

Abstract
Since the first description of apoptosis four decades ago, great efforts have been made to elucidate, both in vivo and in vitro, the molecular mechanisms involved in its regulation. Although the role of cytochrome c during apoptosis is well established, relatively little is known about its participation in signaling pathways in vivo due to its essential role during respiration. To obtain a better understanding of the role of cytochrome c in the onset of apoptosis, we used a proteomic approach based on affinity chromatography with cytochrome c as bait in this study. In this approach, novel cytochrome c interaction partners were identified whose in vivo interaction and cellular localization were facilitated through bimolecular fluorescence complementation. Modeling of the complex interface between cytochrome c and its counterparts indicated the involvement of the surface surrounding the heme crevice of cytochrome c, in agreement with the vast majority of known redox adducts of cytochrome c. However, in contrast to the high turnover rate of the mitochondrial cytochrome c redox adducts, those occurring under apoptosis led to the formation of stable nucleo-cytoplasmic ensembles, as inferred mainly from surface plasmon resonance and nuclear magnetic resonance measurements, which permitted us to corroborate the formation of such complexes in vitro. The results obtained suggest that human cytochrome c interacts with pro-survival, anti-apoptotic proteins following its release into the cytoplasm. Thus, cytochrome c may interfere with cell survival pathways and unlock apoptosis in order to prevent the spatial and temporal coexistence of antagonist signals.
Apoptosis, an event that is both morphologically distinguishable from other types of cell death (e.g. senescence or necrosis) and tightly regulated from a genetic and biochemical point of view, controls tissue homeostasis and eliminates damaged cells in mammals (1). The process is characterized by the co-occurrence of nuclear and cytoplasmic condensation, blebbing of cytoplasmic membranes, and the emergence of apoptotic bodies as a consequence of cell fragmentation (2).
The main processes characterizing apoptosis are driven by a cascade of proteolytic events mediated by caspases (cysteine-dependent aspartate-specific proteases), a subfamily of cysteine proteases (3). Two different pathways—extrinsic, or death receptor-initiated (4), and intrinsic, or mitochondrial (5)—are involved in the activation of these proteolytic events. DNA damage, oxidative stress, and growth factor deprivation are well-known apoptosis inducers that activate the intrinsic pathway (6), involving the permeabilization of the outer mitochondrial membrane. This event occurs prior to the release of pro-apoptotic factors (e.g. apoptosis-inducing factor, cytochrome c (Cc),1 Smac/DIABLO, and HtrA2/Omi) from the mitochondrial intermembrane space into the cytoplasm.
Cc is a well-known heme protein that plays an essential role in homeostasis and apoptosis. With regard to the former, Cc acts as an electron shuttle between complexes III and IV in mitochondrial respiration. In apoptosis, Cc is released from mitochondria into the cytoplasm. During this process, cytosolic Cc and dATP bind to apoptosis protease-activating factor-1 (Apaf-1), forming the apoptosome, a macromolecular platform that in turn leads to the activation of initiator caspases (7, 8).
Under homeostatic conditions, Cc is kept in the mitochondrial space, where its concentration can reach 0.5 to 5 mm (9). Notably, ∼90% of mitochondrial Cc content is sequestered within the cristae of the inner membrane and therefore is unavailable for electron transport (10). Moreover, the interaction of Cc with Apaf-1, leading to apoptosome formation, is one of the earliest events to occur at the onset of apoptosis, a process requiring small amounts of the heme protein because of the amplification of its effect by the proteolytic cascade. The Cc concentration in the intermembrane space is therefore extremely high, leading one to wonder whether Cc is also regulating other processes during programmed cell death (PCD).
Beyond the well-established in vitro role of Cc during apoptosis in mammals, newly proposed putative functions of the heme protein in cell death signaling remain controversial. Some authors suggest that Cc exclusively induces apoptosome formation and caspase activation in the cytosol (11). Others have recently proposed the existence of additional, not yet fully understood Cc functions in the pro-apoptotic response, both in the nucleus (12, 13) and in the endoplasmic reticulum (ER) (14–16).
Even though a function for cytosolic Cc during PCD has been defined only in mammals (8), the mitochondria-to-cytoplasm release of Cc is an evolutionarily conserved event found to occur in yeast (17), plants (18), flies (19), and mammals (20).
The specific role of Cc in apoptosis signaling has not been extensively investigated in vivo because of the difficulty of obtaining Cc knockout mutants. Recently, Vempati et al. (21) were able to produce a Cc knockout mutant in mouse fibroblasts. Intriguingly, this mutant was resistant to pro-apoptotic agents acting through both intrinsic and extrinsic pathways. Meriting particular interest, apoptosis can normally be activated in mutants lacking Apaf-1, the only apoptotic partner of Cc described in vivo thus far (22, 23).
The different phenotypes of Cc and Apaf-1 knockouts and the high concentration of Cc in the mitochondrial intermembrane space cannot be explained if Cc is restrained from interacting with Apaf-1 during PCD. However, these can be easily reconciled if a broader, critical role for Cc in the regulation of life-and-death decisions is considered, as recently proposed by Hüttemann et al. (24).
To facilitate the identification of putative novel Cc interaction partners, here a proteomic approach was developed based on affinity chromatography coupled with MALDI-TOF/TOF. The use of bimolecular fluorescence complementation (BiFC) permitted not only corroboration of the in vivo interaction, but also determination of the in-cell localization of such novel Cc complexes. Moreover, Cc complexes involving ALDOA, ANP32B, eIF2α, SET, STRAP, and YWHAE were modeled in silico using docking algorithms. In vitro validation through surface plasmon resonance (SPR) was performed only for Cc complexes including protein interaction partners expressed in a soluble form, that is, eIF2α, hnRNP C1/C2, HSPA5, SET, and YWHAE. Furthermore, nuclear magnetic resonance (NMR) and isothermal titration calorimetry (ITC) measurements were performed to delve into the complex formation. The qualitative methodological approach proposed herein combines proteomic tools with other complementary techniques, namely, BiFC, SPR, NMR, and ITC, to further corroborate the reliability of the novel Cc-involving protein interactions and to characterize the nature of such complexes from both structural and functional points of view. Two major advantages should be highlighted. First, the synergy among all these techniques reduces the rate of false positives and avoids the need for proteomic replicas, and second, the BiFC experiments, combined with SPR, NMR, and ITC approaches, provided extra information not only on the cellular compartment at which the protein–protein interactions take place, but also on the specificity and lifetime of the Cc-involving complexes.
In addition to the assembly of the apoptotic platform during apoptosis, the results obtained in this study might suggest that Cc is also involved in other signaling pathways, interfering with cell survival and unlocking apoptosis. Indeed, Cc appears to abolish the coexistence of pro-survival and pro-apoptotic signals. The additional functions, besides apoptosome assembly, ascribed to Cc during apoptosis therefore suggest an evolutionarily conserved role for Cc during PCD and might explain its release from mitochondria in the majority of organisms.
EXPERIMENTAL PROCEDURES
Expression and Purification of Cc E104C Mutant
Plasmid pCytH (25), containing the coding region of human Cc, was used as a template to obtain the E104C Cc mutant, in which the glutamate at the C-terminal end was replaced by a cysteine, through site-directed mutagenesis. The oligonucleotides employed to build the E104C species were 5′-gcgaccaattgctgatgaattc-3′ and 3′-cgctggttaacgactacttaag-5′. The E104C Cc was expressed and further purified using ionic exchange chromatography, a process previously described by Rodríguez-Roldán et al. (25).
Jurkat T-cell Cultures and Apoptosis Induction
Jurkat human T-lymphoma cells were cultured in RPMI 1640 (PAA, Uppsala, Sweden) supplemented with 10% heat-inactivated fetal bovine serum (PAA), 2 mm l-glutamine (Invitrogen), 100 units/ml streptomycin (Invitrogen), and 100 μg/ml penicillin (Invitrogen) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. Cells were grown as an exponentially growing confluent monolayer in 645-ml flasks (Nunc, Roskilde, Denmark) with the medium refreshed every 48 h.
Apoptosis was induced in Jurkat human T-lymphoma cells with 10 μm camptothecin (CPT) (Calbiochem) for up to 6 h (26) in order to ensure the release of Cc from mitochondria into the cytosol (27).
DAPI Staining
Non-treated or treated Jurkat T cells with 10 μm CPT were harvested via centrifugation at 1000 × g for 5 min, resuspended in PBS containing 500 ng/ml DAPI, and incubated for 30 min at room temperature. Cells on glass slides were examined under a fluorescence microscope equipped with a DAPI filter, and apoptotic cells were identified by the presence of densely stained granular nuclear apoptotic bodies.
Flow Cytometry Measurements
Jurkat T cells, non-treated or treated with 10 μm CPT, were harvested as described above and fixed in ethanol 70% for 1 h. Cells were resuspended in a propidium iodide solution as previously described (28) to be further incubated for 1 h at 37 °C and analyzed via flow cytometry using a Becton-Dickinson FACS Vantage flow cytometer. Cells with sub-G1 DNA contents were considered to be apoptotic.
DNA Fragmentation Assay
Apoptosis was induced in Jurkat T cells with 10 μm CPT for 0, 6, 10, and 24 h, and the DNA fragmentation assay was carried out as previously described (29). Briefly, cells were harvested via centrifugation at 1000 × g for 5 min, washed twice in PBS, resuspended, and incubated at 37 °C overnight with 500 μl of buffer D (100 mm Tris-HCl, pH 8.5, 5 mm EDTA, 0.2 m NaCl, 0.2% (w/v) SDS, and 0.2 mg/ml proteinase K). Then, 1.5 m NaCl was added and cellular debris was removed via centrifugation at 12,000 × g for 15 min. The DNA present in the supernatant was precipitated with an equal volume of 100% (v/v) ethanol via centrifugation at 12,000 × g for 10 min, washed with 70% ethanol, and incubated for 2 h at 37 °C in buffer E (10 mm Tris-HCl, pH 7.5, 1 mm EDTA, and 100 μg/ml DNase-free RNase A). Then, DNA was loaded onto a 2% agarose gel, and electrophoresis was carried out at 50 V for 2 h in 0.5× Tris borate-EDTA buffer.
Cellular Viability Assay
Cell viability was measured using the trypan blue exclusion assay. Jurkat T cells treated with 10 μm CPT for different times (0, 3, 6, 10, and 24 h) were collected via centrifugation at 1000 × g for 5 min. Cells were resuspended in trypan blue solution and counted with a hemocytometer. The viability percentage of Jurkat T cells was obtained by dividing the number of viable cells by the total number of cells.
Cell Extract Preparation
Cell extracts from 1.8 l of culture with either untreated or 10 μm CPT-treated Jurkat T cells were prepared for affinity chromatography purification. In both cases, cells were harvested via centrifugation at 1000 × g for 5 min, washed twice in PBS, pelleted again, and resuspended to be further lysed via sonication in buffer I (50 mm Tris-HCl, pH 7.5, 50 mm NaCl, 0.25% Triton X-100) supplemented with 1 mm phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 10 μg/ml soybean trypsin inhibitor. Cellular debris was then removed via centrifugation at 20,000 × g for 30 min at 4 °C. Protein aliquots were lyophilized and stored at −80 °C.
Purification by Affinity Chromatography
As previously described by Azzi et al. (30), affinity chromatography was carried out on a column prepared via the covalent linkage of the Cc mutant E104C to the Thiol-Sepharose 4B (TS-4B) matrix (Pharmacia, Uppsala, Sweden). For this preparation, 30 mg of Cc E104C in 50 mm Tris-HCl (pH 7.5), previously treated with 1 mm DTT to prevent the formation of intermolecular disulfide bridges, was added to a suspension of 4 ml of TS-4B (1 g) resuspended in the same buffer. The suspension, following stirring at 4 °C overnight, was then poured into a Poly-Prep Chromatography Column (Bio-Rad) and washed with 5 ml of 1.5 mm 2-mercaptoethanol/50 mm Na acetate (pH 4.5) in order to block unreacted thiol groups. To remove non-covalently bound Cc E104C, the matrix was washed extensively with 30 ml of 50 mm Tris-HCl, pH 7.5, 1 m NaCl, and 1% Triton X-100. Finally, the column was equilibrated in 50 mm Tris-HCl, pH 7.5. As a control, a TS-4B matrix devoid of Cc (blank TS-4B) was prepared following the steps above.
