

Abstract
Recent advances in quantitative proteomic technology have enabled the large-scale validation of biomarkers. We here performed a quantitative proteomic analysis of membrane fractions from colorectal cancer tissue to discover biomarker candidates, and then extensively validated the candidate proteins identified. A total of 5566 proteins were identified in six tissue samples, each of which was obtained from polyps and cancer with and without metastasis. GO cellular component analysis predicted that 3087 of these proteins were membrane proteins, whereas TMHMM algorithm predicted that 1567 proteins had a transmembrane domain. Differences were observed in the expression of 159 membrane proteins and 55 extracellular proteins between polyps and cancer without metastasis, while the expression of 32 membrane proteins and 17 extracellular proteins differed between cancer with and without metastasis. A total of 105 of these biomarker candidates were quantitated using selected (or multiple) reaction monitoring (SRM/MRM) with stable synthetic isotope-labeled peptides as an internal control. The results obtained revealed differences in the expression of 69 of these proteins, and this was subsequently verified in an independent set of patient samples (polyps (n = 10), cancer without metastasis (n = 10), cancer with metastasis (n = 10)). Significant differences were observed in the expression of 44 of these proteins, including ITGA5, GPRC5A, PDGFRB, and TFRC, which have already been shown to be overexpressed in colorectal cancer, as well as proteins with unknown function, such as C8orf55. The expression of C8orf55 was also shown to be high not only in colorectal cancer, but also in several cancer tissues using a multicancer tissue microarray, which included 1150 cores from 14 cancer tissues. This is the largest verification study of biomarker candidate membrane proteins to date; our methods for biomarker discovery and subsequent validation using SRM/MRM will contribute to the identification of useful biomarker candidates for various cancers. Data are available via ProteomeXchange with identifier PXD000851.
Recent advances in proteomic technology have contributed to the identification of biomarkers for various diseases. Improvements in LC-MS technology have led to an increase in the number of proteins that have been identified. In addition, a stable isotopic labeling method using isobaric tag for relative and absolute quantitation (iTRAQ)1 and stable isotope labeling by amino acids in cell culture has enabled the quantitative analysis of multiple samples (1, 2). Therefore, a large number of proteins have already been identified as biomarker candidates; however, only a few of these have been used in practical applications because most have not yet progressed to the validation stage, in which potential biomarker candidates are quantified on a large scale. The validation of biomarker candidates is generally accomplished using Western blotting and enzyme-linked immunosorbent assays (ELISA) if specific and well-characterized antibodies for these candidates are available. However, highly specific antibodies are not currently available for most novel biomarker candidate proteins, and it takes a significant amount of time and money to obtain these antibodies and optimize ELISA assay systems for many candidates; therefore, another validation assay system needs to be developed. Selected (or multiple) reaction monitoring (SRM or MRM) was previously shown to be a potentially effective method for the validation of biomarker candidates (3–5). The SRM/MRM assay can measure multiple targets at high sensitivity and throughput without antibodies; hence, it is useful for initial quantitative evaluations and the large-scale validation of biomarker candidates, which defines validation of hundreds of biomarker candidate proteins simultaneously.
In addition to these technical improvements, the fractionation process also plays an important role in proteome analysis for biomarker discovery. This procedure very effectively analyzes the proteomes of specific cellular compartments or organelles in detail, which reduces sample complexity. The preparation of a membrane fraction was previously shown to be useful for identifying membrane proteins that are generally expressed at relatively low levels. Membrane proteins play critical roles in many biological functions, such as signal transduction, cell-cell interactions, and ion transport, account for ∼38% of all proteins encoded by the mammalian genome and more than one-third of biomarker candidates, and are also potential targets for drug therapy (6, 7). Therefore, membrane proteome analysis is important for biomarker discovery. However, difficulties have been associated with extracting and solubilizing membrane proteins and subsequent protease digestion. Many procedures have consequently been developed to improve the solubilization and digestion of membrane proteins (8–11), and a protocol using phase transfer surfactant (PTS) was shown to be suitable for membrane proteomics using LC-MS/MS (12, 13).
The selection of a control group for comparisons is also important for identifying potential biomarkers. Tissue samples from cancer patients have been used in many studies to discover biomarker candidates by proteomic analysis. Previous studies, including our own, attempted to compare cancer tissues with matched normal tissue (14–17). However, marked differences have been reported in the histology, genetics, and proteomics of normal and cancer tissues, and many biomarker candidates have been identified, by making it difficult to narrow down more reliable candidates for further validation. Lazebnik recently emphasized that the features of malignant, but not benign tumors could be used as a hallmark of cancer (18), and also that premalignant lesions were more appropriate controls for cancer tissue than normal tissue for the identification of biomarker candidates involved in cancer progression. Moreover, comparisons of cancer with and without metastasis may also assist in the discovery of biomarker candidates involved in cancer metastasis. Therefore, the identification of biomarker candidates that can be used to diagnose and determine the prognosis of cancer should become more effective by comparing cancer tissues at different stages, including benign tumors.
We performed a shotgun proteomic analysis of membrane fractions prepared from colorectal cancer tissue and benign polyps in the present study to identify biomarker candidates for the diagnosis and treatment of cancer. We identified a large number of biomarker candidate proteins associated with the progression of colon cancer by using membrane protein extraction with PTS followed by iTRAQ labeling. SRM/MRM confirmed the altered expression of these biomarker candidates, and these results were further verified using an independent set of tissue samples. A protein with uncharacterized function, C8orf55, was also validated with a tissue microarray that included various types of cancers.
EXPERIMENTAL PROCEDURES
Tissue Samples
Tissues from 33 cases of primary colorectal cancer were surgically resected. A total of 16 colon polyps were obtained by endoscopic polypectomy. Written informed consent was obtained from each patient before surgery. The Ethics Committee of Chiba University School of Medicine and our institute approved the protocol. The excised samples were obtained from polyp and cancer tissues within one hour of surgery. All excised tissues were immediately placed in liquid nitrogen and stored at −80 °C for further analyses.
Preparation of Membrane Fractions
Membrane fractions were prepared as previously described (19, 20). Tissue samples were washed twice with ice-cold PBS and then homogenized with a Dounce homogenizer in ice-cold PBS containing a protease inhibitor mixture (Roche Diagnostics, Mannheim, Germany). The homogenate was centrifuged at 1000 × g for 10 min at 4 °C, and the post-nuclear supernatant was centrifuged at 100,000 × g for 1 h at 4 °C. The pellet was suspended in ice-cold 0.1 m Na2CO3 solution and centrifuged at 100,000 × g for 1 h at 4 °C. After its resuspension and centrifugation, the pellet was collected as the membrane fraction. This fraction was solubilized with MPEX PTS reagent solution (GL Science, Tokyo, Japan) at 95 °C for 5 min followed by sonication for 5 min using a Bioruptor sonicator (Cosmo Bio, Tokyo, Japan). After centrifugation at 100,000 × g for 30 min at 4 °C, the supernatant was obtained as a membrane fraction extract and quantified using a DC Protein Assay Kit (Bio-Rad, Hercules, CA, USA). The reference pool was arranged by mixing an equal amount (40 μg) of 18 membrane fraction extracts prepared from the tissues of patients.
