

Background: Ras-GRF1 and Ras-GRF2 are similar exchange factors with different functions in synaptic plasticity.
Results: Chimeras between Ras-GRF proteins reveal that IQ, pleckstrin homology, coiled-coil, and CDC25 domains are most important for signaling specificity.
Conclusion: Signaling specificity of GRF proteins is encoded in a surprisingly small number of their common domains.
Significance: Domains of Ras-GRF proteins have been identified for future studies on signaling specificity.
Keywords: Calcium, Calmodulin (CaM), Mitogen-activated Protein Kinase (MAPK), Ras Protein, Synaptic Plasticity, IQ Motif, LTD, LTP, Ras-GRF1, Ras-GRF2
Abstract
Ras-GRF1 (GRF1) and Ras-GRF2 (GRF2) constitute a family of similar calcium sensors that regulate synaptic plasticity. They are both guanine exchange factors that contain a very similar set of functional domains, including N-terminal pleckstrin homology, coiled-coil, and calmodulin-binding IQ domains and C-terminal Dbl homology Rac-activating domains, Ras-exchange motifs, and CDC25 Ras-activating domains. Nevertheless, they regulate different forms of synaptic plasticity. Although both GRF proteins transduce calcium signals emanating from NMDA-type glutamate receptors in the CA1 region of the hippocampus, GRF1 promotes LTD, whereas GRF2 promotes θ-burst stimulation-induced LTP (TBS-LTP). GRF1 can also mediate high frequency stimulation-induced LTP (HFS-LTP) in mice over 2-months of age, which involves calcium-permeable AMPA-type glutamate receptors. To add to our understanding of how proteins with similar domains can have different functions, WT and various chimeras between GRF1 and GRF2 proteins were tested for their abilities to reconstitute defective LTP and/or LTD in the CA1 hippocampus of Grf1/Grf2 double knock-out mice. These studies revealed a critical role for the GRF2 CDC25 domain in the induction of TBS-LTP by GRF proteins. In contrast, the N-terminal pleckstrin homology and/or coiled-coil domains of GRF1 are key to the induction of HFS-LTP by GRF proteins. Finally, the IQ motif of GRF1 determines whether a GRF protein can induce LTD. Overall, these findings show that for the three forms of synaptic plasticity that are regulated by GRF proteins in the CA1 hippocampus, specificity is encoded in only one or two domains, and a different set of domains for each form of synaptic plasticity.
Introduction
p140 Ras-GRF1 (GRF1) and p130 Ras-GRF2 (GRF2) form a family of calcium/calmodulin (CaM)3-activated exchange factors for both Ras and Rac GTPases (1). The proteins are 71% identical in overall amino acid sequence and their overall structural organizations are quite similar. For example, both contain N-terminal pleckstrin homology (PH), and coiled-coil (CC) domains followed by IQ domains that bind CaM in a calcium-dependent manner. They both also contain Rac-activating Dbl (DH) domains followed by PH domains, C-terminal Ras exchange motifs (REM), and Ras-activating CDC25 domains.
Despite these similarities, GRF1 and GRF2 display distinctly different roles in synaptic plasticity in the CA1 hippocampus. Both proteins mediate NMDA-type glutamate receptor (NMDAR)-induced synaptic plasticity in the CA1 hippocampus but in opposite ways. GRF1 promotes low frequency stimulation-induced LTD (LFS-LTD), whereas GRF2 promotes TBS-LTP (2). GRF1 can promote HFS-LTP in the CA1 beginning at 2 months of age, which involves calcium-permeable AMPA receptor-type glutamate receptors (CP-AMPARs) (3). They also play different roles in hippocampus dependent learning and memory. Although both GRF1 and GRF2 contribute to contextual fear conditioning in 1-month-old mice (4, 5), GRF1 begins to contribute to contextual discrimination at 2 months of age due at least in part to its age-dependent ability to promote HFS-LTP (3).
The specificity underlying how other calcium sensors regulate opposing forms of synaptic plasticity in the hippocampus, such as the LTP-inducing CaM kinase II and LTD-inducing calcineurin, is more obvious because they encode opposing biochemical activities, kinase activity for the former and phosphatase activity for the latter. In contrast, both GRF1 and GRF2 activate the same Ras family GTPases. Although GRF1 and GRF2 have both Rac and Ras activating domains, they do not appear to activate them equally in response to NMDAR stimulation. GRF2 is more effective than GRF1 in mediating NMDAR activation of ERK MAP kinase (MAPK) in brain slices (6).
One possible mechanism by which GRF1 and GRF2 display different signaling specificity is through their association with different neurotransmitter signaling complexes. Multiple forms of NMDARs exist due to differing NR2 subunit isoforms. The dominant NMDARs in the hippocampus contain NR2A and/or NR2B subunits. Receptors containing either or both subunits can generate both LTP and LTD. Interestingly, GRF2 promotes LTP only in response to stimulation of NR2A-containing receptors (2, 3, 7), whereas GRF1 only promotes LTD in response to stimulation of NR2B-containing receptors (2, 8).
To gain more insight into how these two closely related proteins control different forms of synaptic plasticity a set of GRF1/GRF chimeras with different common domains swapped were tested for their ability to promote LTP or LTD and mediate the activation of different MAP kinases. The findings define a surprisingly small number of domains that control functional specificity between GRF1 and GRF2.
EXPERIMENTAL PROCEDURES
Mice
Grf1/Grf2 mice were described previously (9). These mice were maintained on a mixed C57BL/6Jx129 background. Wild-type mice of the same background were also used. All procedures involving mice were in accordance with the animal welfare guidelines of Tufts University.
Extracellular Field Experiments
Experiments were performed as previously described (3). Briefly, mice (1–3 month-old) were anesthetized with halothane and decapitated. Transverse acute hippocampal slices (350 μm) were cut in ice-cold oxygenated sucrose-enhanced artificial cerebrospinal fluid (ACSF) containing 206 mm sucrose, 2 mm KCl, 2 mm MgSO4, 1.25 mm NaH2PO4, 1 mm CaCl2, 1 mm MgCl2, 26 mm NaHCO3, 10 mm d-glucose, pH 7.4. After dissection, slices were incubated in ACSF that contained the following (in mm): 124 NaCl, 2 KCl, 2 MgSO4, 1.25 NaH2PO4, 2 CaCl2, 26 NaHCO3, 10 d-glucose saturated with 95% O2 and 5% CO2 (pH7.4), in which they were allowed to recover for at least 90 min before recording. Recordings were performed in the same solution at room temperature in a chamber submerged in ACSF.
To record field EPSPs (fEPSPs) in the CA1 region of the hippocampus, standard procedures were used. Test stimuli were applied at low frequency (0.05 Hz) at a stimulus intensity that elicited an fEPSP amplitude that was 50% of maximum (∼50–70 μA), and the test responses were recorded for 10 min before the experiment was begun to ensure stability of the response.
To induce LTP, we used two protocols: 1) high frequency stimulation (HFS): two consecutive trains (1 s) of stimuli at 100 Hz separated by 20 s; 2) θ-burst stimulation (TBS): 15 bursts of four pulses at 100 Hz delivered at an interburst interval of 200 ms. To induce LTD, we delivered 900 pulses at 1 Hz. Traces were obtained by pClamp 9.2 and analyzed using the Clampfit 9.2. Data analysis was as follows. The fEPSP magnitude was measured using the initial fEPSP slope and three consecutive slopes (1 min) were averaged and normalized to the mean value recorded 10 min before conditioning stimulus. Data are presented as mean ± S.E. Values expressed here represent 50-min time points after conditioning stimulus was initiated except the percentage of LTD was calculated by averaging the response 35–45 min after induction. The following statistical analysis was carried out: the same time window samples of the control and chimera treatment were compared using paired, two-tailed Student's t test. An effect was considered significant if p < 0.05.