Jurkat T-cell extracts, both untreated and treated with 10 μm CPT, were loaded into the columns, both with and without Cc. The columns were then washed with 30 ml of buffer I and 30 ml of buffer II (50 mm Tris-HCl, pH 7.5, 75 mm NaCl) to remove proteins nonspecifically bound. Proteins interacting with greater strength were then eluted with 30 ml of buffer III (50 mm Tris-HCl, pH 7.5, 300 mm NaCl), collected, lyophilized, and stored at −80 °C before being analyzed using two-dimensional SDS-PAGE.
Four sets of samples were thus obtained: (i) untreated cell extracts loaded into the blank TS-4B column, (ii) untreated cell extracts loaded into the Cc TS-4B column, (iii) apoptotic cell extracts treated with CPT and purified using the blank TS-4B column, and (iv) apoptotic cell extracts treated with CPT and loaded into the Cc TS-4B column.
Two-dimensional SDS-PAGE
The protein samples purified as described above were then analyzed using two-dimensional SDS-PAGE. Isoelectrofocusing was carried out with the PROTEAN Isoelectrofocusing Cell (Bio-Rad) system using 7-cm ReadyStrip IPG Strips (Bio-Rad) with linear pH gradients (pH 3–10). Lyophilized proteins (150 μg) were resuspended in 125 μl of rehydration buffer (7 m urea, 2 m thiourea, 2% CHAPS, 30 mm DTT, 0.5% Bio-Lyte 3/10 ampholyte, and traces of bromphenol blue (Bio-Rad)). The ReadyStrip IPG Strips were rehydrated along with the solution containing the protein mixture for 14 h at room temperature.
Samples were later separated on the ReadyStrip IPG Strips for 16,000 Vh. Isoelectrofocusing gels were then incubated with two equilibration buffers, I (6 m urea, 0.375 m Tris-HCl, pH 8.8, 2% SDS, 20% glycerol, 2% (w/v) DTT) and II (6 m urea, 0.375 m Tris-HCl, pH 8.8, 2% SDS, 20% glycerol, 2.5% (w/v) iodoacetamide), for 15 min. Finally, the isoelectrofocusing gels were loaded onto a 12% polyacrylamide gel (8 cm × 7 cm) that was run at 100 V for 2 h in a Mini-PROTEAN 3 Dodeca Cell (Bio-Rad). The two-dimensional gels were stained using blue silver (31) and analyzed using PDQuest 2D Analysis Software Version 8.0.1 (Bio-Rad).
Protein Preparation for Mass Spectrometry
Gel protein spots of interest were manually excised from micro-preparative gels using pipette tips. The selected proteins were reduced in-gel, alkylated, and digested with trypsin as described by Sechi and Chait (32). Briefly, spots were washed twice with water, shrunk for 15 min with 100% acetonitrile, and dried in a Savant SpeedVac for 30 min. Then, the samples were reduced with 10 mm DTT in 25 mm ammonium bicarbonate for 30 min at 56 °C and subsequently alkylated with 55 mm iodoacetamide in 25 mm ammonium bicarbonate for 15 min in the dark. Finally, samples were digested with 12.5 ng/μl sequencing-grade trypsin (Roche Molecular Biochemicals) in 25 mm ammonium bicarbonate (pH 8.5) at 37 °C overnight.
After digestion, the supernatant was collected, from which 1 μl was spotted onto a MALDI target plate and later air-dried at room temperature. Subsequently, 0.4 μl of a 3 mg/ml α-cyano-4-hydroxy-transcinnamic acid matrix (Sigma) in 50% acetonitrile was added to the dried peptide digest spots and then air-dried at room temperature.
MALDI-TOF-MS
MALDI-TOF-MS analyses were performed in a 4800 Proteomics Analyzer MALDI-TOF/TOF mass spectrometer (Applied Biosystems, Framingham, MA) at the Genomics and Proteomics Center, Complutense University of Madrid. The mass spectrometer was operated in positive reflector mode with an accelerating voltage of 20,000 V. All mass spectra were calibrated internally using peptides from the autodigestion of trypsin.
MALDI-TOF/TOF mass spectrometry analysis yields peptide mass fingerprints, and the peptides observed with a signal-to-noise ratio greater than 10 can be collated and represented as a list of monoisotopic molecular weights. Proteins ambiguously identified by peptide mass fingerprinting were subjected to MS/MS sequencing analyses using the 4800 Proteomics Analyzer (Applied Biosystems). So, from the MS spectra suitable precursors were selected for MS/MS analyses with collision-induced dissociation (atmospheric gas was used), 1 Kv ion reflector mode, and precursor mass windows of ±5 Da. The plate model and default calibration were optimized for the MS/MS spectra processing.
For protein identification, UniProtKB-Swiss-Prot 160909 database v. 57.7 (497,293 sequences, 175,274,722 residues) restricted to human protein was searched using a local license of MASCOT 2.1 through Global Protein Server v. 3.6 from Applied Biosystems. Search parameters were as follows:
Carbamidomethyl cystein as a fixed modification and oxidized methionine as a variable modification
Peptide mass tolerance of 50 to 100 ppm (MS/MS or combined search)
Peptide charge state = +1
One missed trypsin cleavage site
MS/MS fragment tolerance of 0.3 Da
The parameters for the combined search (peptide mass fingerprint and MS/MS spectra) were the same as above.
In all protein identifications, the probability scores were greater than the score fixed by Mascot as significant with a p value of less than 0.05.
Bioinformatics
For protein identification, UniProtKB-Swiss-Prot database v.57.7 restricted to human protein (20,333 sequences) or the NCBI database (10,084,244 sequences) was searched using a local license for MASCOT 2.1. Database search parameters used were the following: trypsin as enzyme; peptide tolerance, 50 ppm for MS analyses and 80 ppm for MS/MS analyses; fragment ion tolerance, 0.3 Da; missed cleavage sites, 1; fixed modification, carbamidomethyl cysteine; and variable modification, methionine oxidation. In all protein identifications, probability scores were greater than the score established by MASCOT as significant, with a p value less than 0.05.
Design of Vectors for BiFC Assays
cDNAs coding for Cc and the proteins identified as Cc potential targets were purchased (GeneService, Nottingham, UK). The cDNAs were amplified and restriction sites required in each case for further cloning were introduced via PCR with the oligonucleotides detailed in the supplemental information (see supplemental Fig. S4 and supplemental Table S1). Whereas Cc cDNA was cloned into the cYFP vector, the cDNAs of the Cc targets were cloned into the nYFP vector.
As discussed by Hu et al. (33), pBiFC-bJunYN155 and pBiFC-bFosYC155 were employed as positive controls, and pBiFC-bJunYN155 and pBiFC-bFosΔZipYC155 were used as negative controls.
BiFC Assays: Human Cell Cultures, Cellular Transfection, and Fluorescence Microscopy
HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) (PAA) supplemented with 2 mm l-glutamine (Invitrogen), 100 units/ml streptomycin (Invitrogen), 100 μg/ml penicillin (Invitrogen), and 10% heat-inactivated fetal bovine serum (PAA) at 37 °C in a humidified atmosphere of 5% CO2 and 95% air. When used for fluorescence microscopy, the HEK293T cells were grown to 80% confluence in 24-well plates with 500 μl of DMEM, and 20-mm coverslips were used. Cells were transiently transfected with the BiFC vectors using Lipofectamine 2000 Transfection Reagent (Invitrogen) following the manufacturer's instructions. In this way, 0.5 μg of DNA per construct was diluted in 50 μl of Opti-MEM medium (Invitrogen) and 2 μl of Lipofectamine was diluted in 50 μl of Opti-MEM medium, and the solutions were incubated separately. Following 5 min of incubation at room temperature, both solutions were mixed and incubated for 20 min at room temperature. Finally, 100 μl of the DNA–Lipofectamine mixture was added to the cells. To favor the protein expression of both constructs, the cells were incubated for 24 h at 37 °C.
Untreated and CPT-treated HEK293T cells on coverslips were mounted in PBS, supplemented with 75% glycerol, and observed under a Leica DM6000 B fluorescence microscope.
Western Blot Analysis
The HEK293T cells transfected with different cDNA vectors were harvested 48 h after transfection via centrifugation at 1500 rpm for 5 min. Total cell extracts were obtained through repeated freeze-thaw cycles. SDS-PAGE was performed using 12% polyacrylamide gels. Proteins were transferred onto nitrocellulose membranes (Bio-Rad) using a Mini Trans-Blot (Bio-Rad) and immunoblotted with a rabbit anti-EGFP polyclonal antibody (1:1000; Biovision Research Products, CA). A horseradish-peroxidase-conjugated goat anti-rabbit IgG (1:12,000; Sigma-Aldrich) was then used for detection. The immunoreactive bands were developed using the ECL Plus Western Blotting Detection System (Amersham Biosciences).
Docking Protocol
A soft docking algorithm implemented in the Biomolecular Complex Generation with Global Evaluation and Ranking (BiGGER) software package (34) was used to determine in silico a model of the complexes between Cc and some novel interaction partners named earlier. For each run, 5000 solutions were generated using a 15° angular step soft dock and a distance of 7 Å. The center of mass for 50 structures with the best global scores was represented. All graphical images of complexes were generated using the UCSF Chimera package.
Cloning, Expression, and Purification of Human Proteins for SPR, ITC, and NMR Measurements
Wild-type human Cc cloned in the pBTR vector under the lac promoter was expressed in the Escherichia coli BL-21(DE3) strain (35). For this, 25 ml of preculture was grown overnight at 37 °C in LB medium supplemented with 100 μg/ml ampicillin. 2.5 ml of preculture was used to inoculate 2.5 l of the same medium in a 5-l Erlenmeyer flask. The culture was shaken at 30 °C for 24 h, after which the cells were harvested at 6000 rpm for 10 min using a Beckman Coulter Avanti J-25 refrigerated centrifuge. Cells were then resuspended in 1.5 mm borate buffer, pH 8.5, sonicated for 4 min, and then centrifuged at 20,000 rpm for 20 min. For NMR measurements, 15N-labeled Cc was produced in minimal media with 15NH4Cl as a nitrogen source. Further purification of wild-type human Cc was carried out as indicated by Rodríguez-Roldán et al. (25).
Proteins interacting with human Cc—ANP32B, eIF2α, hnRNP C1/C2, HSPA5, SET, and YWHAE—were cloned into the pET-28a vector under the T7 promoter using the NdeI-NotI restriction sites. DNA for cloning was obtained via a PCR reaction using the target cDNA sequences, which had been purchased previously (GeneService). DNA inserts coding for Cc targets were ligated into the pET-28a vector. Protein expressions were performed in the E. coli BL-21 (DE3) RIL strain. 250-ml precultures in LB medium supplemented with 50 μg/ml kanamycin were grown overnight and then used to inoculate 2.5-l cultures in 5-l flasks. Following the induction of cultures (1 mm isopropyl 1-thio-â-D-galactopyranoside) and growth at 30 °C for 24 h, cells were harvested at 6000 rpm for 10 min, resuspended in 40 ml of lysis buffer (20 mm Tris-HCl buffer, pH 8, 0.8 m NaCl, 10 mm imidazole, 0.01% phenylmethylsulfonyl fluoride (PMSF), 0.2 mg/ml lysozyme, 5 mm DTT, and 0.02 mg/ml DNase), sonicated for 4 min, and then centrifuged at 20,000 rpm for 20 min. Cc counterparts were purified via affinity chromatography. The above lysate was loaded into an Ni column (Ni Sepharose 6 Fast Flow, GE Healthcare) previously equilibrated with the above buffer, and proteins were eluted via the application of an imidazole gradient from 0 to 300 mm. The purity of Cc targets was checked by running SDS-PAGE. The fractions containing protein were pooled, concentrated by Amicon (10-kDa cutoff membrane) until a protein concentration of 100 μm was reached, and dialyzed against 10 mm HEPES buffer (pH 7.4) for SPR measurements, 5 mm sodium phosphate buffer (pH 6.5) for NMR experiments, or 10 mm sodium phosphate buffer (pH 7.4) for ITC measurements (in all cases, the buffer contained 0.01% PMSF).
