

Background: The genome of the major malaria parasites encodes a single Kinesin-5 homolog.
Results: MMV666693 is a selective allosteric inhibitor of Plasmodium Kinesin-5.
Conclusion: Plasmodium Kinesin-5 is druggable and susceptible to allosteric inhibition.
Significance: This is the first demonstration of allosteric control of a non-human Kinesin-5 by a small chemical and opens the door to new antimalarials.
Keywords: Allosteric Regulation, High Throughput Screening (HTS), Kinesin, Malaria, Plasmodium, MMV666693
Abstract
Plasmodium falciparum and vivax are responsible for the majority of malaria infections worldwide, resulting in over a million deaths annually. Malaria parasites now show measured resistance to all currently utilized drugs. Novel antimalarial drugs are urgently needed. The Plasmodium Kinesin-5 mechanoenzyme is a suitable “next generation” target. Discovered via small molecule screen experiments, the human Kinesin-5 has multiple allosteric sites that are “druggable.” One site in particular, unique in its sequence divergence across all homologs in the superfamily and even within the same family, exhibits exquisite drug specificity. We propose that Plasmodium Kinesin-5 shares this allosteric site and likewise can be targeted to uncover inhibitors with high specificity. To test this idea, we performed a screen for inhibitors selective for Plasmodium Kinesin-5 ATPase activity in parallel with human Kinesin-5. Our screen of nearly 2000 compounds successfully identified compounds that selectively inhibit both P. vivax and falciparum Kinesin-5 motor domains but, as anticipated, do not impact human Kinesin-5 activity. Of note is a candidate drug that did not biochemically compete with the ATP substrate for the conserved active site or disrupt the microtubule-binding site. Together, our experiments identified MMV666693 as a selective allosteric inhibitor of Plasmodium Kinesin-5; this is the first identified protein target for the Medicines of Malaria Venture validated collection of parasite proliferation inhibitors. This work demonstrates that chemical screens against human kinesins are adaptable to homologs in disease organisms and, as such, extendable to strategies to combat infectious disease.
Introduction
Malaria continues to be a major world health problem, with over one-quarter billion new cases a year worldwide. Over the last few years, different strategies and resultant lead compounds to combat this disease have been put forth in the literature. These measures have had some success in practice (reviewed in Refs. 1 and 2), but given that our basic understanding of this infectious organism requires scientific tools that are currently missing, it is not such a surprise that these efforts have not kept pace with growing drug resistance. Hence, the number of effective drugs has been whittled down to a handful of compounds over the past decade (reviewed in Refs. 3 and 4).
New therapeutic strategies to positively impact malaria disease outcomes are urgently needed. Two non-overlapping screening approaches have commonly been used to find new antimalarial candidates. First, recent high throughput screens based on phenotypic assays against living parasites have been successful in identifying lead compounds that effectively halt parasite proliferation (5, 6). However, subsequent development of lead compounds is hampered by lack of information regarding the identity and binding sites of the cellular target(s), necessary to feed structure-activity relationship chemical optimization strategies and inform potential human homolog cross-reactivity.
Second, classic targeted approaches strive to design selective inhibitors to defined target enzymes. Labor-intensive, such strategies have also been restricted mechanistically in the biochemical approach to attacking the parasite. Although promising candidates are being pursued, these varied strategies have not led to new, clinically effective antimalarial therapies due, in part, to the major effort required for tuning candidate structures toward high selectivity of the small chemical inhibitor for the parasite target ortholog. In each case, cross-reactivity to the orthologous mammalian enzyme remains a major concern in preliminary experiments with lead compounds (e.g. see Refs. 7–12). The challenge to develop selective agents with targeted approaches has been a formidable obstacle to overcome in bringing such agents to the clinic.
Existing targeted strategies have also been restricted in choice of cellular target. To date, chemotherapeutic agents targeting the malarial parasite can be sorted into a small number of classes that are directed against limited aspects of the metabolism of this pathogen, such as pyrimidine metabolism (12, 13), folate biosynthesis (10), myristoylation (8), and mitochondrial respiration (9, 14). Missing from the list of current antimalarial drug targets are any therapies directly targeting mitosis. Although elements of cell division have been and continue to be probed for antimalarial potential, including DNA replication (10, 11, 15, 16) and microtubule assembly and function (17–19), specific mitotic targets have not been validated in Plasmodium heretofore. The essential and conserved roles of mitotic enzymes in all eukaryotes argue for the directed development of this class of novel antimalarial candidates. Herein, our goal was to develop second generation small molecule antimalarials that target this underexploited aspect of the Plasmodium life cycle.
As a microtubule cross-linking enzyme, the Kinesin-5 family is required for efficient cell division in all eukaryotes examined and is essential in nearly all (20). The essential Kinesin-5 subfamily mitotic motor proteins bear two important attributes that make them particularly tractable for drug discovery in high throughput screening experiments. Active kinesin motor domain constructs are readily expressed in high yield in bacteria and purified with a small number of steps, which makes this protein target amenable to high throughput screening and further biochemical, biophysical, and cellular study (21–23).
In addition, Kinesin-5 proteins house a druggable allosteric pocket that is conserved within the motor domain and yet variable in sequence across orthologs (20, 24, 25). Human Kinesin-5 inhibitors have been noted for their high degree of specificity for the target enzyme and lack of off-target effects (reviewed in Refs. 26–28). The vast majority of existing drug hits to human Kinesin-5 target the allosteric site, defined by loop-5, and not the highly conserved active site. Furthermore, the poorly conserved residues of loop-5 between paralogs and orthologs confer high selectivity to specific inhibitors, thereby preventing cross-reactivity to other kinesin homologs in different species.
In this work, our approach marries the above two screening approaches; our targeted screen tested, in part, lead compounds that have already been validated as potential antimalarials in phenotypic screens. Recovery of previously validated phenotypic lead compounds as hits in our targeted screen permits rapid confirmation of novel target enzyme importance. Our main hypothesis is that the “druggability” of Kinesin-5 will be conserved in Plasmodium, and screens of such Plasmodium targets will probably recover allosteric inhibitors that exhibit high selectivity and no cross-reactivity with human kinesins. As well as being clinically relevant, new drug leads will also add to the toolkit of probes used to more fully understand the biology of this pathogen.
MATERIALS AND METHODS
Sequence Identification and Phylogenetic Analysis
Plasmodium vivax and Plasmodium falciparum kinesin sequences were identified via the Plasmodium Genomics Resource (PlasmoDB) and cross-referenced to NCBI. The region of the full-length protein corresponding to the motor domain was chosen based on NCBI annotation. The Plasmodium amino acid sequences were analyzed along with >700 kinesin sequences from other taxa that were previously analyzed for kinesin evolutionary relationships (20). A multiple-sequence alignment and unrooted phylogeny were co-calculated using SATé (20, 29). Parameters used in the analysis were as follows: aligner, MAFFT; merger, OPAL; tree estimator, FASTTREE; maximum subproblem size, 200; decomposition, centroid; iteration limit, 20 after last improvement. A postprocessing RAxML search was performed. Run time for the SATé output was 14 days. Following kinesin family identification, sequences for non-Plasmodium taxa were removed. The phylogeny was visualized with FigTree version 1.4.0 (Fig. 1A).
FIGURE 1.

Identification of P. falciparum and P. vivax kinesins. A, left, unrooted, SATé phylogenetic tree of all kinesins in P. falciparum and P. vivax. Sequences are identified as P. falciparum (Pf) or (Pf) or P. vivax (Pv), followed by genbank GI number. Brackets indicate kinesin family affiliation (K number) of each sequence. B, sequence alignment of the motor domains for HsEg5, PvEg5, and PfEg5. Identical residues are shaded gray. Loop-5 and loop-6 segments are marked and shaded in cyan and magenta, respectively, whereas the orthosteric site residues are shaded in green. The Plasmodium motor domains are ≤45% identical to the HsEg5 motor domain, whereas their orthosteric sites are 90% identical to HsEg5. C, homology model of PvEg5 based on the 3HQD structural template (36) with loop-5, loop-6, and the orthosteric site colored as in B. D, x-ray structure of HsEg5 motor domain co-crystalized (3KEN) (34) with inhibitor (gray space-filling representation, S-trityl-l-cysteine), loop-5 (cyan), loop-6 (magenta), and ADP (yellow/red space-filling representation) to illustrate the allosteric loop-5 pocket. E, x-ray structure of HsEg5 (3HQD), colored as in D, with bound AMPPNP (space-filling representation) trapped in a prehydrolysis state.
A homology model of the PvEg5 motor domain was generated using the Swiss-Model homology modeling server (30). An alignment between the PvEg5 motor domain and HsEg5 motor domain was performed with T-Coffee (31). We chose to model PvEg5 against PDB_ID 3HQD (32), a human Kinesin-5 motor domain structure with no gaps and missing the fewest residues (Fig. 1E), and submitted the query to the Swiss-Model server to generate the homology model in Fig. 1C.
Construction of Plasmodium Kinesin-5 Motor Vectors
The synthesized codon-optimized motor domains of P. falciparum and P. vivax Kinesin-5 (PfEg5 residues 1–491 and PvEg5 residues 1–450 respectively) were cloned into Escherichia coli expression vector pET24a to form PfEg5m-pET24a and PvEg5m-pET24a. Both clones were terminated with a tobacco etch virus protease consensus (ENLYFQG) followed by a C-terminal His6 tag. PfEg5-ΔL6 was created by replacing the endogenous loop-6 composed of mostly low complexity sequence (110 amino acids) with a much shorter variant sequence based on loop-6 of the human homolog (Fig. 1, B and C, TDNGTE). All constructs were verified by DNA sequencing.