Hippocampal Brain Slices Preparation for Immunofluorescence
The conditions used to measure MAP kinase activation by TBS stimulation were derived from Dudek and Fields (10). The brain was removed quickly and submerged in ice-cold oxygenated sucrose-replaced ACSF cutting solution (containing the following (in mm): 240 sucrose, 2.5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 25 NaHCO3, 7 MgCl2, pH 7.4). After dissection, slices (350 μm, transverse) were incubated in ACSF that contained the following (in mm): 124 NaCl, 2.8 KCl, 1.5 MgSO4, 1.25 NaH2PO4, 2.5 CaCl2, 26 NaHCO3, 10 d-glucose saturated with 95% O2 and 5% CO2, pH 7.4, in which slices were allowed to recover for at least 2 h before an experiment. Then a single slice was transferred to the recording chamber and submerged continuously by perfusing ACSF at room temperature for HFS-induced p-p38 MAPK. For TBS-induced p-ERK MAPK staining, ACSF at 34 °C was used. We found previously that L-type voltage-dependent calcium channels activate ERK MAPK independently of GRF proteins (data not shown). Thus, 20 μm nifedipine, which blocks these channels, was added to ACSF. Thus, NMDAR-mediated ERK MAPK activation, which we showed previously functions through GRF2, could be specifically activated (7). A unipolar stimulating electrode was placed in the stratum radiatum, and HFS or TBS stimulation was delivered with an intensity of 140 μA, as previously described (10). This intensity was approximately twice that used to generate maximal LTP, and as such generated minimal LTP because cells are already maximally stimulated. However, it was required to ensure that the signal to noise ratio for MAP kinase activation was large enough to quantitatively compare mutant to WT GRFs. Slices were fixed in 4% paraformaldehyde 5 min after TBS or 10 min after HFS stimulation overnight, then replaced with 15% sucrose for 24 h, and recut in a cryostat at 18 μm, and then the sections were blocked with 2% donkey serum in 0.3% Triton X-100 for 1 h at room temperature. The following antibodies were used: anti-p-ERK MAPK or p-p38 MAPK antibodies were obtained from Cell Signaling Technology (MA), and anti-GRF1 (C 18) was obtained from Santa Cruz (CA). Immunofluorescence was performed for double staining for p-ERK MAPK (anti-mouse, 1:400) with GRF1 (anti-rabbit, 1:200), and for single staining for phospho-p38 MAPK (anti-rabbit, 1:300) or GRF1 (anti-rabbit, 1:200) and incubated overnight at 4 °C. The sections were then incubated for 1.5 h at room temperature with a mixture of Alexa 488 and Cy3-conjugated secondary antibody (1:300; Invitrogen). The stained sections were examined with a Nikon (Tokyo, Japan) fluorescence microscope, and images were captured with a CCD spot camera, and subsequently analyzed with ImageJ software (National Institutes of Health). p-p38 MAPK cell counting was performed in the stratum pyramidal region of stimulated regions (up to 200–250 μm from the location of the stimulating electrode), the three most representative sections from each slice were chosen, and labeled cells in a ×20 optic field in each section were counted. For p-ERK MAPK, the average intensity was determined in 20 × 20 pixel areas of the stratum radiatum region. Image intensities from unstimulated regions >200–250 μm from the location of the stimulating electrode that express exogenous GRF proteins from virus infections were subtracted from image intensities from stimulated regions to generate stimulated p-ERK MAPK immunoreactivity values.
Chimera Production
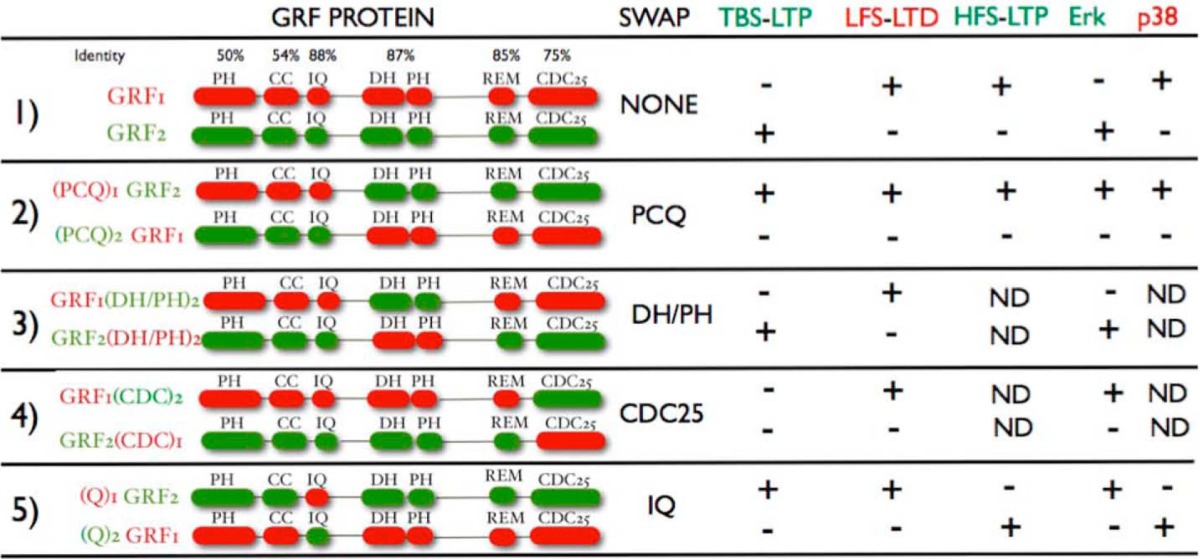
GRF1/GRF2 chimeras (see Table 1) were prepared using overlap PCR with primers that allowed chimera formation with the following amino acid constituents: (PCQ)1GRF2–aa-GRF1(1–235), GRF2(236–1187); (PCQ)2GRF1–aa-GRF2(1–238), GRF2(239–1248); GRF1(IQ)2–aa-GRF2:(205–228), GRF1(1–204)(229–1245); GRF2(IQ)1–aa-GRF1(208-231), GRF2(1–207)(232–1190); GRF1(DH/PH)2–aa-GRF2(236-629), GRF1(1–235)(630–1248); GRF2(DH/PH)1–aa-GRF1(239–629), GRF2(1–238)(630–1187); GRF1(CDC25)2–aa-GRF1(1–1004), GRF2(1005–1245); GRF2(CDC25)1–aa-GRF2(1–949), GRF1(950–1190). DNAs were all sequenced to confirm correct chimeras were made and constructs were expressed in HEK293 cells first to ensure that full-length proteins were generated.
TABLE 1.
Functional domains shared by GRF1 and GRF2 and biological activities of chimeras between them
TBS-LTP-θ-burst stimulation-induced LTP; LFS-LTD, low frequency stimulation-induced LTD; HFS-LTP-high frequency stimulation-induced LTP. (+) fully restored; (−) not fully restored. PH, pleckstrin homology; CC, coiled coil; DH, Dbl-homology; REM, Ras exchange motif.
Viral Vectors
Adenovirus expressing Ras-Grf1, Ras-Grf2, and chimeras between the two of them from the synapsin promoter were generated as described (3). Resultant viral samples were subjected to adenovirus purification using Adenopure kit (Puresyn Inc.). The virus was concentrated by filtration, yielding a concentration greater than 1 × 1011 virus particles/ml. All chimeras generated were expressed at similar levels in lysates of 293 cells.
Stereotactic Surgery
1–3-Month-old double Grf1/Grf2 knock-out or WT mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg), xylazine (10 mg/kg). Once anesthetized, each mouse was placed in a stereotactic frame (myNeuroLab, St. Louis, MO). A surgical incision was made along the midline of the head to expose the skull. Two holes were made in the skull overlying the hippocampus. Coordinates for CA1 injection into the mice were: 2.5-mm posterior to Bregma, ±2.5-mm lateral from the midline, and 1.75-mm below the surface of the skull. Injections were performed with a 10-μl Hamilton syringe fitted with a custom made blunt-ended 30-gauge needle (Hamilton). Each injection consisted of 1 μl of adenovirus expressing various GRF proteins infused at a rate of 0.06 μl/min. An infusion pump controlling the plunger on the Hamilton syringe precisely regulated the rate of injection. The needle was then left in place for 8 min prior to withdrawal from the brain. Electrophysiological recordings began 7–11 days after stereotactic injection of the viral vectors, a date chosen because exogenous GRF proteins accumulate to levels normally found for endogenous proteins in WT animals. After expression, co-staining of GRF with either the neuronal marker, NeuN, or glial marker, GFAP, showed that GRF proteins were found overwhelmingly in neurons, not glial cells, consistent with the neuron-specific synapsin promoter used in these experiments and the known localization of endogenous GRF proteins (11) (data not shown).
Synthetic Peptides
IQ domains of GRF1 (KIKKVQSFLRGWLCRRKWKNIIQ) and GRF2 (KIKKVQSFMRGWLCRRKWKTIVQ) were synthesized by LifeTein LLC. The purity and composition of the peptides was confirmed by mass spectrometry at the Clinical and Translational proteomics core at the Institute of Molecular Medicine, University of Texas at Houston.