SPR Measurements
The formation of complexes between human Cc and its protein interaction partners was assayed with SPR using the BiaCore 3000 and CM4 Sensor Chips. An automated desorption procedure was performed prior to each experiment to ensure the cleanliness of the BiaCore tubing, channels, and sample injection port. The initial electrostatic attraction of Cc to the CM4 Sensor Chip surface was assessed by taking into account its isoelectric point and was optimized to pH 5.8. Cc was then covalently attached to the matrix using standard amine-coupling chemistry, as previously described (36). A reference flow cell was used as a control in which the chip surface was treated as described above, but without the injection of Cc.
The binding measurements were performed at 25 °C using HBS-EP buffer containing 10 mm HEPES, 150 mm NaCl, 3 mm EDTA, and 0.005% surfactant P20 adjusted to pH 7.4. Interactions between Cc and its protein partners were analyzed by passing several concentrations of Cc–protein interaction partners (from 0.1 to 10 μm) over the Cc-modified surface at a flow rate of 10 μl/min. Each concentration was injected at least three times. In each sensogram, the signals from the reference flow cell surface were subtracted. Between injection cycles, bound proteins were removed following the injection of 10 μl of 10 mm NaOH. In each case, after regeneration the SPR signal was returned to baseline by flushing with buffer.
NMR Measurements
All protein samples were concentrated in 5 mm sodium phosphate buffer (pH 6.5) using Millipore (Billerica, MA) 3K NMWL centricons and microcons. Cc–protein partner stock samples ranged in concentration from 200 to 800 μm and were diluted at a final concentration of 50 μm each in Shigemi tubes. Cc was likewise at a final concentration of 50 μm and reduced upon the addition of an aliquot of 0.1 m sodium ascorbate solution up to 4 mm. All NMR samples contained 10% D2O to adjust the lock signal.
NMR experiments were performed in a Bruker Avance 700-MHz spectrometer at 25 °C. The interaction of Cc with its protein partners was followed by acquiring two-dimensional 1H-15N HSQC spectra during the titration of 50 μm 15N-Cc solutions with an increasing amount of each protein partner up to a final Cc:partner molar ratio of 1:1. The pH value of the samples was verified after each titration step. All data processing was performed with Bruker TopSpin 2.0. NMR analyses of chemical-shift perturbations were performed with the SPARKY program.
ITC Measurements
All ITC experiments were performed using an Auto-ITC200 instrument (Microcal, GE Healthcare) at 25 °C by titrating each Cc protein target with Cc. The reference cell was filled with distilled water. The experiments consisted of 10-μl injections of 300 μm Cc in 10 mm sodium phosphate buffer (pH 7.4) into the sample cell, which initially contained 20 μm Cc protein partner solutions in the same buffer. All the solutions were degassed before the titrations were performed. Titrant was injected at appropriate time intervals to ensure that the thermal power signal returned to baseline prior to the next injection. To achieve homogeneous mixing in the cell, the stirring speed was kept constant at 1000 rpm. The data, specifically the heat per injection normalized per mole of injectant versus the molar ratio, were analyzed with Origin 7 (Microcal, Uppsala, Sweden). Calibration and performance tests of the calorimeter were carried out by conducting CaCl2–EDTA titrations with solutions provided by the manufacturer.
RESULTS
Exploring Novel Cc Protein Interaction Partners Using a Proteomic Approach
As explained earlier, the identification of novel Cc protein interaction partners could provide an important starting point for the elucidation of yet unexplored Cc functions during PCD. Thus, to achieve this goal, a proteomic approach combining affinity chromatography with mass spectrometry was used. A C-end cysteine-substituted Cc (E104C) was covalently bound to a thiol-Sepharose 4B (TS-4B) column using a disulfide bridge. The other two Cc cysteines did not interfere, as they bind the heme group (37). A TS-4B column devoid of Cc (blank TS-4B) was used as a control.
Jurkat T-cell cultures grown in RPMI 1640 medium until a final cell density of 0.15 to 1.5 × 106 cells/ml were used to obtain homeostatic and apoptotic cell extracts. As previously indicated by Johnson et al. (26), 10 μm CPT was used to induce apoptosis in the cultures. To ensure that the cultures were undergoing apoptosis, several characteristic hallmarks were tested. Supplemental Fig. S1A shows the DNA “ladder”-shaped fragmentation following 10 h of CPT treatment (29). This fragmentation is in accord with a positive DAPI signal fluorescence for nuclei (supplemental Fig. S1B) and a lower DNA content in apoptotic cells followed by flow cytometry (supplemental Fig. S2). In fact, the population suffering apoptosis upon the addition of CPT was monitored via both flow cytometry and trypan blue dye exclusion (supplemental Fig. S1C). Both analyses indicated that the population of cells undergoing apoptosis increased substantially after 6 h of treatment with CPT. Moreover, they showed that CPT-treated Jurkat T cells were dying specifically as a result of apoptosis, rather than another type of PCD such as necrosis.
Cells were collected for the preparation of extracts 6 h following apoptosis induction, the period when Cc is known to be released from mitochondria into the cytoplasm (38). As approximately only 30% of apoptotic cells were detected via flow cytometry and trypan blue dye exclusion (supplemental Figs. S1C and S2), the data collected can be said to address only the early steps of apoptotic signaling. The extracts from both untreated and CPT-treated Jurkat T cells were loaded into the columns both with and without Cc attached (Fig. 1, left). The putative Cc protein interaction partners were eluted from the Cc TS-4B column by increasing ionic strength, whereas the proteins flowing from the blank TS-4B column served as a control for nonspecific binding. The resulting protein fractions were prepared for two-dimensional SDS-PAGE (Fig. 1, left).
Fig. 1.

Proteomic workflow and two-dimensional SDS-PAGE. Left, scheme of the proteomic workflow used to identify Cc-interacting proteins. Cc interaction partners were purified from untreated and CPT-treated Jurkat T-cell extracts with affinity chromatography, using a column with the E104C mutant covalently bound to the thiol-Sepharose matrix (Cc TS-4B). Samples were resolved via two-dimensional SDS-PAGE, with the resulting spots later identified via MALDI-TOF/TOF. As controls, untreated and CPT-treated cell extracts were loaded into a blank column containing the Sepharose matrix devoid of E104C (blank TS-4B). Right, master gel calculated from the image analysis of gels run under different experimental conditions using PDQuest software (see supplemental data). Red circles represent proteins identified under either apoptotic stimuli or homeostatic and apoptotic conditions. Such proteins were retained in the Cc-bounded TS-4B column, but not in the TS-4B column. Spot numbers are named in supplemental Table S2.
From affinity chromatography, four different protein samples were obtained reflecting different experimental conditions: proteins from (a) untreated and (b) CPT-treated cells purified using the blank TS-4B column, and proteins from (c) untreated and (d) CPT-treated cells purified with the E104C TS-4B column. The mixture of proteins present in each sample was resolved using two-dimensional SDS-PAGE (supplemental Fig. S3). PDQuest 2D Analysis Software version 8.0.1 (Bio-Rad) was then used to analyze the resulting two-dimensional gels, and the spots highlighted in the master gel image (Fig. 1, right) were identified via MALDI-TOF/TOF and a database search. Supplemental Table S2 comprehensively summarizes the putative novel Cc-interacting proteins, along with their cellular localization, identified in CPT-treated cell extracts only or in both untreated and CPT-treated extracts. Through this approach, 21 novel putative Cc interaction partners were identified in homeostatic and/or apoptotic Jurkat T cells, although this list is very likely to contain many false positives (supplemental Data S1).
In Vivo Verification of Cc–Target Interactions: BiFC Assays
In an attempt to reduce the false positive rate and corroborate in vivo the interactions between Cc and its novel putative interaction partners identified in this work, BiFC was employed. This approach permits the analysis of protein–protein interactions in their biological environment, as well as the localization of the protein complexes (39). For this purpose, Cc was fused with the C-end fragment of yellow fluorescent protein (cYFP), whereas its novel protein interaction partners, previously identified in vitro, were fused with the N-end fragment of YFP (nYFP). In each case, coding cDNAs were cloned immediately before the corresponding YFP fragment. The HEK293T cells—rather than Jurkat T cells—were co-transfected because of their more efficient transient transfection and great adherence, allowing one to monitor the changes in cell morphology and protein localization. The interaction of Cc with its counterparts was followed by fluorescence, due to the YFP reconstitution upon Cc-target binding (Fig. 2).
Fig. 2.

BiFC assays and Western blots showing the in vivo interaction of Cc with its potential protein partners. HEK293T cells were transfected with the Cc-cYFP vector, along with another vector containing the N-terminal YFP fragment (nYFP) bound to each Cc protein interaction partner. Images were captured 24 h after transient transfection with Lipofectamine 2000 (Invitrogen) and after 6 h of treatment with 10 μm CPT. Reconstruction of eYFP leads to fluorescence signal emission, indicative of interaction between Cc and its partners. Positive and negative controls were used as described by Hu et al. (39). Scale bar = 5 μm. The expression of Cc interaction partners fused to the nYFP fragment was determined by immunoblotting with a rabbit anti-EGFP polyclonal antibody (BioVision). No bands were observed in the Western blots with non-transfected cells (data not shown).
cDNA coding for human Cc and for 14 out of the 21 potential Cc interaction partners previously identified through the proteomic approach was purchased (Source BioScience, Nottingham, UK). As shown in supplemental Table S1 and supplemental Fig. S4, Cc and the cDNAs of its targets were cloned into vectors containing cYFP and nYFP, respectively (40).
Among the 21 Cc targets, ATP5β, CCT2, NAP1L4, and RPS7 were discarded because they have not been described as apoptosis-related proteins. In addition, cDNAs encoding hnRNP L and RBBP7 were not available from Source BioScience, or the cDNA could not be amplified using PCR. Hsp90B1 was discarded because it has been described in the literature as an Apaf-1 target that does not make direct contact with Cc (41).
YFP fragments are known to complement each other with low efficiency yet still be sufficiently efficient to yield fluorescent complexes even in the absence of a specific interaction (42). To ensure that the interactions involving Cc were not the result of spontaneous YFP complementation, several precise controls were designed based on the expression of two fusion proteins that, being expressed in the same cellular compartment, were unable to interact. These positive vectors, pBiFC-bJunYN155 and pBiFC-bFosYC155, and negative vectors, pBiFC-bJunYN155 and pBiFC-bFosΔZipYC155, were used as controls (33).
Apo-Cc needs to be translocated from the cytosol to mitochondria, where the Cc hemelyase assembles the heme cofactor leading to the formation of the holoprotein. HEK293T cells transfected with both the Cc-cYFP vector and the empty nYFP vector (Fig. 3A) showed a punctuate fluorescence pattern for the distribution of Cc under homeostatic conditions, thereby highlighting its mitochondrial localization (38). Following the treatment of cells with CPT, the fluorescence pattern became diffuse, consistent with the release of Cc from mitochondria into the cytoplasm (Fig. 3B). The expression of the Cc–cYFP fusion protein was confirmed via Western blotting using a rabbit anti-EGFP polyclonal antibody (Fig. 3C).
Fig. 3.
