

Background: The mechanisms through which TRAILR4 interferes with proapoptotic signaling are not conclusively elucidated.
Results: TRAILR4 forms ligand-independent heterodimers with TRAIL death receptors, thereby inhibiting both pro- and anti-apoptotic signaling.
Conclusion: TRAILR4 exerts a dominant negative effect on TRAILR1 through the PLAD-mediated formation of mixed receptor complexes.
Significance: Understanding the mechanism of TRAILR4-mediated apoptosis-inhibition can be advantageous for the development of new TRAIL receptor agonists for cancer therapy.
Keywords: Apoptosis, Fluorescence Resonance Energy Transfer (FRET), Receptor, Signaling, Trail
Abstract
The cytokine TNF-related apoptosis-inducing ligand (TRAIL) and its cell membrane receptors constitute an elaborate signaling system fulfilling important functions in immune regulation and tumor surveillance. Activation of the death receptors TRAILR1 and TRAILR2 can lead to apoptosis, whereas TRAILR3 and TRAILR4 are generally referred to as decoy receptors, which have been shown to inhibit TRAIL-induced apoptosis. The underlying molecular mechanisms, however, remain unclear. Alike other members of the TNF receptor superfamily, TRAIL receptors contain a pre-ligand binding assembly domain (PLAD) mediating receptor oligomerization. Still, the stoichiometry of TRAIL receptor oligomers as well as the issue of whether the PLAD mediates only homotypic or also heterotypic interactions remained inconclusive until now. Performing acceptor-photobleaching FRET studies with receptors 1, 2, and 4, we demonstrate interactions in all possible combinations. Formation of dimers was shown by chemical cross-linking experiments for interactions of TRAILR2 and heterophilic interactions between the two death receptors or between either of the death receptors and TRAILR4. Implications of the demonstrated receptor-receptor interactions on signaling were investigated in suitable cellular models. Both apoptosis induction and activation of the transcription factor NFκB were significantly reduced in the presence of TRAILR4. Our experimental data combined with mathematical modeling show that the inhibitory capacity of TRAILR4 is attributable to signaling-independent mechanisms, strongly suggesting a reduction of signaling competent death receptors through formation heteromeric receptor complexes. In summary, we propose a model of TRAIL receptor interference driven by PLAD-mediated formation of receptor heterodimers on the cell membrane.
Introduction
The tumor necrosis factor superfamily member TNF-related apoptosis-inducing ligand (TRAIL)3 has gained considerable interest in the scientific community during the past decades. This is mainly based on the findings that the ligand TRAIL has been shown to specifically induce apoptosis in many tumor-derived cell lines or primary tumor cells, whereas most normal tissue cells are resistant to the apoptotic effects of TRAIL (1). In the human system TRAIL binds four cell membrane receptors, TRAILR1 to TRAILR4, but also the soluble protein osteoprotegerin (2). Whereas the functional role of TRAIL interaction with osteoprotegerin is controversially discussed (3, 4), the four membrane receptors fall into two classes. TRAILR1 and TRAILR2 each contain a cytoplasmic death domain (DD) and are thus capable to induce the cellular program of apoptosis but also anti-apoptotic signaling. Both signals are believed to be produced from two different, subsequently formed signaling complexes (5, 6). TRAILR3 is a glycosylphosphatidylinositol-anchored protein likely lacking any intracellular signaling capability, and TRAILR4 features a truncated DD with sparsely defined signaling capability in its intracellular part, which is obviously incompetent for apoptosis induction (2). TRAILR3 and TRAIL4 are often referred to as “decoy receptors,” abbreviated as decoy receptor 1 (DcR1) and DcR2 (2).
Most human cell lines coexpress several TRAIL receptors, and thus receptor interference has been proposed to occur at several levels. First, TRAILR3 and TRAILR4 can bind and consume ligand molecules in terms of a classical decoy mechanism. Regarding ligand binding affinities, however, all four TRAIL receptors appear comparable (7–9), arguing for an efficient decoy mechanism only when the respective receptors are present in large excess. In fact, overexpression of TRAILR3 or TRAILR4 has been show to efficiently inhibit TRAILR1- or TRAILR2-mediated apoptosis (10, 11). A second mechanism of receptor interference could occur at the level of signal complex formation. As the ligand TRAIL forms tightly packed homotrimers with three receptor interaction sites between each of its individual subunits (12), it was assumed that ligand-mediated receptor trimerization represents the signal initiating structural event. Being incorporated into such signaling complexes, TRAILR3 or TRAILR4 could interfere, resulting in reduced or prevented signaling. Finally, TRAILR4-derived (more unlikely TRAILR3-derived) signals could interfere with pro-apoptotic signaling of TRAIL death receptors (8).
However, the situation at the level of ligand receptor interaction and signal complex formation is likely to be even more complex. Lenardo and co-workers (13, 14) described a homophilic receptor interaction site, called PLAD (pre-ligand binding assembly domain) that mediates homomultimerization of the respective receptors at the cellular membrane already in the unligated state. The PLAD has been originally detected for the TNF receptor and the CD95/Fas system but later also in the TRAIL receptor system. Remarkably, and in contrast to the TNF receptor system, it was reported that TRAIL receptors are also capable of forming heterooligomers via PLAD interaction (15). This is in agreement with the highly conserved membrane distal partial cysteine-rich domain 1 (CRD1) of these molecules, which probably contains the PLAD interaction motif (12, 15, 16). It was argued that the existence of pre-assembled mixed receptor complexes allows TRAILR4 to act as a regulatory receptor independent from the classical decoy mechanism. In contrast, Micheau and co-workers (17) argued that TRAILR4 becomes recruited into the signaling complex of TRAILR2 in a strictly ligand-dependent manner, whereas TRAILR3, being located in cholesterol enriched membrane microdomains, acts via the decoy mechanism. In addition, overexpression of TRAILR4 has been reported to induce anti-apoptotic activities via stimulation of NFκB (8) or protein kinase B (PKB)/Akt (18), but these signals may be cell-specific as indicated by conflicting results (8, 19).
The stoichiometry reported for PLAD-mediated formation of receptor oligomers is also unresolved. Based on studies with bivalent chemical cross-linking agents, originally a stoichiometry of three was proposed for the TNF receptors and CD95/Fas (13, 14). In our studies, however, using TNF receptor chimeras containing the intracellular signaling domain of CD95/Fas, predominantly homodimer formation was found (20, 21). Deletion studies showed that the PLAD-positive membrane distal CRD of TNFR1 is necessary for high affinity ligand interaction (13). It is evident that the homotrimeric ligand might bind to oligomerized receptors with higher avidity (22) but in addition CRD1 is likely to serve as a scaffold for CRD2, which is involved in ligand interaction (20).
In the present study we confirm that TRAIL receptors 1, 2, and 4 are capable of forming not only homooligomers but also all combinations of heterooligomers in a ligand-independent manner. For both homo- and heterooligomeric complexes the predominant formation of dimers is observed. Using cells coexpressing TRAILR1 and TRAILR4, we demonstrate that the latter efficiently inhibits both pro- and anti-apoptotic signals transduced by TRAILR1. Direct anti-apoptotic signaling mediated by TRAILR4 is excluded experimentally. Mathematical modeling reveals that formation of nonfunctional heteromeric complexes of both TRAIL receptors represents a powerful mechanism of TRAIL receptor interference that is in good accordance with our experimental data. In contrast, a ligand consuming decoy mechanism can be excluded by our simulation studies. Accordingly, the mechanism of TRAILR1 and TRAILR4 interference observed by us is likely based on the formation of signaling incompetent mixed receptor complexes, a process driven by the distribution of homo- and heterodimers present on the cell already in the absence of ligand.
EXPERIMENTAL PROCEDURES
Plasmids and PCR
The expression plasmid pEFpuroTRAILR4 was generated by subcloning TRAILR4 cDNA from the plasmid pCR3 hTRAILR4full (kindly provided by Prof. H. Wajant, University of Wuerzburg, Wuerzburg, Germany) using the restriction endonucleases BamHI and EcoRV (New England Biolabs Inc., Ipswich, MA). Expression plasmids encoding the fusion proteins TRAILR1ΔC-GFP, TRAILR1ΔC-mCherry, TRAILR2ΔC-GFP, TRAILR2ΔC-mCherry, TRAILR4ΔC-GFP, and TRAILR4ΔC-mCherry were produced by PCR cloning. Therefore, the extracellular and transmembrane domains as well as the first 14 intracellular amino acids were amplified from the plasmids pCR3 DR4, pCR3 DR5 (kindly provided by Prof. H. Wajant), or pEFpuroTRAILR4 by standard PCR and subsequently ligated into the expression plasmids peGFP-N1 or pmCherry-N1 (Clontech Laboratories, Mountain View, CA) after double digestion using SacI and AgeI. The expression plasmid coding for TNFR1ΔC-GFP has been described elsewhere (23). For stable expression in mammalian cells, TRAILR2ΔC-GFP and TRAILR4ΔC-GFP were subcloned from pEGFP-N1 into pCDNA3.1+ (Invitrogen) using HindIII and NotI. Generation of TRAILR2ΔC-FLAG was achieved by standard PCR using appropriate primers carrying a nucleotide sequence encoding for a FLAG tag at the 5′-end of the reverse primer. The resulting fragment was ligated into the expression plasmid pEFpuro (24) after restriction endonuclease treatment using BamHI and XbaI. All constructs generated by PCR were verified by DNA sequencing.