Expression and Purification of Kinesin-5 Motor Domains
The E. coli strain BL21DE3 (Invitrogen) was used for expression of P. falciparum and P. vivax protein. One ml of LB, containing 30 μg/ml kanamycin, was inoculated with a single colony to grow at 37 °C for 8 h. Starting with 100 μl of preculture, 100 ml of fresh LB, and 30 μg/ml kanamycin, the culture was grown at 37 °C overnight. Twenty-five ml of overnight culture was used to inoculate 1 liter of TB containing 30 μg/ml kanamycin. The culture was grown for 2.5–3.0 h in TB medium to reach A600 nm 1.5–1.8, at which point, 0.5 mm isopropyl 1-thio-β-d-galactopyranoside was added to induce protein expression for 16 h at 18 °C. Cells were harvested by centrifugation at 3000 × g and washed under osmotic shock conditions to remove the periplasmic fraction (33). The pellet was stored at −80 °C until purification.
Frozen pellets were rapidly resuspended with lysis buffer (75 mm HEPES, 300 mm NaCl, 50 mm imidazole, 0.2 mm ATP, 1 mm MgCl2, 5% glycerol, 1 mm PMSF, 0.04 mg/ml DNase, 0.6 mg/ml lysozyme, pH 7.5, at 4 °C). Cells were lysed by passage through a French press (Emulsiflex). The lysate was clarified by centrifugation at 100,000 × g for 45 min at 4 °C. The supernatant was passed through a 0.22-μm syringe filter.
All Plasmodium Kinesin-5 motors with His6 tag proteins were initially purified using a HisTrap HP column (GE Healthcare). The bound protein was washed with 30 column volumes of His-Buffer A (75 mm HEPES, 300 mm NaCl, 50 mm imidazole, 0.2 mm ATP, 1 mm MgCl2, 5% glycerol, pH 7.5, at 4 °C). Proteins were eluted with 300 mm imidazole in His-Buffer B (75 mm HEPES, 300 mm NaCl, 300 mm imidazole, 0.2 mm ATP, 1 mm MgCl2, 5% glycerol, pH 7.5, at 4 °C). The eluted protein was desalted by passage through a HiPrep 26/10 desalting column (GE Healthcare) equilibrated with desalting buffer (20 mm Tris-HCl, 75 mm NaCl, 0.2 mm ATP, 1 mm MgCl2, 1 mm DTT, 5% glycerol, pH 8.0, at 4 °C). The desalted, partially purified protein was run on a HiTrap HP Q-column (GE Healthcare). The bound protein was washed with 5 column volumes of Q-Buffer A (20 mm Tris, 100 mm NaCl, 1 mm DTT, 0.2 mm ATP, 1 mm MgCl2, 5% glycerol, pH 8.0, at 4 °C). Proteins were gradually eluted with a linear gradient of 0–50% Q-Buffer B (20 mm Tris, 1 m NaCl, 1 mm DTT, 0.2 mm ATP, 1 mm MgCl2, 5% glycerol, pH 8.0, at 4 °C) on an AKTA FPLC system (GE Healthcare). The Plasmodium Kinesin-5 motor protein A280 nm peak was collected and concentrated by centrifugation at 3000 × g for 30 min at 4 °C with an iCONTM concentrator (Pierce). The final protein was estimated to be >90% pure, based on SDS-polyacrylamide gel electrophoresis and Western blotting with His tag antibody, and stored at −80 °C until use. The HsEg5(1–370) motor domain was prepared as described (34).
Chemical Libraries
We obtained the Diversity Set III from the NCI/DTP Open Chemical Repository, which contains a total of 1596 distinct compounds, as a set of microtiter plates with a sample of each compound prepared at 10 mm in 100% DMSO. Similarly, the Medicines for Malaria Venture (MMV)2 box small chemical collection of 400 lead or probe-like compounds arrived dissolved in DMSO at 10 mm each. Upon arrival and prior to assay screen use, NCI and MMV stocks were stored at −20 °C, and diluted in DMSO (ultrapure grade; Sigma) with positive displacement Pipetmen and tips into daughter plates. Once thawed, these daughter plates were not subject to repetitive freeze-thaw cycles in order to maintain the integrity of the compounds. Experiments to determine IC50 values and competition experiments were performed using new compound stocks. Additional Diversity Set III individual compounds were obtained from the NCI/DTP Open Chemical Repository, whereas MMV compounds were obtained from Vitas-M Laboratory Ltd.
NADH-coupled Assay to Monitor Kinesin-5 ATP Hydrolysis
Basal and microtubule (MT)-stimulated ATPase activities of the motor proteins were measured using a coupled pyruvate kinase/lactate dehydrogenase assay in a 96-well plate using a SpectraMax M2E spectrophotometer (Molecular Devices) at 25 °C. Basal ATPase reactions contained 1.25 μm motor, whereas MT-stimulated ATPase reaction mixtures contained 100 nm HsEg5 or 500 nm PvEg5, 10 μm test compounds, and 4 μm tubulin stabilized with 20 μm paclitaxel (Calbiochem). For basal ATPase rates, the test compounds were added to a final concentration of 100 μm; the reaction mixtures contained final concentrations of 1% (v/v) DMSO.
Each mother plate from NCI and MMV contained 80 drug test samples; therefore, the first (A1–H1) and last (A12–H12) columns of each 96-well plate were reserved for control reactions. For PvEg5, negative control samples consisted of two replicates each of complete reaction mixture containing PvEg5, but with mock drug, and complete reaction mixture with mock enzyme and mock drug (negative control or background; gray line in Fig. 3A). For the HsEg5 hit validation assay, reactions were assembled as above while substituting HsEg5 in place of PvEg5. HsEg5 reaction plates contained an additional positive control sample. S-Trityl-l-cysteine, a well characterized and tightly binding inhibitor of HsEg5 (24, 35–37), was used to generate inhibited HsEg5 samples as a positive control, containing the complete reaction mixture, HsEg5 enzyme, and 100 μm S-trityl-l-cysteine.
FIGURE 3.
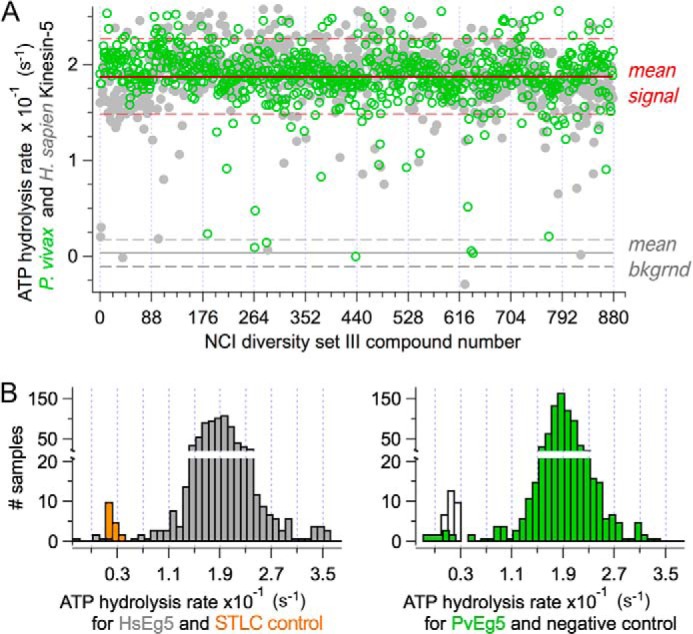
Scatter plot for a replicate set for the small molecule screen. A, scatter plot of basal ATPase activity for PvEg5 (green circles) and HsEg5 (gray circles) challenged by the first 800 compounds (100 μm) from the NCI Diversity Set III. Red line, overall mean enzyme ATPase rate. Dashed red lines, 3 σ distance away from mean. Gray line, background (without motor) signal. Dashed gray lines, 3 σ distance away from mean to mark background levels. B, histograms of the frequency of ATPase rates from scatter plot above showing the distributions of mean HsEg5 ATPase rates (right) and PvEg5 ATPase rates (left) and the mean rates of separation from background levels, respectively.
Both basal and MT-stimulated ATPase reactions reached completion within 5–10 min of initiation, with kinetic measurements of reaction rate complete within the first 2–3 min after initiation. Upon the addition of kinesin, plates were mixed for 5 s and immediately monitored at 340 nm on a SpectraMax M2E spectrophotometer for a total of 5 min, with readings taken every 20 s. Readings were automatically corrected for small changes in total volume using the instrument's path length correction feature. Single 96-well plates containing reaction mixtures were prepared and processed sequentially. Both the PvEg5 and HsEg5 screens were repeated on separate days to verify the recovered hits (see Table 1).
TABLE 1.
Statistical parameters for the NADH-coupled and malachite green assays used in chemical screen for plasmodium Kinesin-5 inhibitors
Z-Factor, signal/noise, and signal/background ratios were for the basal assay condition, in the absence of microtubules. S/N, signal/noise; S/B, signal/background.
| Screen assay | Z-Factor | S/N | S/B | Mean signal | Mean background | n |
|---|---|---|---|---|---|---|
| NADH-coupled | ||||||
| Day 1 | 0.65 | 26 | 27 | 0.18 ± 0.01 | 0.007 ± 0.005 | 20 |
| Day 2 | 0.64 | 18 | 17 | 0.18 ± 0.01 | 0.011 ± 0.010 | 20 |
| Cumulative | 0.64 | 21 | 19 | 0.18 ± 0.01 | 0.009 ± 0.008 | 40 |
| Malachite green | ||||||
| Day 3 | 0.68 | 33 | 44 | 0.17 ± 0.01 | 0.004 ± 0.005 | 40 |
| Day 4 | 0.64 | 25 | 68 | 0.17 ± 0.01 | 0.003 ± 0.003 | 40 |
| Cumulative | 0.66 | 28 | 53 | 0.17 ± 0.01 | 0.003 ± 0.006 | 80 |
To establish a threshold for the sensitivity of both the basal and MT-stimulated ATPase assay, 1.25 μm BSA was substituted for enzyme in mock time course reactions. These control reactions established a noise baseline and never recorded a value in excess of 0.009 s−1 (average = 0.004 ± 0.002 s−1 (n = 6)); control data are independent of, yet consistent with, prior laboratory publications (24, 38). Data were analyzed using IGOR Pro software (Wavemetrics Inc.). The Z-factor statistic was used for judging the quality of the collected data (39). The compounds for which the Plasmodium motor protein basal ATPase activity was reduced by more than 3 times the S.D. of the average uninhibited ATPase activity were considered as potential inhibitors.
