

Background: Cyclic AMP allosterically modulates GNAT-like acyltransferases (KAT) in mycobacteria.
Results: Structures of the protein from M. smegmatis and its mutants are described.
Conclusion: The divergent properties of KATs from M. tuberculosis and M. smegmatis are the result of differential conformational dynamics in the two proteins.
Significance: Apparently similar proteins show subtle differences in their structural features, having evolved based on the needs of the organism.
Keywords: Allosteric Regulation, Crystal Structure, Cyclic AMP (cAMP), Molecular Dynamics, Mycobacteria, Protein Acylation, Acyltransferase
Abstract
Mycobacteria harbor unique proteins that regulate protein lysine acylation in a cAMP-regulated manner. These lysine acyltransferases from Mycobacterium smegmatis (KATms) and Mycobacterium tuberculosis (KATmt) show distinctive biochemical properties in terms of cAMP binding affinity to the N-terminal cyclic nucleotide binding domain and allosteric activation of the C-terminal acyltransferase domain. Here we provide evidence for structural features in KATms that account for high affinity cAMP binding and elevated acyltransferase activity in the absence of cAMP. Structure-guided mutational analysis converted KATms from a cAMP-regulated to a cAMP-dependent acyltransferase and identified a unique asparagine residue in the acyltransferase domain of KATms that assists in the enzymatic reaction in the absence of a highly conserved glutamate residue seen in Gcn5-related N-acetyltransferase-like acyltransferases. Thus, we have identified mechanisms by which properties of similar proteins have diverged in two species of mycobacteria by modifications in amino acid sequence, which can dramatically alter the abundance of conformational states adopted by a protein.
Introduction
It is now appreciated that protein molecules exist in an ensemble of different conformations at equilibrium, and this conformational diversity provides a mechanism that allows multifunctionality as well as regulation of the activity of a protein (1, 2). The presence of multiple domains in a protein further provides an opportunity for allosteric regulation whereby binding of a ligand to one domain can alter the relative ratios of different conformational states of the protein at equilibrium. This “conformational selection” induced by ligand binding can now be reflected in a change in protein activity (3). Therefore, an understanding of the structural features of similar proteins with distinct biochemical properties can provide a molecular appreciation of the means by which the properties of individual proteins are regulated by allosteric effectors.
Cyclic AMP serves as an allosteric regulator for a number of effector proteins with a variety of functions (4, 5). We have recently reported that cAMP can regulate the acylation of lysine residues in mycobacterial proteins by binding to unique cAMP-regulated acyltransferases (6, 7). We called these proteins lysine (K) acyltransferases from Mycobacterium smegmatis (KATms)3 and Mycobacterium tuberculosis (KATmt). Cyclic AMP binds to a classical cyclic nucleotide binding (CNB) domain N-terminal to a GNAT-like acetyltransferase (AT) domain. The two domains are connected to each other by a long linker region of ∼60 residues. Despite an overall similarity in primary amino acid sequence in the CNB domains, KATms has a high affinity for cAMP (100 nm), whereas KATmt shows a 1000-fold lower affinity for cAMP (100 μm). Additionally, the basal acyltransferase activity of KATms is higher and is able to utilize acetyl coenzyme A (acetyl-CoA) to acetylate universal stress protein (USP) even in the absence of cAMP with a ∼2-fold increase in activity in the presence of cAMP. In contrast, cAMP binding to KATmt is essential to allow acetylation of USP (6).
To understand the mechanistic basis of cAMP-regulated acyltransferase activity, we utilized hydrogen deuterium exchange followed by mass spectrometry (MS) approaches and showed that cAMP binding to KATms alters the dynamics of the cAMP binding domain and the interdomain linker (8). We suggested that the linker region assumes a different conformation in the active cAMP-bound state, promoting enhanced catalysis through relief of inhibition. It was not clear at that time whether cAMP binding drives this conformational change as would be expected in the induced fit model or whether cAMP binding shifts the equilibrium to favor the active state. Recently, the crystal structure of KATmt was solved in both the presence and absence of cAMP and revealed that in the absence of cAMP in the structure KATmt adopted an autoinhibited conformation (9). The conformation of KATmt in the absence of cAMP was completely refractory to allowing substrate access to the catalytic site in the AT domain, accounting for the undetectable basal acyltransferase activity in the absence of cAMP. In contrast, upon cyclic AMP binding, a large conformational rearrangement in KATmt releases this autoinhibition with the linker region assisting in inducing the cAMP-mediated conformational switch.
However, we remained intrigued by the significant activity shown by KATms in the absence of cAMP, suggesting that distinct structural features in KATms must account for the reduced extent of autoinhibition of acyltransferase activity in the absence of cAMP. In the current study, we determined the structure of apoKATms and mutants that are compromised in cAMP binding or acyltransferase activity. The structures we describe here, along with confirmatory mutational analysis, identify distinct features that account for the divergent activities of KATms and KATmt in the absence of cAMP. Moreover, structure-driven biochemical analysis elucidated the role of specific residues in the AT domain of KATms that allow acetylation of its substrate, USP, in the absence and presence of cAMP. Finally, molecular dynamics simulations further showed the importance of the distinct amino acid sequence of the flexible regions in the KATms structure that enable sampling of the conformers in the absence of cAMP. Therefore, we have provided a molecular understanding of the basis for the vastly different biochemical properties of KATmt and KATms.
EXPERIMENTAL PROCEDURES
All fine chemicals were from Sigma-Aldrich. Routine bacterial growth medium (Luria Bertani) was purchased from Colloids Impex (India). Middlebrook 7H9 base and 7H10 agar were obtained from BD Biosciences. Restriction enzymes were from MBI Fermentas (Canada) or New England Biolabs. Nickel-nitrilotriacetic acid (Ni-NTA)-agarose was from GE Healthcare. Oligonucleotide primers were synthesized by Xcelris (India) and Macrogen (Korea), and sequences are available on request. Acetyllysine antibody was obtained from Cell Signaling Technology.
Cloning and Mutagenesis
Point mutations in MSMEG_5458 were generated by site-directed mutagenesis as described previously (10). pProMSMEG_5458 (6) was used as the template, and mutations were confirmed by sequencing (Macrogen).
Purification of Wild-type and Mutant KATms
Purification procedures were essentially as described earlier (6) with a few modifications. Cultures of Escherichia coli (either BL21(DE3) or SP850 cyc− cells) harboring the respective plasmids were grown in Terrific Broth, and protein expression was induced following addition of 0.4 mm isopropyl 1-thio-β-d-galactopyranoside. The cell lysate was loaded on Ni-NTA resin (Qiagen), and the hexahistidine-tagged protein was eluted with 50 mm Tris/HCl (pH 8.0), 100 mm NaCl, 300 mm imidazole, 5 mm 2-mercaptoethanol (2-ME), and 10% glycerol. The eluted protein was dialyzed overnight at 4 °C against 50 mm Tris/HCl (pH 8.0), 100 mm NaCl, 5 mm 2-ME, and 10% glycerol in the presence of tobacco etch virus protease to specifically cleave off the hexahistidine tag. Uncleaved protein and tobacco etch virus protease were removed by passing the sample through an Ni-NTA column in 50 mm Tris/HCl (pH 8.0), 100 mm NaCl, 10 mm imidazole, 5 mm 2-ME, and 10% glycerol. For crystallization, the final purification step included gel filtration on a Superdex 200 column (10/300 GL, GE Healthcare) equilibrated in 50 mm Tris/HCl (pH 8.0), 100 mm NaCl, 5 mm 2-ME, and 5% glycerol. Purified proteins were stored at a final concentration of 30 mg/ml at −80 °C. Se-Met-derivatized wild-type KATms was prepared and crystallized in a manner similar to the native protein.