Peptide Labeling with iTRAQ Reagents
iTRAQ labeling was performed as previously described (19–22). The membrane fraction extract (90 μg) for iTRAQ labeling was reduced with 1/20 volume of 100 mm DTT in 50 mm NaHCO3 for 30 min at room temperature (RT) after the addition of bovine serum albumin (BSA) (0.45 μg) as the internal standard. BSA was spiked into each sample and the iTRAQ ratios, 115:114, 116:114, 117:114, of each experiment were normalized according to the iTRAQ ratios of the BSA added to each sample in order to correct for experimental errors, such as tryptic digestion efficiency, and instrumental errors. A 1/20 volume of 550 mm iodoacetic acid in 50 mm NaHCO3 was then used for alkylation for 30 min at RT. The alkylated sample was digested with 1% trypsin overnight at 37 °C and treated using the PTS method (12, 13) to remove the MPEX PTS reagent. This tryptic digest was desalted using C18 stage Tips (23). DTT, which interfered with iTRAQ labeling, was removed using the PTS method and the next stage involved Tip purification. The desalted peptides were then suspended in 30 μl of iTRAQ dissolution buffer and labeled with iTRAQ reagents (Applied Biosystems, Foster City, CA) for 1 h at RT. The tryptic digests of the reference pool, cancer without metastasis, cancer with metastasis, and polyps were labeled with iTRAQ reagents 114, 115, 116, and 117, respectively (supplemental Table S2). The 115:114, 116:114, and 117:114 ratios indicated the relative abundance of proteins in cancer without metastasis, cancer with metastasis, and polyps, respectively, relative to the common reference pool. Therefore, all samples could be compared, even between different experiments. The labeled samples were then pooled and desalted using C18 stage Tips. A total of six 4-plex iTRAQ experiments were performed.
Fractionation with the SCX Column
iTRAQ-labeled peptides were resuspended in buffer A (10 mm KH2PO4 (pH 3) and 25% acetonitrile) and fractionated using a HPLC system (Shimadzu prominence UFLC) with a SCX column (50 × 2.1 mm, 5 μm, 300 Å, ZORBAX 300SCX; Agilent Technology, Santa Clara, CA). Buffer A and buffer B (10 mm KH2PO4 (pH 3), 25% acetonitrile, 1 m KCl) were used in the mobile phase. The loaded peptides were separated at a flow rate of 200 μl/min with a gradient of 0% B for 30 min, 0% to 10% B in 15 min, 10% to 25% B in 10 min, 25% to 40% B in 5 min, 40% to 100% B in 5 min and 100% B for 10 min. The elution was collected every 1 min and desalted using C18 stage Tips. iTRAQ-labeled peptides were divided into 80 fractions by SCX column chromatography. We monitored the concentrations of these fractions by UV spectroscopy and then combined low-concentration fractions, which resulted in 36 fractions. SCX-fractionated peptides were desalted using C18 stage Tips and dissolved in 20 μl of 2% acetonitrile and 0.1% trifluoroacetic acid.
LC-MS/MS
The SCX-fractionated peptides were analyzed by nano-LC-MS/MS using LTQ-Orbitrap XL (Thermo Fisher Scientific, Bremen, Germany) with a nano-LC interface (AMR, Tokyo, Japan), Paradigm MS2 (Michrom Bioresources, Auburn, CA), and HTC PAL autosampler (CTC Analytics, Zwingen, Switzerland). One-quarter or one-fifth of the volume of each SCX fraction was injected into a trap column (0.3 × 5 mm, l-column ODS; Chemicals Evaluation and Research Institute (CERI), Tokyo, Japan) and separated on an analytical column (0.1 × 200 mm in-house developed Tip Column packed with l-column2 C18 particles; CERI). Buffer A (2% acetonitrile, 0.1% formic acid) and buffer B (90% acetonitrile, 0.1% formic acid) were used in the mobile phase, and the injected peptides were eluted using a gradient from 5% to 30% buffer B at a flow rate of 500 nl/min in 145 min. A spray voltage of 2000 V was applied. The MS scan range was m/z 350–1500. The top three precursor ions in the MS scan by Orbitrap were selected for subsequent MS/MS scans by ion trap (CID) and Orbitrap (HCD) in the automated gain control (AGC) mode in which AGC values of 5.00e + 05, 1.00e + 04, and 2.00e + 04 were set for full MS, CID MS/MS, and HCD MS/MS, respectively.
Identification and Quantification of Proteins
Raw data were examined using Proteome Discoverer ver.1.3 (Thermo Fisher Scientific) with Mascot v2.3.1 (Matrix Science, London, UK) against UniProt/SwissProt (release-2010_05), which contained 20,295 sequences of Homo sapiens, following LC-MS/MS analysis. The search parameters were as follows: precursor mass tolerance of 7 ppm, fragment ion mass tolerance of 0.6 Da (CID), and 0.01 Da (HCD), and one missed cleavage was allowed. The carboxymethylation of cysteine, iTRAQ (K), and iTRAQ (N-terminal) was chosen for the fixed modification. iTRAQ (Y) and oxidation (M) were chosen for variable modifications. The false discovery rate (FDR) was calculated by enabling peptide sequence analysis using Percolator. High-confidence peptide identification was obtained by setting a target FDR threshold of <1.0% at the peptide level. A minimum of two peptides meeting the criteria were required for protein identification. Protein quantification was performed using Proteome Discoverer ver.1.3 and the quantitative value was normalized using that of spiked BSA. Unique BSA peptides were examined using Proteome Discoverer ver.1.1 (Thermo Fisher Scientific) against MSIPI-human version 3.67, which contained BSA sequences.
SRM/MRM Analysis
We used SRM/MRM to confirm and further verify the biomarker candidates obtained from iTRAQ. We firstly performed two technical replicates of SRM/MRM for confirmation using the same individual tissue samples as those used in the iTRAQ discovery experiment. Assays were constructed to measure two distinct peptides per-protein and that the individual assays for each of the two peptides are labeled SRM-1 and SRM-2. We then performed two technical replicates of SRM/MRM with one peptide per protein target for verification using a separate tissue sample set from that used in the discovery experiment. Five technical replicates from a tissue sample mixture were used to assess the reproducibility of SRM/MRM. In our experiments, technical replicates were performed as follows; a single sample was fully processed to peptides, and analyzed twice or five times by LC-SRM/MRM method. We did not analyzed process replicates, which includes tryptic digestion and other sample handling steps, in this study.
SRM/MRM was performed as previously described (19, 21). Stable synthetic isotope-labeled peptides (SI peptides) with a C-terminal 15N- and 13C-labeled arginine or lysine residue (isotopic purity >99%) were purchased from Greiner Bio One (Frickenhausen, Germany) (crude purity). The peptide sequence was selected from the unique peptide sequences identified in the iTRAQ experiments. Peptides containing a cysteine residue (Cys) were also used if another adequate sequence peptide could not be detected. If the SI peptide contained a Cys, the peptide was reduced, alkylated, and then used. The SI peptides were divided into four groups and then mixed, and the four mixtures were separately used for SRM/MRM.
The SI peptide mixture was analyzed by the above-mentioned LC-MS/MS method using LTQ Orbitrap-XL to acquire MS data. A preliminary SRM/MRM-transition list for SI peptides was created from the MS data acquired using Pinpoint ver.1.0 (Thermo Fisher Scientific, Bremen, Germany). The SI peptide mixture was then analyzed using a TSQ-Vantage triple quadruple mass spectrometer (Thermo Fisher Scientific, Bremen, Germany) with a nano-LC interface (AMR, Tokyo, Japan), Paradigm MS2 (Michrom Bioresources, Auburn, CA), and HTC PAL autosampler (CTC Analytics, Zwingen, Switzerland). The data obtained were analyzed using Pinpoint software to optimize parameters such as collision energy and acquire the retention times of each SI peptide. The timed-SRM/MRM method (retention time window of ±2 min) was created using these parameters and then optimized. Finally, four optimal transitions per peptide were selected for quantitation using SRM/MRM.
A membrane fraction extract (2 μg) prepared from tissue samples was alkylated with iodoacetamide and then digested as described above for quantitation using SRM/MRM. The digested peptide was dissolved in 2% acetonitrile and 0.1% trifluoroacetic acid, and analyzed using the above-described optimal timed-SRM/MRM method with TSQ-Vantage. We performed a washing step between each LC MS/MS analysis to minimize carry over. The SI peptide mixture was added to the trypsin-digested sample, and the area ratio of the endogenous peptide to the SI peptide was calculated using the transition peak area measured with Pinpoint software. The amount of each SI peptide was adjusted to be similar to the endogenous peptide estimated by the peak area obtained from preliminary SRM/MRM of the sample mixture. The average of these ratios of more than two transitions was first calculated, and the average ratio of two technical replicates of an individual sample was then determined as the relative quantitative value of the target peptide. Statistical analysis of the area ratios was performed using the t test.