Reagents
FLAG-tagged recombinant human TRAIL (TRAIL) was purchased from Enzo Life Sciences (Lörrach, Germany). Cycloheximide and α-FLAG antibody (clone M2) were from Sigma. Bis(sulfosuccinimidyl)-suberate (BS3) was purchased from Pierce. All chemicals were of analytical grade.
Cell Lines and Generation of Stable Transfectants
Mouse embryonic fibroblasts (MFs) generated from TNFR1/TNFR2 double knock-out mice have been described elsewhere (25). HeLa cells had been obtained from the American Type Culture Collection (LGC Standards GmbH, Wesel, Germany). Cells were grown in RPMI 1640 medium containing 2 mm l-glutamine (Invitrogen) supplemented with 5% (v/v) heat-inactivated fetal calf serum (PAN Biotech, Aidenbach, Germany).For generation of stably transfected cells, MFs (3 × 105 cells per well) were seeded in 6-well plates and grown overnight. The following day cells were transfected with the respective expression plasmids using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. HeLa (3 × 105 cells per well) were seeded in 6-well plates and transfected the following day with plasmids pEFpuroTRAILR4 or pCDNA3.1+TRAILR4ΔC-GFP using Effectene (Qiagen, Duesseldorf, Germany) according to the manufacturer's instructions. The day after transfection, cells were selected for stable expression using 2 μg/ml puromycin and/or 200 μg/ml zeocin. After 2–3 weeks cells were sorted by fluorescence-activated cell sorting. Cell sorting was performed two to three times per cell line in total.
Western Blotting
Samples were resolved by Tris/glycine SDS-PAGE and transferred onto nitrocellulose membrane (Whatman, Schleicher & Schuell), which was then blocked using Roche blocking reagent (Roche Diagnostics) or 5% nonfat milk powder (Carl Roth, Karlsruhe, Germany) in PBS containing 0.05% (v/v) Tween 20 for 1 h at room temperature. Cleaved caspase-8 was detected using rabbit α-cleaved Caspase-8 (Cell Signaling Technology, Inc., Danvers, MA) antibody, and for detection of caspase-3, rabbit α-Caspase 3 (Cell Signaling Technology) was used. Proteins fused to FLAG tag or enhanced GFP (eGFP) were detected using rabbit α-DYKDDDDK-tag (Cell Signaling Technology) or mouse α-GFP antibody (Roche Diagnostics), respectively. IκBα and phospho-IκBα were detected using mouse α-IκBα and mouse α-phospho-IκBα, both purchased from Cell Signaling Technology. Horseradish peroxidase-conjugated α-mouse IgG and α-rabbit IgG secondary antibodies were from Dianova (Hamburg, Germany). Mouse α-tubulin-α (NeoMarkers, ThermoFisher Scientific, Waltham, MA) in combination with IRDye800CW-conjugated α-mouse IgG (LI-COR Biotechnology GmbH, Bad Homburg, Germany) was used as loading control.
Cell Death Assay
For cell death assays HeLa cells (2 × 104 cells/well) were grown in 96-well plates overnight. The next day cells were preincubated for 1 h with 20 μm benzyloxycarbonyl-VAD-fluoromethyl ketone (Bachem AG, Bubendorf, Switzerland) or not and then stimulated with serial dilutions of recombinant human TRAIL (previously cross-linked by incubation with 1 μg/ml α-FLAG M2 antibody for 1 h at 37 °C) in combination with 0.5 μg/ml cycloheximide. After 24 h cells were stained with crystal violet (20% methanol, 0.5% crystal violet), and optical density at 550 nm was determined using an ELISA plate reader as described (25).
Chemical Cross-linking
Mouse fibroblasts stably expressing truncated TRAILR variants (1 × 106 cells) were grown overnight in 6-cm cell culture dishes. Chemical cross-linking was performed on ice as described (20). For immunoprecipitation of chemically cross-linked cell surface proteins MFs stably expressing two differently tagged truncated TRAILR variants (3 × 106 cells) were grown overnight in 10-cm cell culture dishes, and cell surface proteins were cross-linked the following day. Cells were washed with ice-cold PBS, harvested, and pelleted. The cells were then lysed in solubilization buffer (50 mm Tris-HCl, 150 mm NaCl, 1 mm EGTA, 1% (v/v) Nonidet P-40, 0.25% (w/v) sodium deoxycholate, pH 7.4) supplemented with complete protease inhibitor mixture (Roche Diagnostics) and 0.5 mm phenylmethylsulfonyl fluoride. Five micrograms of α-FLAG antibody were added and incubated at 4 °C for 2 h before the addition of 25 μl of protein G-Sepharose beads (GE Healthcare) for 16 h on a rotary mixer at 4 °C. Beads were washed with solubilization buffer, and Laemmli sample buffer with 5% (v/v) 2-mercaptoethanol was added.
Flow Cytometry
For FACS analysis 2 × 105 cells and for FACS sorting 3 × 106 cells were resuspended in PBA (PBS supplemented with 0.5% (w/v) BSA and 0.02% (w/v) NaN3) containing primary antibody (α-TRAILR1 MAB347, α-TRAILR2 MAB6311, α-TRAILR3 MAB6302, or α-TRAILR4 MAB 633; all from R&D Systems Inc., Minneapolis, MN). After incubation for 1 h on ice cells were washed in PBA and resuspended in PBA containing the secondary goat α-mouse-IgG+IgM (H+L)-phycoerythrin-conjugated (Dianova). After 45 min of incubation cells were washed again and resuspended in 400 μl of PBA. Cells were analyzed using Beckman Coulter Cytomics FC 500 or sorted using BD Biosciences FACS Vantage SE with FACSDiVa. For sorting, 3 × 104 positive cells were collected in 5 ml of RPMI1640 + 5% FCS containing 100 units/ml penicillin and 100 μg/ml streptomycin. Immediately after sorting, cells were transferred into 6-well plates and cultured at 37 °C and 5% CO2. Results from cytofluorometric analyses are presented as biexponential (logicle) histogram plots.
Acceptor Photobleaching Fluorescence Resonance Energy Transfer (FRET)
MFs (1.5 × 105 cells) were seeded on 18 × 18-mm coverslips and grown overnight. The following day cells were transfected with 0.5 μg of plasmid DNA encoding the receptor-eGFP and 0.25 μg of plasmid DNA encoding the receptor-mCherry fusion protein using Lipofectamine 2000 according to the manufacturer's instructions. After 24 h transfected cells were washed with PBS and fixed using 4% paraformaldehyde in PBS for 15 min at 20–25 °C. Coverslips were then mounted onto microscopy slides using Fluoromount G. Images were acquired using a Zeiss LSM710 confocal laser scanning microscope (Carl Zeiss MicroImaging, Jena, Germany). GFP was excited with the 488 nm line of the argon laser, and fluorescence was detected at 490- 550 nm. mCherry was excited with the 561-nm line of a diode-pumped solid state laser, and fluorescence was detected at 570- 650 nm. The imaging sequence was as follows: first donor and acceptor channels were sequentially scanned at low laser intensity. Subsequently, the acceptor was bleached with the 561-nm laser line at 75% intensity. Finally, donor and acceptor channels were again scanned at low laser intensity. Acquired images were exported to 8-bit TIFF format using Zeiss ZEN 2010 software. For analysis of FRET efficiency, images were imported into Fiji, and FRET efficiency was calculated using the ImageJ plug-in FRETcalc Version 4.0 (26). False-colored FRET images for illustration purposes were calculated using the MATLAB standalone application FRET_Plotter.
Mathematical Modeling
To quantify the effect of the formation of heteromeric receptor complexes on TRAIL signaling, we developed a mechanistic mathematical model of the biochemical reaction network. This model considered the formation of receptor homo- and heterodimers (R1-R3) as well as the binding of TRAIL to receptor dimers (R4-R6). Reactions and parameter values are provided in Table 1.
TABLE 1.