Isothermal Titration Calorimetry (ITC)
ITC measurements were performed in a Microcal VP-ITC instrument in a buffer consisting of 50 mm MOPS, 100 mm KCl, pH 7.2, in the presence of either 1 mm EDTA or 2 mm CaCl2, as described previously (12). Samples were degassed and brought to 25 °C before starting titrations. All titrations were carried out by injecting 5 μl of 150 μm peptide into the cell containing 1.8 ml of 5 μm CaM at 25 °C with constant stirring. Each experiment consisted of 28 injections with a 20-s injection duration and 210-s spacing between injections. The raw data were baseline corrected and integrated peak areas were calculated using Microcal software then plotted as a function of mole ratio. Data were fit with single binding site models to determine the binding stoichiometry (N), association constant (K), enthalpy (ΔH), and entropy (ΔS).
Steady-state Fluorescence
CaM with a single Cys mutation at amino acid 75 was expressed, purified, and labeled with acrylodan (CaM-ACR) as previously described (13). Steady-state fluorescence was accomplished with 150 nm CaM-ACR in a 1-ml cuvette using a PTI Quantamaster fluorimeter. Slit widths were maintained at 1 nm excitation and 5 nm emission with excitation wavelength was set at 375 nm and emission scans were taken from 380 to 580 nm. The base buffer in these experiments was 50 mm MOPS, 100 mm KCl, pH 7.2. Depending on the experiment, GRF1 or GRF2 peptides (1 mm final), chelators, or Ca2+ was added to the cuvette from concentrated stocks to control the reaction conditions as described in the figure legends.
RESULTS
Assay of GRF1 and GRF2 Activities in Vivo
GRF1 and GRF2 functions are remarkably age-dependent, such that they do not contribute significantly to synaptic plasticity in the CA1 region of the hippocampus until mice are ∼3 weeks of age, when the hippocampus begins to contribute to learning and memory (2). At this point GRF1 and GRF2 begin to regulate LFS-LTD and TBS-LTP mediated by NMDARs, respectively. Moreover, GRF1 only begins to contribute to HFS-LTP mediated by CP-AMPARs at 2 months of age when mice reach adulthood (3). This precludes the use of cultured hippocampal neurons to address the role of GRF proteins in synaptic plasticity. Instead we developed an in vivo assay where GRF1, GRF2, and any mutant forms of them are tested for their ability to reconstitute TBS-LTP, LFS-LTD, or HFS-LTP in double Grf1/Grf2 knock-out (DBKO) mice that display none of these forms of synaptic plasticity.
In particular, various GRF proteins are expressed in adenoviral vectors under control of the neuron-specific synapsin promoter. High titer viruses are prepared and stereotactic surgery is used to inject virus specifically into the CA1 region of the hippocampus. Eight days later, when GRFs accumulate to levels similar to those found in WT mice, animals are sacrificed and LTP and/or LTD assays are performed on isolated hippocampal brain slices. Because of the high titer of viruses used (>1011 pfu), most of the principal neurons of the CA1 express GRF proteins. Thus extracellular field recordings, which represent the summed responses from a number of neurons in the vicinity of the recording electrode, can be used to assess synaptic plasticity in regions of the hippocampus where viruses are injected.
Ras-activating CDC25 Domain Is Key for GRF Signaling Specificity Necessary to Induce TBS-LTP Mediated by NMDARs
The first set of chimeras, (PCQ)1GRF2 and (PCQ)2GRF1, where the PH, CC, and IQ domains were swapped (Table 1, see row 2), were tested for their ability to reconstitute TBS-LTP after their re-expression in the CA1 of DBKO mice. Fig. 1A shows that (PCQ)1GRF2 restores LTP in brain slices from DBKO mice to a degree similar to that found in DBKO mice that re-express GRF2 (Fig. 1B) ((PCQ)1GRF2, 150.58 ± 2.26% (n = 8 slices from 5 mice) versus GRF2, 151.75 ± 4.73% (n = 8 slices from 5 mice); p > 0.05). The level of LTP is also similar to that generated by endogenous GRF2 in WT mice (Fig. 1C) (156 ± 6.65% (n = 8 slices from 5 mice). This suggested that TBS-LTP specificity is encoded in the C-terminal DH/PH, REM, and/or CDC25 catalytic domains of GRF2, and that it does not matter which PH, coiled-coil or even calmodulin-binding IQ domains are present in the protein.
FIGURE 1.

The CDC25 domain of GRF2 is critical for GRF proteins to induce TBS-LTP. TBS-LTP was generated in hippocampal brain slices from WT mice or Grf1/Grf2 knock-out (DBKO) mice re-expressing: A, PCQ chimeras, (PCQ)1GRF2 or (PCQ)2GRF1; B, WT GRF2 and control DBKO mice; C, DH/PH chimeras GRF2(DH/PH)1 and GRF1(DH/PH)2; D, CDC25 chimeras GRF2(CDC25)1 and GRF1(CDC25)2. Panels A and B are fEPSP slopes representative of results from at least 6 slices from 3 mice each. Panels C and D quantify average fEPSP slopes for WT mice expressing endogenous GRF2 and various GRF proteins and chimeras. Experiments are from at least 6 slices from 5 mice and data show mean ± S.E. E, functionally inactive GRF1/GRF2 chimeras expressed at similar levels as endogenous WT GRF2 in the CA1. Scale bar, 50 μm. Bar graph represents the average ± S.E. of three tissue sections. n.s., not statistically different.
This conclusion is supported by the results of the opposite chimera, (PCQ)2GRF1, which could not reconstitute LTP in this system (Fig. 1, A and B) ((PCQ)2GRF1, 119.53 ± 3.15% (n = 7 slices from 4 mice) versus GRF2, 151.75 ± 4.73% (n = 8 slices from 5 mice); p < 0.01) despite the fact that it was expressed at levels comparable with endogenous GRF2 in WT mice (Fig. 1E).
To better define which domain in the C terminus encodes TBS-LTP signaling specificity another chimera was made that swapped the Rac-activating DH/PH domains of GRF1 and GRF2 (Table 1, see row 3). Fig. 1C shows that GRF2(DH/PH)1 maintains the ability to promote LTP to a level comparable with re-expressed WT GRF2 even though it contains the DH/PH domain of GRF1 (GRF2(DH/PH)1, 141.74 ± 5.49% (n = 7 slices from 4 mice) versus WT GRF2, 151.75 ± 4.73% (n = 8 slices from 5 mice); p > 0.05). This indicates that specificity for TBS-LTP does not reside in the GRF2 version of the DH/PH domain. Consistent with this conclusion are the results of the opposite chimera GRF1(DH/PH)2, which was not capable of reconstituting LTP any better than that present in uninfected DBKO mice (GRF1(DH/PH)2, 110.42 ± 4.26% (n = 6 slices from 4 mice) versus DBKO, 111.63 ± 2.23% (n = 6 slices from 3 mice); p > 0.05).
These findings pointed to the Ras-activating, catalytic CDC25 domain as most important. To test this hypothesis, another set of chimeras, GRF2(CDC25)1 and GRF1(CDC25)2, were generated (Table 1, see row 4). In contrast to GRF2, re-expressed GRF2(CDC25)1 failed to reconstitute LTP above levels found in mice reconstituted with GRF1 or in control double knock-out mice (Fig. 1D) (GRF2(CDC25)1, 110.22 ± 2.85% (n = 6 slices from 4 mice) versus GRF1, 116.13 ± 4.09% (n = 6 slices from 4 mice); p > 0.05) even though it was expressed at similar levels (Fig. 1E), arguing that specific amino acids of the GRF2 CDC25 are necessary for GRFs to promote NMDAR-mediated LTP. GRF1(CDC25)2 may have some reconstituting activity but it was not statistically different from control DBKO mice (GRF1(CDC25)2, 126.03 ± 5.7% (n = 6 slices from 4 mice) versus DBKO, 111.63 ± 2.23% (n = 6 slices from 3 mice); p > 0.05). Thus, the Ras-activating domain of GRF2 is necessary, but not sufficient for GRF proteins to promote TBS-mediated LTP.
ERK MAPK activity has also been shown to be necessary, but not sufficient, for TBS to induce LTP in the CA1 hippocampus (10, 14). Moreover, ERK MAPK is a known downstream target of GRF2 through its CDC25 domain via activation of Ras proteins. In addition, we showed previously that the contribution of GRF2 to NMDAR-mediated TBS-LTP is via an ERK MAPK-dependent pathway (7), and that GRF2 is more effective than GRF1 in mediating NMDAR-induced ERK MAPK activation (6). Thus, we attempted to test whether the key role of the GRF2 CDC25 domain revealed in LTP studies comparing GRF1(CDC25)2 and GRF2(CDC25)1 (see Fig. 1) is its ability to couple TBS stimulation to ERK MAPK more effectively than the GRF1 CDC25 domain.