We excluded transition peaks with a signal-to-noise ratio <10, which has been used as empirical LOQ (24), and then compared the profile and proportion of the remaining transition peaks between the SI peptide and endogenous peptide to select appropriate peaks for quantitative analysis. The signal-to-noise ratio was identified using Pinpoint software. Removing the outliers of transitions because of interference or co-elution of nonspecific backgrounds was essential to improve accuracy and reliability. Each transition among the samples had to exhibit a similar peak shape to that with the transition of the SI peptide, which resulted in a minimal CV area ratio (CV<35%) between transitions. We confirmed every transition peak by a manual inspection and removed peaks that did not fulfill the above criteria.
Data Analysis
The transmembrane domains of the identified proteins were predicted using the TMHMM program (http://www.cbs.dtu.dk/services/TMHMM/). Candidate proteins were analyzed using ProteinCenter for cellular component annotation (Thermo Fisher Scientific).
Protein Extraction and Western Blotting
Frozen tissue samples were solubilized in lysis buffer (7 m urea, 2 m thiourea, 4% CHAPS, 1% DTT, protease inhibitor mixture; Roche Diagnostics, Mannheim, Germany) using a Bioruptor sonicator (Cosmo Bio, Tokyo, Japan) following centrifugation at 100,000 × g for 30 min at 4 °C. The supernatant proteins were separated by electrophoresis on 5% to 20% precast gradient gels (DRC Co., Ltd., Tokyo, Japan). Proteins were transferred to polyvinylidene fluoride membranes (Millipore, Bedford, MA), and the membranes were then blocked with ImmunoBlock (DS Pharma Biomedical, Osaka, Japan). An anti-ITGA5 antibody (R&D Systems; 1:1000) and anti-C8orf55 antibody (Sigma-Aldrich; 1:1000) were used as primary antibodies. Antigens on the membrane were detected with enhanced chemiluminescence detection reagents (GE Healthcare, Little Chalfont, Buckinghamshire, UK).
Immunohistochemistry
Tissues were fixed on slide glasses with 4% paraformaldehyde for 10 min at 4 °C. After three washes with PBS, the specimens were treated with 0.5% Triton X-100 in PBS followed by blocking with 3% bovine serum albumin in PBS containing 0.1% Tween-20 (PBST) for 1 h. Samples were then incubated with anti-C8orf55 (1:1000) for 1 h. After washing three times with PBST, samples were treated with horseradish peroxidase-conjugated anti-rabbit IgG (GE Healthcare) for 1 h. After another three washes with PBST, the DAKO EnVision/HRP kit (DAKO Japan, Kyoto, Japan) was used to visualize tissue antigens according to the manufacturer's instructions. Tissue sections were counterstained with hematoxylin for 30 s, dehydrated with 100% ethanol and xylene, and coverslips were mounted with Malinol (Mito Pure Chemicals, Tokyo, Japan).
The tissue microarray used in this study (TMA1150) had 1150 cores from 14 common cancer types (100 cases each of lung (squamous cell carcinoma), lung (adenocarcinoma), breast, kidney, biliary tract, thyroid, liver, colon, and stomach cancer; and 50 cases each of prostate, pancreas, bladder, ovary, and uterine body cancer) (25). The normal tissue array used here contained 280 cores from 13 normal tissues (20 or 40 cases each of lung, breast, kidney, biliary tract, thyroid, liver, colon, stomach, prostate, pancreas, bladder, ovary, and uterine body cancer). These tissue arrays were purchased from Pathology Institute Corp. (Toyama, Japan). After sections were deparaffinized and hydrated, antigen retrieval was performed using a pressure chamber (Pascal; DAKO Japan) in which tissues were heated to 125 °C, maintained at this temperature for 1 min, and then cooled to 90 °C. After rinsing, slides were placed in an Autostainer (DAKO Japan) and an Envision+ detection system was used as suggested by the manufacturer's protocol (DAKO Japan). The cores stained with anti-C8orf55 were examined by three of the authors. Staining intensity was recorded using the following scale: 0, no staining, or cytoplasm staining in <10% of tumor cells; 1, faint/barely perceptible cytoplasm staining in >10% of tumor cells (cells exhibited incomplete cytoplasm staining); 2, weak or moderate cytoplasm staining in >10% of tumor cells or strong cytoplasm staining in <30%; and 3, strong cytoplasm staining in >30% of tumor cells.
RESULTS
iTRAQ Analysis of Membrane Proteins Prepared from Colorectal Cancer Tissue and Polyps
We performed shotgun proteomics of colorectal cancer tissue and premalignant lesions using iTRAQ to identify biomarker candidate proteins for colorectal cancer. Six tissues each were collected from patients with colorectal polyps and cancer with and without metastasis to examine the changes in protein expression associated with cancer progression (Supplemental Table 1). We were particularly interested in changes in membrane proteins; therefore, the membrane fraction prepared from these specimens was dissolved in PTS solution and digested with trypsin, followed by the removal of detergents (Fig. 1). Portions of the extracts of all samples were mixed in equal amounts and treated in the same manner to obtain a reference pool. The trypsin-digested reference pool, cancer without metastasis, cancer with metastasis, and polyps were labeled with iTRAQ reagents 114, 115, 116, and 117, respectively (supplemental Table S2). The iTRAQ-labeled peptides were merged in each experimental set, fractionated by SCX chromatography, and analyzed using LC-MS/MS. The ratios 115:114, 116:114, and 117:114 indicated the higher abundance of proteins in cancer without metastasis, cancer with metastasis, and polyps, respectively, than in the same reference pool. The iTRAQ ratios 115:114, 116:114, and 117:114 of each experiment were normalized using the iTRAQ ratios of the BSA added to each sample in order to correct for experimental errors, such as tryptic digestion efficiency, and instrumental errors (supplemental Table S3).
Fig. 1.
Outline of the experimental workflow.
The reproducibility of the sample preparation was demonstrated by labeling membrane fractions from the same tissue (supplemental Fig. S1). The iTRAQ ratios 116:114 of the BSA added were all close to 1, which indicated minimal technical errors including the digestion of proteins with trypsin (supplemental Fig. S1). A total of 5566 unique proteins were identified using six iTRAQ analysis sets (4195–4633 unique proteins in each experiment; supplemental Tables S2–S4). However, data for cancer without metastasis in the 6th iTRAQ set were removed from this list because we could not obtain adequate data for this group, and this was attributed to a failure in iTRAQ labeling. A total of 1567 proteins (28.2%) were predicted to have a transmembrane domain by the TMHMM program (Table I). In addition, 5287 of the 5566 identified proteins were annotated by GO cellular component analysis: 3087 (58.4%) and 652 (12.3%) were predicted to be membrane proteins and extracellular proteins, respectively (Table I). A total of 4747 proteins were quantified with iTRAQ in at least two of the six analysis sets (supplemental Table S3); thus, we investigated changes in the expression of these 4747 proteins with cancer progression. Differences were observed in the expression of 159, 32, or 99 membrane proteins between polyps and cancer without metastasis, cancer with and without metastasis, or polyps and cancer with metastasis, respectively (ratio >2.0, p value <0.1; ratio <0.5, p value <0.1) (Table II). Differences were also noted in the expression of 55, 17, or 37 extracellular proteins between polyps and cancer without metastasis, cancer with and without metastasis, or polyps and cancer with metastasis, respectively. We then focused on extracellular proteins because they are secreted or shed from cancer cells and may be useful markers.