List of biochemical reactions and reaction rates used in our model for receptor-receptor and TRAIL-receptor interaction
(TRAILR1)2, (TRAILR4)2, and TRAILR1-TRAILR4 denote the receptor dimers, and (TRAILR1)2-TRAIL, (TRAILR4)2-TRAIL, and TRAILR1-TRAILR4-TRAIL denote TRAIL receptor complexes. The rates of R6 are estimated as geometric mean of the rates if R4 and R5: kon,6 = (kon,4 × kon,5)1/2, koff,6 = (koff,4 × koff,5)1/2.
| Reactions | kon | koff | |
|---|---|---|---|
| nm−1 × s −1 | s−1 | ||
| R1 | 2 × TRAILR1 ↔ (TRAILR1)2 | 1.15 × 102 | 5.43 × 10−2 |
| R2 | 2 × TRAILR4 ↔ (TRAILR4)2 | 2.42 × 101 | 2.11 × 10−3 |
| R3 | TRAILR1 + TRAILR4 ↔ TRAILR1-TRAILR4 | 1.46 × 101 | 1.13 × 10−3 |
| R4 | (TRAILR1)2 + TRAIL ↔ (TRAILR1)2-TRAIL | 1.43 × 10−4 | 4.13 × 10−3 |
| R5 | (TRAILR4)2 + TRAIL ↔ (TRAILR4)2-TRAIL | 7.04 × 10−5 | 4.38 × 10−3 |
| R6 | TRAILR1-TRAILR4 + TRAIL ↔ TRAILR1-TRAILR4-TRAIL | 1.00 × 10−4 | 4.25 × 10−3 |
To parameterize the model, we mainly used literature data. On- and off-rates of the receptor dimerization reactions and the TRAILR1-TRAIL and TRAILR4-TRAIL interaction have been measured by Lee et al. (7). Using the measured receptor dimerization rates we, however, observed only negligible receptor dimerization. To reproduce our experimental observation, we increased the on-rates till the model predicted roughly 80% of dimerized TRAILR1 in the absence of TRAILR4, which is in agreement with our data. The parameters for TRAILR1-TRAIL and TRAILR4-TRAIL interactions provided a reasonable prediction, and for the binding of TRAIL to TRAILR1-TRAILR4, the geometric average of the previous rates was used. As the reaction rates are not precisely known, we sampled on-rates around the values provided in Table 1 to ensure that the model predictions are robust. TRAILR1 was estimated to be expressed at a level of 104 molecules/cell (4.43 × 10−3 nm as we have 2.67 × 105 cells/ml) and TRAILR4 at 0 molecules/cell (=0 nm) for HeLa cells, 104 molecules/cell (4.43 × 10−3 nm) for HeLa R4 cells, and 105 molecules/cell (4.43 × 10−2 nm) for HeLa R4+ cells.
For the biochemical reaction network defined by R1-R6, the corresponding reaction rate equation has been derived, assuming mass action kinetics. Given this mathematical model we used MATLAB to evaluate the stationary concentration of (TRAILR1)2-TRAIL complexes, which is a measure for TRAIL-induced TRAILR1 signaling. The concentration of (TRAILR1)2-TRAIL complexes has been analyzed for two scenarios, 1) receptor-receptor interaction and decoy action (parameters as stated above) 2) no receptor-receptor interaction and decoy action (k3,on = 0).
RESULTS
TRAIL Receptors 1, 2, and 4 Form Homo- and Heteromeric Complexes in the Absence of Ligand
Interactions between unligated receptors have previously been demonstrated for several members of the TNF receptor superfamily, including TNF receptors (13, 20), CD95/Fas (14, 27), and TRAIL receptors (15). Here we studied TRAIL receptor interaction in the absence of ligand by acceptor-photobleaching FRET analyses. Donor receptors were expressed in immortalized mouse fibroblasts as eGFP fusion proteins with their respective intracellular domain replaced by the fluorescent protein. mCherry was used to replace the cytoplasmic domain in FRET acceptor receptors. All receptor-derived fusion proteins localized predominantly to the plasma membrane, with some fluorescence also detected at intracellular membranes representing most likely the endoplasmic reticulum and the Golgi apparatus (Fig. 1A). Homo- and heterophilic receptor-receptor interactions were studied using TRAILR1-, TRAILR2-, and TRAILR4-derived FRET constructs. As TRAILR3 is anchored within the cell membrane via a glycosylphosphatidylinositol anchor, the approach used here cannot be easily transferred to this receptor. As negative controls TNFR1/TRAILR1 and TNFR1/TRAILR2 FRET pairs were investigated, revealing no significant FRET efficiencies (Fig. 1B). In contrast, all six combinations of the three TRAIL receptors investigated (three homotypic and three heterotypic interactions, Fig. 1B) revealed significant values for FRET efficiencies. These data strongly suggest that the three TRAIL receptor fusion proteins used here are capable to form homo- and heteromers even in the absence of their intracellular signaling domains. Hence, oligomerization of TRAIL receptors is likely to be mediated by their respective extracellular parts, possibly in cooperation with the transmembrane domain, as has been proposed for TRAILR2 by Valley et al. (28).
FIGURE 1.
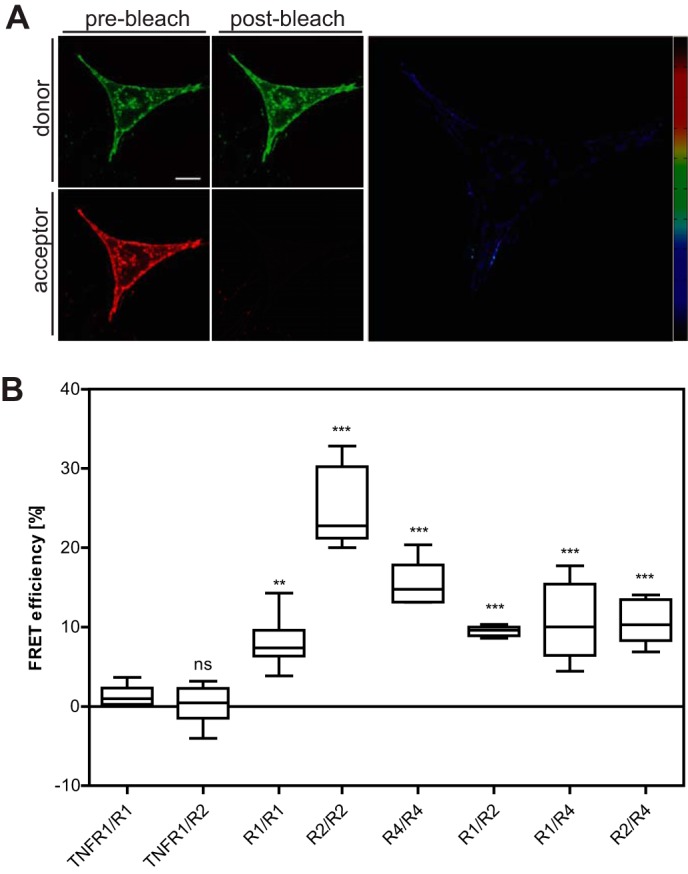
Homo- and heterotypic interactions of TRAIL receptors. A, mouse embryonic fibroblasts from TNFR1−/−/TNFR2−/− mice were transiently transfected with plasmids encoding the fusion proteins TRAILR1ΔC-mCherry and TRAILR4ΔC-GFP. Transfected cells were cultivated for 24 h before being fixed with 4% paraformaldehyde. Images shown represent optical sections obtained by confocal laser scanning microscopy; scale bar = 10 μm. False-colored FRET images at the right were calculated using the MATLAB plugin FRET_Plotter and show relative FRET efficiencies ranging from 0 (black) to 1 (dark red). B, acceptor-photobleaching FRET experiments were performed, and FRET efficiencies were calculated using the ImageJ plug-in FRETcalc. First two groups represent the negative controls (TNFR1ΔC-GFP (TNFR1) in combination with TRAILR1ΔC-mCherry (R1) or TRAILR2ΔC-mCherry (R2)). Values shown were obtained from the analysis of 30 individual cells from 6 independent experiments for each receptor-fusion protein combination. Box and whiskers plot: the box represents the 25th and 75th percentile, and the median is depicted by a horizontal line; whiskers denote 5th and 95th percentile. Significance was tested by one-way analysis of variance and comparison to the TNFR1/R1 negative control using Dunnett's multiple comparison test procedure. ns, not significant; **, p < 0.01; ***, p < 0.001.