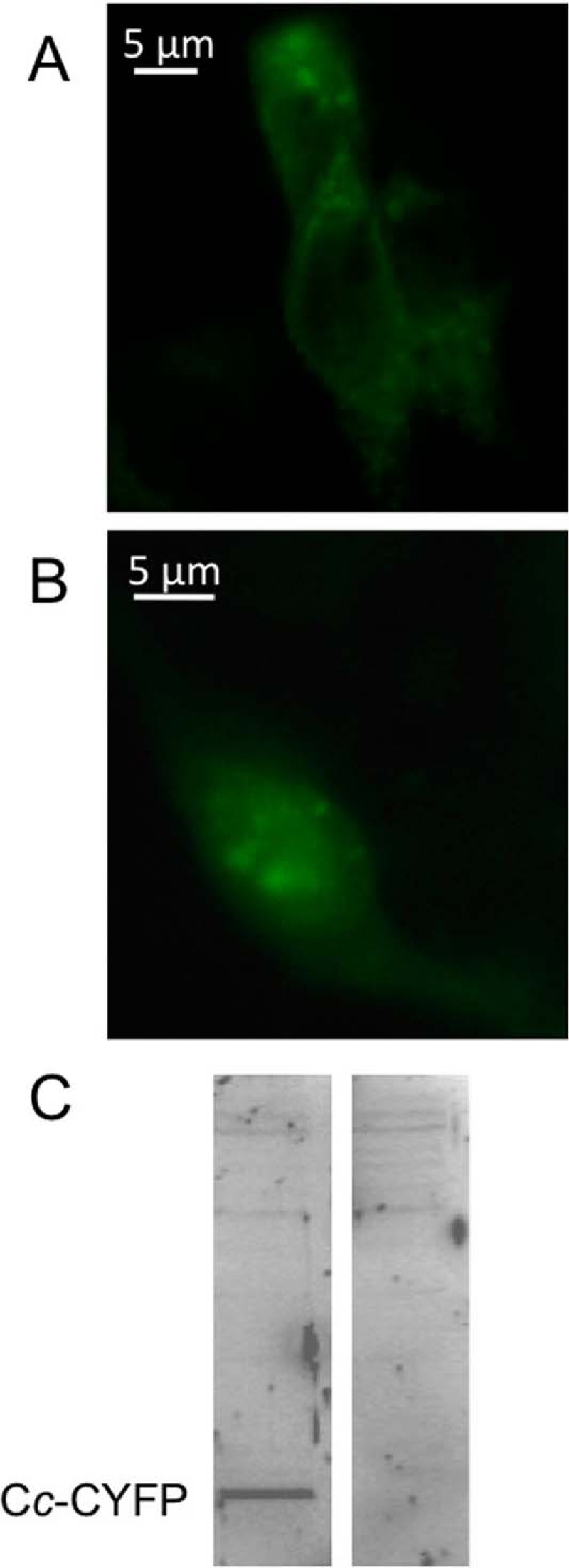
Location of cYFP-tagged Cc under normal and apoptotic conditions. A, homeostatic conditions. Punctuate fluorescence pattern showing the mitochondrial distribution of Cc into HEK293T cells co-transfected with the Cc-cYFP vector and the empty nYFP vector using Lipofectamine 2000 (Invitrogen). B, apoptotic conditions. Diffuse fluorescence pattern showing the cytoplasmic Cc localization of Cc in HEK293T cells transfected as in A, but after 6 h of treatment with 10 μm CPT. C, expression of Cc-cYFP in HEK293T cells. Left, cells transfected with the Cc-cYFP vector. Right, non-transfected cells. The Cc-cYFP fusion protein was immunodetected via Western blotting with an anti-EGFP antibody (BioVision).
BiFC assays were carried out on 14 putative Cc-interacting proteins identified through the proteomic approach. With the exception of CSNKIIβ, CORO1A, TUBB, and MCM7 proteins, the interaction between Cc and the targets was corroborated in vivo (Fig. 2 and Table I). Some of these interactions occur in the cytoplasm, such as those involving ALDOA, eIF2α, MCM6, HSPA5, and YWHAE, whereas others, such as those involving hnRNP C1/C2 and SET, whose YFP fluorescence overlaps with DAPI staining, take place inside the nucleus. Finally, the complexes ANP32B-Cc, NCL-Cc, and STRAP-Cc display nucleo-cytoplasmic localization.
Table I. Human Cc novel confirmed protein targets validated through different orthogonal approaches.
| Protein name | Protein abbreviation | SDS-PAGE and MALDI-TOF/TOF-MS | BiFC | Molecular docking | SPR | NMR | ITC |
|---|---|---|---|---|---|---|---|
| Casein kinase II subunit β | CSNKIIβ | + | - | ||||
| Chaperonin containing TCP1 subunit 2 | CCT2 | + | nd1 | ||||
| Coronin-like protein | CORO1A | + | - | ||||
| Eukaryotic translation initiation factor 2 αa | elF2α | + | + | + | + | + | + |
| Fructose 1,6-bisphosphate aldolase Aa | ALDOA | + | + | + | nd4 | nd4 | nd4 |
| Ribosomal protein S7 | RPS7 | + | nd1 | ||||
| Tubulin β chain | TUBB | + | - | ||||
| Tumor rejection antigen 1 | Hsp90B1 | + | nd2 | ||||
| 14–3-3 epsilona | YWHAE | + | + | + | + | + | + |
| Heterogeneus nuclear ribonucleoprotein C1/C2a | hnRNP C1/C2 | + | + | nd5 | + | - | - |
| Histone-binding protein RBBP7 | RBBP7 | + | nd3 | - | nd3 | nd3 | nd3 |
| Minichromosome maintenance complex 6 | MCM6 | + | + | nd5 | nd4 | nd4 | nd4 |
| Minichromosome maintenance complex 7 | MCM7 | + | - | ||||
| SET nuclear oncogenea | SET | + | + | + | + | + | + |
| Acidic nuclear phosphoprotein 32Ba | ANP32B | + | + | + | - | + | + |
| Heterogeneus nuclear ribonucleoprotein L | hnRNP L | + | nd3 | ||||
| Nucleolin | NCL | + | + | nd5 | nd4 | nd4 | nd4 |
| Nucleosome assembly protein 1-like 4 | NAP1L4 | + | nd1 | ||||
| Ser/Thr kinase receptor associated proteina | STRAP | + | + | + | nd4 | nd4 | nd4 |
| ATP synthase subunit β | ATP5β | + | nd1 | ||||
| Heat shock 70 kDa proteina | HSPA5 | + | + | nd5 | + | + | + |
Notes: nd1, not determined because they are not apoptosis-related proteins; nd2, not determined because the interaction with Cc was previously discarded (41); nd3, not determined because their cDNAs were not available; nd4, not determined because they were not overexpressed as soluble recombinant proteins; nd5, not determined because their PDB coordinates were not available.
a Cc protein interactors validated in at least three out of six techniques.
The lack of interaction between Cc and CSNKIIβ, as demonstrated by the absence of YFP fluorescence, is also shown in Fig. 2. Intriguingly, CSNKIIβ was described as an in vitro Cc interaction partner as early as the 1970s (43, 44). However, such interaction was not detected in vivo with BiFC.
The transient expression of Cc targets fused to nYFP in the BiFC assays was checked by immunoblotting with a rabbit anti-EGFP polyclonal antibody. As illustrated in Fig. 2, all constructs yielded a band of the expected molecular mass for each target.
To double-check that these protein partners were novel and specific for Cc, additional BiFC measurements were performed with Cc from Arabidopsis thaliana, as human and plant Cc are well-conserved hemeproteins with similar electrostatic potential surfaces. As can be seen in supplemental Fig. S5, all the proteins shown to interact with human Cc yielded positive results with plant Cc too, and all of them interacted at the same cell compartment with human and plant Cc. Together, these findings confirm the specificity of all these proteins for the Cc species.
In Silico Modeling of the Cc Complexes
To correlate structural data on the protein interaction partners with in vivo measurements and to determine the three-dimensional structure of the Cc complexes, a set of theoretical computations based on docking algorithms were performed (45). For this purpose, either known structures, as deposited in the PDB database, or homology models were used to set the initial coordinates. In fact, the complexes involving ALDOA (4ALD.pdb) (46), ANP32B (2RR6.pdb) (47), eIF2α (1Q8K.pdb) (48), SET (2E50.pdb) (49), YWHAE (2BR9.pdb) (50), and Cc (1J3S.pdb) (51) were modeled using the PDB coordinates. The structure corresponding to STRAP was obtained via homology comparison using the MODELLER software (52). In order to define the interface with Cc involved in the interaction with the above-mentioned targets, soft docking calculations were performed using BiGGER (34). The Cc complexes with the best global score values are shown in Fig. 4. Interestingly, Cc uses the same surface surrounding the heme crevice (Fig. 4) to interact with all novel interaction partners as earlier studies have shown it to do with its physiological redox partners, namely, cytochrome c1 (53), cytochrome-c peroxidase (54), and cytochrome-c oxidase (55). Fig. 4 shows the distribution of mass centers for Cc with respect to the protein interaction partners. Notably, all of them preferably explored a well-defined area of Cc.
Fig. 4.
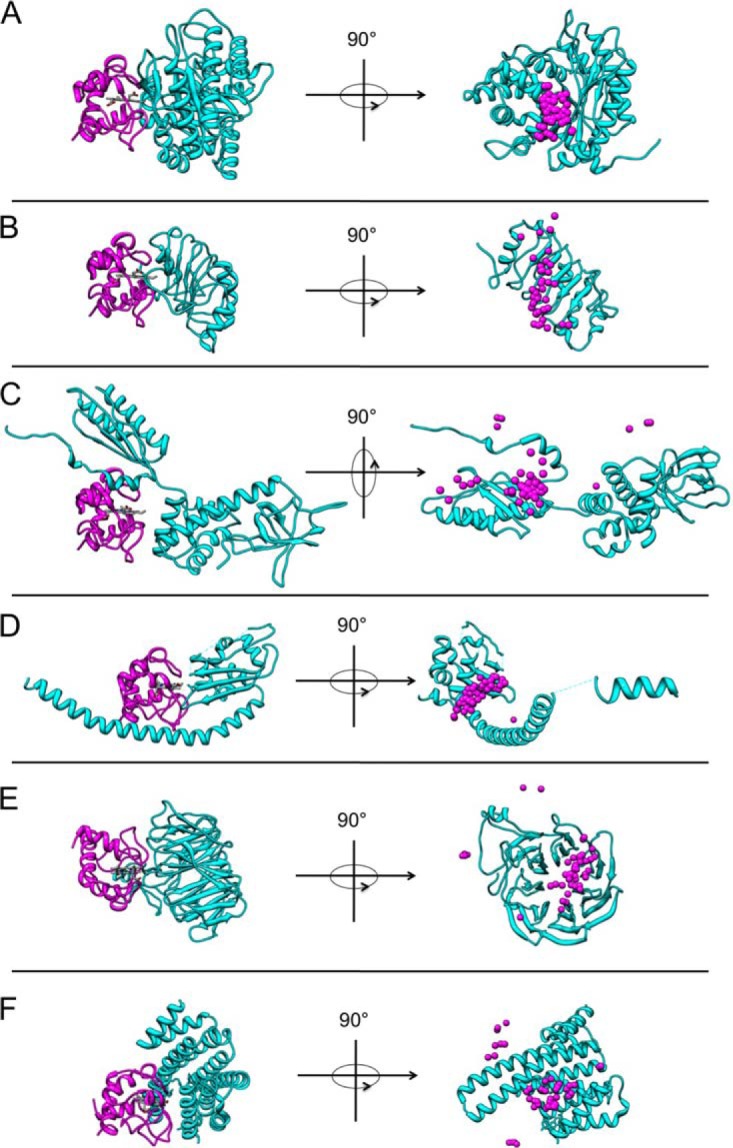
Molecular docking models of Cc complexes. A, CcxALDOA docking model with the best global score (left) in the ratio 1:1 and centers of mass distribution of 50 molecules of Cc with respect to ALDOA (right). Ribbon representation of Cc in magenta, with its heme group in white. The ribbon of ALDOA is colored in cyan. B, CcxANP32B docking model with the best global score values (left) and centers of mass distribution of 50 molecules of Cc with respect to ANP32B (right). Same color-coding as in A. C, CcxeIF2α docking model with the best global score values (left) and centers of mass distribution of 50 molecules of Cc with respect to eIF2α (right). Same color-coding as in A. D, CcxSET docking model with the best global score values (left) and centers of mass distribution of 50 molecules of Cc with respect to SET (right). Same color-coding as in A. E, CcxSTRAP docking model with the best global score values (left) and centers of mass distribution of 50 molecules of Cc with respect to STRAP (right). Same color-coding as in A. F, CcxYWHAE docking model with the best global score values (left) and centers of mass distribution of 50 molecules of Cc with respect to YWHAE (right). Same color-coding as in A.