Crystallization, Data Collection, and Crystal Structure Determination
Crystals were obtained using the hanging drop vapor diffusion technique at 20 °C and with a crystallization drop size of 2 μl with equal volumes of protein:reservoir solution. KATms wild-type Se-Met crystals were grown using a reservoir solution of 0.2 m NaCl, 0.1 m Bis-Tris (pH 6.5), 0.03 m CaCl2, 2% 1,5-diaminopentane dihydrochloride, and 19% PEG 3350. The mutant KATms_R95K formed crystals with a reservoir solution of 0.2 m NaCl, 0.1 m Bis-Tris (pH 6.5), 0.03 m MgCl2, 1.7% 1,5-diaminopentane dihydrochloride, and 21–23% PEG 3350. The mutant KATms_E234A formed crystals with a reservoir solution of 0.2 m NaCl, 0.1 m Bis-Tris (pH 6.5), 0.03 m CaCl2, 2% 1,5-diaminopentane dihydrochloride, and 21% PEG 3350. Several rounds of microseeding were needed to improve the crystals. All crystals were cryoprotected in 25–30% (v/v) ethylene glycol before freezing in liquid nitrogen. Crystals of Se-Met derivatives were additionally soaked with 3.2 mm HgCl2 for 2–4 h before freezing.
Crystallization was attempted in the presence of cAMP and/or acetyl-CoA, but the crystals obtained diffracted poorly. Poor diffraction was also obtained after soaking KATms crystals with cAMP and/or acetyl-CoA.
All x-ray diffraction data sets were collected at the synchrotron Elettra (Trieste, Italy; beamline XRD) at the wavelengths 0.975 Å for the Se-Met derivative of KATms wild type and KATms_E234A mutant and 1.000 Å for KATms_R95K mutant. Diffraction data were processed and scaled using HKL2000 and Scalepack (11). Data statistics are presented in Table 1. Phases for Se-Met crystals were obtained using anomalous data and PHENIX (12). The initial model of KATms wild type was improved with PHENIX Autobuild (12) and Arp/Warp (13, 14) followed by cycles of manual building in O (15). Structures of the KATms mutants R95K and E234A were determined by molecular replacement with Phaser (16) using KATms wild-type structure as the search model followed by cycles of manual building in O. Refinement of all structures was carried out in REFMAC (14, 17). Refinement statistics are given in Table 1. All figures were made in PyMOL. Protein Data Bank codes are listed in Table 1.
TABLE 1.
Crystallographic statistics
The number of crystals for each structure is one.
| KATms_WT | KATms_R95K | KATms_E234A | |
|---|---|---|---|
| Data collection | |||
| Beamline | XRD Elettra | XRD Elettra | XRD Elettra |
| Protein Data Bank code | 4OLL | 4ORF | 4ONU |
| Wavelength (Å) | 0.975 | 1.000 | 0.975 |
| Space group | P212121 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (Å) | 38.30, 82.75, 105.51 | 38.29, 82.85, 104.37 | 38.32, 82.76, 105.97 |
| α, β, γ (°) | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 | 90.0, 90.0, 90.0 |
| Resolution range (Å) | 41.40–1.90 | 44.20–2.00 | 41.40–2.25 |
| High resolution shell (Å) | 1.94–1.90 | 2.05–2.00 | 2.30–2.25 |
| Rmerge | 0.102 (0.668)a | 0.047 (0.648) | 0.111 (0.655) |
| I/σI | 21.9 (3.8) | 26.1 (1.8) | 22.1 (4.3) |
| Completeness (%) | 99.3 (98.4) | 96.9 (96.6) | 99.7 (100.0) |
| Redundancy | 12.8 (11.4) | 4.3 (3.8) | 12.7 (12.0) |
| Refinement | |||
| Rwork (%) | 20.6 | 22.0 | 20.0 |
| Rfree (%)b | 26.3 | 27.6 | 26.3 |
| Number of atoms (protein/water/ions) | 2473/171/3 | 2485/124/3 | 2453/124/2 |
| Average B-factors (protein/water/ions) | 55.3/49.1/59.4 | 58.3/52.8/60.3 | 56.0/45.8/57.1 |
| r.m.s.d. bond length (Å) | 0.018 | 0.017 | 0.015 |
| r.m.s.d. bond angle (°) | 1.9 | 1.8 | 1.7 |
| Ramachandran most favored (%) | 91.4 | 91.9 | 91.7 |
a Values in parentheses are for highest resolution shell.
b Rfree values were calculated from a random set of reflections.
Western Blot Analysis
Protein samples were electrophoresed on a 13.5% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membrane (Immobilon-P, Millipore). The membrane was incubated with primary antibody overnight at 4 °C. KATms and USP polyclonal antisera were generated in the laboratory (6) and used at a dilution of 1:5000. Anti-acetyllysine antibody was used at a dilution of 1:2500. Horseradish peroxidase-conjugated secondary antibody (GE Healthcare) was used and detected by enhanced chemiluminescence (Luminata Crescendo, Millipore).
In Vitro Acetylation Assays
Acetylation assays were carried out in a total reaction volume of 20 μl containing 25 mm Tris/HCl (pH 7.5), 100 mm NaCl, 5 mm EDTA, and 100 μm acetyl-CoA. USP (2 μg) prepared as described (6) was used as a substrate. The reaction was initiated by adding 200 ng of KATms in the presence or absence of cAMP (10 μm). Reactions were carried out at 22 °C for 10 min, terminated by boiling in 4× SDS sample buffer (200 mm Tris/HCl (pH 6.8), 8% SDS, 40% glycerol, 4% 2-ME, 50 mm EDTA, and 0.08% bromphenol blue), and analyzed by immunoblotting with acetyllysine antibody and enhanced chemiluminescence.
Continuous assays were performed as described earlier (6). The assay reaction mixtures contained 0.2 mm NAD, 0.2 mm thiamine pyrophosphate, 5 mm MgCl2, 1 mm DTT, 2.4 mm α-ketoglutarate, 50 μm acetyl-CoA, 50 μm USP, 0.03 unit of α-ketoglutarate dehydrogenase, 190 nm KATms, and 20 mm HEPES (pH 7.5) in a total volume of 100 μl. The reaction was initiated by the addition of USP. The rates were analyzed continuously for 10 min by measuring NADH production at 340 nm using a UV-visible spectrophotometer (TECAN Infinite Pro Series).
Molecular Dynamics
The atomic detail structural models of wild-type KATms_WT and mutant KATms_P170H were constructed by the molecular modeling program CHARMM (18) starting from the crystal structure of the wild-type protein. Residues of the KATms_WT that were not defined in the crystal structure were obtained from on-site energy minimization of the corresponding peptide chains while keeping the rest of the protein structure fixed. The protein was then solvated in an explicit water environment modeled by the TIP3P water model (19) with at least 10-Å padding in each direction from the protein, resulting in an orthogonal periodic box of 92 × 72 × 62 Å (42,448 atoms). In addition, 10 sodium ions were added to achieve electroneutrality of the system. The solvent molecules and the hydrogen atoms in the protein were relaxed by a 2000 step minimization with the backbone atoms restrained at the initial structure. After the relaxation, the system was gradually heated from 0 to 300 K in 200-ps molecular dynamics (MD) simulation in the NVT (constant number of particles, volume, and temperature) ensemble in the absence of any structural restraint. An additional 500-ps MD simulation was performed at constant pressure (1 bar) and at 300 K temperature (constant number of particles, pressure, and temperature (NPT)). At this point, the mutation P170H was introduced in the wild-type structure by replacing the side chain of Pro170 with that of His. Overlapping water molecules were removed before structural optimization of the mutation site. Subsequently, we carried out a 35-ns NPT MD simulation on both systems. All simulations were performed using the CHARMM22 force field (20), and the particle mesh Ewald method (21) was used to treat long range electrostatic interactions with a direct space cutoff of 12 Å.
RESULTS
Overall Structure of KATms
The polypeptide chain of wild-type KATms contains 333 residues that fold into two major domains (Fig. 1A). The N-terminal CNB-like domain comprises residues Val1–Phe139, and the C-terminal AT domain comprises residues Asp203–Val310. The two domains are connected by residues Ile140–Ala202, which we have earlier called the linker peptide (6). The conserved AT domain is followed by an α-helix (α10 or C-terminal helix), a unique extension seen in these acyltransferases, that spatially connects the AT domain with the cAMP binding site in the CNB domain (Figs. 1A and 2).
FIGURE 1.