Table I. Number of predicted membrane proteins.
| Total identified proteins |
5566 |
|
|---|---|---|
| number | % | |
| Number of proteins with transmembrane domains | 1567a | 28.2 |
| GO-annotated | 5287 | 100 |
| Membrane | 3087 | 58.4b |
| Extracellular | 652 | 12.3c |
a Number of proteins with transmembrane domains predicted by TMHMM algorithm.
b,c The ratio of membrane or extracellular proteins to GO-annotated proteins.
Table II. Number of proteins with significant difference in expression. C/P, ratio of cancer without metastasis to polyps. Cm/C, ratio of cancer with metastasis to cancer without metastasis. Cm/P, ratio of cancer with metastasis to polyps. TM + mem, number of proteins with predicted transmembrane domain or annotated as membrane protein. Extra, number of proteins annotated as extracellular protein.
| ratio | p value | C/P |
Cm/C |
Cm/P |
|||
|---|---|---|---|---|---|---|---|
| TM + mem | Extra | TM + mem | Extra | TM + mem | Extra | ||
| > 2.0 | < 0.1 | 108 | 34 | 21 | 8 | 79 | 21 |
| < 0.5 | < 0.1 | 51 | 21 | 11 | 9 | 20 | 16 |
| total | 159 | 55 | 32 | 17 | 99 | 37 | |
Confirmation of Biomarker Candidates by SRM/MRM
Many biomarker candidate proteins have been identified using proteomic analysis; however, most were not validated for the following reasons: (a) the number of candidate proteins was large, (b) specific and well-characterized antibodies for most of these candidates were unavailable for verification by Western blotting, immunostaining, and ELISA, (c) it took too much time and money to optimize these assays, and (d) only a small amount of protein was available to validate biomarker candidates when the protein was prepared from patient tissue, especially the membrane fraction. These difficulties were recently overcome with the SRM/MRM assay, which was shown to be useful for the validation of biomarker candidates because multiple target proteins in a small sample could be analyzed in a single run (3–5). Thus, we used the SRM/MRM method to confirm the results obtained in the iTRAQ experiments and prioritized further validation studies.
In the present study, we selected 105 proteins of the biomarker candidates identified based on the following criteria (Table III): (a) The candidate proteins were quantified in at least two of six iTRAQ experiments. (b) The proteins were predicted to be membrane or extracellular proteins (Human leukocyte antigens were excluded from the candidate list because the proteins were expressed systemically. Proteins such as nuclear or mitochondrial proteins were also excluded.), (c) Differences were observed in the expression of the candidates (ratio >2.0, p value <0.1; ratio <0.5, p value <0.1) between polyps and cancer without metastasis, cancer with and without metastasis, or polyps and cancer with metastasis. Of the selected candidates, 66 proteins were more strongly expressed in nonmetastatic cancer than in polyps, whereas 10 proteins were more strongly expressed in metastatic cancer than in nonmetastatic cancer (Tables 3A and B). Thirteen proteins were more weakly expressed in nonmetastatic cancer than in polyps, whereas six proteins were more weakly expressed in metastatic cancer than in nonmetastatic cancer (Tables 3C and D). Ten proteins were more strongly expressed in metastatic cancer than in polyps (Table 3E).
Table III. List of the proteins analyzed by SRM/MRM and their quantitation data using iTRAQ. p values were calculated by t-test. TM, number of transmembrane domain. C/P, average ratio of cancer without metastasis to polyps. Cm/C, average ratio of cancer with metastasis to cancer without metastasis. Cm/P, average ratio of cancer with metastasis to polyps.
| A. The list of proteins increased in expression between polyps and cancer without metastasis (n = 66) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Protein name | Gene name | TM | GO (mem) | GO (extra) | C/P | p value | Cm/C | p value | Cm/P | p value |
| Q12884 | Seprase | FAP | 1 | mem | 5.98 | <0.01 | 0.67 | 0.190 | 4.03 | 0.029 | |
| P32926 | Desmoglein-3 | DSG3 | 0 | mem | 4.54 | <0.01 | 0.41 | 0.083 | 1.87 | 0.323 | |
| Q6P5W5 | Zinc transporter ZIP4 | SLC39A4 | 7 | mem | 4.35 | 0.075 | 0.42 | 0.189 | 1.84 | 0.217 | |
| Q8NFJ5 | Retinoic acid-induced protein 3 | GPRC5A | 7 | mem | 3.99 | <0.01 | 0.77 | 0.359 | 3.06 | 0.012 | |
| P40199 | Carcinoembryonic antigen-related cell adhesion molecule 6 | CEACAM6 | 0 | mem | 3.69 | 0.029 | 0.85 | 0.690 | 3.12 | 0.031 | |
| O95832 | Claudin-1 | CLDN1 | 4 | mem | 3.47 | 0.054 | 0.51 | 0.180 | 1.77 | 0.127 | |
| Q8TF66 | Leucine-rich repeat-containing protein 15 | LRRC15 | 1 | mem | 3.40 | 0.032 | 0.58 | 0.193 | 1.96 | 0.060 | |
| P24158 | Myeloblastin | PRTN3 | 0 | mem | extra | 3.35 | 0.098 | 0.38 | 0.134 | 1.28 | 0.526 |
| P50150 | Guanine nucleotide-binding protein G(I)/G(S)/G(O) subunit gamma-4 | GNG4 | 0 | mem | 3.31 | 0.074 | 0.77 | 0.570 | 2.56 | 0.051 | |
| P80511 | Protein S100-A12 | S100A12 | 0 | mem | extra | 3.28 | 0.068 | 1.06 | 0.857 | 3.46 | 0.070 |
| P06731 | Carcinoembryonic antigen-related cell adhesion molecule 5 | CEACAM5 | 0 | mem | 3.27 | <0.01 | 0.79 | 0.275 | 2.57 | <0.01 | |
| Q9UKX5 | Integrin alpha-11 | ITGA11 | 1 | mem | 3.23 | <0.01 | 0.62 | 0.081 | 2.00 | 0.016 | |
| Q10588 | ADP-ribosyl cyclase 2 | BST1 | 1 | mem | 3.16 | 0.023 | 0.55 | 0.102 | 1.75 | 0.033 | |
| P08253 | 72 kDa type IV collagenase | MMP2 | 0 | mem | extra | 3.02 | <0.01 | 0.39 | <0.01 | 1.19 | 0.599 |
| Q9H6X2 | Anthrax toxin receptor 1 | ANTXR1 | 1 | mem | 2.98 | <0.01 | 0.70 | 0.027 | 2.09 | <0.01 | |
| Q9Y6I8 | Peroxisomal membrane protein 4 | PXMP4 | 0 | mem | 2.97 | <0.01 | 0.74 | 0.084 | 2.19 | <0.01 | |