In Vitro Validation of Cc Adducts: SPR, NMR, and ITC Measurements
The interaction between Cc and its protein partners was measured in SPR, NMR, and ITC experiments. For SPR spectroscopy, human Cc was immobilized on the sensor surface. Six Cc targets—ANP32B, eIF2α, hnRNP C1/C2, HSPA5, SET, and YWHAE—were overexpressed as soluble recombinant proteins. The SPR sensograms of Cc-binding to YWHAE, eIF2α, hnRNP C1/C2, HSPA5, and SET are shown in Fig. 5, in which the background response was subtracted from the sample sensogram to obtain the actual binding response. The background response was recorded by injecting the analyte through a control or reference flow cell that had no ligand immobilized on the sensor surface. The Cc–ANP32B interaction could not be detected with this technique.
Fig. 5.

SPR measurements. A, sensograms recorded for the binding of human Cc with eIF2α. Three replicate injections were performed for each protein concentration. In each sensogram, the signals from the control surface were subtracted. B, as in A, but for human Cc-hnRNP C1/C2 complex. C, as in A, but for human Cc-HSPA5 complex. D, as in A, but for human Cc-SET complex. E, as in A, but for human Cc-YWHAE complex.
The interactions of Cc with a total of six soluble targets were further analyzed in solution via NMR and ITC. Fig. 6 shows details of the 15N-HSQC spectra of Cc upon binding to eIF2α, ANP32B, HSPA5, SET, YWHAE, and hnRNP C1/C2. Noteworthy, significant CSPs in several Cc amide signals—in particular in that corresponding to the residue glutamate 89—were observed upon binding of Cc to its partners, which is indicative of complex formation in solution of Cc with all but one (hnRNP C1/C2) of the partners. Also, some Cc amide signals broadened beyond the detection limit, thereby suggesting the formation of a long-lived complex, with lifetimes substantially large (in the range of seconds), between Cc and its targets, as inferred from SPR measurements. Furthermore, the ITC thermograms and isotherms revealed that Cc binds to the nucleo/cytoplasmic targets reported herein (Fig. 7)—except for hnRNP C1/C2—with greater affinity (dissociation constant ranging between 1 and 5 μm) than to its mitochondrial redox targets. Interestingly, ANP32B, whose binding to Cc was not detected via SPR, specifically interacted with Cc when analyzed via NMR and ITC (Table I). These structural and functional data suggest that these complexes are highly specific and mainly involve the surface of Cc surrounding the heme cleft, except for the Cc–SET complex, in which additional residues placed at the opposite face of the heme group were also perturbed (Fig. 8). In the latter complex, Cc might be localized in between the two subunits of the SET dimer so as to extend the Cc-interacting surface. Fig. 8 shows not only how relevant the electrostatic interactions are in bringing both proteins sufficiently close to each other, but also how relevant the hydrophobic contacts are in stabilizing a well-defined orientation. Moreover, Cc yields not only specific but also much more stable complexes with its new apoptotic targets relative to those previously described for its well-known respiratory partners (i.e. cytochrome bc1 and cytochrome-c oxidase of the mitochondrial electron transfer chain, with lifetimes of approximately 5 and 1 ms, respectively) (56, 57).
Fig. 6.

Chemical-shift perturbations and line width of the glutamate 89 amide signal in reduced Cc upon binding to its novel protein partners, as observed via NMR spectroscopy. Details of the superimposed 15N-HSQC spectra of 15N-labeled Cc, either free (blue) or upon binding to elF2α, ANP32B, HSPA5, SET, YWHAE, and hnRNP C1/C2 (red). In all cases, the Cc:partner ratio was 1:1. The arrows indicate the direction and magnitude of chemical-shift perturbations.
Fig. 7.

ITC titrations of reduced Cc with its protein partners. The thermograms and binding isotherms (top and bottom, respectively) of Cc with elF2α, ANP32B, HSPA5, SET, YWHAE, and hnRNP C1/C2 are shown. Standard errors are 5% to 10%.
Fig. 8.
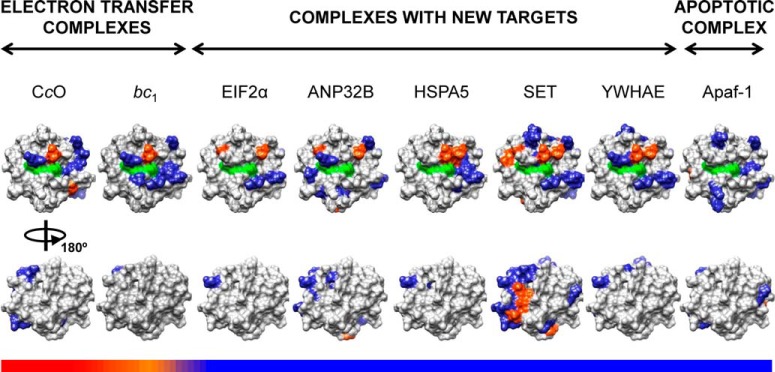
Mapping of Cc upon binding to its well-known and novel targets. Cc residues at the interface area within the corresponding complexes are colored as follows: polar and charged residues in blue, hydrophobic residues in orange. The heme group is marked in green, and the non-interacting residues in gray. Two 180°-rotated surface representations (upper and lower) are depicted for each complex. The horizontal color bars represent the stability and lifetime of the complexes, from highly dynamic/transient (red) to more rigid/stable (blue). Surface representations were generated from the structure of human Cc (PDB entry 1J3S (51)). Cc interfacial residues in the complex with Apaf-1, cytochrome-c oxidase, and cytochrome bc1 were taken from Refs. 7, 55, and 56, respectively.
Network of the Novel Cc-Partner Interactions
In this study, human Cc was identified as a promiscuous protein, able to interact with at least eight confirmed protein targets (Table I) following its release from mitochondria into the cytosol during apoptosis. Thus, Cc appears to be part of a complex regulatory network (Fig. 9).
Fig. 9.

Main functions ascribed to the novel Cc protein partners and network of the resulting interactions. A, diagram with the main functions of novel Cc protein interaction partners identified in vitro through proteomics and confirmed in vivo through BiFC. All targets have been grouped into five functional categories. Several functions can be shared by the same target. B, mapping the interactions between Cc and its novel protein interaction partners, as well as of the latter with the proteins involved in cell death, proliferation, and/or survival, as reported in the literature. Color-coding is the same as in A.
Fig. 9 organizes the main functions of these novel Cc interaction partners in a diagram divided into five main categories:
1. DNA Repair
The SET complex is constituted by SET, pp32, HMG-2, Ape1, NM23-H1, and TREX1 (Fig. 9B) (58). Under homeostatic conditions, this complex is associated with the ER but is mobilized in the nucleus in response to oxidative stress (59). In fact, Ape1, and possibly TREX1 and NM23-H1, seems to be involved in the base excision repair mechanism for single-stranded DNA nicks (60) appearing with oxidative stress (61).
DNA double-strand breaks are generated by exogenous factors (e.g. topoisomerase inhibitors like CPT) and also during DNA replication. Mechanisms for detecting DNA breaks and triggering DNA repair pathways and cell cycle checkpoints have evolved in cells (62, 63). hnRNP C1/C2 has been demonstrated to bind to chromatin in a DNA-damage-dependent manner, as well as to play a role in DNA repair and/or damage response (Fig. 9) (64).
2. Cell Survival Pathways
14–3-3 proteins are involved in the regulation of a wide range of cellular processes, such as the inflammatory response, mitogenic and cell survival signaling, the cell cycle, transcriptional activity, DNA replication, DNA damage, and apoptosis (65). 14–3-3 epsilon (YWHAE), a cytoplasmic member of the family (Fig. 9), has been involved in the regulation of multiple cell survival signaling pathways such as TNF-α/NF-κβ (65, 66) and PI-3K/AKT (67).
STRAP (serine-threonine kinase receptor-associated protein) has been identified as a partner of PDK1 (68). In this study, the co-expression of PDK1 and STRAP suppressed apoptotic cell death. These results suggest that STRAP plays an important role in the modulation of the PDK1-mediated survival signaling pathway. Indeed, STRAP not only positively regulates PDK1, but also regulates the activity of PDK1 downstream targets, activating AKT and inhibiting Bad (69), and thereby promoting cell survival.
HSPA5 is mainly found in the ER, but it is also in the cytoplasm, in the nucleus, and on the cell surface (70–72). During ER stress, HSPA5 is released from the ER into the cytoplasm, interacting with the IKK complex (73) and promoting cell survival and cell proliferation through the NF-κβ signaling pathway (Fig. 9B). Furthermore, HSPA5 physically interacts with Raf-1, known to phosphorylate and inactivate Bad, which results in resistance to apoptosis (74), stabilizes mitochondrial membrane permeability, and inhibits ER stress-induced apoptosis (75).
Hsp70 (Heat shock protein 70) is known to protect cells from apoptosis induced by heat shock, tumor necrosis factor, growth factor withdrawal, oxidative stress, and radiation (76, 77). Recently, NCL has been shown to act as a downstream effector of Hsp70 in protecting cells against oxidative stress-induced apoptosis (78).
3. Metabolic Pathways Blocked during Apoptosis
The inhibition of protein synthesis enhances apoptosis through different stimuli (79). Under apoptotic conditions, eIF2α has been shown to be phosphorylated by PKR. Post-translationally modified eIF2α seems to inhibit translation during apoptosis (80) and to be essential for autophagy initiation (81).
4. Caspase Inhibition
ANP32B (acidic leucine-rich nuclear phosphoprotein 32 B) is involved in gene regulation, acting as a histone chaperone (47) or modulating mRNA trafficking as a HuR ligand (82). In addition, ANP32B can act as a caspase-3 inhibitor (Fig. 9B), as the expression of endogenous ANP32B blocked by a specific siRNA enhances caspase-3 activation and promotes apoptosis (82). In contrast, ANP32B overexpression results in the rescue of Rat1 cells from induced apoptosis (83).
ALDOA (aldolase A) is involved in the conversion of fructose-1,6-bisphosphate to dihydroxyacetonephosphate and glyceraldehyde-3-phosphate (84). The role of glyceraldehyde-3-phosphate as a reversible and noncompetitive inhibitor of caspase-3 has been proposed recently (Fig. 9B) (85).
The ER chaperone protein GRP78 (HSPA5) is involved in the folding and assembly of proteins in the ER (86). Its synthesis can be stimulated by stress conditions that perturb ER function and calcium homeostasis (87). In fact, a subpopulation of HSPA5 exists as an ER transmembrane protein blocking caspase-7 activation (Fig. 9B). Furthermore, HSPA5 forms an inhibitory complex along with caspase-12 (Fig. 9B), preventing its release from the ER (88) and blocking apoptosis.
5. Inhibition of Apoptotic Pathways
Apart from the role of YWHAE and STRAP as positive regulators of cell survival pathways, both are involved in the inhibition of apoptotic signaling pathways through the sequestering of pro-apoptotic proteins (Fig. 9B). Whereas YWHAE interacts with Bad (89, 90), PKC (91), and Ask1 (92), STRAP synergizes with Smad7 in the inhibition of TGF-β signaling, thus blocking apoptosis (93, 94). Moreover, a novel inhibitory function for STRAP regarding Ask1-induced apoptosis has recently been proposed (95).