Crystal structure of KATms_WT. A, ribbon structure of KATms_WT (dark red, CNB domain; cyan, linker peptide; green, AT domain; orange, C-terminal helix). Some residues are shown in sticks (in the CNB domain: carbon, dark red; nitrogen, blue; oxygen, bright red; in AT domain: carbon, green; nitrogen, blue; oxygen, bright red; in the linker peptide: carbon, cyan; nitrogen, blue; oxygen, bright red). The missing part of the linker (linker gap) is marked. Top, schematic representation of the structure with the same color code as in ribbons. B, representation of the electron density in part of the KATms_WT structure (interface among the tip of the C-terminal helix, linker peptide, and CNB domain). KATms is shown in sticks with the same color code as in A. A 2Fo − Fc electron density map is shown as a gray mesh contoured at 1 σ. Water molecules are marked as red spheres. C, superposition of KATms_WT and KATmt_apo (Protein Data Bank code 4AVA) on the CNB domain. KATms is shown in the same colors as in A, and KATmt_apo is shown in gray. Two highly conserved residues of KATms and the N and C termini are marked. Acetyl-CoA from KATmt is shown in sticks (carbon, magenta; oxygen, red; nitrogen, blue; phosphorus, orange; sulfur, yellow). D, superposition of KATms and KATmt-cAMP (Protein Data Bank code 4AVB) on the CNB domain. KATms is shown in the same colors as in A, and KATmt-cAMP is shown in gray. Two highly conserved residues of KATms and the N and C termini are marked. cAMP in KATmt-cAMP is shown in sticks (carbon, light green; oxygen, red; nitrogen, blue; phosphorus, orange).
FIGURE 2.
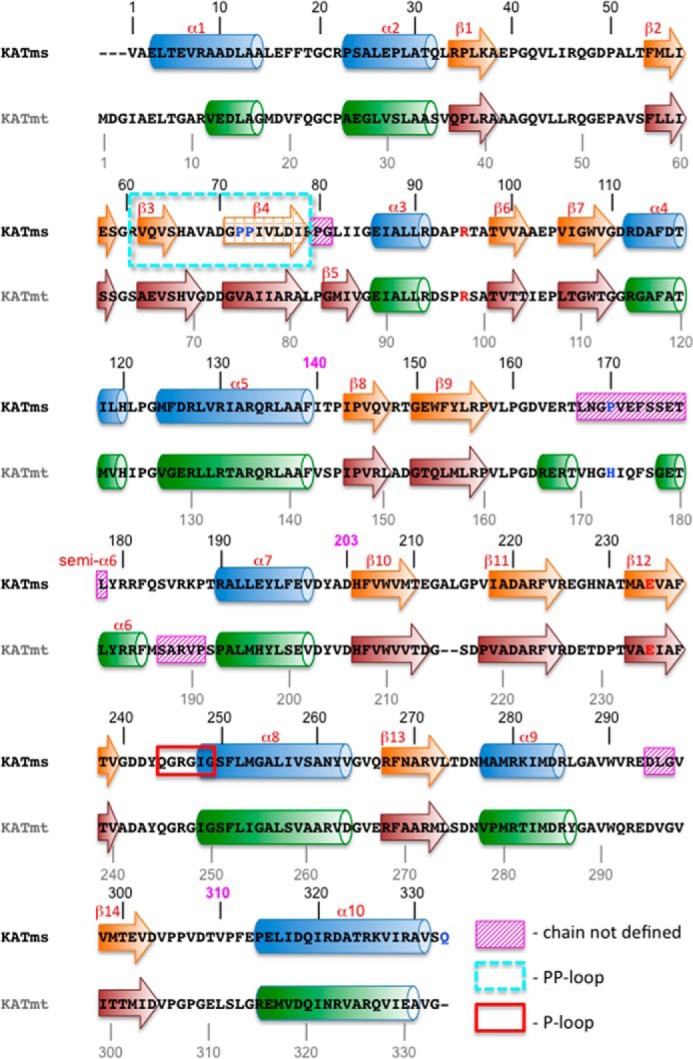
Two-dimensional structural alignment of KATms_WT with KATmt_apo (Protein Data Bank code 4AVA). α-Helices are depicted as cylinders, and β-strands are depicted as arrows and are numbered. The number 140 in magenta above the KATms sequence marks the border the between CNB domain and the linker peptide, 203 marks the border between the linker and the AT domain, and 310 marks the end of the AT domain in KATms. Missing residues in the structures are labeled with magenta hatched rectangles, the PP-loop is labeled with a cyan dashed rectangle, and the P-loop (25) is labeled with a red rectangle. The dashed arrow depicting β4-strand in KATms shows the element that is geometrically close to a β-strand but is not ideal. Deviation from a geometrically ideal structure is also encountered for “semi-α6” in KATms.
The polypeptide chain of KATms shows well defined electron density in the region of the compact CNB and AT domains as well in the C-terminal helix (Fig. 1B). However, electron density is broken at residues Pro80-Gly81 (the hinge at the end of the PP-loop described below), the linker peptide (residues Leu167–Leu178; Figs. 1A and 2), and residues Asp294–Gly296, suggesting high flexibility of these regions. This is in agreement with our earlier studies using hydrogen deuterium exchange followed by MS (8).
Comparison of KATms and KATmt
Polypeptide chains of KATms and KATmt each contain 333 amino acid residues with 60% identity, resulting in similar subdomain structures with high conservation of secondary structure elements (Fig. 2). A structural comparison aligning 277 Cα atoms between KATms and the apo form of KATmt (Protein Data Bank code 4AVA) (22) gave a root mean square deviation (r.m.s.d.) value of 2.5 Å. Although the structure of KATms_WT is generally similar to KATmt_apo, significant structural differences are seen in specific regions of the CNB and AT domains as described later as well as in the conformation of the linker peptide that influences the angle between the two domains (Fig. 1C). As expected, the KATms_WT_apo structure differs more profoundly from the cAMP-bound KATmt (Protein Data Bank code 4AVB) due to a large conformational change in the linker region of KATmt upon cAMP binding and thus the angle between CNB and AT domain but still retains a similar architecture of the individual CNB and AT domains (Fig. 1D).
Structural Features of the CNB Domain and cAMP Binding Site
The extreme N terminus is more structured in KATms than in KATmt with a significantly longer α1 helix (Figs. 1C and 2). The polypeptide chains start to structurally overlap at residues Ala9 and Val12 in KATms and KATmt_apo, respectively. An important structural deviation is seen in the loop between helix α1 and helix α2 probably because of Pro23, a residue that is present only in KATms, whereas other mycobacterial orthologs have an alanine at this position (6).
A second and significant difference between KATms and KATmt structures is located at the β3-turn-β4 motif (Fig. 3A), which forms a cap above the cAMP binding site. This region, including residues Arg61–Glu79 in KATms, contains a pair of proline residues (Pro72 and Pro73) uniquely present in KATms (Fig. 3A). We refer to this loop as the PP-loop. In KATms, the gate into the cAMP binding site is significantly more open than in KATmt_apo (Fig. 3B) with an angle of ∼50° between the PP-loop and the C-terminal helix. In KATmt_apo, this angle is reduced to 30°, and as discussed later, the entry to the cAMP binding pocket in KATmt is additionally blocked by the unique structural twist in the C-terminal helix α10.
FIGURE 3.

PP-loop position relative to the C-terminal helix. A, comparison of KATms_WT with the KATmt_apo structure (superposition on the CNB domain). The ribbon color code is the same as in Fig. 1. The approximate position of a bound cAMP (green sticks as in Fig. 1D) is shown by the superposition of the KATmt-cAMP structure (Protein Data Bank code 4AVB) on the CNB domain of KATms_WT. C termini of KATms (C-KATms) and KATmt_apo (C-KATmt) are marked. Right, multiple sequence alignment of mycobacterial KAT sequences in the PP-loop region (6). Unique KATms prolines (Pro72 and Pro73) are marked in red sticks. B, spatial correlation among the cAMP binding site, PP-loop, and C-terminal (α10) helix. Left panel, KATms_WT; central panel, KATmt_apo (Protein Data Bank code 4AVA); right panel, KATmt-cAMP (Protein Data Bank code 4AVB). KATmt structures are in gray except for the linker region, which is in light green, and the C-terminal helix, which is in light orange. cAMP in the KATmt-cAMP structure is shown in sticks (carbon, green; oxygen, red; nitrogen, blue; phosphorus, orange). The KATms structure is colored as in all figures. An estimation of the angle between the PP-loop and the C-terminal helix is marked with lines, and the size of the angle is in degrees. C, superposition of KATms_WT (color code as above) with KATmt_apo (Protein Data Bank code 4AVA; gray) on the C-terminal helix. KATmt residues are marked with the letters mt. D, superposition of KATms_WT (color code as above) with KATms_R95K (light blue) on the C-terminal helix. Residues in the R95K mutant are marked with a letter k. E, superposition of KATms_WT (color code as above) with KATmt-cAMP (Protein Data Bank code 4AVB; gray) on the C-terminal helix. cAMP is shown in sticks as in B. KATmt residues are marked with the letters mt.