| Q12805 | EGF-containing fibulin-like extracellular matrix protein 1 | EFEMP1 | 0 | mem | extra | 2.97 | <0.01 | 0.57 | 0.037 | 1.71 | 0.040 |
| Q14767 | Latent-transforming growth factor beta-binding protein 2 | LTBP2 | 0 | mem | extra | 2.91 | <0.01 | 0.69 | 0.182 | 2.01 | 0.141 |
| P16444 | Dipeptidase 1 | DPEP1 | 0 | mem | 2.85 | 0.033 | 1.06 | 0.854 | 3.02 | <0.01 | |
| P84157 | Matrix-remodeling-associated protein 7 | MXRA7 | 1 | mem | 2.82 | 0.069 | 0.45 | 0.112 | 1.26 | 0.569 | |
| P11169 | Solute carrier family 2, facilitated glucose transporter member 3 | SLC2A3 | 10 | mem | 2.74 | 0.051 | 0.58 | 0.181 | 1.60 | 0.064 | |
| P08648 | Integrin alpha-5 | ITGA5 | 1 | mem | 2.59 | <0.01 | 0.66 | 0.056 | 1.70 | 0.044 | |
| P55001 | Microfibrillar-associated protein 2 | MFAP2 | 0 | extra | 2.56 | <0.01 | 0.46 | <0.01 | 1.16 | 0.322 | |
| Q9ULK5 | Vang-like protein 2 | VANGL2 | 4 | mem | 2.55 | 0.098 | 0.39 | 0.086 | 1.00 | 0.989 | |
| Q5BJF2 | Transmembrane protein 97 | TMEM97 | 4 | mem | 2.54 | <0.01 | 0.73 | 0.250 | 1.85 | 0.040 | |
| Q07075 | Glutamyl aminopeptidase | ENPEP | 1 | mem | 2.53 | <0.01 | 0.70 | 0.201 | 1.77 | 0.104 | |
| Q9UGT4 | Sushi domain-containing protein 2 | SUSD2 | 1 | mem | 2.46 | 0.013 | 0.58 | 0.066 | 1.43 | 0.062 | |
| Q8N6Q3 | CD177 antigen | CD177 | 0 | mem | 2.45 | 0.031 | 0.50 | 0.055 | 1.23 | 0.378 | |
| P07093 | Glia-derived nexin | SERPINE2 | 0 | mem | extra | 2.43 | 0.059 | 0.85 | 0.699 | 2.06 | 0.132 |
| Q96KR6 | Transmembrane protein C20orf108 | C20orf108 | 3 | mem | 2.39 | 0.020 | 0.73 | 0.287 | 1.75 | 0.041 | |
| P09619 | Beta-type platelet-derived growth factor receptor | PDGFRB | 1 | mem | 2.38 | <0.01 | 0.85 | 0.423 | 2.01 | 0.014 | |
| Q7L4E1 | Protein FAM73B | FAM73B | 0 | mem | 2.34 | <0.01 | 0.50 | <0.01 | 1.17 | 0.289 | |
| O75954 | Tetraspanin-9 | TSPAN9 | 4 | mem | 2.31 | <0.01 | 0.70 | 0.088 | 1.61 | <0.01 | |
| Q9Y625 | Glypican-6 | GPC6 | 0 | mem | extra | 2.31 | <0.01 | 0.63 | 0.055 | 1.45 | 0.179 |
| Q8IUS5 | Epoxide hydrolase 4 | EPHX4 | 1 | mem | 2.29 | 0.043 | 1.13 | 0.614 | 2.59 | <0.01 | |
| P36269 | Gamma-glutamyltransferase 5 | GGT5 | 1 | mem | 2.28 | <0.01 | 0.71 | 0.172 | 1.63 | 0.047 | |
| Q8IWU6 | Extracellular sulfatase Sulf-1 | SULF1 | 0 | extra | 2.28 | <0.01 | 0.82 | 0.445 | 1.88 | 0.074 | |
| Q6ZMP0 | Thrombospondin type-1 domain-containing protein 4 | THSD4 | 0 | extra | 2.26 | 0.042 | 0.59 | 0.278 | 1.35 | 0.656 | |
| P21730 | C5a anaphylatoxin chemotactic receptor | C5AR1 | 7 | mem | 2.22 | 0.090 | 0.42 | 0.065 | 0.93 | 0.776 | |
| P35555 | Fibrillin-1 | FBN1 | 0 | mem | extra | 2.22 | 0.039 | 0.38 | 0.022 | 0.84 | 0.363 |
| P98095 | Fibulin-2 | FBLN2 | 0 | extra | 2.20 | <0.01 | 0.68 | 0.206 | 1.49 | 0.300 | |
| P31997 | Carcinoembryonic antigen-related cell adhesion molecule 8 | CEACAM8 | 0 | mem | extra | 2.20 | 0.090 | 0.51 | 0.118 | 1.11 | 0.592 |
| Q14766 | Latent-transforming growth factor beta-binding protein 1 | LTBP1 | 0 | mem | extra | 2.19 | <0.01 | 0.62 | 0.015 | 1.35 | 0.087 |
| Q99720 | Sigma non-opioid intracellular receptor 1 | SIGMAR1 | 1 | mem | 2.19 | <0.01 | 0.86 | 0.439 | 1.88 | <0.01 | |
| P50281 | Matrix metalloproteinase-14 | MMP14 | 1 | mem | extra | 2.19 | <0.01 | 0.70 | 0.096 | 1.53 | 0.078 |
| P02786 | Transferrin receptor protein 1 | TFRC | 1 | mem | extra | 2.18 | <0.01 | 1.09 | 0.579 | 2.38 | <0.01 |
| P31431 | Syndecan-4 | SDC4 | 1 | mem | extra | 2.16 | 0.082 | 0.55 | 0.143 | 1.20 | 0.172 |
| Q9UBG0 | C-type mannose receptor 2 | MRC2 | 1 | mem | 2.15 | <0.01 | 0.68 | 0.078 | 1.47 | 0.230 | |
| Q9P121 | Neurotrimin | NTM | 0 | mem | 2.15 | 0.058 | 0.56 | 0.082 | 1.20 | 0.368 | |
| P09486 | SPARC | SPARC | 0 | extra | 2.14 | <0.01 | 0.85 | 0.321 | 1.81 | 0.025 | |
| P05106 | Integrin beta-3 | ITGB3 | 1 | mem | 2.13 | 0.023 | 0.70 | 0.245 | 1.49 | 0.165 | |
| P04792 | Heat shock protein beta-1 | HSPB1 | 0 | mem | 2.11 | <0.01 | 1.34 | 0.421 | 2.83 | 0.035 | |
| Q9NVM1 | Protein FAM176B | FAM176B | 1 | mem | 2.08 | 0.046 | 1.03 | 0.951 | 2.13 | 0.276 | |
| P08514 | Integrin alpha-IIb | ITGA2B | 1 | mem | 2.08 | 0.083 | 0.88 | 0.759 | 1.83 | 0.130 | |
| Q8WUY1 | UPF0670 protein C8orf55 | C8orf55 | 1 | extra | 2.07 | <0.01 | 1.40 | 0.158 | 2.90 | <0.01 | |
| P12314 | High affinity immunoglobulin gamma Fc receptor I | FCGR1A | 1 | mem | 2.07 | <0.01 | 0.68 | 0.105 | 1.41 | 0.149 | |
| P04216 | Thy-1 mem glycoprotein | THY1 | 0 | mem | 2.06 | <0.01 | 0.77 | 0.092 | 1.59 | 0.023 | |
| P08174 | Complement decay-accelerating factor | CD55 | 0 | mem | extra | 2.05 | <0.01 | 1.04 | 0.879 | 2.13 | 0.020 |
| Q96HV5 | Transmembrane protein 41A | TMEM41A | 6 | mem | 2.04 | <0.01 | 0.76 | 0.060 | 1.54 | <0.01 | |
| Q9ULS5 | Transmembrane and coiled-coil domains protein 3 | TMCC3 | 2 | mem | 2.04 | 0.040 | 0.61 | 0.195 | 1.25 | 0.434 | |
| Q01628 | Interferon-induced transmembrane protein 3 | IFITM3 | 2 | mem | 2.04 | 0.021 | 1.07 | 0.770 | 2.18 | <0.01 | |
| P04920 | Anion exchange protein 2 | SLC4A2 | 11 | mem | 2.04 | 0.044 | 0.82 | 0.441 | 1.66 | <0.01 | |
| Q9Y289 | Sodium-dependent multivitamin transporter | SLC5A6 | 14 | mem | 2.03 | <0.01 | 0.65 | 0.030 | 1.31 | 0.086 | |
| P30273 | High affinity immunoglobulin epsilon receptor subunit gamma | FCER1G | 1 | mem | 2.02 | <0.01 | 0.71 | 0.140 | 1.43 | 0.141 | |
| P08473 | Neprilysin | MME | 1 | mem | 2.01 | 0.097 | 0.87 | 0.626 | 1.74 | 0.030 | |
| P13688 | Carcinoembryonic antigen-related cell adhesion molecule 1 | CEACAM1 | 1 | mem | extra | 2.00 | 0.014 | 0.74 | 0.169 | 1.48 | 0.173 |
| B. The list of proteins increased in expression between cancer without and with metastasis (n = 10) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Protein name | Gene name | TM | GO (mem) | GO (extra) | C/P | p value | Cm/C | p value | Cm/P | p value |