The role of ERK MAPK in mediating TBS-LTP in the CA1 is based mainly on the fact that inhibitors of the ERK MAPK pathway block TBS-LTP (14–16). In related studies demonstrating that TBS increases ERK MAPK activity, the effects were quite small (∼20% increase) (16), or TBS was used at an intensity greater than that used to promote LTP to detect a clear signal (10).
When we stimulated hippocampal brain slices with standard TBS conditions that induce LTP, we also detected only a small increase in p-ERK MAPK in a minority of stratum rediatum (dendrites) or stratum pyramidale (somata) that were too few to permit statistically reliable comparisons between chimeras (data not shown). Thus, we used the previously described TBS protocol (10) (2 times the intensity used for maximal LTP induction, even though it maximally stimulated cells so that LTP could no longer be measured).
Brain slices from DBKO mice re-expressing GRF2, GRF2(CDC25)1, or GRF1(CDC25)2 were stained with activation specific p-ERK MAPK antibodies 5 min after this enhanced TBS. Fig. 2 shows that despite the high stimulation intensity used in these experiments, brain slices from DBKO mice failed to generate detectable activation of ERK MAPK. This indicates that all of the ERK MAPK-promoting activity induced by this protocol was generated by GRF proteins, and not unrelated proteins. Moreover, re-expression of GRF2 restored TBS-induced ERK MAPK activation in samples from DBKO mice to a level similar to that found in samples from WT mice, where signaling occurs through endogenous GRF2. In contrast, GRF2(CDC25)1 did not reconstitute ERK MAPK activation, consistent with its inability to restore TBS-LTP. Thus, the GRF2, but not GRF1, CDC25 domain is capable of transmitting this TBS signal to ERK MAPK.
FIGURE 2.

Activation of ERK MAPK is associated with the presence of the CDC25 domain of GRF2 and is necessary but not sufficient to promote TBS-LTP by GRF proteins. A, hippocampal brain slices from WT mice (WT), DBKO mice expressing GRF2, or the chimeric GRF proteins indicated and DBKO mice (DBKO), were exposed to TBS (140 μA) and then 5 min later samples were frozen and immunostained with anti-p-ERK MAPK antibodies. Representative p-ERK MAPK signals from slices are shown (top) (*, shows approximate stimulating electrode position; ▾, shows background area chosen at least 200 μm from electrode where GRF proteins are expressed, but stimulated cells are not observed). B, representative images of stimulated brain slices stained with GRF antibodies. C, quantification of fluorescence intensity in A and B. The left bar graph represents the signals obtained from samples described in A. The signal represents that from the stimulated area minus the signal from the neighboring unstimulated area both of which express exogenous GRF proteins. This value was then compared with that obtained with brain slices from WT mice expressing endogenous GRF2. The final results are expressed as fold-change compared with WT. The right bar graph represents the signals obtained from the GRF immunostaining. All data are the average ± S.E. of at least 3 independent experiments; **, p ≤ .01; n.s., not statistically different. Scale bar, 50 μm.
Interestingly, GRF1(CDC25)2 was also capable of restoring TBS-induced ERK MAPK activation in brain slices from DBKO mice (Fig. 2), even though it could not restore TBS-LTP (Fig. 1). This finding is consistent with previous studies showing that elevated ERK MAPK activity is necessary, but not sufficient, to induce TBS-LTP (10). Even taking into account that the HFS protocol used to stimulate ERK MAPK used a higher intensity than that used to promote LTP, the findings obtained with this set of chimeras support the idea that the GRF2 CDC25 domain encodes specificity for TBS-LTP induction by GRF proteins because of its ability to mediate activation of ERK MAPK more effectively than the GRF1 CDC25 domain.
These findings also indirectly show that the REM domains of GRFs (Table 1, see row 1), which influences CDC25 domain function in the SOS Ras exchange factor (17), do not contribute to signaling specificity associated with TBS-LTP. This is because GRF1(CDC25)2, which has the REM domain of GRF1, is just as potent in promoting ERK MAPK signaling as GRF2.
N-terminal PH and/or Coiled-coil Domains Are Key for GRF Signaling Specificity Necessary to Mediate HFS-LTP Induced by CP-AMPARs
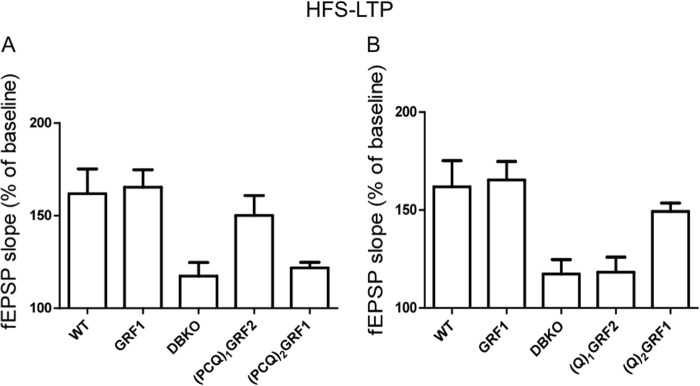
We showed previously that GRF1, but not GRF2, can promote HFS-LTP that engages CP-AMPARs in the CA1 hippocampus of mice at least 2 months of age (3). Thus, GRF1/GRF2 chimeras were tested using a high frequency stimulation LTP paradigm. First, we tested the PCQ motif swap mutants and found that (PCQ)1GRF2 was able to promote this form of LTP almost as well as GRF1 did, even though the entire C terminus was from inactive GRF2 (Fig. 3A) ((PCQ)1GRF2, 150.23 ± 10.70% (n = 8 slices from 5 mice) versus WT GRF1, 165.41 ± 9.38% (n = 8 slices from 6 mice) p > 0.05; or endogenous GRF1 in WT mice, 161.76 ± 4.73% (n = 10 slices from 6 mice); p > 0.05). This implied that the N terminus of GRFs encodes specificity for HFS-LTP induction. This conclusion was supported by results using the opposite chimera, (PCQ)2GRF1, which supported HFS-LTP no better than control DBKO mice (Fig. 3A) ((PCQ)2GRF1, 121.82 ± 2.96% (n = 9 slices from 5 mice) versus DBKO, 117.42 ± 7.37% (n = 7 slices from 5 mice). Interestingly, this finding is the opposite of that for TBS-LTP described above that uses NMDARs, but not CP-AMPARs, where the C termini of GRFs encode signaling specificity.
FIGURE 3.
The PH and/or coiled-coil domains of GRF1 determine whether GRF proteins induce HFS-LTP. A and B, HFS-LTP was generated in hippocampal brain slices from WT mice and DBKO mice re-expressing indicated GRF proteins or various GRF1/GRF2 chimeras. Data show quantified average fEPSP slopes from at least 6 slices from 4 mice and data show mean ± S.E.
To better define which domains within the PCQ motif are most important for GRF specificity in mediating HFS-LTP, (Q)1GRF2 and(Q)2GRF1, a chimera pair with IQ domains swapped was assayed (Table 1, see row 5). Interestingly, unlike (PCQ)1GRF2, (Q)1GRF2 did not restore HFS-LTP in the CA1 hippocampus, showing that the IQ domain of GRF1 was not sufficient to induce HFS-LTP and that the PH and/or coiled-coil domains of GRF1 are important (Fig. 3B) ((Q)1GRF2, 118.33 ± 7.64% (n = 6 slices from 4 mice) versus WT GRF1, 165.41 ± 9.38% (n = 8 slices from 6 mice); p < 0.01). The protein was functional because it restored LFS-LTD (see Fig. 5 below). In contrast, the opposite chimera (Q)2GRF1 did reconstitute HFS-LTP to levels almost as well as exogenous or endogenous WT GRF1 in this system, demonstrating that a specific form of the IQ domain is not required to support HFS-LTP (Fig. 3B) ((Q)2GRF1, 149.33 ± 4.24% (n = 6 slices from 4 mice) versus WT GRF1, 165.41 ± 9.38% (n = 8 slices from 6 mice); or endogenous GRF1 in WT mice, 161.76 ± 4.73% (n = 10 slices from 6 mice), p > 0.05). Together these finding indicate that the PH, and/or coiled-coil domains, but not the IQ motif, of GRF1 are mainly responsible for encoding specificity that allows GRFs to promote HFS-LTP that is mediated by CP-AMPARs.
FIGURE 5.