In KATmt, the β3-turn-β4 region equivalent to the PP-loop moves significantly inward upon cAMP binding and forms a caplike feature above the cAMP binding site (Figs. 1, C and D, and 3B). In KATms, the PP-loop is in an open position in the cAMP-free structure but may move inward and become fixed following cAMP binding. This hypothesis is indeed supported by results of previous hydrogen deuterium exchange followed by MS showing a significantly decreased deuterium exchange in this region of KATms in the presence of cAMP (8).
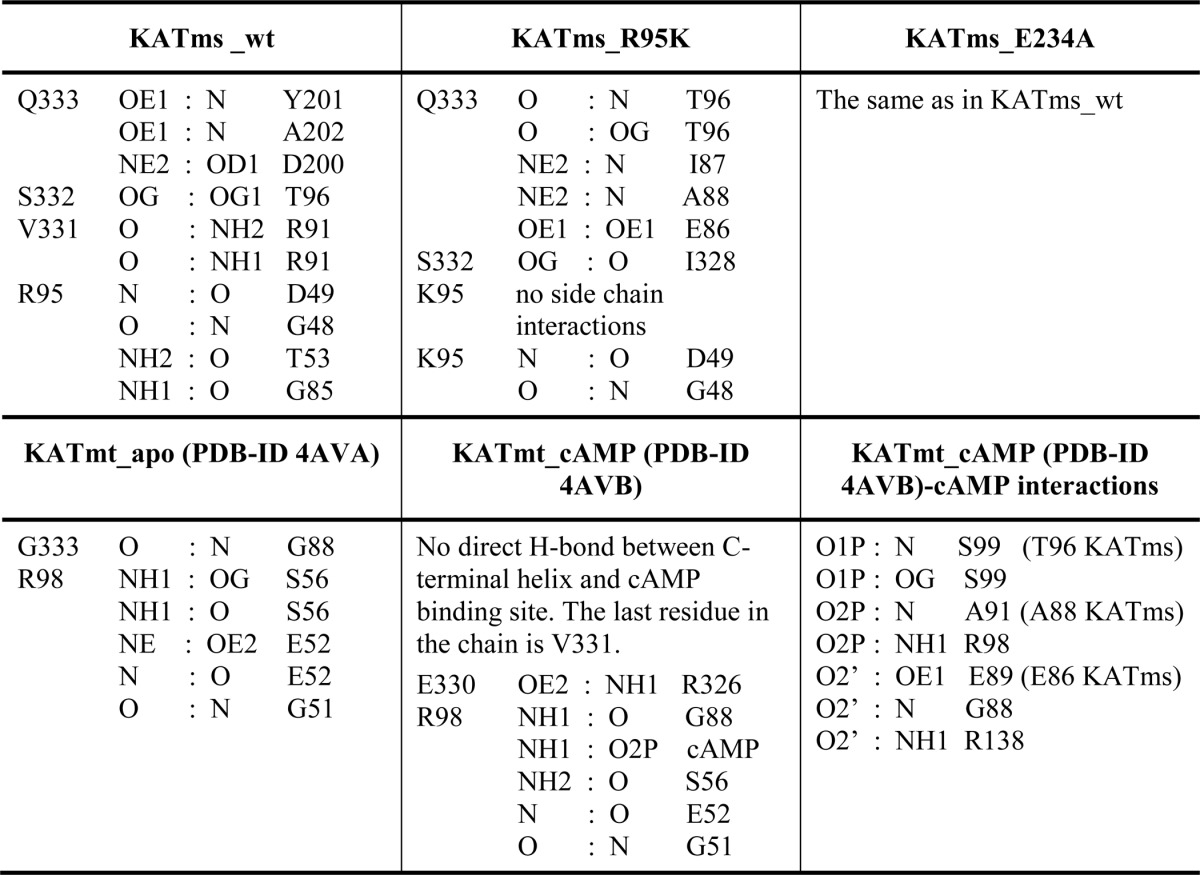
An important conserved residue in the CNB domain of KATms is Arg95, which was shown to contribute significantly to cAMP binding and following mutation abolished cAMP-mediated activation of acyltransferase activity (6). In KATmt-cAMP, the Nη1 atom of the corresponding Arg98 side chain is within hydrogen bonding distance with O2P of the phosphate moiety of the bound cAMP, but mutation of Arg98 to Ala did not abolish cAMP-mediated acetylation of USP (9). To elucidate the role of this residue, we determined the structure of the R95K mutant of KATms. Although the overall structure of KATms_R95K is very similar to KATms wild type, the mutation causes a conformational rearrangement at the cAMP binding site that now prevents access of cAMP. Although the CNB domains of KATms_WT and KATms_R95K superimpose closely with an r.m.s.d. of 0.6 Å, a significant deviation is seen in the PP-loop between KATms_WT and KATms_R95K (Fig. 3D) with the PP-loop of the R95K mutant moving slightly closer to the cAMP binding site. Moreover, we were surprised to find clearly defined electron density for showing that the final residues of the C-terminal helix of KATms_R95K take a different turn than that seen in KATms_WT, closing the cAMP binding site and making contacts with residues that at equivalent positions in KATmt-cAMP are involved in cAMP binding (Fig. 3D). For example, in the KATmt-cAMP structure, peptide nitrogens of Ala91 and Ser99 interact with the phosphate moiety of cAMP, and Glu89 interacts with the ribose ring (Table 2). However, in the KATms_R95K mutant, the carbonyl oxygen of Gln333 of the twisted C-terminal helix α10 makes hydrogen bonds to the peptide bond nitrogen as well as to Oγ atom of Thr96 (equivalent to Ser99 in KATmt), and the Gln333 Nϵ2 atom forms hydrogen bonds to the peptide bond nitrogen of Ala88 (equivalent to Ala91), thus interfering with cAMP binding (Table 2). Additionally, the side chain of Gln333 in KATms_R95K forms a hydrogen bond to the amide nitrogen of Ile87, which is also situated in the cAMP binding site, as well as to the side chain of Glu86 whose equivalent in KATmt-cAMP, Glu89, forms a hydrogen bond with a ribose moiety of cAMP (Table 2). Therefore, it can be hypothesized that this strong engagement of the C-terminal residues with residues in the cAMP binding site in the KATms_R95K mutant contributes significantly to the low binding of cAMP as determined before for KATms_R95K (6). Thus, we suggest that the loss of interactions between the Arg side chain and the rest of the cAMP binding site upon mutation to Lys facilitates the inward turn and subsequent interactions of the C-terminal helix. In concert with this structural change, the PP-loop moves toward the center of the molecule, additionally closing the cAMP binding site albeit to a lesser extent than seen in KATmt_apo (Fig. 3, C and D).
TABLE 2.
Polar interactions at the interface of the cAMP binding site and the C-terminal helix in KATms and KATmt
The Linker Peptide
The conserved fold of the CNB domain ends after helix α5 (Ile140) and is connected to the AT domain (starting at Asp203) with a 60-residue-long linker peptide (Fig. 2). The beginning of the linker peptide is well structured into the β8-strand-turn-β9-strand element, which after residue Val158 continues into a random coil structure defined in electron density up to residue Thr166. The electron density then breaks for 12 residues of sequence 167LNGPVEFSSETL178 and continues again with the residue Tyr179 at the beginning of a short semi-α6-helical fold, which then continues into helix α7 followed by the AT domain (Figs. 1A, 2, and 4A).
FIGURE 4.