TRAILR2 Predominantly Forms Homodimers in the Absence of a Functional Cytoplasmic Domain
Conflicting reports exist concerning the exact stoichiometry of ligand-independent TNFR1 complexes. Formation of homotrimers (13), but also more recently the predominant formation of homodimers, has been reported (20, 21, 29). In the case of TRAIL receptors, Sachs and co-workers (28) proposed TRAILR2 homotrimer formation in the absence of ligand, but receptor dimerization after ligation, partially driven by interactions of the transmembrane domains. Our FRET data indicated that TRAILR2 might form homomeric receptor complexes with particularly high efficiency as compared with the other TRAIL receptor members. We, therefore, decided to investigate the stoichiometry of receptor homomers in this particular system. Chemical cross-linking experiments were performed using a FLAG-tagged TRAILR2 variant consisting of the complete extracellular and transmembrane domains of the wild type receptor yet retaining only the first 14 intracellular amino acids, with the cytoplasmic death domain being deleted and replaced by the FLAG epitope tag (amino acid sequence DYKDDDDK). The resulting TRAILR2ΔC-FLAG fusion protein was stably expressed in mouse embryonic fibroblasts, and cell surface expression was verified by flow cytometry (Fig. 2A). Cells were treated with dilutions of the amine-reactive and non-membrane permeable cross-linker BS3 at 4 °C, i.e. under conditions of highly reduced membrane fluidity. Western blot analyses revealed efficient homodimer formation of TRAILR2ΔC-FLAG (Fig. 2B). In a concentration-dependent manner a strong specific band with an apparent molecular mass of ∼55 kDa could be detected in addition to the band representing the monomeric receptor variant with a calculated molecular mass of 27 kDa. Higher oligomers representing e.g. trimers or tetramers could not be detected even at the highest BS3 concentration used. The observed near to quantitative formation of homodimeric complexes at cross-linker concentrations of ≥500 μm indicates that homodimerization of TRAILR2 at the plasma membrane is strongly favored. The very strong appearance of the presumed homodimeric band at high cross-linking efficiencies in comparison to the relatively weak monomeric band in the absence of cross-linker is likely to be explained by an avidity effect of the IgG antibody used for immunoblot detection.
FIGURE 2.

A truncated variant or TRAILR2 predominantly forms homodimers on the plasma membrane. A, cell surface expression of a FLAG-tagged truncated version of human TRAILR 2 in MF cells (filled gray histogram). Isotype controls are shown as black histograms. B, MF TRAILR2ΔC-FLAG were incubated with increasing concentrations of the chemical cross-linker BS3. Whole cell lysates were resolved by reducing SDS-PAGE, and Western blot analysis was performed using a FLAG-tag-specific antibody. Precise non-stained molecular weight markers are given on the left. Shown is one representative experiment of three. C, cell surface proteins were immunostained with mouse α-TRAILR2 antibody (red histogram) or the respective IgG2B isotype control antibody (black histogram) followed by incubation with α-mouse IgG-PE conjugated secondary antibody (top histogram, showing total expression of both TRAILR2 variants) or left unstained (bottom histogram, showing fluorescence emission of TRAILR2ΔC-GFP; the inset shows fluorescence in FL1 channel of untransfected parental MF cells). Schematic depiction of the TRAILR variants was employed for the analysis of receptor/receptor interaction and flow cytometric analysis of cell surface expression of the receptor variants in the TRAILR2ΔC-FLAG and TRAILR2ΔC-GFP coexpressing cell line. PE, phycoerythrin. D, MFs stably expressing TRAILR2ΔC-FLAG and TRAILR2ΔC-GFP were incubated with 500 μm BS3 on ice for 30 min or left untreated. Whole cell lysate was subjected to immunoprecipitation using α-FLAG M2 antibody or IgG1 isotype control antibody and protein G-Sepharose beads. Precipitated proteins were subjected to SDS-PAGE and Western blot analysis using α-GFP antibody. Precise non-stained molecular weight markers are given on the left. Results shown are representative of three independent experiments. CRD, cysteine rich domain; S, stalk region; TM, transmembrane domain.
To confirm ligand-independent dimerization of the death receptor TRAILR2 in an additional approach, immunoprecipitation experiments of BS3-cross-linked cell surface proteins were performed. Immortalized mouse fibroblasts were engineered to stably coexpress TRAILR2ΔC-FLAG and TRAILR2ΔC-GFP. Expression of the two different receptor fusion proteins was verified by flow cytometry. In doing so, expression of TRAILR2ΔC-GFP was analyzed in fluorescence channel 1, whereas the signal emanating from immunostaining of both TRAILR2 variants at the cell surface using TRAILR2 specific monoclonal antibody followed by phycoerythrin-conjugated secondary antibody was measured in fluorescence channel 2 (Fig. 2C). Western blot analysis of the immunoprecipitates revealed a BS3-dependent band with an apparent molecular mass of ∼80 kDa (Fig. 2D, rightmost lane). The apparent molecular weight of this protein complex supports the notion that it constitutes a dimeric receptor complex containing one of each TRAILR2 fusion proteins, TRAILR2ΔC-FLAG (27 kDa) and TRAILR2ΔC-GFP (53 kDa).
TRAILR4 Efficiently Antagonizes TRAILR1-induced Apoptosis
To investigate how TRAILR4 might interfere with TRAILR1 signaling, TRAILR4 was ectopically expressed in HeLa cells, yielding the cell line HeLa R4. Wild type HeLa cells expressed considerable amounts of TRAILR1 but only comparably low amounts of TRAILR2 on their cell surface, whereas both decoy receptors could not be detected by flow cytometry (Fig. 3A). In HeLa R4 cells expression of TRAILR4 (MFI 1767 ± 196 AU, mean ± S.D., n = 7) was comparable with that of TRAILR1 (Fig. 3B; MFI 2163 ± 389 AU). A second cell line, HeLa R4+, generated by several rounds of fluorescence-activated cell sorting showed a stable and high expression of TRAILR4 (MFI 6949 ± 490 AU), whereas the expression level of TRAILR1 (MFI 1837 ± 214 AU) was largely unchanged (Fig. 3C). Cytotoxicity assays using antibody-cross-linked soluble TRAIL in combination with cycloheximide showed that cells overexpressing TRAILR4 were significantly protected against TRAIL-induced apoptosis compared with parental HeLa cells (Fig. 3D). These experiments also revealed a clear dose-response relationship of TRAILR4-mediated interference with TRAILR1 signaling in this cellular model. Cotreatment with the pan-caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone completely inhibited TRAIL-induced cell death, confirming apoptotic cell death upon TRAIL treatment (data not shown).
FIGURE 3.

Overexpression of TRAILR4 interferes with TRAIL-induced cell death. A, cytofluorometric analysis of TRAIL receptor cell surface expression in HeLa cells. B, cell surface expression of TRAILR4 in HeLa R4 cells, which express TRAILR4 at a level comparable to TRAILR1. C, HeLa R4+ cells show a significantly stronger expression of TRAILR4. D, HeLa (squares), HeLa R4 (circles), or HeLa R4+ (triangles) cells were treated with serial dilutions of antibody-cross-linked sTRAIL (TRAIL) in combination with 0.5 μg/ml cycloheximide. After 24 h cell viability was determined by crystal violet staining. Shown are mean values ± S.D. calculated from nine independent experiments each performed in triplicate. Significance was tested using two-way analysis of variance in combination with Bonferroni post-test. ***, p < 0.001 was considered significant.
To investigate the potential involvement of TRAILR4-activated PKB/Akt pathways in the observed inhibitory effects (18), the abundance of phosphorylated Akt (at serine residue 473) in HeLa wild type, HeLa R4, and HeLa R4+ cells was determined by Western blotting. Basal levels of phosphorylated Akt were low and comparable in all three cell lines. In addition, TRAIL treatment for up to 120 min did not result in any detectable changes in Akt phosphorylation (data not shown). Furthermore, potential differences in intracellular signaling between HeLa and HeLa R4 cells were assessed using the PathScan Intracellular Signaling Array kit, which allows for the parallel detection of 18 signaling molecules when becoming cleaved or phosphorylated. The results showed that basal phosphorylation levels were similar for most targets in these two cell lines. Significant changes in response to TRAIL treatment were only observed for the phosphorylation of Bad and for poly(ADP-ribose) polymerase cleavage at aspartate 214. As expected, the latter response to TRAIL treatment was stronger in HeLa as compared with HeLa R4 (data not shown). Together, no evidence was found for the involvement of TRAILR4-derived anti-apoptotic signals, which could interfere with TRAILR1-mediated apoptosis induction.
A TRAILR4 Mutant with Truncated Signaling Domain Efficiently Interferes with TRAILR1-induced Apoptosis
To confirm that TRAILR4-derived intracellular signals do not play a major role in the protection against TRAILR1-induced apoptosis, HeLa cells overexpressing a truncated, hence signaling-incapable variant of this receptor, were generated. This truncated receptor variant TRAILR4ΔC consisted of the complete extracellular and transmembrane domains of the wild type receptor, thus retaining the capability to bind its ligand and to homo- and heteromerize with other TRAIL receptors via the PLAD. All but the first 14 intracellular amino acids had been deleted and were replaced by the eGFP to ensure this receptor was incapable of transducing intracellular signals. Stable HeLa transfectants were produced expressing TRAILR4ΔC at comparably high levels (MFI 4155 ± 660 AU), whereas expression of the other three TRAIL receptors was essentially unchanged (Fig. 4A). Cytotoxicity assays using antibody-cross-linked soluble TRAIL in combination with the protein synthesis inhibitor cycloheximide revealed significantly reduced apoptotic cell death in HeLa cells positive for this TRAILR4 mutant as compared with parental cells (Fig. 4B). Again, pretreatment of HeLa R4ΔC cells with the pan-caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone efficiently blocked the residual TRAIL-mediated cytotoxic effects (Fig. 4B).
FIGURE 4.