The IQ domain of GRF1 is necessary and sufficient for GRF proteins to induce LFS-LTD. LFS-LTD was generated in hippocampal brain slices from DBKO mice re-expressing various GRF proteins and chimeras. Panels A and B show fEPSP slopes representative of results from at least 6 slices from 3 mice each. Panels C and D show quantified average fEPSP slopes from at least 5 slices from 3 mice and data show mean ± S.E. *, p ≤ 0.05; ***, p ≤ 0.001; n.s., not statistically different; inset, a comparison of the IQ motif sequences between GRF1 and GRF2 with differences marked.
We showed previously that CP-AMPARs promote HFS-LTP through the GRF1 effector, p38 MAPK. Thus, we next tested whether the ability to link HFS to p38 MAPK activation was correlated with an ability of the chimera to mediate HFS-LTP. Brain slices from DBKO mice expressing these chimeras were stained with activation-specific p-p38 MAPK antibodies 10 min after HFS stimulation (again with an intensity that is ∼2 times greater than that used to generate LTP to yield a p-p38 MAPK signal that was strong enough above background to compare GRF proteins). Fig. 4 shows that no signal is obtained in brain slices from DBKO mice, indicating that even under these high intensity conditions, all p38 MAPK activation generated occurs through GRF proteins. Moreover, as expected, GRF1 expression restored HFS-induced p38 MAPK activation to levels comparable with seen in WT mice that occurs through endogenous GRF1. (Q)2GRF1, which restored HFS-LTP, also restored p38 MAPK activation to levels comparable with re-expressed GRF1. In contrast, (Q)1GRF2, which did not restore HFS-LTP, also did not restore p38 MAPK activation. Thus, even though the stimulation protocol for p38 MAPK and LTP formation were not the same, these findings support the idea that the PH and/or coiled-coils of GRF1 encode specificity for HFS-LTP by virtue of their ability to promote p38 MAPK activation better than the comparable domains in GRF2.
FIGURE 4.

Activation of p38 MAPK mediated by the PH and/or coiled-coil domains of GRF1 is necessary and sufficient to induce HFS-LTP by GRF proteins. Hippocampal brain slices from WT mice or DBKO mice expressing the indicated WT and chimeric GRF proteins were exposed to HFS stimulation (140 μA) and then 10 min later samples were frozen and immunostained with anti-p-p38 MAPK antibodies. Representative signals from slices are shown (left) and then quantified for phospho-p38 MAPK-stained cells (right). Data represent the mean ± S.E. for at least three independent experiments; *, p ≤ 0.05; **, p ≤ 0.01; n.s., not statistically different; scale bar, 50 μm.
CaM Binding IQ Domain Is Key for GRF Signaling Specificity Necessary to Mediate LFS-LTD Induced by NMDARs
We showed previously that GRF1, but not GRF2, mediates LFS-LTD in the CA1 (2). Thus, we began studying GRF domain contribution to specificity for this form of LTD induction by comparing the biological activities of (PCQ)1GRF2 and (PCQ)2GRF1 chimeras. (PCQ)1GRF2 was able to restore LTD in the CA1 of DBKO mice to levels comparable with re-expressed WT GRF1 (Fig. 5, A and B) ((PCQ)1GRF2, 47.20 ± 8.57% (n = 6 slices from 4 mice); WT GRF1, 50.90 ± 6.19% (n = 5 slices from 4 mice); p > 0.05) indicating that the N-terminal PH, coiled-coil and/or IQ motifs encode specificity for LTD. It did not matter which C-terminal DH/PH or CDC25 domains were present. This conclusion was reinforced by experiments with (PCQ)2GRF1 (Fig. 5A) ((PCQ)2GRF1, 86.22 ± 4.63% (n = 6 slices from 4 mice) versus WT GRF1, 50.90 ± 6.19% (n = 5 slices from 4 mice); p < 0.05), which failed to reconstitute LTD.
To begin to better define which of the three N-terminal domains was most important for LTD, (Q)1GRF2 and (Q)2GRF1, which have only their IQ motifs switched, were compared. Remarkably, (Q)1GRF2 was almost as effective as GRF1 (Fig. 5D) ((Q)1GRF2, 65.07 ± 5.10% (n = 6 slices from 4 mice) versus WT GRF1, 50.90 ± 6.19% (n = 5 slices from 4 mice); p > 0.05) and (PCQ)1GRF2 (Fig. 5C) in the ability to reconstitute LTD. This is despite the fact that 22 of the 25 amino acids in the IQ domains of GRF1 and GRF2 are identical. In other words, only 3 amino acid changes in its IQ motif can endow GRF2 with the ability to promote LTD (Fig. 5D). Moreover, the opposite chimera, (Q)2GRF1, failed to significantly promote LTD above baseline levels in GRF2 reconstituted mice and control DBKO mice (Fig. 5D) ((Q)2GRF1, 83.65 ± 6.04% (n = 6 slices from 4 mice) versus WT GRF2, 93.40 ± 2.29% (n = 5 slices from 4 mice); p > 0.05; and versus DBKO, 91.15 ± 2.55% (n = 5 slices from 3 mice); p > 0.05). Interestingly, this protein still retained the ability to promote HFS-LTP (see Fig. 3B). Together these findings support the idea that the IQ motif of GRF1 is both necessary and sufficient to encode specificity in GRF proteins to mediate NMDAR induction of LTD. These findings imply that the IQ motif of GRF1 can encode specificity for both responding to the correct upstream calcium signal that is destined to promote LTD and also engaging the proper downstream signaling cascade necessary for LTD.
To assess the possibility that the IQ domains of GRF1 and GRF2 provide distinct CaM-binding properties that could account for the appropriate response to upstream calcium signals, we quantified the affinities of peptides containing the two IQ motifs for calcium/CaM. Synthetic peptides representing the amino acids switched in the GRF chimeras (GRF1p and GRF2p) were synthesized and analyzed for their binding to CaM. Using steady-state fluorescence, GRF1p and GRF2p were demonstrated to interact with CaM labeled with the fluorescent dye acrylodan (CaM-ACR) (Fig. 6). CaM-ACR increases in fluorescence when bound to Ca2+ and addition of GRF1p or GRF2p produced a further increase in fluorescence intensity and induced a blue shift in the emission spectra indicative of binding. Interactions with apo-CaM were assessed directly by reversing the order of addition; GRF1p or GRF2p were added to CaM-ACR in the presence of EDTA and shifts in the spectrum were again detected indicative of binding. These results showed qualitatively that the IQ domains of GRF1 and GRF2 bind to both apo-CaM and Ca2+/CaM. To determine whether the peptides were binding in a 1:1 complex and to quantify the differences in the binding affinity, ITC was employed (Fig. 7). From these data, we can conclude that both peptides bind to apo-CaM at a 1:1 ratio and that the affinity of the IQ domain from GRF1 is significantly stronger than the binding of that from GRF2 (∼396 and 665 nm, respectively). These affinities are comparable with those of the IQ motif of neurogranin (18), which is thought to bind to apo-CaM and act as a CaM buffer in the synapse, implying that GRF1 and GRF2 are also pre-bound to apo-CaM in neurons.
FIGURE 6.

Fluorescence measurement of GRF peptide binding to CaM. Steady-state fluorescence was accomplished in a PTI fluorimeter as described under “Experimental Procedures.” Excitation was at 375 nm and emission scans were collected from 380 to 580, respectively. For panels A and B, each experiment began by adding CaM-ACR to 150 nm final concentration (black squares) to buffer containing 100 μm EGTA, scans were taken, and then Ca2+ was added to 400 μm final concentration (black circles). Then either the GRF1 (GRF1p; panel A) or GRF2 (GRF2p; panel B) peptide was added and traces were again collected (black triangles). Finally, EDTA to a final concentration of 10 mm was added (decreasing free Ca2+ to <5 nm) and traces were taken. Note the blue shift in the emission spectra both when Ca2+ is added and again with peptide along with increased fluorescence intensities. The addition of EDTA largely abrogated the fluorescence intensity but there was a persistent blue shift in the emission spectra indicating the peptides were interacting with apo-CaM. To more directly investigate this possibility, the impact of peptide binding to apo-CaM was addressed. Fluorescence scans of 150 nm CaM-ACR (black squares) in the presence of 1 mm EDTA were followed by the addition of 1 μm GRF1p (panel C) or 1 μm GRF2p (panel D) and scans again taken (black circles). Note the blue shift in the spectra indicating binding of the peptides to apo-CaM, but only a modest intensity increase was evident. Ca2+ was then added to 1.6 mm (to give 400 μm final; black triangles) and scans were again taken. Notice that there is no further blue shift in the spectra but a significant increase in intensity is evident with Ca2+ (black triangles). Following addition of EDTA, to 10 mm final concentration (inverted black triangles), the fluorescence intensity returned to that slightly below those before the addition of Ca2+ (black circles) but note that there remains a blue shift in the spectra consistent with binding to apo-CaM.