| Q96HR9 | Receptor expression-enhancing protein 6 | REEP6 | 2 | mem | 1.13 | 0.651 | 3.18 | 0.070 | 3.61 | 0.035 | |
| P05451 | Lithostathine-1-alpha | REG1A | 0 | extra | 0.20 | 0.164 | 3.08 | <0.01 | 0.60 | 0.379 | |
| Q8N323 | Protein FAM55A | FAM55A | 1 | extra | 0.22 | 0.102 | 2.98 | 0.057 | 0.65 | 0.416 | |
| O95395 | Beta-1,3-galactosyl-O-glycosyl-glycoprotein beta-1,6-N-acetylglucosaminyltransferase 3 | GCNT3 | 1 | mem | 0.82 | 0.595 | 2.85 | 0.086 | 2.33 | 0.089 | |
| O95994 | Anterior gradient protein 2 homolog | AGR2 | 0 | extra | 0.44 | 0.012 | 2.56 | 0.094 | 1.12 | 0.727 | |
| Q9NRD8 | Dual oxidase 2 | DUOX2 | 6 | mem | 0.43 | 0.081 | 2.51 | 0.045 | 1.07 | 0.843 | |
| Q8TD06 | Anterior gradient protein 3 homolog | AGR3 | 0 | extra | 0.51 | 0.028 | 2.49 | 0.017 | 1.26 | 0.301 | |
| Q09327 | Beta-1,4-mannosyl-glycoprotein 4-beta-N-acetylglucosaminyltransferase | MGAT3 | 1 | mem | 0.89 | 0.694 | 2.24 | 0.046 | 1.99 | 0.024 | |
| Q9Y5L3 | Ectonucleoside triphosphate diphosphohydrolase 2 | ENTPD2 | 2 | mem | extra | 1.36 | 0.369 | 2.09 | 0.028 | 2.83 | <0.01 |
| Q8NCC5 | Sugar phosphate exchanger 3 | SLC37A3 | 12 | mem | 0.94 | 0.838 | 2.06 | 0.016 | 1.92 | 0.030 | |
| C. The list of proteins decreased in expression between polyps and cancer without metastasis (n = 13) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Protein name | Gene name | TM | GO (mem) | GO (extra) | C/P | p Value | Cm/C | p Value | Cm/P | p Value |
| A8K7I4 | Calcium-activated chloride channel regulator 1 | CLCA1 | 0 | mem | extra | 0.14 | <0.01 | 0.85 | 0.673 | 0.12 | <0.01 |
| Q01524 | Defensin-6 | DEFA6 | 0 | extra | 0.18 | 0.053 | 0.69 | 0.428 | 0.12 | 0.043 | |
| Q9Y6R7 | IgGFc-binding protein | FCGBP | 0 | mem | extra | 0.23 | <0.01 | 1.12 | 0.830 | 0.25 | <0.01 |
| Q6ZMB0 | UDP-GlcNAc:betaGalbeta-1,3-N-acetylglucosaminyltransferase 6 | B3GNT6 | 1 | mem | 0.26 | <0.01 | 1.10 | 0.713 | 0.29 | <0.01 | |
| Q02817 | Mucin-2 | MUC2 | 0 | extra | 0.26 | <0.01 | 1.31 | 0.537 | 0.34 | <0.01 | |
| Q07654 | Trefoil factor 3 | TFF3 | 0 | extra | 0.32 | <0.01 | 1.14 | 0.651 | 0.37 | <0.01 | |
| Q9HC84 | Mucin-5B | MUC5B | 0 | extra | 0.36 | 0.038 | 1.29 | 0.352 | 0.47 | 0.042 | |
| P27216 | Annexin A13 | ANXA13 | 0 | mem | 0.37 | <0.01 | 1.24 | 0.574 | 0.46 | 0.018 | |
| P24588 | A-kinase anchor protein 5 | AKAP5 | 0 | mem | 0.43 | 0.017 | 1.12 | 0.606 | 0.48 | 0.011 | |
| Q7Z3J2 | UPF0505 protein C16orf62 | C16orf62 | 0 | mem | 0.46 | <0.01 | 1.39 | 0.072 | 0.65 | <0.01 | |
| P13727 | Bone marrow proteoglycan | PRG2 | 0 | extra | 0.48 | 0.045 | 0.89 | 0.695 | 0.43 | 0.021 | |
| Q6UXG2 | UPF0577 protein KIAA1324 | KIAA1324 | 1 | mem | 0.49 | 0.051 | 1.25 | 0.390 | 0.61 | 0.085 | |
| Q9Y2J2 | Band 4.1-like protein 3 | EPB41L3 | 0 | mem | 0.49 | 0.062 | 1.35 | 0.086 | 0.66 | 0.167 | |
| D. The list of proteins decreased in expression between cancer without and cancer with metastasis (n = 6) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Protein name | Gene name | TM | GO (mem) | GO (extra) | C/P | p Value | Cm/C | p Value | Cm/P | p Value |
| P08123 | Collagen alpha-2(I) chain | COL1A2 | 0 | mem | 1.87 | 0.086 | 0.34 | 0.010 | 0.63 | 0.213 | |
| O75015 | Low affinity immunoglobulin gamma Fc region receptor III-B | FCGR3B | 1 | mem | 2.14 | 0.236 | 0.34 | 0.057 | 0.73 | 0.673 | |
| P02452 | Collagen alpha-1(I) chain | COL1A1 | 0 | mem | extra | 1.86 | 0.109 | 0.36 | 0.025 | 0.66 | 0.252 |
| P02461 | Collagen alpha-1(III) chain | COL3A1 | 0 | mem | 1.70 | 0.182 | 0.39 | 0.039 | 0.66 | 0.152 | |
| Q15063 | Periostin | POSTN | 0 | mem | 1.57 | 0.214 | 0.43 | <0.01 | 0.67 | 0.406 | |
| O43934 | UNC93-like protein MFSD11 | MFSD11 | 10 | mem | 1.27 | 0.434 | 0.45 | 0.067 | 0.58 | 0.110 | |
| E. The list of proteins increased in expression between polyps and cancer with metastasis (n = 10) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Accession | Protein name | Gene name | TM | GO (mem) | GO (extra) | C/P | p Value | Cm/C | p Value | Cm/P | p Value |
| P21589 | 5′-nucleotidase | NT5E | 2 | mem | 1.69 | 0.104 | 1.41 | 0.447 | 2.39 | 0.082 | |
| Q92968 | Peroxisomal membrane protein PEX13 | PEX13 | 0 | mem | 1.35 | 0.012 | 1.73 | 0.031 | 2.34 | 0.012 | |
| O43291 | Kunitz-type protease inhibitor 2 | SPINT2 | 1 | mem | extra | 1.63 | <0.01 | 1.39 | 0.419 | 2.27 | 0.087 |
| Q8N4S7 | Progestin and adipoQ receptor family member 4 | PAQR4 | 3 | mem | 1.82 | <0.01 | 1.23 | 0.342 | 2.25 | 0.019 | |
| Q8NBM4 | Ubiquitin-associated domain-containing protein 2 | UBAC2 | 4 | mem | 1.95 | <0.01 | 1.14 | 0.441 | 2.22 | <0.01 | |
| Q96CP7 | TLC domain-containing protein 1 | TLCD1 | 5 | mem | 1.33 | 0.288 | 1.67 | 0.226 | 2.21 | 0.100 | |
| P05546 | Heparin cofactor 2 | SERPIND1 | 0 | extra | 2.15 | 0.124 | 0.99 | 0.978 | 2.13 | 0.024 | |
| P11166 | Solute carrier family 2, facilitated glucose transporter member 1 | SLC2A1 | 12 | mem | 1.92 | <0.01 | 1.10 | 0.716 | 2.11 | 0.031 | |
| Q9BQD7 | Protein FAM173A | FAM173A | 1 | mem | 1.55 | <0.01 | 1.36 | 0.236 | 2.11 | 0.050 | |
| Q96B21 | Transmembrane protein 45B | TMEM45B | 5 | mem | 1.29 | 0.132 | 1.61 | 0.100 | 2.07 | 0.018 | |
One or two peptide sequences corresponding to the 105 candidate proteins were selected as target sequences for SRM/MRM. We performed two technical replicates for each analysis. SI peptides were synthesized (supplemental Table S5) and spiked into the sample as an internal standard in SRM/MRM. Four transitions per peptide were selected based on precursor and product ion intensities, and parameters such as collision energy were optimized (supplemental Table S6).