Structural relationship of the linker region with the AT domain. A, the fold of the linker peptide in KATms in comparison with KATmt_apo (Protein Data Bank code 4AVA) with superposition on the CNB domain. KATms is in the same colors as in Fig. 1. KATmt_apo is in gray except for its linker peptide, which is shown in light yellow. Some secondary structure elements that are mentioned in the text are marked. The KATmt residue names have mt next to their number. Acetyl-CoA from KATmt_apo is shown in sticks (carbon, magenta; oxygen, red; phosphorus, orange; nitrogen, blue; sulfur, yellow). B, left, upper panel, multiple sequence alignment of mycobacterial KATs showing the amino acid sequences in the region of the flexible linker, including P170, in KATms. Lower panel, multiple sequence alignment of mycobacterial KATs showing the part of the AT domain with the unique residues Asn269 and Ala278 in M. smegmatis. Right, upper panel, surface of the AT domain (green) of KATms_WT. The surface of the linker residues seen in the KATms structure is in cyan, and residues Ala278 and Gln244 of KATms are in yellow. The linker loop of KATmt_apo (Protein Data Bank code 4AVA) (gray ribbon) with His173 in sticks is shown (H173mt) as well as acetyl-CoA (magenta sticks; with superposition of KATms_WT and KATmt_apo on the AT domain). Lower panel, surface of KATmt_apo (gray) showing the more open binding site for acetyl-CoA in contrast to the closed binding site in KATms. The surface of His173 of KATmt_apo is in bright green. C, superposition of KATms_WT (color code as in Fig. 1A) and KATmt_apo (gray except for the linker peptide, which is in bright yellow) on the AT domain. Acetyl-CoA from KATmt_apo is shown in sticks colored as in A. D, the same as in C with a closer look into the acetyl-CoA binding site; Glu234 and Asn269 of KATms_WT are shown in sticks. His173 from KATmt_apo and acetyl-CoA are shown in sticks as well. E, superposition of KATms_WT and KATmt_apo (Protein Data Bank code 4AVA) on the AT domain showing the hydrogen bond pattern of KATmt_apo with acetyl-CoA. KATmt is in gray except for the linker peptide (bright yellow), and His173 of KATmt is shown in sticks. The water molecule coordinated by Glu235 and His173 of KATmt is shown as a red sphere. Hydrogen bonds between KATmt protein residues and acetyl-CoA are shown as dashed red lines. Selected residues named in text are marked. KATmt residues have the extension mt. F, superposition of KATms_WT and KATmt-cAMP (Protein Data Bank code 4AVB) on the AT domain showing the hydrogen bond pattern of KATmt-cAMP with acetyl-CoA. The color code is the same as in E.
The general fold of the part of KATms linker peptide that is defined by the electron density matches relatively well with the equivalent region in KATmt_apo (Fig. 4A). However, superposition of the CNB domains of KATms and KATmt_apo reveals a structural deviation starting at the final turn of the α5 helix. This is followed by a prominent difference in the angle of the β8-strand-turn-β9-strand motif between the two structures (Fig. 4A). Although in KATms the linker residues Leu167–Leu178 are not seen, the corresponding residues in KATmt_apo are defined and make crucial contacts with the AT domain and acetyl-CoA (Fig. 4). Interestingly, residues 187SARVP191 in KATmt_apo are not defined (Fig. 2), whereas the corresponding region in KATms is defined and is exposed to solution.
The region of the linker peptide from Leu167 to Leu178 (KATms numbering) is relatively well conserved in mycobacteria (Fig. 4B). Importantly, however, Pro170 in KATms seems to be unique because it is replaced with histidine in other mycobacterial KATs, including KATmt. In KATmt_apo, the respective His173 forms a stacking interaction of the imidazole side chain with the planar acetyl group of acetyl-CoA and a hydrogen bond between the imidazole Nϵ2 atom and carbonyl oxygen of Arg272 residing in the AT domain. These interactions enable the linker peptide to play an autoinhibitory role in KATmt_apo (Ref. 9 and Fig. 4), although upon cAMP binding to KATmt, His173 moves far out of the substrate binding site, enabling acetylation of the protein substrate (Fig. 4F).
The structure of KATms confirms the role of His173 in autoinhibition of KATmt as well in binding of acetyl-CoA. The replacement of this His with a structurally distinct proline residue in KATms disables not only the interaction with potentially bound acetyl-CoA but may be also one of the reasons for very high structural flexibility of this region of the linker peptide in KATms. It is important to note that in KATmt acetyl-CoA is engaged by the linker peptide in either the absence (interaction with His173 in KATmt_apo) or presence of cAMP (interaction with the α6-helix in KATmt-cAMP). Therefore, the high flexibility of the linker and loss of the His side chain may account for the absence of the naturally bound acetyl-CoA in structures of KATms and consequently the absence of the autoinhibitory effect of the linker in KATms.
AT Domain and Acetyl-CoA Binding Site
The AT domain of KATms (residues Asp203–Val310) and KATmt_apo (residues Asp206–Leu311) superimpose very well with an r.m.s.d. value of 1.1 Å. Deviations are seen in loops Glu211–Ile218 of KATms (KATmt Asp214–Val219) and Glu226–Ala230 (KATmt Asp227–Pro231). In both cases, the loops in KATms lack proline residues that are conserved in other mycobacteria (Pro218 in KATmt corresponds to Val217 in KATms, and Pro231 in KATmt corresponds to Ala230 in KATms), which may account for the different twists of these two loops.
There is an interesting detail in the acetyl-CoA binding site in KATms (Fig. 4). The acetyl-CoA taken from the KATmt_apo structure fits relatively well into the binding channel site in KATms, indicating that the site is prebuilt in mycobacterial KATs. However, the binding channel in KATms is locked by the bridge formed by residues Gln244 and Ala278 (Fig. 4, B and E), which may contribute, along with the flexibility of the linker peptide discussed above, to preventing binding of acetyl-CoA to this conformation in KATms. Although Gln244 is conserved among other mycobacterial KATs, a Pro is found as the equivalent residue to Ala278 in other KATs (Fig. 4B). The presence of Ala278 instead of Pro may influence the shape and flexibility of the acetyl-CoA binding site in KATms.
The GNAT family of acyltransferases contains a conserved glutamate in the vicinity of the acetyl-CoA binding site (6). Catalysis in GNATs is believed to occur by direct nucleophilic attack on a substrate Lys residue by the conserved glutamate (23), potentially Glu234 in KATms and Glu235 in KATmt, located in the vicinity of the acetyl-CoA as well as the protein substrate binding site (Fig. 4, E and F). However, mutation of Glu234 to Ala in KATms reduced basal acyltransferase activity, which was increased in the presence of cAMP (6), clearly indicating that Glu234 was not the sole determinant for acyltransferase activity. We determined the structure of the E234A mutant protein and observed no dramatic change in the conformation of the acyltransferase domain. Indeed, in contrast to the KATms_R95K structure, the structure of KAT_E234A superposed almost completely with the KATms_WT structure with an overall r.m.s.d. of 0.4 Å (315 Cα atoms aligned). Therefore, we looked for an additional explanation that would account for the low catalytic activity of the E234A mutant protein in the absence of cAMP and that allowed recovery of catalytic activity on binding cAMP. The side chain of Asn269 is positioned close to the side chain of Glu234 (∼3.5 Å) (Fig. 4D). Residues in other mycobacterial KATs corresponding to Asn269 are either Ala (KATmt) or Ser (Fig. 4B). Asn269 is positioned relatively far from the putative acetyl-CoA binding site, suggesting that following cAMP binding the conformational change that occurs may bring Asn269 closer to the active site, thereby assisting efficient catalysis.
C-terminal Helix
In general, the C-terminal α10-helix in KATms is less conserved in sequence in comparison with other mycobacterial KATs (8). In KATmt, the C-terminal helix was shown to play the role of a steric latch that contributes to the autoinhibition of KATmt in the absence of cAMP (9). Extending the length of this helix by a single amino acid results in considerable acyltransferase activity of KATmt even in the absence of cAMP. The amino acid alignment of mycobacterial KATs suggests that KATms is longer by one residue at the C terminus (6). Structural alignment of KATms_WT with KATmt (Fig. 2) revealed that the α10-helix in KATms is indeed longer by one residue at the C terminus than that of KATmt. This results in a different structure of the final twist of this helix and alters its structural relationship to the cAMP binding site and the PP-loop (Fig. 3 and Table 2). In contrast to the KATmt_apo structure, the final residue of α10-helix, Gln333 in KATms_WT, does not protrude into the cAMP binding site (Fig. 3, A and C) but rather engages its side chain with hydrogen bond interactions to the residues Asp200, Tyr201, and Ala202 in the hinge between the linker peptide and AT domain (Table 2). This structural nuance may also contribute to the angular difference between the CNB and AT domains between KATms and KATmt and, thus, the higher affinity for cAMP binding to KATms than KATmt (6).