Malachite Green Assay to Monitor Kinesin-5 ATP Hydrolysis
The protocol used to measure kinesin ATPase activity was a modification of the malachite green assay kit protocol (BioAssay Systems). The reaction mixture contained a 300 nm concentration of the kinesin protein and 100 μm Mg-ATP in 1× TAM buffer (50 mm Tris-HCl (pH 7.4), 2 mm MgCl2) for basal ATPase reactions, whereas MT-stimulated ATPase reaction mixtures contained 10 nm HsEg5 or 100–250 nm PvEg5 and 1 μm tubulin stabilized with 20 μm paclitaxel (Calbiochem). The ATPase reaction was conducted at 25 °C for 0 and 20 min and was stopped by the addition of malachite green reagent. The amount of PvEg5 in the secondary assays was altered to ensure that the IC50 was reached within the linear range for the assay. Formation of inorganic phosphate was monitored spectrophotometrically at A620 nm. Inorganic phosphate concentration generated in the reaction mixture with time was calculated using a standard curve. In addition, each kinesin protein was tested for linear response to phosphate production and to length of reaction time. Hydrolysis rates shown are averages and S.E. values from triplicate experiments.
IC50 Calculation
Normalized percentage inhibition of ATPase activity was plotted as a function of compound concentration. Data were fit to a sigmoidal curve for non-linear regression analysis using Igor Pro software (Wavemetrics, Inc.). No constraints were placed on the top, bottom, or Hill slope of the curve fit in the graphing software. Compounds that did not reach a maximal inhibition plateau could not, therefore, have IC50 values determined by this analysis.
Microtubule Co-sedimentation Assay
The Plasmodium protein (2 μm final) was mixed with paclitaxel-stabilized bovine brain MTs (5 μm tubulin final), 2 mm AMPPNP in BRB80 buffer (80 mm Pipes, pH 6.8, 1 mm EGTA, and 1 mm MgCl2) and incubated at 25 °C for 10 min. The samples were centrifuged at 100,000 × g for 40 min at 25 °C to separate pellet (MT-binding Plasmodium protein) and supernatant (free Plasmodium protein) fractions. The pellet fractions were washed with BRB80 containing 20 μm paclitaxel, and bound motor was eluted from the pellet via a 1-h incubation with 2 mm ATP. The samples were subsequently centrifuged at 100,000 × g for 40 min at 25 °C and pellet, and supernatant fractions were prepared for SDS-PAGE. Densitometry of Coomassie Blue-stained proteins was used to determine the relative amounts of Plasmodium protein in the supernatant and pellet fractions for each sample.
Lineweaver-Burk Analysis
To determine the mode of basal inhibition of the enzyme with respect to ATP substrate, PvEg5 (300 nm) activity was measured by testing fixed drug concentrations (0, 25, and 75 μm) against varying MgATP concentrations (0, 6.25, 12.5, 25, 50, 100, and 150 μm) using the malachite green assay (above). The resulting data for the 3–4 highest MgATP concentrations were analyzed by double-reciprocal plots. The double-reciprocal plots were generated with Igor Pro software (Wavemetrics Inc.). The x and y coordinates of the intersection from the three fitted lines, corresponding to the three concentrations of inhibitor, denote the value of −1/Km and 1/Vmax, respectively.
Double-reciprocal plot analysis to determine the mode of inhibition of PvEg5 with respect to tubulin concentration was performed as above. The reaction mixtures contained 50 nm motor, 100 μm MgATP, and drug held fixed at several concentrations (0, 25, and 75 μm) over a range of tubulin concentrations (0, 0.01, 0.02, 0.04, 0.08, 0.16, 0.32, 0.64, 1.25, and 2.5 μm). The resulting data for the 4–5 highest tubulin concentrations were analyzed by double-reciprocal plots, as described above.
RESULTS
Plasmodium Genomes Have a Single Candidate Kinesin-5 Motor Protein
Critical for all eukaryotic cells, kinesin family members carry out distinct essential roles in the cell, including microtubule depolymerization, microtubule assembly, and cargo transport. However, to our knowledge, a complete bioinformatic analysis of Plasmodium kinesins has not been reported. The genome sequences of a series of three extant strains of P. falciparum and P. vivax were examined for kinesin family members using the existing annotation; protein pattern motif searching tools, including kinesin patterns in InterPro; and BLAST searches with human kinesins. In total, the P. falciparum genome contains 10 putative kinesins, whereas P. vivax contains 9 candidate kinesins (Fig. 1A). Thus, Plasmodium cells have less than a quarter of the kinesins found in humans, whose genome contains 45 of these motor proteins. In addition, despite their close taxonomic relationship, P. vivax was found to contain an additional kinesin not present in P. falciparum.
We queried the phylogenetic organization of these Plasmodium sequences against a parent kinesin family tree of 78 taxa. The nine candidate kinesins common between both Plasmodium species fall into six different families with different cellular roles (Fig. 1A) (reviewed in Ref. 40). These are Kinesin-6 (spindle assembly and cytokinesis), Kinesin-7 (kinetochore-MT attachment and chromosome congression), Kinesin-5 (spindle pole separation and spindle bipolarity), Kinesin-19 (unknown function), Kinesin-8 (chromosome congression), and Kinesin-13 (kinetochore-MT error correction and chromosome segregation). P. vivax was found to contain a Kinesin-4 representative, which is absent in P. falciparum and is thought to be involved in chromosome positioning. We note that in other eukaryotic systems, Kinesin-6, -7, -8, and -13 proteins have established roles in altering microtubule dynamics. Interestingly, there are no Plasmodium counterparts to the canonical Kinesin-1, which moves cellular cargo over vast distances, and only Kinesin-19 is hypothesized to be a processive kinesin (20).
Both P. falciparum and P. vivax contain a single Kinesin-5 homolog, hereafter termed PfEg5 and PvEg5, respectively (Fig. 1, A and B). Candidate Plasmodium Kinesin-5 proteins contain the active site elements (P-loop, switch I, and switch II sequences) with absolute identity (green highlight in Fig. 1B). They also contain the requisite sequence elements for microtubule interaction (20). Notably, these Plasmodium kinesins have loop-5 sequences that are longer than and divergent from the human ortholog. The P. falciparum and P. vivax proteins have 42 and 41 residues in loop-5, respectively, compared with only 21 in HsEg5 (Fig. 1B). For loop-5 alone, there is 65% identity between the Plasmodium Kinesin-5 proteins but no significant sequence identity between the human and Plasmodium loop-5 segments. Thus, we speculate that it is feasible to identify compounds that would selectively affect Plasmodium Kinesin-5 motor domains via allosteric mechanisms, and these compounds would not alter human Kinesin-5 behavior.
Native P. vivax Kinesin-5 (PvEg5) and modified P. falciparum Kinesin-5 (PfEg5) proteins were bacterially expressed and purified. A prerequisite for conducting the proposed high throughput screening effort is the availability of protein in high purity and yield. We synthesized codon-optimized ORFs for the motor domains of both P. falciparum and vivax enzymes, PfEg5(1–506) and PvEg5(1–450), respectively. C-terminal His6-tagged motor domain constructs of both P. falciparum Kinesin-5 (PFC077c, PfEg5) and P. vivax Kinesin-5 (PVX-095355, PvEg5) were synthesized, cloned into pET24a, and expressed. Although the native PvEg5 expression readily produced soluble protein, we were unable to produce any significant soluble amount of PfEg5.
The P. falciparum genome is extremely AT-rich, with stretches of low complexity that manifest in stretches of Asp/Lys commonly inserted within most proteins, typically in domain boundaries or external loops (41–43). The P. vivax genome, although also AT-rich, suffers from fewer Asp/Lys insertions. Not surprisingly, the motor domain sequences of PvEg5 and PfEg5 reveal stretches of low complexity Asp/Lys-rich sequence inserted within both loop-5 and loop-6 of the motor domain (Fig. 1, B and C). Although loop-5 forms a critical drug-binding component of the putative Kinesin-5 allosteric site (Fig. 1, D and E), there is no evidence to date that loop-6 plays a critical role in kinesin motor allostery or function. However, Asp/Lys insertions are often found to be problematic for bacterial protein expression and can cause aggregation and precipitation of expressed proteins. To circumvent solubility issues with PfEg5, we engineered a variant enzyme with the loop-6 sequence deleted and replaced with a short cognate sequence derived from HsEg5 (PfEg5-ΔL6). Given that the PvEg5 motor domain contains a shorter loop-6 (62 residues; Fig. 1, B and C) than PfEg5 (111 residues; Fig. 1B) and it did not adversely impact bacterial protein expression, the loop-6 in PvEg5 was left intact.
Protein purification required sequential nickel affinity and ion exchange column purification procedures. Final PvEg5 and PfEg5-ΔL6 products (Fig. 2A) migrated at the expected molecular masses of 53 and 46 kDa, respectively. They also had greater than 90% purity, as determined by densitometry of SDS-PAGE. Importantly for our purposes, yields of purified protein were high and amenable for high throughput screens; the average yield of PvEg5 was 3 mg/liter of medium, whereas PfEg5-ΔL6 cultures returned 1 mg/liter of medium.
FIGURE 2.