A truncated, signaling-incapable TRAILR4 variant confers protection from TRAILR1-induced apoptosis. A, flow cytometric analysis of TRAIL receptor cell surface expression in HeLa R4ΔC cells. B, HeLaR4ΔC (diamonds) in comparison to HeLa cells (squares) were treated with serial dilutions of antibody-cross-linked sTRAIL (TRAIL) in combination with 0.5 μg/ml cycloheximide. After 24 h cell viability was determined by crystal violet staining. Shown are the mean values ± S.D. calculated from nine independent experiments each performed in triplicate. Significance was tested using two-way analysis of variance in combination with the Bonferroni post-test. ***, p < 0.001 was considered significant. Open diamonds show viability of HeLa R4ΔC cells preincubated with the pan-caspase inhibitor benzyloxycarbonyl-VAD-fluoromethyl ketone (20 μm) before TRAIL treatment (n = 3). C, caspase cleavage in HeLa wild type cells and cells overexpressing TRAILR4. Cells were treated with 300 ng/ml antibody-cross-linked sTRAIL for the indicated times or left untreated. Total cell extracts were subjected to SDS-PAGE and Western blot analysis using antibodies directed against cleaved caspase-8 or caspase-3. Tubulin-α was used as loading control. Blots shown are representative of three independent experiments.
Data obtained with the TRAILR4ΔC positive HeLa cells strongly suggested inhibitory effects at the level of signaling complex formation, i.e. at a level above initiator caspase activation. We, therefore, compared activation kinetics of the initiator caspase-8 and the effector caspase-3 by Western blotting in all four cell lines. TRAIL treatment induced cleavage of caspase-8 in all four investigated cell lines. The intermediate caspase-8 fragments p43/41 could be detected in all cell lines after 2 h of stimulation, albeit the amounts were reduced in cells expressing full-length or the truncated variant of TRAILR4 (Fig. 4C). Caspase-8 cleavage products p43/41 were detectable in all cell lines for the further duration of the experiment and showed maximal abundance 6 h after stimulation. At all time points investigated, wild type HeLa cells showed the highest levels of the caspase-8 p43/41 cleavage products. The final caspase-8 cleavage fragment, p18, was detected in HeLa cells after 4 h of TRAIL stimulation. In HeLa R4 and HeLa R4ΔC cells caspase-8 processing to the p18 fragment was considerably reduced. Even after 6 h of TRAIL treatment, active caspase-8 could not be detected in HeLa R4+ cells. As an expected consequence of the reduced initiator caspase activation, cleavage of the effector caspase-3 was also strongly reduced in all TRAILR4 (mutant) positive HeLa cells. Complete activation of caspase-3, i.e. cleavage into the p19 and p17 fragments, became apparent in HeLa cells after 4 and 6 h of TRAIL treatment, whereas in HeLa R4+ cells these products were not detectable. These data are in accordance with the expression levels of the receptors at the cell surface as determined by flow cytometry, showing that HeLa R4 (Fig. 3B) and HeLa R4ΔC (Fig. 4A) cells exhibit a lower membrane expression of their respective receptor (derivative) as compared with HeLa R4+ cells (Fig. 3C). Together, activation of both procaspase-8 and procaspase-3 could be shown to be significantly reduced in response to TRAIL treatment in HeLa cells engineered to express either full-length or a truncated variant of TRAILR4.
Interference of TRAILR4 with TRAILR1-mediated Activation of the Transcription Factor NFκB
Besides the induction of apoptosis, TRAILR1 and TRAILR2 are known to also activate additional, non-apoptotic, signaling pathways like the mitogen-activated protein (MAP) kinases, the transcription factor nuclear factor of κB (NFκB), or PKB/Akt (30, 31). Importantly, these signals are likely to be (mainly) produced by a secondary signaling complex (complex II), formed after internalization of the membrane-associated proapoptotic signaling complex I (death-inducing signaling complex (DISC)) (30). We, therefore, investigated whether TRAILR4 also interferes with complex II-derived signals of TRAILR1. Activation of NFκB in the four HeLa (-derived) cell lines was investigated by analyzing the phosphorylation of the NFκB inhibitor IκBα. In HeLa cells phosphorylation of IκBα became apparent after 30 min of treatment with 300 ng/ml antibody-cross-linked sTRAIL (Fig. 5A). The level of phospho-IκBα increased with time reaching its maximum after 120 min of TRAIL stimulation. Kinetics of IκBα phosphorylation in the HeLa-derived, TRAILR4-expressing cell line HeLa R4 was similar with the maximum level of phospho-IκBα detected after 90 min. However, the level of IκBα phosphorylation was reduced in comparison to the parental cell line (Fig. 5B). In HeLa R4+ cells, overexpressing TRAIL receptor 4 at a considerably higher level than HeLa R4, phosphorylation of IκBα was extremely weak even at the later time points. Similar data were obtained using HeLa R4ΔC cells expressing high levels of a truncated TRAILR4 variant (Fig. 5, A and B). These results demonstrate that interference of TRAILR4 with TRAILR1 affects both pro- and anti-apoptotic signals produced by different, subsequently formed signaling complexes.
FIGURE 5.
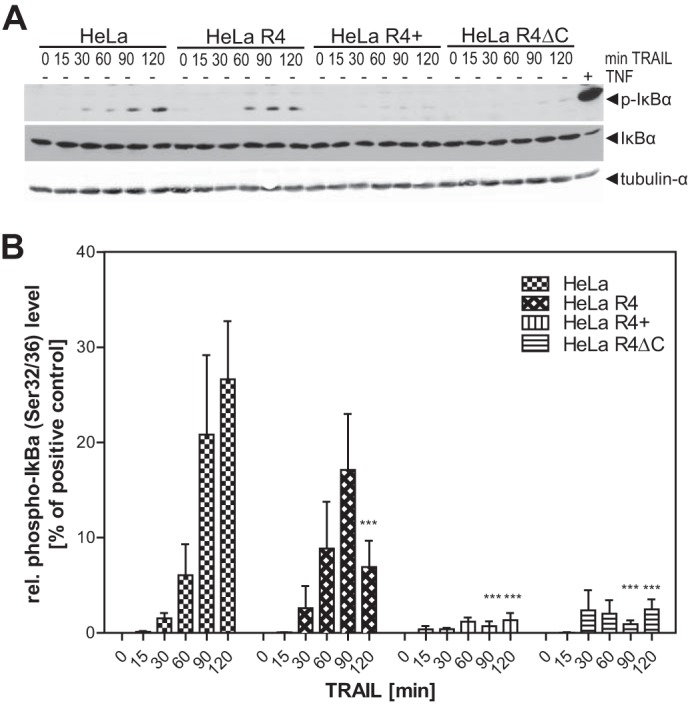
Interference of TRAILR4 with TRAILR1-induced phosphorylation of IκBα. A, cells were treated with 300 ng/ml antibody-cross-linked sTRAIL for the indicated time points. As the positive control, wild type HeLa cells were treated with 10 ng/ml TNF for 5 min (rightmost lane). Cell lysates were subjected to Western blot analysis using a phospho-IκBα (Ser-32/36)-specific antibody. Blots were then reprobed for total IκBα. Tubulin-α was used as the loading control. B, relative phospho-IκBα band intensities were densitometrically quantified and normalized to the tubulin-α loading control. The normalized phospho-IκBα level of TNF-treated HeLa cells (i.e. the positive control) was set to 100%, and all other values were normalized to this control. ***, p < 0.001 was considered to be significant as determined by two-way analysis of variance and Bonferroni post-test in comparison to the respective time point of TRAIL treated HeLa cells. Values shown are the mean ± S.E. (n = 5).
Mathematical Modeling of TRAILR4-mediated Receptor Interference
To facilitate a more quantitative evaluation of the efficiencies of the various proposed modes of interaction by which TRAILR4 could interfere with TRAILR1 signaling, we developed a mathematical model for TRAILR1-TRAILR4 interplay. Clearly, establishment of a mathematical model quantitatively describing the interference of (potential) pro- and anti-apoptotic intracellular signals of these two receptors is far beyond the scope of this work. Yet as this mechanism could be conclusively excluded by our experiments, a theoretical analysis of intracellular signaling was not essential. We, therefore, decided to mathematically describe only membrane processes schematically depicted in Fig. 6, namely the decoy mechanism and receptor-receptor interactions, to estimate their impact on the relative signaling strength. The resulting mathematical model has been parameterized with own data and literature data. TRAILR1 was estimated to be expressed at a level of 10 000 molecules per cell by comparison of flow cytometric data with other cellular systems expressing known amounts of receptors. For HeLa R4 cells the same number of TRAILR4 was assumed; for HeLa R4+ cells a 10-fold excess, i.e. 100,000 molecules per cell at the cell surface. Initially, all affinity values for the molecular interactions were taken from literature (7). However, using these affinity values for receptor-receptor interactions, only marginal dimerization of receptors was found in our mathematical model. Rather, the vast majority of the molecules existed as monomers. This can be easily understood, because Lee et al. (7) had used the extracellular domains of the TRAIL receptors in a classic plasmon resonance study, i.e. with one partner immobilized to a carrier and the interacting molecule present in solution, allowing the latter to freely diffuse in all three dimensions, to freely rotate, etc. Cell-bound TRAIL receptors, in contrast, are fixed by their transmembrane domains in the plasma membrane in a more or less directed and parallel aligned manner. Diffusion is only possible within the level of the plasma membrane, strongly increasing their chance for proper PLAD-mediated interactions. We, therefore, increased the kon values for receptor-receptor interactions, thereby lowering the KD values until ∼80% of all TRAILR1 molecules (in the absence of TRAILR4) molecules were incorporated in dimers. Increasing the kon values for TRAILR4 interactions by the same factor revealed that ∼90% of TRAILR4 was complexed due to its somewhat higher affinity (7) in a respective virtual TRAILR4-positive cell lacking TRAILR1. The relative distribution of TRAILR1 monomers, TRAILR1 homodimers, and TRAILR1/TRAILR4 heterodimers predicted from the model in the absence of ligand is shown in Fig. 7A. To account for parameter uncertainties, we considered not only the scaled version of the parameters determined by Lee et al. (7) but sampled parameters in their proximity. All qualitative results stated below hold under parameter uncertainties, and quantitative results show little variation.