In KATmt_apo, the cAMP binding site is closed by the protrusion of the tip of the C-terminal helix and the lid formed by the PP-loop, thus preventing access of cAMP. Therefore, this structure must breathe to capture cAMP when available. For KATmt, it was shown that to bind cAMP the C-terminal helix and the PP-loop have to be pulled apart with the PP-loop now shielding cAMP within the CNB domain (Fig. 3, B, C, and E, and Ref. 9). A similar series of steps may take place in KATms as suggested by data obtained in the presence of cAMP using hydrogen deuterium exchange followed by MS (8). However, our structure shows that because of a wider opening of the CNB domain due to the more open conformation of the PP-loop and C-terminal helix (Fig. 3, A, B, and C) and the highly flexible linker peptide KATms can achieve a productive conformation in the absence of cAMP due to its more breathable structure.
Structure-based Mutational Analysis of KATms
In the absence of structures of KATms bound to either cAMP and/or acetyl-CoA, we resorted to mutational analysis to identify structural changes that may occur in KATms following binding of cAMP and acetyl-CoA. We reasoned that mutation of Pro170 (analogous to His173 in KATmt) to a Lys residue would provide an acetylatable residue in the vicinity of the active site. Although KATms contains four Lys residues, none of them appeared to be acetylated in the protein as purified from E. coli (Fig. 5A). However, we observed that KATms P170K was robustly acetylated as purified, indicating the close positioning of Lys170 to the AT domain (Fig. 5A). Based on the conformational changes upon cAMP binding seen in KATmt (9), the linker peptide of KATms containing Lys170 would move away from the active site in the presence of cAMP and be more surface-exposed. To monitor this structural change, we tested the ability of the P170K mutant protein to be deacetylated by the sirtuin enzyme Rv1151c (7). We hypothesized that in the presence of cAMP the outward movement of acetylated Lys170 would allow greater access of Rv1151c and, therefore, more efficient deacetylation. As shown in Fig. 5A, right panel, KATms_Lys170 was marginally deacetylated in the absence of cAMP but was almost completely deacetylated by Rv1151c when bound to cAMP. Therefore, we can conclude that the conformational change that occurs in KATms on cAMP binding may indeed be similar to that seen in KATmt.
FIGURE 5.

Critical role of Pro170 in KATms. A, purified wild-type KATms or mutant KATms_P170K was analyzed by Western blot analysis with acetyllysine antibody either directly or after incubation with cAMP and acetyl-CoA. KATms_P170K protein was acetylated as purified from E. coli, suggesting autoacetylation as a result of close proximity of Lys170 to the AT domain (left panel). Data shown are representative of assays performed twice. Incubation with the sirtuin enzyme of cAMP-free or cAMP-bound KATms_P170K resulted in efficient deacetylation only in the presence of cAMP (right panel), demonstrating that a conformational change had occurred in KATms following cAMP binding. Data shown are representative of assays performed twice. B, either wild-type KATms or the P170H mutant protein was incubated with USP and acetyl-CoA in either the absence or presence of cAMP, and acetylation of USP was monitored by Western blot analysis. C, either wild-type KATms or P170H mutant was incubated with [3H]cAMP in the presence of varying concentrations of unlabeled cAMP as indicated. Data shown are the mean ± S.D. (error bars) of assays repeated thrice. Inset, binding assays using purified protein (1 μg for KATms_WT (black bars) or 5 μg for KAT_ms_P170H (white bars)) in the absence and presence of 1 mm unlabeled cAMP. Values shown are bound cpm of a representative experiment repeated thrice.
We then mutated Pro170 to a His residue that is found in KATmt, predicting that this mutation may now allow autoinhibitory interactions with the linker region in KATms. In agreement with our predictions, this single mutation resulted in almost complete loss of basal AT activity in KATms, whereas the addition of cAMP restored activity to levels seen in the wild-type protein (Table 3 and Fig. 5B). We also monitored cAMP binding affinity with the P170H protein and found that the IC50 showed an increased affinity for cAMP (Ki, 28.3 ± 24.5 versus 144.4 ± 10.9 nm for wild type; Fig. 5C), although the fraction of protein capable of binding cAMP was much lower (Fig. 5C, inset). Therefore, this single residue change appeared to contribute significantly to altering the biochemical properties of KATms.
TABLE 3.
Activities of wild-type and mutant KATms proteins
Continuous monitoring of enzymatic activity was performed as described in the text using equal amounts of individual proteins (500 ng). Values shown represent the mean ± S.D. of assays performed thrice.
| Protein | NADH formed |
|
|---|---|---|
| −cAMP | +cAMP | |
| nmol/ml/min | ||
| Wild type | 19.9 ± 4.6 | 27 ± 4.9 |
| P170H | 1 ± 0.3 | 26.9 ± 5.1 |
| E234A | 1.8 ± 1.3 | 22.2 ± 7.4 |
| N269A | 15 ± 6.8 | 17.7 ± 6.1 |
| E234A/N269A | 4.8 ± 2.9 | 8.2 ± 4.5 |
| R95K/E234A | 0.1 ± 0.1 | 0.1 ± 0.01 |
An important finding that emerged from our earlier mutational analysis as well as the structure of KATms was the putative role for Asn269 in the acetylation reaction in the presence of cAMP. To confirm this, we generated the N269A mutant protein and the double mutant E234A/N269A and compared their activities with the wild type and the E234A mutant. As we had reported earlier, the basal activity of the E234A mutant protein was low with significant activity seen in the presence of cAMP (6). The N269A protein retained high activity in the absence of cAMP (Table 3 and Fig. 6A) presumably because of the presence of Glu234 but showed only a marginal increase in activity in the presence of cAMP, emphasizing the role of Asn269 in the acetylation reaction only in the presence of cAMP. The E234A/N269A double mutant protein showed low activity both in the absence and presence of cAMP.
FIGURE 6.

Role of Glu234 and Asn269 in the AT domain of KATms. A, acetylation of USP was monitored in the presence of either wild-type or mutant proteins as indicated by Western blot analysis using an acetyllysine antibody. Data shown are representative of assays performed twice. B, acetylation of USP was monitored in the presence of wild-type or mutant KATmt proteins as indicated in the presence or absence of cAMP. Data shown are representative of assays repeated twice.
To confirm that Asn269 assists in the enzymatic reaction in the presence of cAMP, we generated the R95K/E234A protein where binding of cAMP is prevented by the R95K mutation. This protein showed very low activity in both the absence and presence of cAMP (Table 3). KATmt contains an Ala at an equivalent position to Asn269. Therefore, we mutated Glu235 in KATmt to an Ala and monitored activity in either the presence or absence of cAMP. The mutant protein showed poor activity in either the presence or absence of cAMP (Fig. 6B), once again reiterating the importance of Asn269 in KATms.
Molecular Dynamics Simulation
Our results so far demonstrate that the structure of KATms has regions of high flexibility that account for its high basal acyltransferase activity. Therefore, we performed MD simulations on KATms_WT and a model where Pro170 was mutated to His to identify regions of the molecule that contribute most to the dynamics of the structure. Structural changes during 35-ns MD simulation of KATms_WT and KATms_P170H, monitored as r.m.s.d. relative to the crystal structure of the wild-type protein, are shown in Fig. 7A. Following the equilibration phase (∼10 ns) when both undergo major structural relaxations, the systems reached a relatively stable phase for the simulation times greater than 15 ns. The average r.m.s.d. of the backbone per residue was calculated relative to the averaged structure of each protein over the last 20 ns of trajectories. Pronounced peaks were observed for residues 169–178 (residues within the linker peptide) of the wild-type protein and for residues 66–76 (residues within the PP-loop) of the KATms_P170H mutant (Fig. 7B).
FIGURE 7.

Molecular dynamics simulations of KATms. A, snapshot of the superimposed KATms_WT (red) and KATms_P170H mutant (blue) structures at the end of MD simulation. Enhanced segments (described under “Results”) shown in green for KATms_WT and purple for KATms_P170H mutant (mut) are selected to describe relative structural changes of functional loops during the simulation. B, average r.m.s.d. per residue of Cα atoms relative to the averaged structure during the final 20 ns of the MD simulation is shown. Black bars indicate the positions of the PP-loop (residues 61–79), linker peptide (residues 140–202), and the α10-helix (residues 314–331). C, distances between centers of mass of selected segments depicted in green and purple as defined in A as a function of simulation time.