Purification and characterization of PvEg5 and PfEg5-ΔL6. A, left, fractions from PvEg5 purification; right, fractions from PfEg5-ΔL6 purifications. Lanes 1 and 2 of both panels show elution fractions from nickel column purification and desalting steps, respectively. Lane 3 in both panels shows S-column elution fraction for each motor, with over 90% purity. B, basal (black) and MT-stimulated (gray) ATPase activity for motor domains of PvEg5, PfEg5-ΔL6, and HsEg5. C, Coomassie-stained SDS-PAGE of microtubule co-sedimentation assays and molecular weight markers (M). Microtubules were incubated with Plasmodium motor domains treated with AMPPNP, ATP, and ADP, respectively. Insoluble pellet (P) fractions were separated from the soluble supernatant (S) fractions. The top gel shows PfEg5-ΔL6 partitioning into the microtubule pellet fractions or soluble fractions depending on nucleotide treatment as marked. The second gel shows the partitioning behavior of the treated PfEg5-ΔL6 motor domain without any added microtubules. The third and fourth gels similarly show treated PvEg5 motor domain with and without added microtubules, respectively. Error bars, S.D.
As anticipated for a kinesin motor protein capable of mechanotransduction, both the purified Plasmodium proteins were capable of ATP hydrolysis. First, the basal ATPase activity of wild type PvEg5 was 0.17 ± 0.01 s−1; modified PfEg5-ΔL6 had a basal ATPase rate of 0.17 ± 0.02 s−1 (Fig. 2B). These basal catalytic rates were comparable with the 0.18 s−1 rate of the native human Eg5 motor in our hands (24, 34, 38, 44). Moreover, equivalent rates were obtained with either the NADH-coupled assay, which monitors the decay of the 340-nm absorbance of NADH upon ADP production, or with the malachite green assay, which monitors dye interaction with phosphate ion and is monitored instead at 620 nm. Second, like human Eg5, the Plasmodium enzymes also exhibited characteristic stimulation of ATPase rates in the presence of microtubules (Fig. 2B), albeit lower than the human homolog. Possible reasons for lower -fold MT enhancement of Plasmodium proteins are that Plasmodium motors are inherently slower than their human counterparts or they are not activated to the same extent by bovine microtubules as they would be by Plasmodium microtubules.
Third, Plasmodium enzymes demonstrated expected kinesin-microtubule interaction that is dependent on nucleotide state. In microtubule pelleting assays, both PvEg5 and PfEg5-ΔL6 proteins bound to microtubules in the presence of AMPPNP (Fig. 2C); this non-hydrolyzable analog of ATP elicits the tightly bound prehydrolysis state of Kinesin-5 (32, 45, 46). In microtubule release assays, the tight kinesin-microtubule interaction was interrupted upon incubation with ATP and released into the soluble fraction (Fig. 2C). Thus, we conclude that in vitro behaviors of these expressed and purified proteins are consistent with their identification as Plasmodium kinesin motor proteins.
Method Assessment for in Vitro Screen Discovery of Plasmodium Kinesin-5 Inhibitors
High throughput screening allows a laboratory to quickly conduct thousands to millions of chemical tests and thus rapidly identify compounds that serve as starting points for drug design and as research tools for biology. Prerequisites of automated data processing, liquid handling devices, and sensitive detectors were already in hand; this study was performed manually and not automated with robotics. A total of 1596 compounds from the NCI Diversity Set III chemical library and 400 lead compounds comprising the open-access MMV box collection were tested in this study. The NCI Diversity Set III compounds comprise a set of structurally well characterized and relatively rigid compounds that were selected on the basis of probing wide and distinct ranges of chemical space. The 400 MMV box compounds, on the other hand, have been shown to possess potent antimalarial activity against blood stage parasites of P. falciparum while not adversely affecting the growth of human cultured epidermal kidney cell lines (47). No target enzymes have yet been identified for any of the malaria box compounds, and no prior kinesin has formally been used to interrogate these specific chemical libraries. For each library, compounds in provided mother stock plates were carried into daughter plates for daily experiments. Assay plates were identical copies of the daughter plates.
Because kinesin motor proteins catalyze ATP hydrolysis and transduce its energy to force and motion along the microtubule, our screen was a measurement of kinesin catalysis by UV-visible absorbance in microplate format. Two different assays were used in our screen: one that monitors ADP product formation and another that measures Pi concentration. The first is the NADH-coupled ATPase assay (22), which was successfully utilized in previous screens for human Eg5 inhibitors (35, 38, 48, 49) and has long been used in our laboratory (24, 32, 34, 38, 44). This assay monitors ADP concentration through a coupled reaction that results in the oxidation of NADH to NAD+, detectable by decreasing absorbance at 340 nm. Although the NADH-coupled ATPase assay showed robust signal/noise and strong Z-factor statistics for our Kinesin-5 proteins (Table 1), the inclusion of pyruvate kinase and lactate dehydrogenase (ATP regeneration system) in this assay leaves open the possibility of the occurrence of false positives evolving instead from assay component enzymes.
Furthermore, we found that ∼20% of the compounds exhibit significant absorbance at the assay wavelength of 340 nm upon systematic evaluation of all mother plates. We speculate that this is a common occurrence in high throughput screens, given that aromatic ring structures are frequently part of the chemical scaffold in candidate compound libraries. Electron delocalization across aromatic rings may be altered when compounds bind to a protein surface and thereby modify their absorbance spectrum. As such, overlapping absorbance at 340 nm and changes in compound contribution as a function of protein binding will confound interpretation of kinesin catalytic activity. This unavoidable assay interference suggests that more than one type of assay employing different detection wavelengths is recommended in such screens.
Alternatively, the effects of test compounds on kinesin proteins were assayed using a colorimetric malachite green ATPase activity assay, in which the dye interaction with phosphate ion is monitored at 620 nm (50–52). By directly monitoring Pi formation, this assay avoids nonspecific effects that are inherently possible with the NADH-coupled assay. However, the limited dynamic range of the dye color reaction to free Pi makes this assay particularly susceptible to contaminating Pi entering the assay via nucleotide used during enzyme purification or from buffer contamination. Nonetheless, with experimental care, this assay also showed comparably strong Z-factor statistics (Table 1) and was much less affected by test compound absorption at 620 nm. We found the two alternative ATPase assays to be complementary, and they permitted us to discover compounds that would have been excluded by either assay used singly.
Identification of Small Molecule Inhibitors of Plasmodium Kinesin-5
Using the 1996 compounds from the two chemical libraries, we simultaneously screened their effect on human Kinesin-5 motor domain ATPase activity in parallel with our Plasmodium kinesins in three steps. The first step consisted of measuring the effect of each compound on a single kinesin motor. Our high throughput screen employed the requisite negative controls and triplicate measurements on separate days. Moreover, this classic one-drug one-assay screening was expanded to include two different methods of ATPase detection and two independent protein purifications apiece of human Kinesin-5 and P. vivax Kinesin-5 for a total of >14,000 individual ATPase assays. Because this number of assays required 45–50 mg of purified motor/ortholog, this primary screen did not employ PfEg5-ΔL6, due to its lower protein yield upon bacterial culture and purification.
A sample data set is shown in Fig. 3A; ATP hydrolysis rates of PvEg5 and HsEg5 in the presence of a small molecule compound at 100 μm are shown in open green circles and closed gray circles, respectively. This strategy allowed us to control for several potential confounding variables and sources of false positives in one step. For example, we were able to immediately distinguish compounds that exhibited selectivity for either enzyme. In addition, this screening strategy also allowed us to eliminate compounds that were effective inhibitors but nonspecific, such as compounds that may chelate the essential Mg2+ cofactor of all kinesins.
For this primary basal ATPase assay screen, the histogram representation of the number of Eg5 samples within the binned range of 0.01 ATPase rates (Fig. 3B) clearly shows that negative control rates were separated from median Eg5 rates by greater than 10 S.D. values in our experiments. We chose a 4σ cut-off for the identification of hits to minimize the likelihood of false positives and to produce a manageable number of hits. Cumulatively, our primary screen provided 56 inhibitors of either human or P. vivax Kinesin-5 proteins or of both; this hit rate approaches 2.8%.
Positive hits from the primary screen were subsequently challenged to inhibit the microtubule-stimulated ATPase activity of PvEg5 and HsEg5 in a secondary screen. This step is necessary because inhibition of MT-stimulated ATPase activity is probably required to inhibit the biological outcome of kinesin motor activity. Although we originally intended to use the NADH-coupled assay for this secondary screen, we instead chose to rely on the dye-based malachite green assay due to reduced interference from compound absorbance. Because the ATPase activity of motors increases in the presence of microtubules, both the Plasmodium motor and test compound concentrations were reduced to 250 nm and 10.0–1.25 μm, respectively, to maintain comparable molar ratios with the basal ATPase assay conditions, to fall within the linear range of our malachite green assay, and to span hit compound IC50 range. In general, inhibition of basal ATPase activity is always correlated with comparable inhibition of MT-stimulated ATPase activity (data not shown). Hits recovered from our secondary screen fell into three classes (Table 2). We recovered three inhibitors that had apparent selectivity for PvEg5 (NSC19063, NSC99796, and MMV666693) and one compound that was modestly more selective for HsEg5 (NSC80141). Six compounds inhibited both PvEg5 and HsEg5 (NSC70931, NSC44750, NSC92937, NSC228150, NSC129260, and NSC65248).
TABLE 2.