FIGURE 7.

The IQ domain of GRF1 binds to apo-calmodulin with higher affinity than the IQ domain of GRF2. ITC was accomplished on a VP-ITC unit at 25 °C as described under “Experimental Procedures.” Reactions were accomplished in 50 mm MOPS, 100 mm KCl with either 2 mm Ca2+ (panels A and B) or 1 mm EDTA (panels C and D). The reaction cell contained 5 μm CaM and the injection pipette was filled with GRF1 peptide (GRF1p; panels A and C) or GRF2 peptide (GRF2p; panels B and D). For Ca2+/CaM reactions (panels A and B) the peptide concentration was 75 μm in the pipette and for the apo-CaM conditions (panels C and D) the peptide was at 150 μm. The recorded heat signatures (all were exothermic) were normalized to the mole of injectant and the resulting data were fit with a single site-binding model using Microcal software. E, table summarizing the values from fits to the data. Kd was calculated as 1/Ka.
Extending these studies, we determined that the affinity of both peptides for Ca2+/CaM was much higher than for apo-CaM, consistent with previous findings that GRF proteins preferentially bind CaM in a calcium-dependent manner (19). Quantitatively, the affinity of the IQ domain of GRF2 for Ca2+/CaM was slightly greater than that for GRF1 (∼15 and 29 nm, respectively). However, with binding this tight, the differences between the absolute values from ITC should be interpreted with some caution. Moreover, the affinity differences of Ca2+/CaM for CaMKII, a mediator of LTP, and calcineurin, a mediator of LTD, are much larger. Thus, the Ca2+/CaM affinity differences reported here between GRF1 and GRF2 are not likely to be physiologically significant.
Overall, these findings imply that intrinsic differences in affinities of the IQ motifs of GRF1 and GRF2 are most significant in binding to apo-CaM. This would lead to greater probability of CaM occupancy of the IQ domain of GRF1 relative to GRF2 in the basal state and could account for the ability of GRF1, but not GRF2, to mediate the low levels of calcium influx associated with LFS-LTD.
DISCUSSION
This study explored how two closely related GRF family members could influence different types of synaptic plasticity. It reveals which of the six functional domains shared by GRF1 and GRF2 are most important for their ability to mediate different forms of synaptic plasticity induced by either NMDA or CP-AMPA glutamate receptors. Overall, these findings show that for the three types of synaptic plasticity that are regulated by GRF proteins in the CA1 hippocampus, TBS-LTP by GRF2, HFS-LTP by GRF1, and LFS-LTD by GRF1, specificity is encoded in only one or two of their common domains, and a different set of domains for each form of synaptic plasticity. In particular, the GRF2 CDC25 domain is required for a GRF protein to induce TBS-LTP. The GRF1 IQ domain is required for a GRF protein to induce LFS-LTD, and GRF1 PH/CC domains are required for a GRF protein to induce HFS-LTP.
These findings are surprising for multiple reasons. First, one could have reasonably predicted that because none of the domains shared by GRF1 and GRF2 are identical they all would contribute to signaling specificity that distinguishes GRF1 from GRF2, and that all chimeras generated would be defective for all three forms of synaptic plasticity studied. Almost the opposite is true in that only one or at most two domains were found to encode specificity for a particular type of synaptic plasticity. In fact, in one case, LFS-LTD, the IQ domain of GRF1 inserted into the GRF2 sequence was sufficient to endow the protein with the ability to promote LTD. Another surprise was that it was not possible to predict which domain swaps would be most potent in changing signaling specificity based on the divergence in sequence identity. For example, (IQ)1 and (IQ)2 are the most homologous of all the domain pairs studied, and yet swapping the former into GRF2, which involved only 3 amino acid changes, has the ability to convert GRF2 into an LTD-inducing protein. In contrast, the PH/CC domains are the least conserved between GRF1 and GRF2, and yet swapping them has no effect on TBS-LTP or LFS-LTD. However, they do play a role in another form of synaptic plasticity, because the PH/CC domains of GRF2 could not substitute for their counterparts in GRF1 for the induction of HFS-LTP.
For both forms of LTP, we have connected a specific downstream function as a mediator of domain-regulated signaling specificity. For TBS-LTP in the CA1 mediated by GRF2, ERK MAPK activation is known to be necessary, but not sufficient (10, 14). Our data imply that the GRF2 CDC25 domain contributes to signaling specificity required for TBS-LTP induction because its ability to promote ERK MAPK activation is better than that of the same domain in GRF1. First, the presence of the GRF2 CDC25 domain in a chimera and the ability of the chimera to promote stimulus-induced activation of ERK MAPK are both necessary for TBS-LTP induction. Second, whereas necessary, the presence of both the GRF2 CDC25 domain and an ability of the chimera to promote ERK MAPK activation are not sufficient to induce TBS-LTP. However, an important caveat is that to generate a strong enough ERK MAPK signal to compare wild-type and mutant GRF proteins, we had to increase the intensity of the TBS to double the levels that are used to generate LTP. Thus, it is possible that other ERK MAPK-activating signaling pathways not involved in promoting LTP are engaged under these conditions. However, it is unlikely because such signals must have emerged from the GRF2 CDC25 domain, as no ERK MAPK signal is detectable when this stimulation paradigm is used with DBKO mice unless a GRF protein containing that domain is reintroduced into them.
The CDC25 domain is the catalytic domain that directly activates Ras proteins, which are known to stimulate the Raf kinase/ERK MAPK signaling cascade. Thus, it was not surprising for us to discover that specificity of GRF signaling to promote ERK MAPK-dependent TBS-LTP could be accounted for our finding that the GRF2 CDC25 domain is more efficient in promoting ERK MAPK activation by TBS than the GRF1 CDC25 domain. Nevertheless, an alternative explanation for specificity was possible, based on the fact that GRF1, but not GRF2, is constitutively associated with NMDARs containing NR2B subunits through an NR2B-binding site on GRF1 (8). This subset of NMDARs is also associated with Syn-GAP, a negative regulator of active Ras (20). Thus, the GRF1 CDC25 domain could be just as potent as the GRF2 CDC25 in activating Ras and ERK MAPK, if not for the negative effects of being associated with NR2B-containing NMDARs. However, this potential mechanism, based on differential localization of GRF1 and GRF2 in the synapse does not appear to be most important because the chimera, GRF2(CDC25)1, cannot mediate TBS-induced ERK MAPK activation and TBS-LTP induction even though it does not contain the NR2B-binding site from GRF1. Also, GRF1(CDC25)2 can still restore ERK MAPK activation by TBS in DBKO mice even though it contains the NR2B binding site of GRF1. Finally, the observation that GRF1(CDC25)2 restores TBS-induced ERK MAPK activation, but not LTP, argues that GRF2 contributes another unidentified function for LTP induction encoded presumably by a region(s) of the protein not investigated here.
For HFS-LTP mediated by GRF1 in mice at least 2 months of age, p38 MAPK activation is known to be required (3). Our data imply that the GRF1 PH/CC domains contribute to signaling specificity of GRF proteins needed for HFS-LTP by regulating the activity of this kinase. This is because the presence of these domains in a chimera and the ability of the chimera to activate p38 MAP kinase were both necessary and sufficient to promote HFS-LTP. Again, the caveat is the same as in the ERK MAPK experiments. A p38 MAP kinase signal unrelated to HFS-LTP could have been introduced, because we needed to use intensities of HFS that were twice that used to generate maximal LTP to generate a strong enough signal to compare chimeras. However, this also appears unlikely, as this HFS paradigm did not activate p38 MAP kinase in mice lacking GRF proteins and it was only detectable when a GRF protein with the PH and coiled-coil of GRF1 was re-introduced into them.
Unlike the case of the CDC25 domains and ERK MAPK activation, it is not obvious how the PH and/or coiled-coil domains from GRF1 can be more effective than those from GRF2 in promoting p38 MAP kinase activation required for HFS-LTP, because the domain of GRF proteins that is known to activate p38 MAP kinase is the Rac GTPase-activating DH domain.