We next analyzed the inter-residual distance as a function of time during the simulation. We first defined representative reference points as centers of mass of the following segments: 68–74 (PP-loop) and 322–329 (α10-helix) for the “PP-loop opening” of the protein and 172–175 (linker peptide) and 294–296 (AT domain) for the “linker peptide opening” (Fig. 7A). Time evolution of representative distances as a function of time (Fig. 7C) demonstrated a larger opening (distance, ∼16 Å in the mutant versus ∼12 Å in the wild type) of the loop 68–74 relative to the α10-helix (PP-loop opening) in the KATms_P170H mutant. The opening of the substrate binding site or the linker peptide opening was smaller than that of the wild type (distance, ∼24 versus ∼27 Å, respectively). Furthermore, these significant movements of the PP-loop and the (modeled) linker peptide appear to be synchronized. In the wild-type protein, the PP-loop moves toward the cAMP binding domain and the C-terminal helix during the simulation, whereas the linker peptide moves away from the substrate binding site. This would allow KATms to adopt a conformation that enables the acetylation of the protein substrate even in the absence of cAMP.
The PP-loop of the P170H mutant protein oscillated significantly during the simulation as seen from pronounced peaks for residues 66–76 (Fig. 7B). Moreover, the PP-loop of mutated KATms turned out to be more open relative to the KATms_WT during MD simulation as revealed by larger distances of the PP-loop opening (Fig. 7C). As a consequence, mutated KATms is thus expected to bind cAMP with even higher affinity than KATms_WT and therefore supports our experimental data (Fig. 5C). Simultaneously, the linker peptide seems to tend to close the protein substrate binding site, which is in agreement with lower acyltransferase activity in the absence of cAMP for the P170H mutant (Table 3 and Fig. 5B). Other parts of the structure stayed relatively stable during the MD simulations (Fig. 7B).
DISCUSSION
In this study, we identified critical differences in the structures of KATms and KATmt that now translate to dramatic differences in their biochemical properties. In contrast to KATmt, a significant fraction of KATms must exist in an active conformer, thus accounting for significant acyltransferase activity in the absence of cAMP. As we have shown here, single amino acid changes can have profound effects in determining the distribution of conformational ensembles of a protein.
KATms and KATmt share 60% identity in amino acid sequence and, therefore, similarity of the overall fold between their crystal structures. However, detailed inspection of the structural elements and spatial relation between them reveals (i) differences in the angle between the CNB and AT domains, (ii) a substantially more open access to the cAMP binding site in KATms, (iii) the absence of acetyl-CoA in the AT domain of KATms, and (iv) a longer C-terminal helix with a distinct final twist in KATms. The crucial structural elements that contribute to these differences are the PP-loop of the CNB domain, the linker peptide, and the C-terminal helix. Interestingly, sequence alignment of KATs from diverse mycobacterial species (6) shows that many residues that contribute to the unique properties of KATms are also unique to this protein.
The C-terminal helix attached to the AT domain is a unique structural element present in mycobacterial KATs. A salt bridge is formed between Asp73 and Arg326 from the C-terminal helix in KATmt, thereby closing access to the cAMP binding site. We suggest that this engagement of the C-terminal helix with the CNB domain partly contributes to the low affinity of KATmt for cAMP. In KATms, the C-terminal helix is one residue longer than that of KATmt, resulting in a dramatic change of the twist of the final turn of the helix that loosens its spatial interaction with the cAMP binding site. This lengthening of the helix results in the engagement of its tip residue, Gln333, with the body of the enzyme at the junction between the linker peptide and the AT domain. Thus, this contributes to further uncoupling of the already open conformation of the PP-loop, resulting in a distance between the corresponding residues Asp70 and Arg325 in KATms that is too large to allow the salt bridge formation seen in KATmt. Consequently, the KATms cAMP binding site is significantly less occluded than in KATmt, resulting in a 1000-fold higher affinity for cAMP in KATms (6).
Another striking difference between KATms and KATmt is the role of the highly conserved Arg in the cAMP binding site that anchors cAMP via interaction of its side chain with the phosphate group of the nucleotide (9). In KATms, Arg95 seems to be crucial for cAMP binding because the KATms_R95K mutant does not bind cAMP and shows no enhancement of acetylation in the presence of cAMP (6). This mutation also structurally blocks the cAMP binding site consequent to interaction with the C-terminal helix, resulting in a different angle between CNB and AT domains and the movement of the PP-loop closer to the nucleotide binding site than that seen in the wild-type structure. In contrast, mutation of the conserved Arg98 in KATmt to Lys or Ala did not completely abolish cAMP-regulated acyltransferase activity (Fig. 6B and Ref. 9, respectively). A plausible explanation could again be found in the shorter C-terminal helix in KATmt, which, because of its less flexible final turn, cannot lock the KATmt_R98K mutant at the interface with the CNB domain to the extent seen in KATms, thereby allowing a significant fraction of protein to adopt the cAMP-binding, active conformation.
The regulatory role of the linker peptide as seen in KATmt is reduced in KATms by the presence of Pro170 (His173 in KATmt), which contributes to high flexibility of the central part of the linker peptide of the KATms apo form. As a consequence, there is no occlusion of the protein substrate binding site in KATms as seen in KATmt via interaction of His173 with acetyl-CoA. Consequently, KATms can catalyze the transfer of the acyl group to the protein substrate in the absence of cAMP. However, the linker peptide of KATms must still contribute to the rotational dynamics of the two conserved domains that bring the enzyme into a conformation that allows cAMP binding and consequently increased AT activity. The conformational flexibility of the linker peptide noted in KATmt (9) was confirmed by biochemical analysis of KATms_P170K mutant (Fig. 5A).
Mutation of Pro170 to His abolished acyltransferase activity of KATms in the absence of cAMP, generating a cAMP-dependent protein acyltransferase as is KATmt. The presence of the imidazole side chain in KATms_P170H, therefore, may now help engage the linker region with Glu234 of KATms, thus preventing substrate binding. However, it is interesting that although the P170H protein as purified contained a large fraction that was unable to bind cAMP the affinity of the fraction that retained cAMP binding was even higher that of the wild-type KATms. This can be explained by the presence of the longer C-terminal helix and more open PP-loop of KATms. Furthermore, the interaction of the linker peptide with the AT domain in KATms_P170H could result in an energetically more stable structure than that of the wild-type protein, thereby diminishing the fraction of protein present in an active conformation with open cAMP and protein substrate binding sites.
The relevance of the Pro170 in KATms was further supported by the results of molecular dynamics studies on KATms_WT and KATms_P170H, which showed prominent differences in PP-loop and substrate binding site flexibility between the two proteins. This confirms our hypothesis that Pro170 is one of the most crucial residues that enables KATms structural flexibility in the absence of cAMP, and the presence of a histidine residue as seen in KATmt allows activity of the enzyme only in the presence of cAMP.
Both available KATmt structures contain acetyl-CoA bound to the protein as purified from E. coli (9). In contrast, none of the KATms structures presented here have bound acetyl-CoA. The stacking interaction of His173 of the linker peptide with acetyl-CoA in KATmt could account for its continued presence in the AT domain even during purification procedures. Such an interaction is not possible in KATms due to the flexible linker and absence of the conserved histidine. Comparison of the acetyl-CoA binding site in KATms with that seen in KATmt and other GNAT superfamily members showed that although the acetyl-CoA binding site is basically prebuilt in KATms the entrance of the binding cleft is blocked by a bridge formed between residue Ala278 and Gln244. Thus, some motion will be required in the AT domain to allow acetyl-CoA binding during the enzymatic reaction. Recently solved structures of a GNAT member from Pseudomonas aeruginosa (24) in the absence or presence of acetyl-CoA reveal that a slight broadening of the cleft between the conserved loop (also called P-loop in GNATs (Fig. 2 and Ref. 25)) preceding α8-helix and the neighboring α9-helix (KATms annotation) in the AT domain occurs upon acetyl-CoA binding. Therefore, breathing of the KATms structure could pull apart the bridge between Gln244 of the P-loop and Ala278 of α9-helix and allow acetyl-CoA to slide into the binding cleft.