Selection of hit compounds and their normalized inhibitory activity in MT-stimulated ATPase assays
Shown are the averages and S.D. values of 6–12 independent measurements. STLC, which binds to the loop-5 pocket, is not expected to inhibit PvEg5, which has a different loop-5 sequence.
| Compound (12.5 μm) | PvEg5 |
HsEg5 |
||
|---|---|---|---|---|
| MT-stimulated rate | Normalized rate | MT-stimulated rate | Normalized rate | |
| s−1 | % | s−1 | % | |
| DMSO | 1.21 ± 0.10 | 6.49 ± 0.31 | ||
| STLC | 1.04 ± 0.04 | 86 ± 5 | 1.43 ± 0.13 | 22 ± 3 |
| NSC19063 | 0.06 ± 0.03 | 5 ± 1 | 3.53 ± 0.01 | 54 ± 1 |
| NSC99796 | 0.03 ± 0.03 | 2 ± 2 | 4.92 ± 0.18 | 76 ± 3 |
| MMV666693 | 0.69 ± 0.16 | 57 ± 13 | 7.06 ± 0.74 | 109 ± 11 |
| NSC80141 | 0.46 ± 0.09 | 38 ± 8 | 0.29 ± 0.20 | 4 ± 4 |
| NSC70931 | 0.06 ± 0.01 | 5 ± 1 | 0.98 ± 0.47 | 15 ± 7 |
| NSC44750 | 0.06 ± 0.07 | 5 ± 5 | 1.01 ± 1.01 | 15 ± 8 |
| NSC92937 | 0.02 ± 0.03 | 2 ± 2 | 0.69 ± 0.61 | 11 ± 9 |
| NSC228150 | 0.08 ± 0.04 | 6 ± 3 | 1.93 ± 0.03 | 30 ± 1 |
| NSC129260 | 0.50 ± 0.13 | 42 ± 11 | 2.42 ± 0.58 | 37 ± 9 |
| NSC65248 | 0.66 ± 0.15 | 55 ± 13 | 4.36 ± 0.17 | 67 ± 3 |
The third and last step reported herein consisted of IC50 determination to gauge the potency of positives from the secondary screen and analysis of selectivity ratios (SRs) to quantify whether a compound has different potency on two targets. We chose one compound from each of the three different classes of recovered inhibitors for these analyses: NSC44750, which inhibited both human and Plasmodium Kinesin-5; NSC80141, a potent inhibitor of HsEg5 that can also inhibit Plasmodium Kinesin-5, albeit to a much lower extent; and MMV666693, which inhibited PvEg5 more strongly than HsEg5. Dose-response curves were measured for each compound against PvEg5 and HsEg5 (Fig. 4). Importantly, these data were obtained on independent syntheses of compound material; solid powder stocks were obtained from NCI and from commercial vendors. All three compounds exhibited the expected inhibition patterns (Fig. 4); this was a key test in demonstrating that the primary results derived from the original microplate-based library were repeatable.
FIGURE 4.

Examples of dose-response curves from the three classes of hit compounds. Shown are chemical structures (left panels) and dose-response curves of both the basal (middle panels) and MT-stimulated (right panels) ATPase assays for Plasmodium-specific inhibitor MMV666693 effects on PvEg5 (green triangles, IC50 = 12.5 μm), PfEg5-ΔL6 (blue triangles, IC50 = 23.4 μm), and HsEg5 (open circles) (A), HsEg5-selective inhibitor NSC80141 effects on PvEg5 (green triangles, IC50 = 769 nm) and HsEg5 (open circles, IC50 = 99 nm) (B), and non-selective inhibitor NSC44750 effects on PvEg5 (green triangles, IC50 = 75 nm) and HsEg5 (open circle, IC50 = 176 nm) (C). Error bars, S.D.
The IC50 values for inhibition of basal ATP hydrolysis were determined via measurement of catalytic rates as a function of inhibitor concentration (Fig. 4, center panels). Values calculated for MMV666693 were 13.4 and 45.0 μm for PvEg5 and PfEg5-ΔL6, respectively. The HsEg5 IC50 value for MMV666693 could not be calculated because there was no apparent inhibition of the human ortholog. Calculated IC50 values for NSC80141 were 27.3 and 9.4 μm for PvEg5 and HsEg5, respectively. The IC50 values for NSC44750 were nearly equivalent for the human and Plasmodium kinesin proteins: 2.4 μm for PvEg5 and 5.1 μm for HsEg5.
To determine the IC50 values for inhibition of MT-stimulated activity, ATPase rates in the presence of microtubules were measured as a function of inhibitor concentration (Fig. 4, right panels). The MMV666693 IC50 values were 12.5 and 23.4 μm for PvEg5 and PfEg5-ΔL6, respectively; as seen for basal ATPase activity, there was no apparent inhibition of HsEg5 MT-stimulated activity by the MMV box compound. For both NSC80141 and NSC44750, the median inhibitory concentration against Kinesin-5 proteins decreased to the nanomolar range in the presence of microtubules. For NSC80141, HsEg5 exhibited an IC50 of 99 nm, whereas PvEg5 had an 8-fold higher IC50 value of 769 nm. For NSC44750, PvEg5 and HsEg5 had measured IC50 values of 75 and 176 nm, respectively.
Because the fundamental objective of this work is to discover inhibitors that are solely selective for Plasmodium Kinesin-5, we calculated SR values, which are quantitative comparisons of potencies between off-target and target. Here, our reports for the selectivity ratio of a particular compound are the IC50 for HsEg5 divided by the IC50 for Plasmodium Kinesin-5. In cases where HsEg5 did not exhibit any inhibition by the compound, we use a conservative estimate of 1500 μm as its IC50 value. Using the data above, selectivity ratios toward the Plasmodium enzyme for NSC44850 are 2.1 and 2.3 for the absence and presence of microtubules, respectively; for NSC80141, they are 0.3 and 0.1 for basal and MT-simulated conditions, respectively. The average selectivity ratio is 40 for a compound that differentiates between off-target and target variants (53), and our values for these two compounds fall below threshold.
In contrast, MMV666693 had selectivity ratios of 111.9 and 33.3 for PvEg5 and PfEg5-ΔL6, respectively, in the basal condition. Microtubule-stimulated assays also result in SR values for MMV666693 in a similar range: 120 for PvEg5 and 64.1 for PfEg5-ΔL6. For either the basal or MT-stimulated catalysis, SR values for MMV666693 indicate that this compound can highly differentiate between human and Plasmodium Kinesin-5 proteins. In addition, comparison of other screens (53) shows that 80% confidence in target versus off-target inhibition is associated with an SR of >110.
Biochemical Mode of Action for MMV666693 against Plasmodium Kinesin-5
Following the above three steps in our screening protocol, we then investigated whether MMV666693 can compete with substrate for the active, or orthosteric, site in P. vivax motor domains. We measured the effects of increasing concentrations of ATP on the inhibitory activity of the MMV box compound in the absence of microtubules. Double reciprocal plot analysis of these data (Fig. 5A) demonstrated that the data were linear over the concentration range examined. Both Km and Vmax were significantly changed by increasing concentrations of inhibitor. MMV666693 exhibited mixed inhibition with MgATP in binding PvEg5, in the absence of microtubules. Linear mixed-type inhibition is a form of noncompetitive inhibition; MMV666693 binds to an allosteric site. The compound may bind to PvEg5, regardless of whether the Plasmodium motor domain has substrate bound or not. Thus, this inhibitor does not compete and does not bind to the PvEg5 active site.
FIGURE 5.

Double-reciprocal plot analysis of selective inhibitor MMV666693. A, double-reciprocal plot analysis of PvEg5 to determine the mode of inhibition with respect to ATP. We find that both Km and Vmax vary at different inhibitor concentrations, supporting an allosteric mixed mode model of inhibition. Km values are 33 ± 5, 66 ± 26, and 110 ± 75 for 0, 25, and 75 μm inhibitor, respectively. Vmax values are 0.22 ± 0.02, 0.19 ± 0.07, and 0.11 ± 0.08 for 0, 25, and 75 μm inhibitor, respectively. B, double-reciprocal plot analysis of PvEg5 to determine the mode of inhibition with respect to microtubules. Similar to the results with ATP, we find that both Km and Vmax vary at different inhibitor concentrations, again supporting an allosteric mixed mode model of inhibition. Km values are 0.03 ± 0.01, 0.20 ± 0.07, and 0.40 ± 0.13 for 0, 25, and 75 μm inhibitor, respectively. Vmax values are 1.5 ± 0.1, 1.0 ± 0.3, and 0.3 ± 0.1 for 0, 25, and 75 μm inhibitor, respectively.
Likewise, to determine if MMV666693 competes with microtubules for binding to Plasmodium kinesins, MT-stimulated ATPase assays were conducted at different MMV666693 concentrations for several MT (tubulin) concentrations. Increasing concentration of the compound decreased the apparent maximum enzyme reaction rate. In a Lineweaver-Burk plot (Fig. 5B), the resulting data were consistent with the conclusion that MMV666693 also demonstrated mixed inhibition with tubulin for PvEg5. Together, these data show that MMV666693 is an allosteric inhibitor that does not compete with either the active site or the MT-binding site within the Plasmodium motor domain. However, the presence of MMV666693 resulted in change in the apparent affinity of substrate or microtubules.
DISCUSSION
The goal of this study was to discover drugs that block the Plasmodium Kinesin-5 function and yet do not affect the human motor protein. The antimalarial drug pipeline is in clear need of new candidates to bolster a shrinking pool of effective medicines. This need has prompted a series of phenotype-based screens designed to identify compounds that inhibit parasite proliferation in vitro. The hundreds of effective compounds uncovered could potentially revitalize the antimalarial drug pipeline. For example, open access to the MMV box catalyzes research on putative new antimalarials in the laboratory and in the clinic. However, a major challenge with the development and optimization of these compounds lies in the unknown identity of most of the target enzymes affected by these compounds. Indeed, without knowledge of the target and the target binding site, it will be challenging to improve or tune their potency. For example, controversy concerning the mechanism of action of artemisinins, the current last line of defense against the parasite, has hampered efforts to design effective analogs or variants that might circumvent recently detected resistance (54, 55).
Alternatively, targeted approaches to discover new drug candidates have been reported; a well characterized essential Plasmodium enzyme is chosen and inhibitors are designed that can distinguish the Plasmodium enzyme orthosteric site from that of any human homologs. This tactic has most recently been taken with key enzymes of the myristoylation (8), glycolytic (56), or pentose phosphate pathway (57) of the parasite as the main targets. However, the targeted orthosteric sites are often highly conserved in homologs and present a major challenge to drug designers to utilize often subtle structural differences to successfully create highly selective inhibitors that can avoid cross-reactivity.