We excluded transition peaks with a signal-to-noise ratio <10, which has been used as empirical LOQ (24), and then compared the profile and proportion of the remaining transition peaks between the SI peptide and endogenous peptide to select appropriate peaks for quantitative analysis. Removing the outliers of transitions because of interference or co-eluting nonspecific backgrounds was essential to improve accuracy and reliability. Each transition among the samples had to exhibit a similar peak shape to that with the transition of the SI peptide, which resulted in a minimal CV area ratio (CV<35%) between transitions. We confirmed every transition peak by a manual inspection and removed the peaks that did not conform to the above criteria, which led to accurate and significant quantitation (supplemental Fig. S2).
We obtained the average of these ratios of more than two transitions as the relative quantitative value of the target peptide. Statistical analysis of the area ratios was performed using the t test. In addition, if the expression of one of the two peptides of proteins was significantly different between the sample groups, we considered the protein to be differentially expressed. Using the SRM/MRM method, 172 peptides from 98 proteins were quantified in more than three samples from polyps and cancer with or without metastasis (supplemental Table S7). Significant differences (ratio >2.0, p value <0.1; ratio <0.5, p value <0.1) in at least one of the targeted peptides were detected in 69 proteins (supplemental Fig. S3, supplemental Table S7).
The expression of ITGA5, GPRC5A, PDGFRB, and TFRC was shown to be different in colorectal or other cancer tissues (26–29). The results of iTRAQ and SRM/MRM on these proteins are shown in Fig. 2A. The expression of these proteins showed very similar patterns on iTRAQ and SRM/MRM (supplemental Fig. S4). Furthermore, changes in the expression of ITGA5 were confirmed by Western blotting (Fig. 2B). The similar results obtained by SRM/MRM and iTRAQ were further verified by Western blotting, which indicated that the SRM/MRM assay can be used to confirm the candidates identified in the discovery phase.
Fig. 2.

Representative results obtained with iTRAQ and SRM/MRM in the discovery and confirmation steps. A, The iTRAQ and SRM/MRM data for ITGA5, GPRC5A, PDGFRB, and TFRC are shown. P, polyp. C, cancer without metastasis. Cm, cancer with metastasis. Area ratio, the ratio of the peak area of the endogenous peptide to that of the SI peptide. Assays were constructed to measure two distinct peptides per-protein listed in supplemental Table S5 and that the individual assays for each of the two peptides are labeled SRM-1 and SRM-2. B, Western blotting analysis of polyps and cancer with and without metastasis using an anti-ITGA5 antibody.
Verification of Biomarker Candidates by SRM/MRM
We verified 69 confirmed proteins in an independent set of patient samples (polyps (n = 10), cancer without metastasis (n = 10), and cancer with metastasis (n = 10)) (Table IV, supplemental Table S1, S9, supplemental Fig. S5). We performed five technical replicates using sample mixtures prepared from patient tissue samples to evaluate the reproducibility of our SRM/MRM assay, and obtained high reproducibility (CV<11%) (supplemental Table S8). We did not analyzed process replicates, therefore the actual experimental variability is likely higher than shown by the technical replicate performance owing to variability in digestion and other sample handling steps. The expression levels of a total of 20 proteins: GPRC5A, PRTN3, CEACAM5, ANTXR1, PXMP4, SLC2A3, ENPEP, PDGFRB, GGT5, MMP14, TFRC, MRC2, SPARC, HSPB1, FCGR1A, THY1, TMEM41A, SLC4A2, FCER1G, and CEACAM1, were significantly higher in cancer without metastasis than in polyps (ratio >2.0, p value <0.05). In addition, the expression levels of 10 proteins: ITGA11, BST1, LTBP2, ITGA5, TMEM97, TSPAN9, SIGMAR1, C8orf55, UBAC2, and SERPIND1, were significantly higher in cancer without or with metastasis than in polyps (ratio >1.7, p value <0.05). The expression levels of another five proteins: CEACAM6, LRRC15, GPC6, C5AR1, and TLCD1, were markedly higher in cancer tissues than in polyps. The expression levels of eight proteins: CLCA1, FCGBP, B3GNT6, MUC2, ANXA13, AKAP5, PRG2, and KIAA1324, were lower in cancer with and without metastasis than in polyps (ratio >0.5, p value <0.05). The expression of EPB41L3 was also shown to be lower in cancer tissues than in polyps. This verification step as well as the discovery step revealed that the expression levels of ITGA5, GPRC5A, PDGFRB, and TFRC were markedly higher in cancer tissues than in polyps (Fig. 3). Overall, the expression patterns of 47 out of 69 confirmed proteins were similar between the confirmation and verification analyses.
Table IV. SRM/MRM analysis of biomarker candidate proteins. p values were calculated by t-test. C/P, average ratio of cancer without metastasis to polyps. Cm/C, average ratio of cancer with metastasis to cancer without metastasis. Cm/P, average ratio of cancer with metastasis to polyps.