Another important difference between KATms and KATmt lies in the vicinity of the highly conserved Glu235 residue in KATmt (Glu234 in KATms). We show that mutation of Glu235 in KATmt abolished catalytic activity in the presence or absence of cAMP, whereas in KATms, the E234A mutant protein retained activity in the presence of cAMP. This indicates that Glu234 may not act as a general base during catalysis in KATms and may play a role in functional substrate binding. Because of structural similarity, a similar function could be proposed for Glu235 in KATmt. Indeed, His173 in the linker region of KATmt_apo probably mimics the position of Lys of the substrate (9), and it superimposes quite well with the substrate cis-4-hydroxy-l-proline seen in the structure of another GNAT acetyltransferase, Mpr1 (Protein Data Bank code 3W6X (26)). Glu235 in KATmt_apo coordinates a water molecule, which in turn is hydrogen-bonded to His173 (Fig. 4E). This water remains at this position after cAMP binding (Fig. 4F) and, thus, is probably involved in coordination of the protein substrate. In Mpr1, Asn135 is found at an equivalent position to Glu235 in KATmt and was shown to be important for substrate recognition (26). Conversely, the residue critical for catalysis was found to be Asn178, which is located on the other side of the acetyl-CoA binding cleft. It was proposed that the side chain of Asn178 stabilizes the thiolate anion of acetyl-CoA after the degradation of the tetrahedral intermediate and that this thiolate anion could be protonated by the water molecule that is coordinated by the Asn178 side chain (26). KATmt and KATms both have asparagine at a position equivalent to Asn178 (Asn277 in KATmt and Asn276 in KATms) (Fig. 4, E and F). In fact, in the KATmt structure, Asn277 does offer an H-bond to acetyl-CoA oxygen O5P and coordinates a water molecule and, thus, perhaps performs a role similar to that of Asn178 in Mpr1.
In contrast to KATmt, the E234A mutation in KATms was almost as active as the wild-type protein in the presence of cAMP. We show here that Asn269 is required to support the catalytic reaction in the absence of the side chain of Glu234 following the conformational rearrangement that occurs in cAMP-bound KATms. Asn269 in the wild-type protein might correctly orient and locate Glu234 in a manner similar to that seen in Mpr1 where Asn172 assists the function of Asn135 (present at a position equivalent to Glu234 in KATms) by maintaining its correct orientation and location (26). Thus, rather than in catalysis, both Glu234 and Asn269 in KATms may have a role to play in substrate positioning and perhaps substrate-induced conformational changes that are unique to KATms.
In conclusion, our structural analyses of wild-type and mutant KATms proteins reveal important differences that can account for the distinct biochemical properties of KATmt and KATms. Although our results clearly show that cAMP-free KATmt and KATms show unique structural features, we predict that the conformations of cAMP-bound structures are also likely to be subtly different. Clearly, these proteins have evolved to suit the distinct environments that M. tuberculosis and M. smegmatis inhabit and the demands placed on them to regulate their biology by cAMP and protein acylation.
Acknowledgments
We acknowledge the help of Davor Obradović with preparation and crystallization of proteins. We thank the staff at the synchrotron facility Elettra in Trieste, Italy (beamline XRD) for support with data collection and Gregor Gunčar for assistance with the structure determination.
This work was supported by the Slovenian Research Agency (to M. P., K. R., and F. M.), the Department of Science and Technology Indo-Slovene Joint Science and Technology Program (to M. P. and S. S. V), and an Intensification of Research in High Priority Area proposal (to S. S. V).
The atomic coordinates and structure factors (codes 4OLL, 4ORF, and 4ONU) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- KATms
- lysine acyltransferase from M. smegmatis
- KATmt
- lysine acyltransferase from M. tuberculosis
- GNAT
- Gcn5-related N-acetyltransferase
- CNB
- cyclic nucleotide binding
- AT
- acetyltransferase
- USP
- universal stress protein
- Ni-NTA
- nickel-nitrilotriacetic acid
- 2-ME
- 2-mercaptoethanol
- Bis-Tris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MD
- molecular dynamics
- r.m.s.d.
- root mean square deviation
- KAT
- lysine acyltransferase.
REFERENCES
- 1. Caetano-Anollés G., Wang M., Caetano-Anollés D., Mittenthal J. E. (2009) The origin, evolution and structure of the protein world. Biochem. J. 417, 621–637 [DOI] [PubMed] [Google Scholar]
- 2. James L. C., Tawfik D. S. (2003) Conformational diversity and protein evolution—a 60-year-old hypothesis revisited. Trends Biochem. Sci. 28, 361–368 [DOI] [PubMed] [Google Scholar]
- 3. Wrabl J. O., Gu J., Liu T., Schrank T. P., Whitten S. T., Hilser V. J. (2011) The role of protein conformational fluctuations in allostery, function, and evolution. Biophys. Chem. 159, 129–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berman H. M., Ten Eyck L. F., Goodsell D. S., Haste N. M., Kornev A., Taylor S. S. (2005) The cAMP binding domain: an ancient signaling module. Proc. Natl. Acad. Sci. U.S.A. 102, 45–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rehmann H., Wittinghofer A., Bos J. L. (2007) Capturing cyclic nucleotides in action: snapshots from crystallographic studies. Nat. Rev. Mol. Cell Biol. 8, 63–73 [DOI] [PubMed] [Google Scholar]
- 6. Nambi S., Basu N., Visweswariah S. S. (2010) cAMP-regulated protein lysine acetylases in mycobacteria. J. Biol. Chem. 285, 24313–24323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nambi S., Gupta K., Bhattacharyya M., Ramakrishnan P., Ravikumar V., Siddiqui N., Thomas A. T., Visweswariah S. S. (2013) Cyclic AMP-dependent protein lysine acylation in mycobacteria regulates fatty acid and propionate metabolism. J. Biol. Chem. 288, 14114–14124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nambi S., Badireddy S., Visweswariah S. S., Anand G. S. (2012) Cyclic AMP-induced conformational changes in mycobacterial protein acetyltransferases. J. Biol. Chem. 287, 18115–18129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee H. J., Lang P. T., Fortune S. M., Sassetti C. M., Alber T. (2012) Cyclic AMP regulation of protein lysine acetylation in Mycobacterium tuberculosis. Nat. Struct. Mol. Biol. 19, 811–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shenoy A. R., Visweswariah S. S. (2003) Site-directed mutagenesis using a single mutagenic oligonucleotide and DpnI digestion of template DNA. Anal. Biochem. 319, 335–336 [DOI] [PubMed] [Google Scholar]
- 11. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 12. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Perrakis A., Morris R., Lamzin V. S. (1999) Automated protein model building combined with iterative structure refinement. Nat. Struct. Biol. 6, 458–463 [DOI] [PubMed] [Google Scholar]
- 14. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones T. A., Zou J. Y., Cowan S. W., Kjeldgaard M. (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. A 47, 110–119 [DOI] [PubMed] [Google Scholar]
- 16. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Vagin A. A., Steiner R. A., Lebedev A. A., Potterton L., McNicholas S., Long F., Murshudov G. N. (2004) REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr. D Biol. Crystallogr. 60, 2184–2195 [DOI] [PubMed] [Google Scholar]
- 18. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 20. MacKerell A. D., Jr., Bashford D., Bellott M., Dunbrack R. L., Jr., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., 3rd, Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 21. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 22. Krissinel E., Henrick K. (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60, 2256–2268 [DOI] [PubMed] [Google Scholar]
- 23. Dyda F., Klein D. C., Hickman A. B. (2000) GCN5-related N-acetyltransferases: a structural overview. Annu. Rev. Biophys. Biomol. Struct. 29, 81–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Majorek K. A., Kuhn M. L., Chruszcz M., Anderson W. F., Minor W. (2013) Structural, functional, and inhibition studies of a Gcn5-related N-acetyltransferase (GNAT) superfamily protein PA4794: a new C-terminal lysine protein acetyltransferase from Pseudomonas aeruginosa. J. Biol. Chem. 288, 30223–30235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vetting M. W., de Carvalho L. P., Yu M., Hegde S. S., Magnet S., Roderick S. L., Blanchard J. S. (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch. Biochem. Biophys. 433, 212–226 [DOI] [PubMed] [Google Scholar]
- 26. Nasuno R., Hirano Y., Itoh T., Hakoshima T., Hibi T., Takagi H. (2013) Structural and functional analysis of the yeast N-acetyltransferase Mpr1 involved in oxidative stress tolerance via proline metabolism. Proc. Natl. Acad. Sci. U.S.A. 110, 11821–11826 [DOI] [PMC free article] [PubMed] [Google Scholar]