The three nucleases that form the SET complex—Ape1, NM23-H1, and TREX1—are activated during PCD by the cleavage of the inhibitor protein SET by granzyme A or K, causing single-stranded DNA damage (61, 96, 97).
DISCUSSION
In vivo functions displayed by known components of apoptotic signaling pathways such as Bcl-2 proteins, caspases, and Apaf-1 have been defined through genetic analyses (2). However, this approach has hardly been used in the case of Cc because of its essential requirement during mitochondrial respiration, with the exception of two reports leading to different conclusions (21, 98). In a study by Li et al. (98), a Cc knockout cell line was obtained that showed resistance to inducers for the apoptotic intrinsic pathway but an increased sensitivity to extrinsic ones. However, Vempati et al. (21) indicated that the Cc knockout cell line developed by Li's group expressed a testis-specific Cc isoform. They then removed both Cc isoforms and observed that the cell line was resistant to both apoptotic intrinsic and extrinsic pathway inducers.
Cell fate is known to be regulated by a fine-tuned balance between pro-apoptotic and pro-survival signaling pathways. The data obtained in this study revealed the interaction of human Cc with eight novel partners, extensively considered as pro-survival, anti-apoptotic proteins (Fig. 9). Some of the novel Cc interaction partners identified in the study are involved in DNA damage response/repair mechanisms, protein synthesis, survival signaling pathways, and processes known to be essential during homeostatic cell growth. Other Cc targets block apoptosis by inhibiting caspases or impairing apoptotic signals. It is worth mentioning that some of these novel Cc counterparts—namely, SET, YWHAE, STRAP, and HSPA5—act either in cell survival or in apoptosis.
Intriguingly, Cc offers the surface surrounding the heme pocket for interaction with all partners reported in this study, as inferred from the docking calculations. In fact, the area matches that used by Cc to bind the vast majority of its known redox partners (53, 54, 99–102). The in vitro interactions of some of these Cc complexes—namely, those involving eIF2α, hnRNP C1/C2, ANP32B, HSPA5, SET, and YWHAE—have been mostly corroborated through a combination of SPR, NMR, and ITC approaches (Figs. 5–8). These measurements revealed that the bimolecular complexes formed by Cc with all its novel nuclear or cytoplasmic targets are specific and rather stable, as is the well-known interaction of Cc with Apaf-1 to form the apoptosome (Fig. 8). Interestingly, the surfaces used by Cc (mainly residues surrounding the heme cleft) to bind to Apaf-1 and the novel counterparts reported herein are very similar, and resemble that reported for the well-known Cc-involving electron transfer complexes. Unlike such novel stable complexes, the redox interactions of mitochondrial Cc with cytochrome bc1 or cytochrome-c oxidase are transient and highly dynamic, as the electron transfer reactions demand. This finding suggests that the balance between cell life and death is closely related to transient and stable protein–protein interactions, respectively.
The biointeractomic scaffold hovering around Cc occurs in different cellular compartments, as indicated by BiFC assays (Fig. 2). Along with the intrinsic apoptotic route that leads to apoptosome formation and caspase activation, Cc interacts with other proteins located in the cytoplasm such as YWHAE, STRAP, ALDOA, and eIF2α, as well as with those translocated to the cytoplasm after an apoptotic stimulus such as HSPA5. Notably, some nuclear protein targets of Cc—namely, hnRNP C1/C2, SET, and ANP32B—have also been identified. All of these are involved in DNA damage response, DNA replication, and transcriptional regulation. To the knowledge of the authors, the present study represents the first time that nuclear protein interaction partners of Cc have been proposed, explaining the nuclear localization of the heme protein, as observed previously (12, 13, 103).
These new Cc-interacting targets suggest additional functions for Cc beyond electron transfer reaction inside the mitochondria and apoptosome formation under cytosolic PCD stimuli. Cc might play a key role in a complex signaling network by disrupting cell survival and unlocking apoptotic pathways. In support of this model, the Drosophila Cc distal knockout mutant displays a profound delay of apoptosis, even if the heme protein is not essential for the assembly of an apoptosome-like structure (19, 104).
Nevertheless, evidence of new targets and potential new functions ascribed to Cc during apoptosis allows one to understand the different phenotypes of Cc and Apaf-1 knockouts (21–23), which cannot be explained if Cc is restricted to binding Apaf-1 during apoptosis. In contrast, both phenotypes could be easily reconciled if Cc plays a broader and crucial role as a PCD signaler, unlocking apoptosis and inhibiting cell survival.
In summary, the present study suggests a multifunctional action of Cc in response to PCD stimuli. The findings can open up new ways to understand the process in organisms that lack Apaf-1 or whose apoptosome is devoid of Cc. Given that Cc is highly concentrated in the mitochondrial intermembrane space and that its release into the cytoplasm is an evolutionarily well-conserved event, it is likely that Cc regulates other metabolic processes during PCD, as has been similarly corroborated in plants (105).
Supplementary Material
Acknowledgments
We thank Dr. R. Rodríguez (cicCartuja, Seville, Spain) for her contribution to mass spectrometry analysis and Dr. A. Orea (cicCartuja, Seville, Spain) for her support with fluorescence microscopy. We thank Dr. Manuel Angulo and Dr. Encarnación Zafra for assistance in the NMR data collection at the CITIUS NMR Facility at the University of Seville. nYFP and cYFP vectors were kindly provided by Dr. P. Ciruela (Barcelona, Spain), and positive and negative controls (pBiFC-bJunYN155, pBiFC-bFosYC155 and pBiFC-bFosΔZipYC155) for BiFC assays were kindly offered by Dr. T. K. Kerppola (Ann Arbor, MI). The proteomic analyses were carried out at the Proteomics Facility UCM-FPCM, a member of the ProteoRed Network. We thank Blas Moreno-Beltrán for his critical reading of the manuscript.
Footnotes
Author contributions: J.M., I.D., K.G., J.A.N., M.H., A.D., and M.A.D. designed research; J.M. and K.G. performed research; J.M. and A.V. contributed new reagents or analytic tools; J.M., I.D., K.G., S.J., R.B., A.V., A.D., and M.A.D. analyzed data; J.M., I.D., K.G., A.D., and M.A.D. wrote the paper; S.J. and R.B. took SPR measurements.
* We are grateful to both the Spanish Ministry of Science and Innovation (BFU2009–07190 and BFU2012–31670) and the Regional Government of Andalusia (BIO198) for their financial support. Additional funding for J. Martínez-Fábregas was provided by a Spanish Ministry of Science and Innovation FPI grant (BES-2007–16156). Support for the recording of SPR measurements at Saarland University (Saarbrücken, Germany) was provided to K. González-Arzola through an IUBMB Wood-Whelan Fellowship. Thanks are due for financial support in the form of access to the Bio-NMR Research Infrastructure co-funded under the 7th Framework Programme of the EC (FP7/2007–2013) grant agreement 261863 and project contract RII3-026145.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- Cc
- Cytochrome c
- Apaf-1
- apoptosis protease-activating factor-1
- BiFC
- bimolecular fluorescence complementation
- CPT
- camptothecin
- DAPI
- 4′,6-diamidino-2-phenylindole
- EGFP
- enhanced green fluorescent protein
- ER
- endoplasmic reticulum
- ITC
- isothermal titration calorimetry
- NMR
- nuclear magnetic resonance
- PCD
- programmed cell death
- SPR
- surface plasmon resonance
- TS-4B
- Thiol-Sepharose 4B
- YFP
- yellow fluorescent protein.
REFERENCES
- 1. Jiang X., Wang X. (2004) Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 73, 87–106 [DOI] [PubMed] [Google Scholar]
- 2. Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nuñez G., Benedict M. A., Hu Y., Inohara N. (1998) Caspases: the proteases of the apoptotic pathway. Oncogene 17, 3237–3245 [DOI] [PubMed] [Google Scholar]
- 4. Krammer P. H. (2000) CD95's deadly mission in the immune system. Nature 407, 789–795 [DOI] [PubMed] [Google Scholar]
- 5. Suen D. F., Norris K. L., Youle R. J. (2008) Mitochondrial dynamics and apoptosis. Genes Dev. 22, 1577–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chipuk J. E., Green D. R. (2005) Do inducers of apoptosis trigger caspase-independent cell death? Nat. Rev. Mol. Cell Biol. 6, 268–275 [DOI] [PubMed] [Google Scholar]
- 7. Yu X., Acehan D., Ménétret J. F., Booth C. R., Ludtke S. J., Riedl S. J., Shi Y., Wang X., Akey C. W. (2005) A structure of the human apoptosome at 12.8 Å resolution provides insights into this cell death platform. Structure 13, 1725–1735 [DOI] [PubMed] [Google Scholar]
- 8. Li P., Nijhawan D., Budihardjo I., Srinivasula S. M., Ahmad M., Alnemri E. S., Wang X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/Caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489 [DOI] [PubMed] [Google Scholar]
- 9. Forman H. J., Azzi A. (1997) On the virtual existence of superoxide anions in mitochondria: thoughts regarding its role in pathophysiology. FASEB J. 11, 374–375 [DOI] [PubMed] [Google Scholar]
- 10. Delivani P., Martin S. J. (2006) Mitochondrial membrane remodeling in apoptosis: an inside story. Cell Death Differ. 13, 2007–2010 [DOI] [PubMed] [Google Scholar]
- 11. Solary E., Giordanetto F., Kroemer G. (2008) Re-examining the role of cytochrome c in cell death. Nat. Genet. 40, 379–380 [DOI] [PubMed] [Google Scholar]
- 12. Nur-E-Kamal A., Gross S. R., Pan Z., Balklava Z., Ma J., Liu L. F. (2004) Nuclear translocation of cytochrome c during apoptosis. J. Biol. Chem. 279, 24911–24914 [DOI] [PubMed] [Google Scholar]
- 13. Ruíz-Vela A., González de Buitrago G., Martínez-A. C. (2002) Nuclear Apaf-1 and cytochrome c redistribution following stress-induced apoptosis. FEBS Lett. 517, 133–138 [DOI] [PubMed] [Google Scholar]
- 14. Boehning D., Patterson R. L., Sedaghat L., Glebova N. O., Kurosaki T., Snyder S. H. (2003) Cytochrome c binds to inositol (1,4,5) triphosphate receptors, amplifying calcium-dependent apoptosis. Nat. Cell Biol. 5, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 15. Boehning D., van Rossum D. B., Patterson R. L., Snyder S. H. (2005) A peptide inhibitor of cytochrome c/inositol 1,4,5-triphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc. Natl. Acad. Sci. U.S.A. 102, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szado T., Vanderheyden V., Parys J. B., De Smedt H., Rietdorf K., Kotelevets L., Chastre E., Khan F., Landegren U., Söderberg O., Bootman M. D., Roderick H. L. (2008) Phosphorylation of inositol 1,4,5-triphosphate receptors by protein kinase B/Akt inhibits Ca2+ release and apoptosis. Proc. Natl. Acad. Sci. U.S.A. 105, 2427–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giannattasio S., Atlante A., Antonacci L., Guaragnella N., Lattanzio P., Passarella S., Marra E. (2008) Cytochrome c is released from coupled mitochondria of yeast en route to acetic acid-induced programmed cell death and can work as an electron donor and a ROS scavenger. FEBS Lett. 582, 1519–1525 [DOI] [PubMed] [Google Scholar]