Our screen is inspired by previous screens for inhibitors of human Kinesin-5, a popular target for antimitotic cancer chemotherapy development over the past decade due, in large measure, to the ease of finding highly selective drugs. By chemically aiming at its unique allosteric site, Kinesin-5 inhibitors take advantage of a built-in selectivity wherein they are unlikely to find corresponding binding sites in any other kinesin. Furthermore, our laboratory has shown that, without the structural constraints of an orthosteric site, the allosteric site of homologs differs significantly, thus providing built in target selectivity, whereas the allosteric mechanism mediated by the allosteric site itself is conserved (24). Additionally, recent work indicates that human Kinesin-5 may harbor additional allosteric sites that may increase the likelihood of the discovery of selective agents (49, 58). By extension, we expect Plasmodium Kinesin-5 to offer at least one distinct allosteric site together with a conserved essential cellular function in mitosis and, therefore, to represent a good starting point for our targeted screen.
Translational Applications of the Plasmodium Kinesin-5 Screen Hits
We were able to identify three different classes of Plasmodium Kinesin-5 inhibitors. The first class of inhibitors includes compounds selective for Plasmodium proteins; these can serve as candidate starting points for clinical purposes and as probes for understanding cellular networks in which these kinesins participate. Compounds that inhibit both human and Plasmodium proteins in our screen probably bind to a common site on the motor domain and elicit parallel challenges to mechanotransduction; these compounds are not leads for clinical purposes but are useful research tools to define common versus unique elements of kinesin function and of mitosis across diverse eukaryotes. Last, although we utilized HsEg5 as a control for the selectivity of the Plasmodium protein inhibitors, the third class of compounds, found to be selective for HsEg5, are nonetheless candidates themselves as human antimitotic therapies.
Our discussion focuses on the first class of inhibitors identified, which consists of three compounds. These are MMV66693, an oxazine derivative (Table 2 and Fig. 4A, top left); NSC99796, a furobenzopyranone (Table 2); and NSC19063, a purine derivative (Table 2). Having recovered MMV66693 as a selective hit from the Malaria for Medicine Venture collection was fortuitous and immediately validated the strategic utility of merging leads from phenotypic screens with targeted approaches.
MMV666693 represents, to our knowledge, the first malaria box compound to formally have its protein target identified. MMV666693 (Mr 309.3) is a member of the “druglike” compound subset rather than the “probelike” subset in this collection. Furthermore, this compound has been reported to be active against the P. falciparum 3D7 asexual parasite (EC50 of 23.8 nm against P. falciparum 3D7 (47) and EC50 of 36.2 nm against P. falciparum K1 (ChEMBL-NTD repository)); it inhibited 96% of parasite proliferation at 5 μm. Importantly, there is a good correlation between our in vitro work herein and published in vivo data on MMV666693. As such, the existing cellular data in combination with our in vitro work also suggest that MMV666693 would be equally potent against the P. vivax asexual parasite.
Second, MMV666693 showed no potential to inhibit HsEg5 within the range of its solubility (>500 μm). This finding is supported by earlier work that showed this compound has no deleterious effect upon the growth and proliferation of HEK 293 cells in vitro (47). If this compound could cross-react with the human homolog, we would instead expect to see the classic Kinesin-5 loss of function mitotic catastrophe in the HEK 293 cells with subsequent cell cycle arrest with monopolar spindles (59). This match between in vivo and in vitro data from independent groups also indicates that we will see similar phenotypic effects on Plasmodium cells for the other two compounds identified in our screen.
The ability of MMV666693 to inhibit both Plasmodium motors, but not the human homolog, argues that the site of interaction is probably composed of sequences that are not conserved within HsEg5. For example, residues comprising the orthosteric sites of both Plasmodium and human Kinesin-5 are >90% identical and, thus, not favored for contributing to differential binding of a compound between these two homologs. The competition experiments in Fig. 5 support this conclusion. We found that inhibition of the Plasmodium motors by MMV666693 is not competitive with substrate ATP or with microtubules. Taken together, these data argue that MMV666693 does not target either the orthosteric site or other conserved elements, including the microtubule-binding site, but rather probably targets an additional allosteric site. Candidate allosteric sites include the loop-5 pocket, which is 65% identical between the Plasmodium motors, and loop-6; others have been suggested for Kinesin-5 via computational prediction (60). The minimal reduction in efficacy of the compound to PfEg5-ΔL6 compared with PvEg5 disfavors loop-6 as the binding site for MMV666693. Further experiments are under way to determine if the loop-5 pocket serves as the allosteric binding site for MMV666693.
Beyond direct determination of the allosteric binding site of MMV666693, there are still many open questions to be answered for this first set of Plasmodium inhibitors. For example, it remains to be determined whether the Diversity Set compounds from NCI (NSC99796 and NSC19063) will be equally potent in a parasite phenotypic assay. Such studies on the malarial parasitic organism will have to test more than one stage of the Plasmodium life cycle. Beyond their asexual antimalarial activity, a recent report (61) showed that MMV compounds have activity against both early and late stage gametocytes. Of interest, MMV666693 has an EC50 of ∼400 nm against late (IV-V) stage gametocyte activity and of 2000 nm against early (I–III) stage gametocyte activity. Current criteria for potential transmission-blocking drugs posit that inhibition of the late stage gametocytes is equal to or better than targeting asexual stages. Several groups have pointed out that activity against early or late stage gametocytes does not guarantee that a compound will have transmission-blocking ability in the field or within in vivo models (47, 61).
Although all three inhibitors have reasonable SR ratios for Plasmodium versus human kinesins, the biochemical mode of action may or may not be equivalent. In the future, we will test whether NSC99796 and NSC19063 are competitive inhibitors for the active/MT sites or if they are allosteric inhibitors. Although MMV66693 and NSC99796 both showed high selectivity and NSC19063 showed moderate selectivity for the Plasmodium enzymes over the human homolog, there is little chemical resemblance between the inhibitors; a pharmacophore model should be determined when a larger chemical space has been sampled.
Research Tools for Plasmodium Motor Proteins
The discovery of Plasmodium-specific inhibitors of cell proliferation provides new tools for studying motor driven processes, including mitosis, in this infectious disease organism. Although the morphology and structure of Plasmodium cell division was established over 30 years ago (reviewed in Ref. 62), knowledge of the molecular components, assembly, and regulation of the mitotic apparatus in this organism is only recently beginning to be elucidated (for examples, see Refs. 63 and 64). Small molecule effectors of kinesin motor proteins, in addition to new clinical treatments for malaria, establish a toolkit of probes that will permit the dissection of mitotic mechanisms in this organism.
Compared with 45 kinesins in human cells, the nine kinesins of Plasmodium could feasibly all be targeted in our screen in the future. A battery of selective Plasmodium inhibitors would provide an unprecedented tool chest of probes to uncover the functions of the kinesin superfamily in this organism. In addition to Kinesin-5, the P. falciparum and P. vivax genomes contain a Kinesin-7 homolog. For example, another mitotic kinesin, Kinesin-7, participates in kinetochore attachment and spindle function; the P. falciparum and P. vivax genomes contain a Kinesin-7 homolog. The report of an allosteric inhibitor of human Kinesin-7, CENP-E, from a recent chemical screen (65), argues that such a toolbox of small molecule probes is possible. With a limited number of kinesins and a subset that are required for proliferation, our targeted strategy also remains an attractive option for the future development of additional drug candidates.
Plasmodium-specific inhibitors of Kinesin-5 also can serve as tools to understand motor proteins in this eukaryote and allow comparison for evolutionary divergence of sequence, function, and regulation across different taxa. For example, we highlight that the correspondence between IC50 and EC50 values for human-specific Kinesin-5 inhibitors are not the same as observed for Plasmodium-specific inhibitors. Ispinesib, a human Kinesin-5 inhibitor and candidate chemotherapeutic treatment in Phase II clinical trials (20), has nearly identical inhibitory values between in vitro MT-stimulated conditions (1.7 nm) and human cell lines (1.2–9.5 nm). In contrast, there is a 1000-fold difference between these measurements for the Plasmodium Kinesin-5 inhibitor; MMV666693 was more potent against the P. falciparum parasite (23.8–36.2 nm) than our in vitro data would suggest (23.4 μm).
There are several possible explanations for this difference between cellular and biochemical measurements for Plasmodium and human Kinesin-5 proteins. The 3-fold order of magnitude change in response may simply result from use of a chimeric P. falciparum protein in our in vitro experiments that is modified enough not to reflect normal function in the cell or from our use of non-native tubulin in our assays. However, the IC50 for MMV666690 for a native P. vivax kinesin did not differ greatly. Alternatively, it may be that there are a lesser number of motor proteins involved in Plasmodium mitosis, lower redundancy in function, and thereby fewer measures in the cell to compensate for loss of Kinesin-5 function. Although we are not aware of any successful knock-out experiments directed at Plasmodium Kinesin-5, it has been shown to be essential for cell division in the vast majority of eukaryotes and has only been shown to share some redundant function with Kinesin-12 motor proteins. However, our analysis does not find any potential homologs of Kinesin-12 in this organism.
In summary, our report is the first inhibitor screen for Plasmodium Kinesin-5 proteins, and this is the first demonstration that this approach can be applied and extended to non-human kinesins. MMV666693 is a unique tool to further study Plasmodium kinesin mechanotransduction at the atomic level. It can also be used to determine the full range of potential cellular functions of Kinesin-5 in this organism; with a limited set of kinesins, less specialized roles for each family member may be manifest. The distinct advantages of the built-in selectivity of allosteric Kinesin-5 inhibitors identify the kinesin family as a promising target for the development of near term therapeutics.
Acknowledgments
We are indebted to NCI, National Institutes of Health, and the Medicines for Malaria Venture for providing public access to the compounds screened in this work. We thank Matthew Dean for assistance with developing protein purification strategies for Plasmodium kinesins and Hoang Nyugen for assistance with initial inhibitor screening.