One possible explanation is suggested by previous studies on these domains in both GRF1 and a related Rac exchange factor Tiam1, which has a similar PH/CC motif at its N terminus. The GRF1 study showed that these domains influence the activation process of GRF1 without affecting CaM binding (21). The Tiam1 study showed that its N-terminal PH and coiled-coil domains bind to a set of scaffolds, one of which promotes p38 MAP kinase activation by its DH (22–24). Thus, the PH and coiled-coil domains of GRF1 could be more effective than their GRF2 counterparts by binding a scaffold protein that promotes p38 activation, a possibility presently under investigation.
The downstream signaling pathway activated by GRF1 to promote NMDAR-mediated LTD has not been clearly elucidated, so that how the specific IQ domain of GRF1 could encode this form of signaling specificity more effectively than that of GRF2 is less well understood. Some, but not all, reports showed that inhibitors of p38 block LTD induction in the CA1, suggesting that GRF1 activation of p38 could be involved in this process as well (2, 25–27). However, stimuli that activate LTD did not lead to p38 activation (data not shown). Thus, at best p38 may play a permissive role in this process. Instead, the well characterized LTD promoters, the protein phosphatases, calcineurin and protein phosphatase 1, are instead likely to be involved (28). Because we found previously that the PH, coiled-coil and IQ domains function together to properly localize GRF1 in the cell (21), the specific sequences of the GRF1 IQ motif may target the protein through its effects on the structure of the entire motif to a key cellular site that engages one these phosphatases.
The discovery that the specificity for the ability of the GRF proteins to promote NMDAR-mediated LTD resides predominantly in the subtle difference (only 3 amino acids) between the GRF1 and GRF2 IQ domains raised the obvious possibility that (IQ)1 and (IQ)2 have different affinities for CaM. Our ITC data with peptides representing the IQ domains of both proteins suggest the following potential explanation to describe how the detected differences in CaM binding lead to differences in NMDAR-mediated LTD. At rest, CaM is largely in the apo state and whereas GRF1 and GRF2 both bind to apo-CaM, GRF1 does so ∼1.7 times better. The pre-association of CaM has been proposed to increase the speed and probability of CaM activation of ion channels (29). Thus, the higher affinity between IQ1 and apo-CaM than IQ2 and apo-CaM may prime GRF1 to respond more effectively to lower levels of Ca2+ produced during the induction of LTD. Because GRF2 has a weaker binding affinity for apo-CaM, the probability of association is lower and therefore its response to a rise in Ca2+ would be less robust. An interesting parallel exists between the CaM-binding properties of the IQ domains of GRF1 and GRF2 and the IQ domain in the NR1 subunit of the NMDA receptor (30) although GRFs bind ∼4–5 times tighter to apo-CaM. Interestingly, GRF1 and GRF2 are different from their counterpart regulators of NMDAR-mediated LTD and LTP, calcineurin and CaMKII, respectively. Neither of the latter is predicted to bind apo-CaM in cells. Instead, they display a greater than 2 orders of magnitude difference in their affinities for Ca2+/CaM (31) that potentially explains their differential responses to calcium in vivo.
Aligning the amino sequences of the GRF IQ domains with that of neuromodulin and neurogranin whose crystal structure complexed with CaM is known suggests which of the three amino acid differences that distinguish IQ1 from IQ2 may account for their affinity differences with apo-CaM (32). The C-terminal amino acids of the IQ motifs of these proteins that align with Asn-Ile-Ile and Thr-Ile-Val of GRF1 and GRF2 respectively, do not directly contact CaM. In contrast, the N-terminal Phe of neuromodulin and neurogranin does align, and mutating this residue blocks CaM binding. These findings suggest that the bulkier Met found in the comparable position of GRF2 compared with the Leu found in GRF1 is the likely difference that destabilizes the interaction of the former protein with apo-CaM.
It is also possible that CaM bound to GRF proteins has activities beyond modulating the Ca2+ responsiveness of GRF1 and GRF2. Apo-CaM has been implicated as an important structural protein that can associate domains or subunits of ion channels together (29). Thus, preferential association of pre-bound CaM to GRF1 could also contribute to downstream signaling specificity that allows GRF1 but not GRF2 to promote LTD.
Finally, a particularly interesting chimera is (PCQ)1GRF2 because it can mediate all three forms of synaptic plasticity normally regulated by GRF1 and GRF2 in the CA1 hippocampus. This raises the interesting question of why two GRF proteins exist, if one form can accomplish all of its tasks. A plausible answer is that domains that do not appear to contribute to the specific functions of GRF proteins studied here may do so when GRF1 and GRF2 play other roles in different regions of the brain. For example, GRF1 has been shown to contribute to the late stages of adult neurogenesis in the dentate gyrus of the hippocampus (33). It also plays a role in visual recognition memory and perirhinal cortex synaptic plasticity (34). In addition, GRF1 functions through ERK MAPK regulation in mediating dopamine signaling in the striatum (35). GRF2 also functions to regulate dopamine secretion via ERK MAPK regulation in cells of the nucleus accumbens to influence alcohol consumption (36). In each of these cases, and as new functions for GRF proteins are discovered, the distinction between GRF1 and GRF2 functions may be found to be encoded in different domains than those found here for synaptic plasticity in the CA1 hippocampus.
Overall, we have defined a small number of functional domains among those common to GRF1 and GRF2, including the N-terminal PH, coiled-coil, and IQ motif and C-terminal CDC25 domains, which endow each exchange factor with signaling specificity to carry out their specific roles in three forms of synaptic plasticity in the CA1 region of the hippocampus. These findings will focus future studies on these particular domains to reveal the detailed mechanism underlying GRF signaling specificity. These findings also highlight the versatility of multidomain protein families in that they can empower different domains with signaling specificity in different settings, expanding the functional potential of a pair of protein family members.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 MH083324 (to L. A. F.) and RO1 GM097553 (to M. N. W.) and Grant P30 NS047243 through the Tufts Center for Neuroscience Research.
- CaM
- calmodulin
- CP-AMPARs
- calcium permeable 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid receptors
- GRF1
- Ras guanine nucleotide releasing factor 1
- RAS-GRF2
- Ras guanine nucleotide releasing factor 2
- NMDAR
- N-methyl-d-aspartate receptor
- LTP
- long term potentiation
- LTD
- long term depression
- fEPSP
- field excitatory postsynaptic potential
- TBS
- θ-burst stimulation
- HFS
- high frequency stimulation
- DH
- Dbl homology
- PH
- pleckstrin homology
- REM
- Ras exchange motif
- PCQ
- PH/coiled-coil/IQ motif
- ACSF
- artificial cerebrospinal fluid
- ITC
- isothermal titration calorimetry
- DBKO
- double Grf1/Grf2 knock-out
- aa
- amino acid
- LFS
- low frequency stimulation.