FIGURE 6.
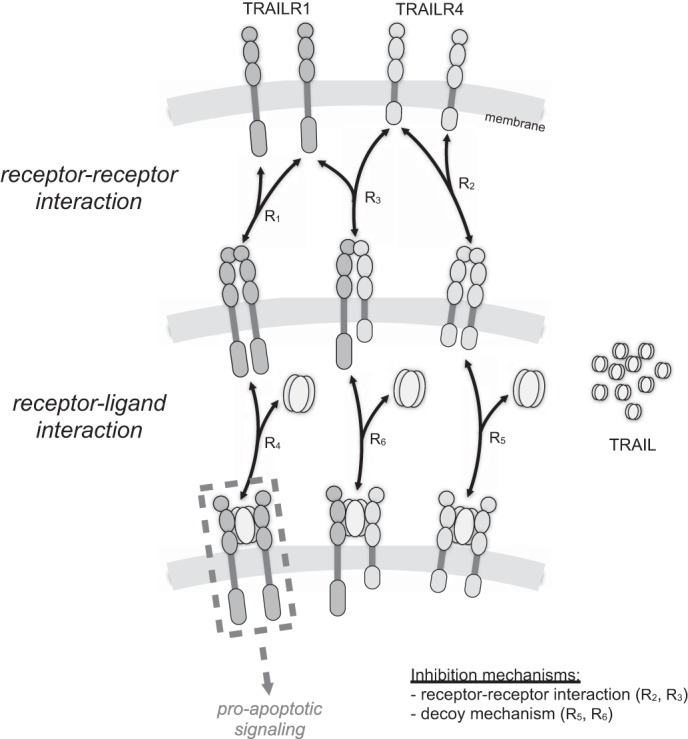
Mathematical model for receptor-receptor and receptor-TRAIL interactions. Receptor-receptor and receptor-ligand interactions have been described by six reversible biochemical reactions. The resulting biochemical reaction network accounts for the formation of homo- and heteromeric receptor complexes as well as for a decoy mechanism via TRAILR4. We derived the corresponding ordinary differential equation model assuming mass action kinetics. Parameters for the model have been determined using literature and own experimental data.
FIGURE 7.
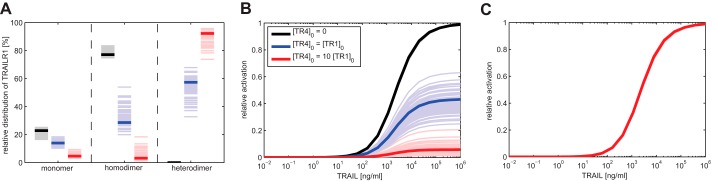
Effect of decoy mechanism on TRAIL signaling is negligible. A, relative distribution of TRAILR1 in three different receptor complexes as predicted by mathematical modeling for HeLa (expressing only TRAILR1; black), HeLa R4 (expressing TRAILR1 and TRAILR4 at comparable levels; blue), and HeLa R4+ cells (expressing ∼10-fold higher levels of TRAILR4 than TRAILR1; red) in the absence of ligand. B, dose response for the predicted proapoptotic activity of TRAIL, represented by the abundance of (TRAILR1)2-TRAIL complexes. C, dose response for the predicted proapoptotic activity of TRAIL, assuming that no receptor heterodimers are formed and that TRAILR4 merely inhibits apoptosis induction through a decoy mechanism. Lines showing relative receptor activation for the three cell lines are virtually identical and, therefore, indistinguishable. Bold lines represent the prediction for the parameter values stated under “Results.” Thin lines correspond to further parameter samples and are used to illustrate the effects of uncertainties in receptor-receptor interaction kinetics.
We assumed that TRAILR1 homodimers are capable of inducing intracellular signals after ligation, whereas TRAILR4 homodimers and receptor heterodimers are not. The addition of ligand was assumed to lead to the incorporation of ligated receptor dimers into larger ligand-receptor clusters, thereby converting TRAILR1 homodimers into an active signaling state. This model of death receptor signal initiation by changes in the backbone conformation of the molecules was recently proposed for TNFR1 by Sachs and co-workers (29) and is in excellent agreement with our own experimental data obtained in a chimeric TNFR/CD95 system (20, 21). At high TRAIL concentrations the majority of the initial receptor monomers was efficiently included in ligand-receptor clusters. For HeLa R4 and HeLa R4+ cells, the relative amount of TRAILR1 homodimers is reduced to 44 and 8%, respectively, as compared with wild type HeLa cells lacking TRAILR4 (Fig. 7B). Accordingly, TRAILR1 receptors in HeLa R4 and HeLa R4+ cells were predicted to initiate intracellular signaling with the correspondingly reduced strength. Albeit the strength of a receptor-induced signal cannot be necessarily translated directly into the magnitude of an induced cellular response, these results are in good accordance with our presented experimental data (Figs. 3D and 5B). In summary, the mathematical model confirms that interference of TRAILR4 with TRAILR1 at the level of signal complex formation can represent a very efficient mechanism under the here used conditions.
Our mathematical model also allowed us to quantify the effects of the classical decoy mechanism. Setting the interaction between TRAILR1 and TRAILR4 to zero, ligand-consuming effects by TRAILR4 could be estimated and were found to be totally negligible (Fig. 7C). Clearly, our model only describes the initial situation after addition of ligand, thereby disregarding ligand consumption by receptor internalization and recovery of the latter by de novo synthesis. However, in our assay systems ligand is available in a large excess as compared with the receptor numbers. We calculated that at the highest TRAIL concentration used in our experiments (300 ng/ml) the ratio of TRAIL molecules available per TRAILR4 molecule is ∼3000 in HeLa R4 cells or ∼300 in HeLa R4+ cells. Even if TRAILR4 would be continuously internalized with ligand and fully replaced by newly synthesized molecules, thereby consuming one TRAIL molecule per receptor and hour, the residual ligand concentration at the end of a typical cytotoxicity assay would still be >90% of its initial concentration, allowing full activation of the death receptor(s).
DISCUSSION
Stoichiometry of PLAD Interaction
Homo-oligomerization of TNF receptor family members in absence of ligand has been reported for CD95/Fas and the TNF and TRAIL receptors (7, 13–15, 20, 21, 27, 28, 32). Reports concerning the stoichiometry of these complexes, however, have been contradictory. Formation of dimers, trimers, and higher order oligomers has been suggested based on cross-linking studies with bifunctional chemical reagents. As the experimental conditions and the cross-linking agents used in these studies differ considerably, it is hard to evaluate these conflicting data. Based on our results from FRET studies we suggested that among the TRAIL receptors, TRAILR2 might form ligand-independent homooligomers with particularly high efficiency (Fig. 1B). For detailed cross-linking studies we, therefore, used mutants of this receptor where the cytoplasmic part had been replaced by a FLAG tag or eGFP, implying that interactions mediated by the intracellular part should be canceled out. In fact, at higher BS3 concentrations formation of the presumed TRAILR2ΔC-FLAG protein homodimer cross-linking product in the cell membrane was near to total as indicated by the disappearance of the band representing the monomer (Fig. 2B). Moreover, no band likely to represent cross-linked homotrimers of the receptor construct appeared even at the highest BS3 concentration. Thus, it can be assumed that ligand-independent pre-assembly of the TRAIL receptor constructs used in our studies strongly favors the formation of receptor homodimers.
