Brahmer J. R., Chang I., Darbonne W. C., Graham R. A., Zerivitz K. L., Low J. A., Von Hoff D. D. (2011) Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced, or metastatic solid tumors. Clin. Cancer Res. 17, 2502–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rudin C. M., Hann C. L., Laterra J., Yauch R. L., Callahan C. A., Fu L., Holcomb T., Stinson J., Gould S. E., Coleman B., LoRusso P. M., Von Hoff D. D., de Sauvage F. J., Low J. A. (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N. Engl. J. Med. 361, 1173–1178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Griffith T. S., Brunner T., Fletcher S. M., Green D. R., Ferguson T. A. (1995) Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 270, 1189–1192 [DOI] [PubMed] [Google Scholar]
- 61. Bennett M. W., O'Connell J., Houston A., Kelly J., O'Sullivan G. C., Collins J. K., Shanahan F. (2001) Fas ligand upregulation is an early event in colonic carcinogenesis. J. Clin. Pathol. 54, 598–604 [DOI] [PMC free article] [PubMed] [Google Scholar]