This work was supported, in whole or in part, by National Institutes of Health Grants R01GM066328 (to E. W.) and R01GM097350 (to S. K.). This work was also supported by the Louisiana State University School of Graduate Studies (to J. R.) and the LSU School of Medicine (to L. L.).
- MMV
- Medicines for Malaria Venture
- MT
- microtubule
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- SR
- selectivity ratio.
REFERENCES
- 1. Anthony M. P., Burrows J. N., Duparc S., Moehrle J. J., Wells T. N. (2012) The global pipeline of new medicines for the control and elimination of malaria. Malar. J. 11, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Biamonte M. A., Wanner J., Le Roch K. G. (2013) Recent advances in malaria drug discovery. Bioorg. Med. Chem. Lett. 23, 2829–2843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen I., Eastman R., Lanzer M. (2011) Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 585, 1551–1562 [DOI] [PubMed] [Google Scholar]
- 4. Cohen J. M., Smith D. L., Cotter C., Ward A., Yamey G., Sabot O. J., Moonen B. (2012) Malaria resurgence: a systematic review and assessment of its causes. Malar. J. 11, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gamo F.-J., Sanz L. M., Vidal J., de Cozar C., Alvarez E., Lavandera J.-L., Vanderwall D. E., Green D. V., Kumar V., Hasan S., Brown J. R., Peishoff C. E., Cardon L. R., Garcia-Bustos J. F. (2010) Thousands of chemical starting points for antimalarial lead identification. Nature 465, 305–310 [DOI] [PubMed] [Google Scholar]
- 6. Guiguemde W. A., Shelat A. A., Bouck D., Duffy S., Crowther G. J., Davis P. H., Smithson D. C., Connelly M., Clark J., Zhu F., Jiménez-Díaz M. B., Martinez M. S., Wilson E. B., Tripathi A. K., Gut J., Sharlow E. R., Bathurst I., El Mazouni F., Fowble J. W., Forquer I., McGinley P. L., Castro S., Angulo-Barturen I., Ferrer S., Rosenthal P. J., Derisi J. L., Sullivan D. J., Lazo J. S., Roos D. S., Riscoe M. K., Phillips M. A., Rathod P. K., Van Voorhis W. C., Avery V. M., Guy R. K. (2010) Chemical genetics of Plasmodium falciparum. Nature 465, 311–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang Y., Evans G. B., Clinch K., Crump D. R., Harris L. D., Fröhlich R. F., Tyler P. C., Hazleton K. Z., Cassera M. B., Schramm V. L. (2013) Transition state analogues of Plasmodium falciparum and human orotate phosphoribosyltransferases. J. Biol. Chem. 288, 34746–34754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rackham M. D., Brannigan J. A., Moss D. K., Yu Z., Wilkinson A. J., Holder A. A., Tate E. W., Leatherbarrow R. J. (2013) Discovery of novel and ligand-efficient inhibitors of Plasmodium falciparum and Plasmodium vivax N-myristoyltransferase. J. Med. Chem. 56, 371–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nilsen A., LaCrue A. N., White K. L., Forquer I. P., Cross R. M., Marfurt J., Mather M. W., Delves M. J., Shackleford D. M., Saenz F. E., Morrisey J. M., Steuten J., Mutka T., Li Y., Wirjanata G., Ryan E., Duffy S., Kelly J. X., Sebayang B. F., Zeeman A.-M., Noviyanti R., Sinden R. E., Kocken C. H., Price R. N., Avery V. M., Angulo-Barturen I., Jiménez-Díaz M. B., Ferrer S., Herreros E., Sanz L. M., Gamo F.-J., Bathurst I., Burrows J. N., Siegl P., Guy R. K., Winter R. W., Vaidya A. B., Charman S. A., Kyle D. E., Manetsch R., Riscoe M. K. (2013) Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med. 5, 177ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yuthavong Y., Tarnchompoo B., Vilaivan T., Chitnumsub P., Kamchonwongpaisan S., Charman S. A., McLennan D. N., White K. L., Vivas L., Bongard E., Thongphanchang C., Taweechai S., Vanichtanankul J., Rattanajak R., Arwon U., Fantauzzi P., Yuvaniyama J., Charman W. N., Matthews D. (2012) Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. U.S.A. 109, 16823–16828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Coteron J. M., Marco M., Esquivias J., Deng X., White K. L., White J., Koltun M., El Mazouni F., Kokkonda S., Katneni K., Bhamidipati R., Shackleford D. M., Angulo-Barturen I., Ferrer S. B., Jiménez-Díaz M. B., Gamo F.-J., Goldsmith E. J., Charman W. N., Bathurst I., Floyd D., Matthews D., Burrows J. N., Rathod P. K., Charman S. A., Phillips M. A. (2011) Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 54, 5540–5561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Booker M. L., Bastos C. M., Kramer M. L., Barker R. H., Jr., Skerlj R., Sidhu A. B., Deng X., Celatka C., Cortese J. F., Guerrero Bravo J. E., Crespo Llado K. N., Serrano A. E., Angulo-Barturen I., Jiménez-Díaz M. B., Viera S., Garuti H., Wittlin S., Papastogiannidis P., Lin J. W., Janse C. J., Khan S. M., Duraisingh M., Coleman B., Goldsmith E. J., Phillips M. A., Munoz B., Wirth D. F., Klinger J. D., Wiegand R., Sybertz E. (2010) Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J. Biol. Chem. 285, 33054–33064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hyde J. E. (2007) Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr. Drug Targets 8, 31–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fry M., Pudney M. (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharm. 43, 1545–1553 [DOI] [PubMed] [Google Scholar]
- 15. Vyas V. K., Ghate M. (2011) Recent developments in the medicinal chemistry and therapeutic potential of dihydroorotate dehydrogenase (DHODH) inhibitors. Mini Rev. Med. Chem. 11, 1039–1055 [DOI] [PubMed] [Google Scholar]
- 16. Imwong M., Russell B., Suwanarusk R., Nzila A., Leimanis M. L., Sriprawat K., Kaewpongsri S., Phyo A. P., Snounou G., Nosten F., Renia L. (2011) Methotrexate is highly potent against pyrimethamine-resistant Plasmodium vivax. J. Infect. Dis. 203, 207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dempsey E., Prudêncio M., Fennell B. J., Gomes-Santos C. S., Barlow J. W., Bell A. (2013) Antimitotic herbicides bind to an unidentified site on malarial parasite tubulin and block development of liver-stage Plasmodium parasites. Mol. Biochem. Parasitol. 188, 116–127 [DOI] [PubMed] [Google Scholar]
- 18. Chakrabarti R., Rawat P. S., Cooke B. M., Coppel R. L., Patankar S. (2013) Cellular effects of curcumin on Plasmodium falciparum include disruption of microtubules. PLoS One 8, e57302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kappes B., Rohrbach P. (2007) Microtubule inhibitors as a potential treatment for malaria. Future Microbiol. 2, 409–423 [DOI] [PubMed] [Google Scholar]
- 20. Wojcik E. J., Buckley R. S., Richard J., Liu L., Huckaba T. M., Kim S. (2013) Kinesin-5: cross-bridging mechanism to targeted clinical therapy. Gene 531, 133–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stock M. F., Hackney D. D. (2001) Expression of kinesin in Escherichia coli. Methods Mol. Biol. 164, 43–48 [DOI] [PubMed] [Google Scholar]
- 22. Huang T. G., Hackney D. D. (1994) Drosophila kinesin minimal motor domain expressed in Escherichia coli: purification and kinetic characterization. J. Biol. Chem. 269, 16493–16501 [PubMed] [Google Scholar]
- 23. Gilbert S. P., Johnson K. A. (1993) Expression, purification, and characterization of the Drosophila kinesin motor domain produced in Escherichia coli. Biochemistry 32, 4677–4684 [DOI] [PubMed] [Google Scholar]
- 24. Liu L., Parameswaran S., Liu J., Kim S., Wojcik E. (2011) Loop 5-directed compounds inhibit chimeric Kinesin-5 motors: implications for conserved allosteric mechanisms. J. Biol. Chem. 286, 6201–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maliga Z., Mitchison T. J. (2006) Small-molecule and mutational analysis of allosteric Eg5 inhibition by monastrol. BMC Chem. Biol. 6, 2–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. El-Nassan H. B. (2013) Advances in the discovery of kinesin spindle protein (Eg5) inhibitors as antitumor agents. Eur. J. Med. Chem. 62, 614–631 [DOI] [PubMed] [Google Scholar]
- 27. Gartner M., Sunder-Plassmann N., Seiler J., Utz M., Vernos I., Surrey T., Giannis A. (2005) Development and biological evaluation of potent and specific inhibitors of mitotic kinesin Eg5. Chembiochem 6, 1173–1177 [DOI] [PubMed] [Google Scholar]
- 28. Sarli V., Giannis A. (2008) Targeting the kinesin spindle protein: basic principles and clinical implications. Clin. Cancer Res. 14, 7583–7587 [DOI] [PubMed] [Google Scholar]
- 29. Liu K., Warnow T. J., Holder M. T., Nelesen S. M., Yu J., Stamatakis A. P., Linder C. R. (2012) SATe-II: very fast and accurate simultaneous estimation of multiple sequence alignments and phylogenetic trees. Syst. Biol. 61, 90–106 [DOI] [PubMed] [Google Scholar]
- 30. Schwede T., Kopp J., Guex N., Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Notredame C., Higgins D. G., Heringa J. (2000) T-coffee: a novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 [DOI] [PubMed] [Google Scholar]
- 32. Parke C. L., Wojcik E. J., Kim S., Worthylake D. K. (2010) ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J. Biol. Chem. 285, 5859–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Magnusdottir A., Johansson I., Dahlgren L.-G., Nordlund P., Berglund H. (2009) Enabling IMAC purification of low abundance recombinant proteins from E. coli lysates. Nat. Methods 6, 477–478 [DOI] [PubMed] [Google Scholar]