By coexpressing both the FLAG- and the eGFP-tagged versions of TRAILR2, we were able to perform additional control experiments. After chemical cross-linking, receptor complexes were immunoprecipitated with a FLAG-specific antibody and analyzed using an α-eGFP antibody. This revealed a strong band with the expected molecular mass of the heterodimer (∼80 kDa) comprising one molecule of each TRAILR2ΔC-FLAG (27 kDa) and TRAILR2ΔC-eGFP (53 kDa) (Fig. 2D). Again, no evidence for cross-linking products with a higher molecular weight was obtained. The same approach was repeated using cells coexpressing two different types of TRAIL receptors fused either to eGFP or a FLAG epitope tag (i.e. TRAILR1-FLAG and TRAILR2ΔC-eGFP, etc.). This approach yielded precisely the same results; that is to say the exclusive detection of dimeric TRAIL receptor complexes (data not shown), indicating that not only homotypic yet also heterotypic receptor-receptor interactions result in the (predominant) dimerization of TRAIL receptors.
The chemical BS3 is a cross-linking agent that is water-soluble but not membrane-permeable, hence being potentially reactive with the extracellular N terminus of TRAILR2 plus six available lysine residues present in the extracellular part. Presuming the receptors preferentially form homotrimers or even higher oligomers in the plasma membrane, it appears likely that aggregates higher than dimers would have been formed and detected in our cross-linking experiments. In fact, when we cross-linked soluble TNF with BS3, representing a homotrimer (3 × 17 kDa) also containing 6 lysine residues per monomer, we readily detected a strong band of the supposed TNF homotrimer (data not shown). This indicates that an underestimation of the number of receptor molecules constituting the pre-assembled oligomers is unlikely. Together, all experiments we performed with the aim to investigate the stoichiometry of TRAIL receptor homo- and heteromers strongly suggest the preferential existence as homo- and heterodimers on the cell membrane. This implication is in accordance with recent data obtained with TNF receptor/Fas chimeric molecules (20, 21, 32) as well as the recently proposed model by Sachs and co-workers (29) for signal initiation by wild type TNFR1.
Strikingly, however, Sachs and co-workers (28) also performed BS3 cross-linking experiments with TRAILR2, albeit in its wild type form, and demonstrate the predominant formation of homotrimers. Differences in the experimental design that might account for the conflicting results include the cell lines as well as the receptor constructs used. Valley et al. (28) used the hematopoietic human lines Jurkat and BJAB, which might be both capable of producing the ligand TRAIL, which could form endogenous receptor complexes on the cell surface. Indeed, in our hands Jurkat cells are positive for membrane TRAIL expression as determined by flow cytometry (data not shown). Another obvious difference in both studies is the use of wild type TRAILR2 by Valley et al. (28) versus constructs lacking the functional intracellular part in our studies. Notably, Valley et al. (28) point out that in fact TRAILR2 interacts with its cytoplasmic parts in the absence of ligand.
Molecular Mechanisms and Quantitative Aspects of TRAILR1/TRAILR4 Interference
As recently discussed by Kimberley and Screaton (2), four different mechanisms might determine the interference of the so-called decoy receptors with TRAILR1 (and TRAILR2) in a nonexclusive manner. The first is the classical decoy receptor mechanism based on competition of both receptors for their shared ligand. Efficiency of this molecular mechanism is governed by the respective ligand binding affinities. However, the ligand binding affinities of TRAIL decoy receptors are comparable to those of the death receptors (7, 9, 33), arguing against a high efficacy of the decoy mechanism. In fact, our model predicts virtually no decoy-mediated interference of an even 10-fold excess of TRAILR4 on signaling of TRAILR1 at the initial phase of a standard cytotoxicity assay (Fig. 7C). Because of the large excess of ligand this situation will not change over many hours, arguing that the decoy mechanism of TRAIL receptor interference might play only a role in particular situations like very low ligand concentrations and very high expression rates of decoy receptors. Clearly, the decoy mechanism is insufficient to explain the molecular mechanism underlying the efficient inhibitory effects of TRAILR4 on TRAILR1 signaling in our cellular system.
A second possible mechanism is based on the reported anti-apoptotic signaling capacity of TRAILR4, e.g. via activation of NFκB or PKB/Akt (8, 18, 34). This potential mechanism could also be excluded in our experiments by using a TRAILR4 variant lacking the intracellular signaling domain. This receptor variant strongly interfered with TRAILR1-mediated induction of apoptosis (Fig. 4B) and activation of NFκB (Fig. 5, A and B). Moreover, using our HeLa R4 and HeLa R4+ cells we could not detect any TRAIL-induced phosphorylation of PKB/Akt (data not shown), confirming that TRAILR-mediated intracellular signaling might be highly cell type-specific as indicated by the inconsistent reports (8, 18, 19, 34–36). Furthermore, using a commercial intracellular signaling antibody array no signals, like activation of PKB/Akt, ERK1/2, or SAPK/JNK were detected, which could be attributed to TRAILR4. However, poly(ADP-ribose) polymerase cleavage, likely to be mediated by TRAILR1, could be easily detected in this assay (data not shown).
Excluding signaling by TRAILR4 and the decoy mechanism, we therefore propose that TRAILR4 forms mixed signaling complexes together with TRAILR1, which have altered, reduced, or even no signaling capability. Mixed complexes might be either formed in a strictly ligand-dependent manner (the third possible mechanism of receptor interference proposed by Kimberley and Screaton (2)), might be guided by the preformed mixture of PLAD-mediated homo- and heterodimers of receptors (mechanism four), or their production might be even more complex (22). By all means, all PLAD-PLAD affinities appear to be comparable (7) as is the case for ligand binding affinities of the four membrane receptors (7, 9, 33). Accordingly, one has to assume that after ligand binding TRAIL death and decoy receptors occur on the plasma membrane in a complex conglomerate of homo- and heteromers within ligand-receptor clusters.
In good agreement with the predictions from our mathematical model, where the distribution of PLAD-mediated homo- and heteromers (Fig. 7A) was translated into TRAILR1-mediated signaling strength as a function of the ligand concentration (Fig. 7B), we demonstrate that TRAILR4 is a very efficient negative regulator of both apoptosis induction (Fig. 3D) and activation of NFκB (Fig. 5, A and B). It appears likely that the interference between TRAILR4 and TRAILR2 might be guided by similar mechanisms. The functional role of TRAILR3 could be similar as compared with TRAILR4, although its differential distribution on the cellular membrane must be taken into account. Accordingly, the TRAIL signaling system in human cells is of exceptional complexity. For example, in cells coexpressing all 4 transmembrane TRAIL receptors, 10 different combinations of receptor dimers are possible. Whereas the signaling pattern of death receptor homodimers is fairly well elucidated, the signaling capability of heteromers like TRAILR1/R2 but also TRAILR1/R4 and TRAILR2/R4 awaits further investigation.
Acknowledgments
We thank Jessica Tepperink and Beate Schmieder for excellent technical assistance, Harald Wajant for plasmids pCR3 DR4, pCR3 DR5, and pCR3 TRAILR4, Felix Neugart for the MATLAB plugin FRET_Plotter, and Fabian Theis for helpful comments regarding the mathematical modeling.
This work was supported in part by the German Research Foundation (DFG) within the Cluster of Excellence in Simulation Technology (EXC 310/1) at the University of Stuttgart, German Federal Ministry of Education and Research (BMBF) Grant PREDICT 0316186A (to S. N. and P. S.), and by the BMBF within Virtual Liver Project Grant 0315766 and LungSys II Grant 0316042G (to J. H.).
- TRAIL
- TNF-related apoptosis-inducing ligand
- PLAD
- pre-ligand binding assembly domain
- DD
- death domain
- DR
- death receptor
- NFκB
- nuclear factor κ-light-chain-enhancer of activated B cell
- BS3
- bis(sulfosuccinimidyl)-suberate
- MFI
- mean fluorescence intensity
- AU
- arbitrary units
- PKB
- protein kinase B
- MF
- mouse fibroblasts
- eGFP
- enhanced GFP.