- 18. Balk J., Leaver C. J., McCabe P. F. (1999) Translocation of cytochrome c from the mitochondria to the cytosol occurs during heat-induced programmed cell death in cucumber plants. FEBS Lett. 463, 151–154 [DOI] [PubMed] [Google Scholar]
- 19. Arama E., Bader M., Srivastava M., Bergmann A., Steller H. (2006) The two Drosophila cytochrome c proteins can function in both respiration and caspase activation. EMBO J. 25, 232–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bossy-Wetzel E., Newmeyer D. D., Green D. R. (1998) Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vempati U. D., Diaz F., Barrientos A., Narisawa S., Mian A. M., Millán J. L., Boise L. H., Moraes C. T. (2007) Role of cytochrome c in apoptosis: increased sensitivity to tumor necrosis factor alpha is associated with respiratory defects but not with lack of cytochrome c release. Mol. Cell. Biol. 27, 1771–1783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Marsden V. S., O'Connor L., O'Reilly L. A., Silke J., Metcalf D., Ekert P. G., Huang D. C., Cecconi F., Kuida K., Tomaselli K. J., Roy S., Nicholson D. W., Vaux D. L., Bouillet P., Adams J. M., Strasser A. (2002) Apoptosis initiated by Bcl-2-regulated caspases activation independently of the cytochrome c/Apaf-1/caspases-9 apoptosome. Nature 419, 634–637 [DOI] [PubMed] [Google Scholar]
- 23. Shawgo M. E., Shelton S. N., Robertson J. D. (2009) Caspase-9 activation by the apoptosome is not required for Fas-mediated apoptosis in type II Jurkat cells. J. Biol. Chem. 284, 33447–33455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hüttemann M., Pecina P., Rainbolt M., Sanderson T. H., Kagan V. E., Samavati L., Doan J. W., Lee I. (2011) The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: from respiration to apoptosis. Mitochondrion 11, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodríguez-Roldán V., García-Heredia J. M., Navarro J. A., Hervás M., De la Cerda B., Molina-Heredia F. P., De la Rosa M. A. (2006) A comparative kinetic analysis of the reactivity of plant, horse and human cytochrome c towards cytochrome c oxidase. Biochem. Biophys. Res. Commun. 346, 1108–1113 [DOI] [PubMed] [Google Scholar]
- 26. Johnson N., Ng T. T., Parkin J. M. (1997) Camptothecin causes cell cycle perturbations within T-lymphoblastoid cells followed by dose dependent induction of apoptosis. Leuk. Res. 21, 961–972 [DOI] [PubMed] [Google Scholar]
- 27. Goldstein J. C., Waterhouse N. J., Juin P., Evan G. I., Green D. R. (2000) The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2, 156–162 [DOI] [PubMed] [Google Scholar]
- 28. Nicoletti I., Migliorati G., Pagliacci M. C., Grignani F., Riccardi C. (1991) A rapid and simple method for measuring thymocite apoptosis by propidium iodide staining and flow cytometry. J. Immunol. Methods 139, 271–279 [DOI] [PubMed] [Google Scholar]
- 29. Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. (1996) Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 86, 147–157 [DOI] [PubMed] [Google Scholar]
- 30. Azzi A., Bill K., Broger C. (1982) Affinity chromatography purification of cytochrome c binding enzymes. Proc. Natl. Acad. Sci. U.S.A. 79, 2447–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Candiano G., Bruschi M., Musante L., Santucci L., Ghiggeri G. M., Carnemolla B., Orecchia P., Zardi L., Righetti P. G. (2004) Blue silver: a very sensitive colloidal Coomassie G-250 staining for proteome analysis. Electrophoresis 25, 1327–1333 [DOI] [PubMed] [Google Scholar]
- 32. Sechi S., Chait B. T. (1998) Modification of cysteine residues by alkylation. A tool in peptide mapping and protein identification. Anal. Chem. 70, 5150–5158 [DOI] [PubMed] [Google Scholar]
- 33. Hu C. D., Chinenov Y., Kerppola T. K. (2002) Visualization of interactions among bZip and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell. 9, 789–798 [DOI] [PubMed] [Google Scholar]
- 34. Palma P. N., Krippahl L., Wampler J. E., Moura J. J. (2000) BiGGER: a new (soft) docking algorithm for predicting protein interactions. Proteins 39, 372–384 [PubMed] [Google Scholar]
- 35. Olteanu A., Patel C. N., Dedmon M. M., Kennedy S., Linkoff M. W., Minder C. M., Potts P. R., Deskmukh M., Pielak G. J. (2003) Stability and apoptotic activity of recombinant human cytochrome c. Biochem. Biophys. Res. Commun. 312, 733–740 [DOI] [PubMed] [Google Scholar]
- 36. Janocha S., Bichet A., Zöllner A., Bernhardt R. (2011) Substitution of lysine with glutamic acid at position 193 in bovine CYP11A1 significantly affects protein oligomerization and solubility but not enzymatic activity. Biochim. Biophys. Acta 1814, 126–131 [DOI] [PubMed] [Google Scholar]
- 37. Allen J. W., Tomlinson E. J., Hong L., Ferguson S. J. (2002) The Escherichia coli cytochrome c maturation (Ccm) system does not detectably attach heme to single cysteine variants of an apopcytochrome c. J. Biol. Chem. 277, 33559–33563 [DOI] [PubMed] [Google Scholar]
- 38. Goldstein J. C., Muñoz-Pinedo C., Ricci J. E., Adams S. R., Kelekar A., Schuler M., Tsien R. Y., Green D. R. (2005) Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 12, 453–462 [DOI] [PubMed] [Google Scholar]
- 39. Hu C. D., Grinberg A. V., Kerppola T. K. (2006). Visualization of protein interactions in living cells using bimolecular fluorescence complementation (BiFC) analysis. Curr. Protoc. Cell Biol. Chapter 21, Unit 21.3 [DOI] [PubMed] [Google Scholar]
- 40. Gandia J., Galino J., Amaral O. B., Soriano A., Lluís C., Franco R., Ciruela F. (2008) Detection of higher-order G protein-coupled receptor oligomers by a combined BRET-BiFC technique. FEBS Lett. 582, 2979–2984 [DOI] [PubMed] [Google Scholar]
- 41. Pandey P., Saleh A., Nakazawa A., Kumar S., Srinivasula S. M., Kumar V., Weichselbaum R., Nalin C., Alnemri E. S., Kufe D., Kharbanda S. (2000) Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 19, 4310–4322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kerppola T. K. (2006) Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 1, 1278–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Taborsky G. (1970) Interaction of cytochrome c and the phosphoprotein phosvitin. Formation of a complex with an intact 695-m.mu. absorption band. Biochemistry 9, 3768–3774 [DOI] [PubMed] [Google Scholar]
- 44. Yoshimura T., Matsushima A., Aki K. (1979) Interaction of cytochrome c with the phosphoprotein phosvitin. Biochim. Biophys. Acta 581, 361–324 [DOI] [PubMed] [Google Scholar]
- 45. Mintseris J., Wiehe K., Pierce B., Anderson R., Chen R., Janin J., Weng Z. (2005) Protein-protein docking benchmark 2.0: an update. Proteins 60, 214–216 [DOI] [PubMed] [Google Scholar]
- 46. Dalby A., Dauter Z., Littlechild J. A. (1999) Crystal structure of human muscle aldolase complexed with fructose 1,6-bisphosphate: mechanistic implications. Protein Sci. 8, 291–297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tochio N., Umehara T., Munemasa Y., Suzuki T., Sato S., Tsuda K., Koshiba S., Kigawa T., Nagai R., Yokoyama S. (2010) Solution structure of histone chaperone ANP32B: interaction with core histones H3-H4 through its acidis concave domain. J. Mol. Biol. 401, 97–114 [DOI] [PubMed] [Google Scholar]
- 48. Ito T., Marintchev A., Wagner G. (2004) Solution structure of human initiation factor eIF2alpha reveals homology to the elongation factor eEF1B. Structure 12, 1693–1704 [DOI] [PubMed] [Google Scholar]
- 49. Muto S., Senda M., Akai Y., Sato L., Suzuki T., Nagai R., Senda T., Horikoshi M. (2007) Relationship between the structure of SET/TAF-Iβ/INHAT and its histone chaperone activity. Proc. Natl. Acad. Sci. U.S.A. 104, 4285–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yang X., Lee H. W., Sobott F., Papagrigoriou E., Robinson C. V., Grossmans J. G., Sundström M., Doyle D. A., Elkins J. M. (2006) Structural basis for protein-protein interactions in the 14–3-3 protein family. Proc. Natl. Acad. Sci. U.S.A. 103, 17237–17242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Jeng W. Y., Shiu J. H., Tsai Y. H., Chiang W. J. (2002). Expression and characterization of recombinant human cytochrome c in E. coli. J. Bioenerg. Biomembr. 34 (6), 423–431 [DOI] [PubMed] [Google Scholar]
- 52. Dolan M. A., Noah J. W., Hurt D. (2012) Comparison of common homology modelling algorithms: application of user-defined alignments. Methods Mol. Biol. 857, 399–414 [DOI] [PubMed] [Google Scholar]
- 53. König B. W., Osheroff N., Wilms J., Muijsers A. O., Dekker H. L., Margoliash E. (1980) Mapping of the interaction domain for purified cytochrome c1 on cytochrome c. FEBS Lett. 111, 395–398 [DOI] [PubMed] [Google Scholar]
- 54. Worrall J. A., Kolczak U., Canters G. W., Ubbink M. (2001) Interaction of yeast iso-1-cytochrome c with cytochrome c peroxidase investigated by [15N, 1H] heteronuclear NMR spectroscopy. Biochemistry 40, 7069–7076 [DOI] [PubMed] [Google Scholar]
- 55. Sakamoto K., Kamiya M., Imai M., Shinzawa-Itoh K., Uchida T., Kawano K., Yoshikawa S., Ishimori K. (2011) NMR basis for interprotein electron transfer gating between cytochrome c and cytochrome c oxidase. Proc. Natl. Acad. Sci. U.S.A. 108, 12271–12276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lange C., Hunte C. (2002) Crystal structure of the yeast cytochrome bc1 complex with its bound substrate cytochrome c. Proc. Natl. Acad. Sci. U.S.A. 99, 2800–2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Li Y., Naqui A., Frey T. G., Chance B. (1987) A new procedure for the purification of monodisperse highly active cytochrome c oxidase from bovine heart. Biochem. J. 242, 417–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yan N., Regalado-Magdos A. D., Stiggelbout B., Lee-Kirsch M. A., Lieberman J. (2010) The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol. 11, 1005–1013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martinvalet D., Zu P., Lieberman J. (2005) Granzyme A induces caspase-independent mitochondrial damage, a required first step for apoptosis. Immunity 22, 355–370 [DOI] [PubMed] [Google Scholar]
- 60. Demple B., DeMott M. S. (2002) Dynamics and diversions in base excision DNA repair of oxidized abasic lesions. Oncogene 21, 8926–8934 [DOI] [PubMed] [Google Scholar]
- 61. Chowdhury D., Beresford P. J., Zhu P., Zhang D., Sung J. S., Demple B., Perrino F. W., Lieberman J. (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142 [DOI] [PubMed] [Google Scholar]