- 34. Wojcik E. J., Dalrymple N. A., Alford S. R., Walker R. A., Kim S. (2004) Disparity in allosteric interactions of monastrol with Eg5 in the presence of ADP and ATP: a difference FT-IR investigation. Biochemistry 43, 9939–9949 [DOI] [PubMed] [Google Scholar]
- 35. DeBonis S., Skoufias D. A., Lebeau L., Lopez R., Robin G., Margolis R. L., Wade R. H., Kozielski F. (2004) In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol. Cancer Ther. 3, 1079–1090 [PubMed] [Google Scholar]
- 36. Debonis S., Skoufias D. A., Indorato R.-L., Liger F., Marquet B., Laggner C., Joseph B., Kozielski F. (2008) Structure-activity relationship of S-trityl-l-cysteine analogues as inhibitors of the human mitotic kinesin Eg5. J. Med. Chem. 51, 1115–1125 [DOI] [PubMed] [Google Scholar]
- 37. Skoufias D. A., DeBonis S., Saoudi Y., Lebeau L., Crevel I., Cross R., Wade R. H., Hackney D., Kozielski F. (2006) S-Trityl-l-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 281, 17559–17569 [DOI] [PubMed] [Google Scholar]
- 38. Learman S. S., Kim C. D., Stevens N. S., Kim S., Wojcik E. J., Walker R. A. (2009) NSC 622124 inhibits human Eg5 and other kinesins via interaction with the conserved microtubule-binding site. Biochemistry 48, 1754–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang J. H., Chung T. D., Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 40. Lawrence C. J., Dawe R. K., Christie K. R., Cleveland D. W., Dawson S. C., Endow S. A., Goldstein L. S., Goodson H. V., Hirokawa N., Howard J., Malmberg R. L., McIntosh J. R., Miki H., Mitchison T. J., Okada Y., Reddy A. S., Saxton W. M., Schliwa M., Scholey J. M., Vale R. D., Walczak C. E., Wordeman L. (2004) A standardized kinesin nomenclature. J. Cell Biol. 167, 19–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muralidharan V., Oksman A., Pal P., Lindquist S., Goldberg D. E. (2012) Plasmodium falciparum heat shock protein 110 stabilizes the asparagine repeat-rich parasite proteome during malarial fevers. Nat. Commun. 3, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zilversmit M. M., Volkman S. K., DePristo M. A., Wirth D. F., Awadalla P., Hartl D. L. (2010) Low-complexity regions in Plasmodium falciparum: missing links in the evolution of an extreme genome. Mol. Biol. Evol. 27, 2198–2209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Xue H. Y., Forsdyke D. R. (2003) Low-complexity segments in Plasmodium falciparum proteins are primarily nucleic acid level adaptations. Mol. Biochem. Parasitol. 128, 21–32 [DOI] [PubMed] [Google Scholar]
- 44. Kim E. D., Buckley R., Learman S., Richard J., Parke C., Worthylake D. K., Wojcik E. J., Walker R. A., Kim S. (2010) Allosteric drug discrimination is coupled to mechanochemical changes in the Kinesin-5 motor core. J. Biol. Chem. 285, 18650–18661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kapoor T. M., Mitchison T. J. (2001) Eg5 is static in bipolar spindles relative to tubulin: evidence for a static spindle matrix. J. Cell Biol. 154, 1125–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lockhart A., Cross R. A. (1996) Kinetics and motility of the Eg5 microtubule motor. Biochemistry 35, 2365–2373 [DOI] [PubMed] [Google Scholar]
- 47. Spangenberg T., Burrows J. N., Kowalczyk P., McDonald S., Wells T. N., Willis P. (2013) The open access malaria box: a drug discovery catalyst for neglected diseases. PLoS One 8, e62906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang L., Jiang C., Liu F., You Q.-D., Wu W.-T. (2008) Cloning, enzyme characterization of recombinant human Eg5 and the development of a new inhibitor. Biol. Pharm. Bull. 31, 1397–1402 [DOI] [PubMed] [Google Scholar]
- 49. Rickert K. W., Schaber M., Torrent M., Neilson L. A., Tasber E. S., Garbaccio R., Coleman P. J., Harvey D., Zhang Y., Yang Y., Marshall G., Lee L., Walsh E. S., Hamilton K., Buser C. A. (2008) Discovery and biochemical characterization of selective ATP competitive inhibitors of the human mitotic kinesin KSP. Arch. Biochem. Biophys. 469, 220–231 [DOI] [PubMed] [Google Scholar]
- 50. Baykov A. A., Evtushenko O. A., Avaeva S. M. (1988) A malachite green procedure for orthophosphate determination and its use in alkaline phosphatase-based enzyme immunoassay. Anal. Biochem. 171, 266–270 [DOI] [PubMed] [Google Scholar]
- 51. Henkel R. D., VandeBerg J. L., Walsh R. A. (1988) A microassay for ATPase. Anal. Biochem. 169, 312–318 [DOI] [PubMed] [Google Scholar]
- 52. Kodama T., Fukui K., Kometani K. (1986) The initial phosphate burst in ATP hydrolysis by myosin and subfragment-1 as studied by a modified malachite green method for determination of inorganic phosphate. J. Biochem. 99, 1465–1472 [DOI] [PubMed] [Google Scholar]
- 53. Goedken E. R., Devanarayan V., Harris C. M., Dowding L. A., Jakway J. P., Voss J. W., Wishart N., Jordan D. C., Talanian R. V. (2012) Minimum significant ratio of selectivity ratios (MSRSR) and confidence in ratio of selectivity ratios (CRSR): quantitative measures for selectivity ratios obtained by screening assays. J. Biomol. Screen. 17, 857–867 [DOI] [PubMed] [Google Scholar]
- 54. Tilley L., Charman S. A., Vennerstrom J. L. (2012) in Neglected Diseases and Drug Discovery, pp. 33–64, RSC Drug Discovery Series No. 14, Royal Society of Chemistry, London [Google Scholar]
- 55. O'Neill P. M., Barton V. E., Ward S. A. (2010) The molecular mechanism of action of artemisinin: the debate continues. Molecules 15, 1705–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Harris M. T., Walker D. M., Drew M. E., Mitchell W. G., Dao K., Schroeder C. E., Flaherty D. P., Weiner W. S., Golden J. E., Morris J. C. (2013) Interrogating a hexokinase-selected small-molecule library for inhibitors of Plasmodium falciparum hexokinase. Antimicrob. Agents Chemother. 57, 3731–3737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Preuss J., Maloney P., Peddibhotla S., Hedrick M. P., Hershberger P., Gosalia P., Milewski M., Li Y. L., Sugarman E., Hood B., Suyama E., Nguyen K., Vasile S., Sergienko E., Mangravita-Novo A., Vicchiarelli M., McAnally D., Smith L. H., Roth G. P., Diwan J., Chung T. D., Jortzik E., Rahlfs S., Becker K., Pinkerton A. B., Bode L. (2012) Discovery of a Plasmodium falciparum glucose-6-phosphate dehydrogenase 6-phosphogluconolactonase inhibitor (R,Z)-N-((1-ethylpyrrolidin-2-yl)methyl)-2-(2-fluorobenzylidene)-3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazine-6-carboxamide (ML276) that reduces parasite growth in vitro. J. Med. Chem. 55, 7262–7272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Indorato R.-L., DeBonis S., Kozielski F., Garcia-Saez I., Skoufias D. A. (2013) STLC-resistant cell lines as tools to classify chemically divergent Eg5 targeting agents according to their mode of action and target specificity. Biochem. Pharm. 86, 1441–1451 [DOI] [PubMed] [Google Scholar]
- 59. Mayer T. U., Kapoor T. M., Haggarty S. J., King R. W., Schreiber S. L., Mitchison T. J. (1999) Small molecule inhibitor of mitotic spindle bipolarity identified in a phenotype-based screen. Science 286, 971–974 [DOI] [PubMed] [Google Scholar]
- 60. Zhang W. (2011) Exploring the intermediate states of ADP-ATP exchange: a simulation study on Eg5. J. Phys. Chem. B 115, 784–795 [DOI] [PubMed] [Google Scholar]
- 61. Duffy S., Avery V. M. (2013) Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar. J. 12, 408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Francia M. E., Striepen B. (2014) Cell division in apicomplexan parasites. Nat. Rev. Microbiol. 12, 125–136 [DOI] [PubMed] [Google Scholar]
- 63. Arnot D. E., Ronander E., Bengtsson D. C. (2011) The progression of the intra-erythrocytic cell cycle of Plasmodium falciparum and the role of the centriolar plaques in asynchronous mitotic division during schizogony. Int. J. Parasitol. 41, 71–80 [DOI] [PubMed] [Google Scholar]
- 64. Mahajan B., Selvapandiyan A., Gerald N. J., Majam V., Zheng H., Wickramarachchi T., Tiwari J., Fujioka H., Moch J. K., Kumar N., Aravind L., Nakhasi H. L., Kumar S. (2008) Centrins, cell cycle regulation proteins in human malaria parasite Plasmodium falciparum. J. Biol. Chem. 283, 31871–31883 [DOI] [PubMed] [Google Scholar]
- 65. Wood K. W., Lad L., Luo L., Qian X., Knight S. D., Nevins N., Brejc K., Sutton D., Gilmartin A. G., Chua P. R., Desai R., Schauer S. P., McNulty D. E., Annan R. S., Belmont L. D., Garcia C., Lee Y., Diamond M. A., Faucette L. F., Giardiniere M., Zhang S., Sun C.-M., Vidal J. D., Lichtsteiner S., Cornwell W. D., Greshock J. D., Wooster R. F., Finer J. T., Copeland R. A., Huang P. S., Morgans D. J., Jr., Dhanak D., Bergnes G., Sakowicz R., Jackson J. R. (2010) Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc. Natl. Acad. Sci. U.S.A. 107, 5839–5844 [DOI] [PMC free article] [PubMed] [Google Scholar]