REFERENCES
- 1. Johnstone R. W., Frew A. J., Smyth M. J. (2008) The TRAIL apoptotic pathway in cancer onset, progression, and therapy. Nat. Rev. Cancer 8, 782–798 [DOI] [PubMed] [Google Scholar]
- 2. Kimberley F. C., Screaton G. R. (2004) Following a TRAIL: update on a ligand and its five receptors. Cell Res. 14, 359–372 [DOI] [PubMed] [Google Scholar]
- 3. Zinonos I., Labrinidis A., Lee M., Liapis V., Hay S., Ponomarev V., Diamond P., Findlay D. M., Zannettino A. C., Evdokiou A. (2011) Anticancer efficacy of Apo2L/TRAIL is retained in the presence of high and biologically active concentrations of osteoprotegerin in vivo. J. Bone Miner. Res. 26, 630–643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Toni E. N., Thieme S. E., Herbst A., Behrens A., Stieber P., Jung A., Blum H., Göke B., Kolligs F. T. (2008) OPG is regulated by β-catenin and mediates resistance to TRAIL-induced apoptosis in colon cancer. Clin. Cancer Res. 14, 4713–4718 [DOI] [PubMed] [Google Scholar]
- 5. Varfolomeev E., Maecker H., Sharp D., Lawrence D., Renz M., Vucic D., Ashkenazi A. (2005) Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 280, 40599–40608 [DOI] [PubMed] [Google Scholar]
- 6. Jin Z., El-Deiry W. S. (2006) Distinct signaling pathways in TRAIL- versus tumor necrosis factor-induced apoptosis. Mol. Cell. Biol. 26, 8136–8148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee H. W., Lee S. H., Lee H. W., Ryu Y. W., Kwon M. H., Kim Y. S. (2005) Homomeric and heteromeric interactions of the extracellular domains of death receptors and death decoy receptors. Biochem. Biophys. Res. Commun. 330, 1205–1212 [DOI] [PubMed] [Google Scholar]
- 8. Degli-Esposti M. A., Dougall W. C., Smolak P. J., Waugh J. Y., Smith C. A., Goodwin R. G. (1997) The novel receptor TRAIL-R4 induces NF-κB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 7, 813–820 [DOI] [PubMed] [Google Scholar]
- 9. Truneh A., Sharma S., Silverman C., Khandekar S., Reddy M. P., Deen K. C., McLaughlin M. M., Srinivasula S. M., Livi G. P., Marshall L. A., Alnemri E. S., Williams W. V., Doyle M. L. (2000) Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J. Biol. Chem. 275, 23319–23325 [DOI] [PubMed] [Google Scholar]
- 10. Pan G., Ni J., Wei Y.-F., Yu G.-l., Gentz R., Dixit V. M. (1997) An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 [DOI] [PubMed] [Google Scholar]
- 11. Degli-Esposti M. A., Smolak P. J., Walczak H., Waugh J., Huang C. P., DuBose R. F., Goodwin R. G., Smith C. A. (1997) Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J. Exp. Med. 186, 1165–1170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hymowitz S. G., Christinger H. W., Fuh G., Ultsch M., O'Connell M., Kelley R. F., Ashkenazi A., de Vos A. M. (1999) Triggering cell death: the crystal structure of apo2L/TRAIL in a complex with death receptor 5. Mol. Cell 4, 563–571 [DOI] [PubMed] [Google Scholar]
- 13. Chan F. K., Chun H. J., Zheng L., Siegel R. M., Bui K. L., Lenardo M. J. (2000) A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288, 2351–2354 [DOI] [PubMed] [Google Scholar]
- 14. Siegel R. M., Frederiksen J. K., Zacharias D. A., Chan F. K., Johnson M., Lynch D., Tsien R. Y., Lenardo M. J. (2000) Fas preassociation required for apoptosis signaling and dominant inhibition by pathogenic mutations. Science 288, 2354–2357 [DOI] [PubMed] [Google Scholar]
- 15. Clancy L., Mruk K., Archer K., Woelfel M., Mongkolsapaya J., Screaton G., Lenardo M. J., Chan F. K. (2005) Preligand assembly domain-mediated ligand-independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 102, 18099–18104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mongkolsapaya J., Grimes J. M., Chen N., Xu X.-N., Stuart D. I., Jones E. Y., Screaton G. R. (1999) Structure of the TRAIL-DR5 complex reveals mechanisms conferring specificity in apoptotic initiation. Nat. Struct. Biol. 6, 1048–1053 [DOI] [PubMed] [Google Scholar]
- 17. Mérino D., Lalaoui N., Morizot A., Schneider P., Solary E., Micheau O. (2006) Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell. Biol. 26, 7046–7055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lalaoui N., Morlé A., Mérino D., Jacquemin G., Iessi E., Morizot A., Shirley S., Robert B., Solary E., Garrido C., Micheau O. (2011) TRAIL-R4 promotes tumor growth and resistance to apoptosis in cervical carcinoma HeLa cells through AKT. PLoS ONE 6, e19679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harper N., Farrow S. N., Kaptein A., Cohen G. M., MacFarlane M. (2001) Modulation of tumor necrosis factor apoptosis-inducing ligand-induced NF-κB activation by inhibition of apical caspases. J. Biol. Chem. 276, 34743–34752 [DOI] [PubMed] [Google Scholar]
- 20. Branschädel M., Aird A., Zappe A., Tietz C., Krippner-Heidenreich A., Scheurich P. (2010) Dual function of cysteine rich domain (CRD) 1 of TNF receptor type 1: conformational stabilization of CRD2 and control of receptor responsiveness. Cell. Signal. 22, 404–414 [DOI] [PubMed] [Google Scholar]
- 21. Boschert V., Krippner-Heidenreich A., Branschädel M., Tepperink J., Aird A., Scheurich P. (2010) Single chain TNF derivatives with individually mutated receptor binding sites reveal differential stoichiometry of ligand receptor complex formation for TNFR1 and TNFR2. Cell. Signal. 22, 1088–1096 [DOI] [PubMed] [Google Scholar]
- 22. Winkel C., Neumann S., Surulescu C., Scheurich P. (2012) A minimal mathematical model for the initial molecular interactions of death receptor signaling. Math. Biosci. Eng. 9, 663–683 [DOI] [PubMed] [Google Scholar]
- 23. Gerken M., Krippner-Heidenreich A., Steinert S., Willi S., Neugart F., Zappe A., Wrachtrup J., Tietz C., Scheurich P. (2010) Fluorescence correlation spectroscopy reveals topological segregation of the two tumor necrosis factor membrane receptors. Biochim. Biophys. Acta 1798, 1081–1089 [DOI] [PubMed] [Google Scholar]
- 24. Huang D. C., Cory S., Strasser A. (1997) Bcl-2, Bcl-x(L) and adenovirus protein E1B19kD are functionally equivalent in their ability to inhibit cell death. Oncogene 14, 405–414 [DOI] [PubMed] [Google Scholar]
- 25. Krippner-Heidenreich A., Tübing F., Bryde S., Willi S., Zimmermann G., Scheurich P. (2002) Control of receptor-induced signaling complex formation by the kinetics of ligand/receptor interaction. J. Biol. Chem. 277, 44155–44163 [DOI] [PubMed] [Google Scholar]
- 26. Stepensky D. (2007) FRETcalc plugin for calculation of FRET in non-continuous intracellular compartments. Biochem. Biophys. Res. Commun. 359, 752–758 [DOI] [PubMed] [Google Scholar]
- 27. Papoff G., Hausler P., Eramo A., Pagano M. G., Di Leve G., Signore A., Ruberti G. (1999) Identification and characterization of a ligand-independent oligomerization domain in the extracellular region of the CD95 death receptor. J. Biol. Chem. 274, 38241–38250 [DOI] [PubMed] [Google Scholar]
- 28. Valley C. C., Lewis A. K., Mudaliar D. J., Perlmutter J. D., Braun A. R., Karim C. B., Thomas D. D., Brody J. R., Sachs J. N. (2012) Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces death receptor 5 networks that are highly organized. J. Biol. Chem. 287, 21265–21278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lewis A. K., Valley C. C., Sachs J. N. (2012) TNFR1 signaling is associated with backbone conformational changes of receptor dimers consistent with overactivation in the R92Q TRAPS mutant. Biochemistry 51, 6545–6555 [DOI] [PubMed] [Google Scholar]
- 30. Gonzalvez F., Ashkenazi A. (2010) New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 29, 4752–4765 [DOI] [PubMed] [Google Scholar]
- 31. Azijli K., Weyhenmeyer B., Peters G. J., de Jong S., Kruyt F. A. (2013) Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell. Death. Differ. 20, 858–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Richter C., Messerschmidt S., Holeiter G., Tepperink J., Osswald S., Zappe A., Branschädel M., Boschert V., Mann D. A., Scheurich P., Krippner-Heidenreich A. (2012) The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol. Cell. Biol. 32, 2515–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tur V., van der Sloot A. M., Reis C. R., Szegezdi E., Cool R. H., Samali A., Serrano L., Quax W. J. (2008) DR4-selective tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) variants obtained by structure-based design. J. Biol. Chem. 283, 20560–20568 [DOI] [PubMed] [Google Scholar]
- 34. Hu W.-H., Johnson H., Shu H.-B. (1999) Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-κB and JNK activation and apoptosis through distinct pathways. J. Biol. Chem. 274, 30603–30610 [DOI] [PubMed] [Google Scholar]
- 35. Meng R. D., McDonald E. R., 3rd, Sheikh M. S., Fornace A. J., Jr., El-Deiry W. S. (2000) The TRAIL decoy receptor TRUNDD (DcR2, TRAIL-R4) is induced by adenovirus-p53 overexpression and can delay TRAIL-, p53-, and KILLER/DR5-dependent colon cancer apoptosis. Mol. Ther. 1, 130–144 [DOI] [PubMed] [Google Scholar]
- 36. Marsters S. A., Sheridan J. P., Pitti R. M., Huang A., Skubatch M., Baldwin D., Yuan J., Gurney A., Goddard A. D., Godowski P., Ashkenazi A. (1997) A novel receptor for Apo2L/TRAIL contains a truncated death domain. Curr. Biol. 7, 1003–1006 [DOI] [PubMed] [Google Scholar]