- 62. Sancar A., Lindsey-Boltz L. A., Unsal-Kaçmaz K., Linn S. (2004) Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 73, 39–85 [DOI] [PubMed] [Google Scholar]
- 63. Jackson S. P. (2002) Sensing and repairing DNA double-strand breaks. Carcinogenesis 23, 687–696 [DOI] [PubMed] [Google Scholar]
- 64. Lee S. Y., Park J. H., Kim S., Park E. J., Yun Y., Kwon J. (2005) A proteomics approach for the identification of nucleophosmin and heterogeneous nuclear ribonucleoprotein C1/C2 as chromatin-binding proteins in response to DNA double-strand breaks. Biochem. J. 388, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zuo S., Xue Y., Tang S., Yao J., Du R., Yang P., Chen X. (2010) 14–3-3 epsilon dynamically interacts with key components of mitogen-activated protein kinase signal module for selective modulation of the TNF-α-induced time course-dependent NF-κβ activity. J. Proteome Res. 9, 3465–3478 [DOI] [PubMed] [Google Scholar]
- 66. Franzoso G., Zazzeroni F., Papa S. (2003) JNK: a killer on a transcriptional leash. Cell Death Differ. 10, 13–15 [DOI] [PubMed] [Google Scholar]
- 67. Tzivion G., Gupta V. S., Kaplun L., Balan V. (2006) 14–3-3 proteins as potential oncogenes. Semin. Cancer Biol. 16, 203–213 [DOI] [PubMed] [Google Scholar]
- 68. Seong H. A., Jung H., Choi H. S., Kim K. T., Ha H. (2005) Regulation of transforming growth factor-β signaling and PDK1 kinase activity by physical interaction between PDK1 and serine-threonine kinase receptor-associated protein. J. Biol. Chem. 280, 42897–42908 [DOI] [PubMed] [Google Scholar]
- 69. Chen Y., Rodrik V., Foster D. A. (2005) Alternative phospholipase D/mTOR survival signal in human breast cancer cells. Oncogene 24, 672–679 [DOI] [PubMed] [Google Scholar]
- 70. Mintz P. J., Kim J., Do K. A., Wang X., Zinner R. G., Cristofanilli M., Arap M. A., Hong W. K., Troncoso P., Logothetis C. J., Pasqualini R., Arap W. (2003) Fingerprinting the circulating repertoire of antibodies from cancer patients. Nat. Biotechnol. 21, 57–63 [DOI] [PubMed] [Google Scholar]
- 71. Shin B. K., Wang H., Yim A. M., Le Naour F., Brichory F., Jang J. H., Zhao R., Puravs E., Tra J., Michael C. W., Misek D. E., Hanash S. M. (2003) Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J. Biol. Chem. 278, 7607–7616 [DOI] [PubMed] [Google Scholar]
- 72. Bukau B., Horwich A. L. (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92, 351–366 [DOI] [PubMed] [Google Scholar]
- 73. Shkoda A., Ruiz P. A., Daniel H., Kim S. C., Rogler G., Sartor R. B., Haller D. (2007) Interleukin-10 blocked endoplasmic reticulum stress in intestinal epithelial cells: impact on chronic inflammation. Gastroenterology 132, 190–207 [DOI] [PubMed] [Google Scholar]
- 74. Jin S., Zhuo Y., Guo W., Field J. (2005) p21-activated kinase 1 (Pak1)-dependent phosphorylation of Raf-1 regulates its mitochondrial localization, phosphorylation of BAD, and Bcl-2 association. J. Biol. Chem. 280, 24698–24705 [DOI] [PubMed] [Google Scholar]
- 75. Shu C. W., Sun F. C., Cho J. H., Lin C. C., Liu P. F., Chen P. Y., Chang M. D., Fu H. W., Lai Y. K. (2007) GRP78 and Raf-1 cooperatively confer resistance to endoplasmic reticulum stress-induced apoptosis. J. Cell. Physiol. 215, 627–635 [DOI] [PubMed] [Google Scholar]
- 76. Arya R., Mallik M., Lakhotia S. C. (2007) Heat shock genes—integrating cell survival and death. J. Biosci. 32, 595–610 [DOI] [PubMed] [Google Scholar]
- 77. Zhao Y., Wang W., Qian L. (2007) Hsp70 may protect cardiomyocytes from stress-induced injury by inhibiting Fas-mediated apoptosis. Cell Stress Chap. 12, 83–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Jiang B., Zhang B., Liang P., Song J., Deng H., Tu Z., Deng G., Xiao X. (2010) Nucleolin/C23 mediates the antiapoptotic effect of heat shock protein 70 during oxidative stress. FEBS J. 277, 642–652 [DOI] [PubMed] [Google Scholar]
- 79. Polunovsky V. A., Wendt C. H., Ingbar D. H., Peterson M. S., Bitterman P. B. (1994) Induction of endothelial cell apoptosis by TNF-α: modulation by inhibitors of protein synthesis. Exp. Cell Res. 214, 584–594 [DOI] [PubMed] [Google Scholar]
- 80. Saelens X., Kalai M., Vandenabeele P. (2001) Translation inhibition in apoptosis: caspase-dependent PKR activation and eIF2-α phosphorylation. J. Biol. Chem. 276, 41620–41628 [DOI] [PubMed] [Google Scholar]
- 81. Kouroku Y., Fujita E., Tanida I., Ueno T., Isoai A., Kumagai H., Ogawa S., Kaufman R. J., Kominami E., Momoi T. (2007) ER stress (PERK/eIF2-α phosphorylation) mediates the polyglutamine-induced LC3 conversion, an essential step for autophagy formation. Cell Death Differ. 14, 230–239 [DOI] [PubMed] [Google Scholar]
- 82. Shen S. M., Yu Y., Wu Y. L., Cheng J. K., Wang L. S., Chen G. Q. (2010) Downregulation of ANP32B, a novel substrate of caspase-3, enhances caspase-3 activation and apoptosis induction in myeloid leukemic cells. Carcinogenesis 31, 419–426 [DOI] [PubMed] [Google Scholar]
- 83. Sun W., Kimura H., Hattori N., Tanaka S., Matsuyama S., Shiota K. (2006) Proliferation related acidic leucine-rich protein PAL31 functions as a caspase-3 inhibitor. Biochem. Biophys. Res. Commun. 342, 817–823 [DOI] [PubMed] [Google Scholar]
- 84. Yao D. C., Tolan D. R., Murray M. F., Harris D. J., Darras B. T., Geva A., Neufeld E. J. (2004) Hemolytic anemia and severe rhabdomyolysis caused by compound heterozygous mutations of the gene for erythrocyte/muscle isozyme of aldolase, ALDOA (Arg303X/Cys338Tyr). Blood 103, 2401–2403 [DOI] [PubMed] [Google Scholar]
- 85. Jang M., Kang H. J., Lee S. Y., Chung S. J., Kang S., Chi S. W., Cho S., Lee S. C., Lee C. K., Park B. C., Bae K. H., Park S. G. (2009) Glyceraldehyde-3-phosphate, a glycolytic intermediate, plays a key role in controlling cell fate via inhibition of caspase activity. Mol. Cell 28, 559–563 [DOI] [PubMed] [Google Scholar]
- 86. Hsu W. C., Wang H. K., Lee L. C., Fung H. C., Lin J. C., Hsu H. P., Wu Y. R., Ro L. S., Hu F. J., Chang Y. T, Chen G. J., Chen C. M. (2008) Promoter polymorphisms modulating HSPA5 expression may increase susceptibility to Taiwanese Alzheimer's disease. J. Neural Transm. 115, 1537–1543 [DOI] [PubMed] [Google Scholar]
- 87. Lee A. S. (2001) The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem. Sci. 26, 504–510 [DOI] [PubMed] [Google Scholar]
- 88. Rao R. V., Peel A., Logvinova A., del Rio G., Hermel E., Yokota T., Goldsmith P. C., Ellerby L. M., Ellerby H. M., Bredesen D. E. (2002) Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 514, 122–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Won J., Kim D. Y., La M., Kim D., Meadows G. G., Joe C. O. (2003) Cleavage of 14–3-3 protein by caspase-3 facilitates Bad interaction with Bcl-X(L) during apoptosis. J. Biol. Chem. 278, 19347–19351 [DOI] [PubMed] [Google Scholar]
- 90. Zhang L., Chen J., Fu H. (1999) Suppression of apoptosis signal-regulating kinase 1-induced cell death by 14–3-3 proteins. Proc. Natl. Acad. Sci. U.S.A. 96, 8511–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Supekova L., Pezacki J. P., Su A. I., Loweth C. J., Riedl R., Geierstanger B., Schultz P. G., Wemmer D. E. (2002) Genomic effects of polyamide/DNA interactions on mRNA expression. Chem. Biol. 9, 821–827 [DOI] [PubMed] [Google Scholar]
- 92. Subramanian R. R., Zhang H., Wang H., Ichijo H., Miyashita T., Fu H. (2004) Interaction of apoptosis signal-regulating kinase 1 with isoforms of 14–3-3 proteins. Exp. Cell Res. 294, 581–591 [DOI] [PubMed] [Google Scholar]
- 93. Datta P. K., Moses H. L. (2000) STRAP and Smad7 synergize in the inhibition of transforming growth factor β signaling. Mol. Cell. Biol. 20, 3157–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kleeff J., Ishiwata T., Maruyama H., Friess H., Truong P., Büchler M. W., Falb D., Korc M. (1999) The TGF-beta signaling inhibitor Smad7 enhances tumorigenicity in pancreatic cancer. Oncogene 18, 5363–5372 [DOI] [PubMed] [Google Scholar]
- 95. Jung H., Seong H. A., Manoharan R., Ha H. (2010) Serine-threonine kinase receptor-associated protein inhibits apoptosis signal-regulating kinase 1 function through direct interaction. J. Biol. Chem. 285, 54–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Fan Z., Beresford P. J., Oh D. Y., Zhang D., Lieberman J. (2003) Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 112, 659–672 [DOI] [PubMed] [Google Scholar]
- 97. Zhao T., Zhang H., Guo Y., Zhang Q., Hua G., Lu H., Hou Q., Liu H., Fan Z. (2007) Granzyme K cleaves the nucleosome assembly protein SET to induce single-stranded DNA nicks of target cells. Cell Death Differ. 14, 489–499 [DOI] [PubMed] [Google Scholar]
- 98. Li K., Li Y., Shelton J. M., Richardson J. A., Spencer E., Chan Z. J., Wang X., Williams R. S. (2000) Cytochrome c deficiency causes embryonic lethality and attenuates stress-induced apoptosis. Cell 12, 389–399 [DOI] [PubMed] [Google Scholar]
- 99. Crowley P. B., Rabe K. S., Worrall J. A., Canters G. W., Ubbink M. (2002) The ternary complex of cytochrome f and cytochrome c: identification of a second binding site and competition for plastocyanin binding. Chembiochem 3, 526–533 [DOI] [PubMed] [Google Scholar]
- 100. Worrall J. A., Reinle W., Bernhardt R., Ubbink M. (2003) Transient protein interactions studied by NMR spectroscopy: the case of cytochrome c and adrenodoxin. Biochemistry 42, 7068–7076 [DOI] [PubMed] [Google Scholar]
- 101. Ubbink M., Bendall D. S. (1997) Complex of plastocyanin and cytochrome c characterized by NMR chemical shift analysis. Biochemistry 36, 6326–6335 [DOI] [PubMed] [Google Scholar]
- 102. Volkov A. N., Ferrari D., Worrall J. A., Bonvin A. M., Ubbink M. (2005) The orientations of cytochrome c in the highly dynamic complex with cytochrome b5 visualized by NMR and docking using HADDOCK. Protein Sci. 14, 799–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Godoy L. C., Muñoz-Pinedo C., Castro L., Cardaci S., Schonhoff C. M., King M., Tórtora V., Marín M., Miao Q., Jiang J. F., Kapralov A., Jemmerson R., Silkstone G. G., Patel J. N., Evans J. E., Wilson M. T., Green D. R., Kagan V. E., Radi R., Mannick J. B. (2009) Disruption of the M80-Fe ligation stimulates the translocation of cytochrome c to the cytoplasm and nucleus in nonapoptotic cells. Proc. Natl. Acad. Sci. U.S.A. 106, 2653–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Mendes C. S., Arama E., Brown S., Scherr H., Srivastava M., Bergman A., Steller H., Mollereau B. (2006) Cytochrome c-d regulates developmental apoptosis in the Drosophila retina. EMBO Rep. 7, 933–939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Martínez-Fabregas J., Díaz-Moreno I., González-Arzola K., Janocha S., Navarro J. A., Hervás M., Bernhardt R., Díaz-Quintana A., De la Rosa M. A. (2013) New Arabidopsis thaliana cytochrome c partners: a look into the elusive role of cytochrome c in programmed cell death in plants. Mol. Cell. Proteomics 12, 3666–3676 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.