REFERENCES
- 1. Feig L. A. (2011) Regulation of neuronal function by Ras-GRF exchange factors. Genes Cancer 2, 306–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Li S., Tian X., Hartley D. M., Feig L. A. (2006) Distinct roles for Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) and Ras-GRF2 in the induction of long-term potentiation and long-term depression. J. Neurosci. 26, 1721–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jin S. X., Arai J., Tian X., Kumar-Singh R., Feig L. A. (2013) Acquisition of contextual discrimination involves the appearance of a Ras-GRF1/p38 MAP kinase-mediated signaling pathway that promotes LTP. J. Biol. Chem. 288, 21703–21713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Arai J. A., Li S., Hartley D. M., Feig L. A. (2009) Transgenerational rescue of a genetic defect in long-term potentiation and memory formation by juvenile enrichment. J. Neurosci. 29, 1496–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. d'Isa R., Clapcote S. J., Voikar V., Wolfer D. P., Giese K. P., Brambilla R., Fasano S. (2011) Mice lacking Ras-GRF1 show contextual fear conditioning but not spatial memory impairments: convergent evidence from two independently generated mouse mutant lines. Front. Behav. Neurosci. 5, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tian X., Gotoh T., Tsuji K., Lo E. H., Huang S., Feig L. A. (2004) Developmentally regulated role for Ras-GRFs in coupling NMDA glutamate receptors to Ras, Erk and CREB. EMBO J. 23, 1567–1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jin S. X., Feig L. A. (2010) Long-term potentiation in the CA1 hippocampus induced by NR2A subunit-containing NMDA glutamate receptors is mediated by Ras-GRF2/Erk map kinase signaling. PLoS One 5, e11732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Krapivinsky G., Krapivinsky L., Manasian Y., Ivanov A., Tyzio R., Pellegrino C., Ben-Ari Y., Clapham D. E., Medina I. (2003) The NMDA receptor is coupled to the ERK pathway by a direct interaction between NR2B and RasGRF1. Neuron 40, 775–784 [DOI] [PubMed] [Google Scholar]
- 9. Li S., Tian X., Hartley D. M., Feig L. A. (2006) The environment versus genetics in controlling the contribution of MAP kinases to synaptic plasticity. Curr. Biol. 16, 2303–2313 [DOI] [PubMed] [Google Scholar]
- 10. Dudek S. M., Fields R. D. (2001) Mitogen-activated protein kinase/extracellular signal-regulated kinase activation in somatodendritic compartments: roles of action potentials, frequency, and mode of calcium entry. J. Neurosci. 21, RC122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zippel R., Gnesutta N., Matus-Leibovitch N., Mancinelli E., Saya D., Vogel Z., Sturani E. (1997) Ras-GRF, the activator of Ras, is expressed preferentially in mature neurons of the central nervous system. Brain Res. Mol. Brain Res. 48, 140–144 [DOI] [PubMed] [Google Scholar]
- 12. Forest A., Swulius M. T., Tse J. K., Bradshaw J. M., Gaertner T., Waxham M. N. (2008) Role of the N- and C-lobes of calmodulin in the activation of Ca2+/calmodulin-dependent protein kinase II. Biochemistry 47, 10587–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Waxham M. N., Tsai A. L., Putkey J. A. (1998) A mechanism for calmodulin (CaM) trapping by CaM-kinase II defined by a family of CaM-binding peptides. J. Biol. Chem. 273, 17579–17584 [DOI] [PubMed] [Google Scholar]
- 14. Selcher J. C., Weeber E. J., Christian J., Nekrasova T., Landreth G. E., Sweatt J. D. (2003) A role for ERK MAP kinase in physiologic temporal integration in hippocampal area CA1. Learn Mem. 10, 26–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. English J. D., Sweatt J. D. (1997) A requirement for the mitogen-activated protein kinase cascade in hippocampal long term potentiation. J. Biol. Chem. 272, 19103–19106 [DOI] [PubMed] [Google Scholar]
- 16. Winder D. G., Martin K. C., Muzzio I. A., Rohrer D., Chruscinski A., Kobilka B., Kandel E. R. (1999) ERK plays a regulatory role in induction of LTP by θ frequency stimulation and its modulation by β-adrenergic receptors. Neuron 24, 715–726 [DOI] [PubMed] [Google Scholar]
- 17. Freedman T. S., Sondermann H., Friedland G. D., Kortemme T., Bar-Sagi D., Marqusee S., Kuriyan J. (2006) A Ras-induced conformational switch in the Ras activator Son of sevenless. Proc. Natl. Acad. Sci. U.S.A. 103, 16692–16697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoffman L., Chandrasekar A., Wang X., Putkey J. A., Waxham M. N. (2014) Neurogranin alters the structure and calcium-binding properties of calmodulin. J. Biol. Chem. 289, 14644–14655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farnsworth C. L., Freshney N. W., Rosen L. B., Ghosh A., Greenberg M. E., Feig L. A. (1995) Calcium activation of Ras mediated by the neuronal exchange factor Ras-GRF. Nature 376, 524–527 [DOI] [PubMed] [Google Scholar]
- 20. Kim M. J., Dunah A. W., Wang Y. T., Sheng M. (2005) Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46, 745–760 [DOI] [PubMed] [Google Scholar]
- 21. Buchsbaum R., Telliez J. B., Goonesekera S., Feig L. A. (1996) The N-terminal pleckstrin, coiled-coil, and IQ domains of the exchange factor Ras-GRF act cooperatively to facilitate activation by calcium. Mol. Cell. Biol. 16, 4888–4896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Buchsbaum R. J., Connolly B. A., Feig L. A. (2002) Interaction of Rac exchange factors Tiam1 and Ras-GRF1 with a scaffold for the p38 mitogen-activated protein kinase cascade. Mol. Cell. Biol. 22, 4073–4085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Buchsbaum R. J., Connolly B. A., Feig L. A. (2003) Regulation of p70 S6 kinase by complex formation between the Rac guanine nucleotide exchange factor (Rac-GEF) Tiam1 and the scaffold spinophilin. J. Biol. Chem. 278, 18833–18841 [DOI] [PubMed] [Google Scholar]
- 24. Connolly B. A., Rice J., Feig L. A., Buchsbaum R. J. (2005) Tiam1-IRSp53 complex formation directs specificity of rac-mediated actin cytoskeleton regulation. Mol. Cell. Biol. 25, 4602–4614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bolshakov V. Y., Carboni L., Cobb M. H., Siegelbaum S. A., Belardetti F. (2000) Dual MAP kinase pathways mediate opposing forms of long-term plasticity at CA3-CA1 synapses. Nat. Neurosci. 3, 1107–1112 [DOI] [PubMed] [Google Scholar]
- 26. Zhu J. J., Qin Y., Zhao M., Van Aelst L., Malinow R. (2002) Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell 110, 443–455 [DOI] [PubMed] [Google Scholar]
- 27. Peineau S., Nicolas C. S., Bortolotto Z. A., Bhat R. V., Ryves W. J., Harwood A. J., Dournaud P., Fitzjohn S. M., Collingridge G. L. (2009) A systematic investigation of the protein kinases involved in NMDA receptor-dependent LTD: evidence for a role of GSK-3 but not other serine/threonine kinases. Mol. Brain 2, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Malenka R. C., Bear M. F. (2004) LTP and LTD: an embarrassment of riches. Neuron 44, 5–21 [DOI] [PubMed] [Google Scholar]
- 29. Saimi Y., Kung C. (2002) Calmodulin as an ion channel subunit. Annu. Rev. Physiol. 64, 289–311 [DOI] [PubMed] [Google Scholar]
- 30. Akyol Z., Bartos J. A., Merrill M. A., Faga L. A., Jaren O. R., Shea M. A., Hell J. W. (2004) Apo-calmodulin binds with its C-terminal domain to the N-methyl-d-aspartate receptor NR1 C0 region. J. Biol. Chem. 279, 2166–2175 [DOI] [PubMed] [Google Scholar]
- 31. Quintana A. R., Wang D., Forbes J. E., Waxham M. N. (2005) Kinetics of calmodulin binding to calcineurin. Biochem. Biophys. Res. Commun. 334, 674–680 [DOI] [PubMed] [Google Scholar]
- 32. Kumar V., Chichili V. P., Zhong L., Tang X., Velazquez-Campoy A., Sheu F. S., Seetharaman J., Gerges N. Z., Sivaraman J. (2013) Structural basis for the interaction of unstructured neuron specific substrates neuromodulin and neurogranin with calmodulin. Sci. Rep. 3, 1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Darcy M. J., Trouche S., Jin S. X., Feig L. A. (2014) Age-dependent role for ras-GRF1 in the late stages of adult neurogenesis in the dentate gyrus. Hippocampus 24, 315–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Silingardi D., Angelucci A., De Pasquale R., Borsotti M., Squitieri G., Brambilla R., Putignano E., Pizzorusso T., Berardi N. (2011) ERK pathway activation bidirectionally affects visual recognition memory and synaptic plasticity in the perirhinal cortex. Front. Behav. Neurosci. 5, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fasano S., D'Antoni A., Orban P. C., Valjent E., Putignano E., Vara H., Pizzorusso T., Giustetto M., Yoon B., Soloway P., Maldonado R., Caboche J., Brambilla R. (2009) Ras-guanine nucleotide-releasing factor 1 (Ras-GRF1) controls activation of extracellular signal-regulated kinase (ERK) signaling in the striatum and long-term behavioral responses to cocaine. Biol. Psychiatry 66, 758–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Stacey D., Bilbao A., Maroteaux M., Jia T., Easton A. C., Longueville S., Nymberg C., Banaschewski T., Barker G. J., Büchel C., Carvalho F., Conrod P. J., Desrivières S., Fauth-Bühler M., Fernandez-Medarde A., Flor H., Gallinat J., Garavan H., Bokde A. L., Heinz A., Ittermann B., Lathrop M., Lawrence C., Loth E., Lourdusamy A., Mann K. F., Martinot J. L., Nees F., Palkovits M., Paus T., Pausova Z., Rietschel M., Ruggeri B., Santos E., Smolka M. N., Staehlin O., Jarvelin M. R., Elliott P., Sommer W. H., Mameli M., Müller C. P., Spanagel R., Girault J. A., Schumann G., and IMAGEN Consortium (2012) RASGRF2 regulates alcohol-induced reinforcement by influencing mesolimbic dopamine neuron activity and dopamine release. Proc. Natl. Acad. Sci. U.S.A. 109, 21128–21133 [DOI] [PMC free article] [PubMed] [Google Scholar]