| Gene name | C/P | p Value | Cm/C | p Value | Cm/P | p Value |
|---|---|---|---|---|---|---|
| FAP | 1.59 | 0.515 | 2.21 | 0.052 | 3.52 | 0.198 |
| GPRC5A | 4.31 | 0.040 | 1.30 | 0.514 | 5.59 | <0.01 |
| CEACAM6 | 13.41 | 0.096 | 0.87 | 0.822 | 11.61 | <0.01 |
| LRRC15 | 2.51 | 0.084 | 1.83 | 0.237 | 4.59 | 0.037 |
| PRTN3 | 2.68 | 0.014 | 1.67 | 0.098 | 4.47 | <0.01 |
| CEACAM5 | 7.29 | 0.044 | 0.85 | 0.737 | 6.22 | <0.01 |
| ITGA11 | 1.90 | 0.019 | 0.82 | 0.408 | 1.55 | 0.066 |
| BST1 | 1.93 | 0.012 | 1.84 | 0.064 | 3.55 | <0.01 |
| MMP2 | 1.18 | 0.601 | 1.11 | 0.761 | 1.30 | 0.396 |
| ANTXR1 | 3.23 | <0.01 | 1.08 | 0.818 | 3.48 | <0.01 |
| PXMP4 | 2.29 | <0.01 | 0.80 | 0.385 | 1.82 | 0.025 |
| EFEMP1 | 1.30 | 0.478 | 1.04 | 0.900 | 1.35 | 0.358 |
| LTBP2 | 1.83 | 0.036 | 1.10 | 0.676 | 2.02 | <0.01 |
| SLC2A3 | 3.56 | 0.030 | 0.92 | 0.817 | 3.28 | <0.01 |
| ITGA5 | 1.83 | <0.01 | 1.79 | 0.162 | 3.28 | 0.031 |
| MFAP2 | 1.41 | 0.394 | 1.17 | 0.496 | 1.65 | 0.128 |
| TMEM97 | 2.00 | <0.01 | 0.83 | 0.411 | 1.67 | 0.064 |
| ENPEP | 3.83 | <0.01 | 1.13 | 0.445 | 4.32 | <0.01 |
| CD177 | 1.17 | 0.581 | 1.42 | 0.224 | 1.66 | 0.144 |
| C20orf108 | 1.23 | 0.368 | 0.94 | 0.823 | 1.16 | 0.560 |
| PDGFRB | 2.22 | <0.01 | 1.00 | 0.995 | 2.22 | <0.01 |
| FAM73B | 1.22 | 0.207 | 0.51 | <0.01 | 0.62 | 0.013 |
| TSPAN9 | 1.75 | <0.01 | 0.99 | 0.968 | 1.74 | <0.01 |
| GPC6 | 1.89 | 0.072 | 1.20 | 0.614 | 2.26 | 0.044 |
| GGT5 | 2.06 | 0.034 | 1.24 | 0.432 | 2.56 | <0.01 |
| C5AR1 | 1.48 | 0.120 | 1.49 | 0.167 | 2.21 | 0.016 |
| FBN1 | 1.37 | 0.443 | 1.34 | 0.257 | 1.84 | 0.072 |
| FBLN2 | 1.75 | 0.102 | 1.02 | 0.946 | 1.79 | 0.087 |
| SIGMAR1 | 1.74 | <0.01 | 0.98 | 0.914 | 1.71 | 0.013 |
| MMP14 | 2.43 | <0.01 | 1.00 | 0.988 | 2.42 | <0.01 |
| TFRC | 2.32 | 0.018 | 1.01 | 0.973 | 2.35 | 0.027 |
| MRC2 | 2.09 | <0.01 | 1.13 | 0.631 | 2.36 | <0.01 |
| SPARC | 2.49 | <0.01 | 0.82 | 0.317 | 2.03 | 0.027 |
| HSPB1 | 2.73 | 0.016 | 1.50 | 0.231 | 4.10 | <0.01 |
| C8orf55 | 1.92 | <0.01 | 0.74 | 0.123 | 1.42 | 0.024 |
| FCGR1A | 2.47 | <0.01 | 1.40 | 0.277 | 3.45 | <0.01 |
| THY1 | 2.14 | <0.01 | 1.00 | 0.983 | 2.15 | <0.01 |
| TMEM41A | 2.04 | <0.01 | 0.90 | 0.593 | 1.84 | <0.01 |
| SLC4A2 | 2.41 | <0.01 | 0.92 | 0.746 | 2.21 | 0.014 |
| FCER1G | 2.23 | <0.01 | 0.97 | 0.888 | 2.17 | <0.01 |
| MME | 5.21 | 0.058 | 0.97 | 0.959 | 5.05 | 0.058 |
| CEACAM1 | 5.95 | 0.025 | 0.83 | 0.646 | 4.92 | <0.01 |
| REEP6 | 1.21 | 0.509 | 0.97 | 0.934 | 1.18 | 0.608 |
| GCNT3 | 1.75 | 0.078 | 1.46 | 0.306 | 2.55 | 0.063 |
| AGR3 | 0.20 | <0.01 | 1.89 | 0.073 | 0.38 | 0.021 |
| ENTPD2 | 1.11 | 0.800 | 0.88 | 0.778 | 0.98 | 0.942 |
| CLCA1 | 0.17 | 0.022 | 1.32 | 0.739 | 0.22 | 0.019 |
| FCGBP | 0.22 | <0.01 | 1.15 | 0.782 | 0.25 | <0.01 |
| B3GNT6 | 0.32 | <0.01 | 1.48 | 0.359 | 0.48 | 0.036 |
| MUC2 | 0.14 | <0.01 | 2.02 | 0.279 | 0.29 | 0.013 |
| TFF3 | 0.33 | 2.80 | 0.93 | |||
| ANXA13 | 0.23 | <0.01 | 1.41 | 0.259 | 0.32 | <0.01 |
| AKAP5 | 0.19 | 0.016 | 0.83 | 0.487 | 0.16 | 0.013 |
| C16orf62 | 0.76 | 0.442 | 0.59 | 0.45 | ||
| PRG2 | 0.34 | 0.018 | 1.10 | 0.744 | 0.38 | 0.021 |
| KIAA1324 | 0.32 | <0.01 | 1.13 | 0.657 | 0.36 | <0.01 |
| EPB41L3 | 0.55 | 0.060 | 0.67 | 0.142 | 0.37 | <0.01 |
| COL1A2 | 1.55 | 0.438 | 1.22 | 0.650 | 1.90 | 0.031 |
| COL1A1 | 1.39 | 0.590 | 1.19 | 0.737 | 1.65 | 0.093 |
| COL3A1 | 1.24 | 0.642 | 1.34 | 0.517 | 1.67 | 0.212 |
| POSTN | 0.90 | 0.687 | 1.94 | 0.018 | 1.75 | 0.033 |
| NT5E | 1.05 | 0.802 | 1.21 | 0.473 | 1.27 | 0.329 |
| PEX13 | 1.73 | <0.01 | 0.81 | 0.224 | 1.40 | 0.025 |
| UBAC2 | 1.87 | <0.01 | 0.95 | 0.834 | 1.78 | 0.044 |
| TLCD1 | 2.13 | <0.01 | 0.76 | 0.335 | 1.63 | 0.113 |
| SERPIND1 | 1.57 | 0.018 | 1.21 | 0.371 | 1.90 | <0.01 |
| SLC2A1 | 2.57 | 0.175 | 1.18 | 0.758 | 3.03 | 0.051 |
| FAM173A | 1.18 | 0.433 | 0.81 | 0.259 | 0.96 | 0.860 |
| TMEM45B | 1.35 | 0.255 | 0.97 | 0.918 | 1.30 | 0.400 |
Fig. 3.
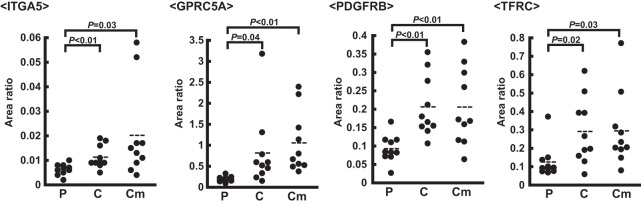
Representative results of SRM/MRM in the verification step. The SRM/MRM data of ITGA5, GPRC5A, PDGFRB, and TFRC are shown. P, polyp. C, cancer without metastasis. Cm, cancer with metastasis. Area ratio, the ratio of the peak area of the endogenous peptide to that of the SI peptide.
Further Validation of C8orf55 by Western Blotting and Immunohistochemistry
We focused on C8orf55 among the biomarker candidates that displayed significant differences in SRM/MRM because it has not been previously reported as a biomarker candidate for cancer and a specific antibody against this protein was available. C8orf55 (also called THEM6) is a 208-amino acid protein that has one predicted transmembrane domain in the N-terminal region; however, its function is unknown. iTRAQ and subsequent confirmation using the SRM/MRM assay revealed that the expression of C8orf55 was up-regulated with cancer progression (Fig. 4A). Furthermore, in the verification step, the expression of this protein was higher in cancer without metastasis than in polyps (ratio = 1.92, p value<0.01). Western blotting was also performed to verify these changes in expression levels (Fig. 4B). Immunohistochemical analysis of colorectal cancer tissue showed that the expression of C8orf55 was high in cancer cells, but was negligible in normal cells (Fig. 4C). These results indicated that the expression of C8orf55 increased in a stepwise fashion with cancer progression.
Fig. 4.

Validation of the biomarker candidate C8orf55. A, The iTRAQ and SRM/MRM data of C8orf55. P, polyp. C, cancer without metastasis. Cm, cancer with metastasis. Area ratio, the ratio of the peak area of the endogenous peptide to that of the SI peptide. Assays were constructed to measure two distinct peptides per-protein listed in supplemental Table S5 and that the individual assays for each of the two peptides are labeled SRM-1 and SRM-2. B, Western blotting analysis of polyps and cancer with and without metastasis using an anti-C8orf55 antibody. C, Immunohistochemical staining of colorectal cancer tissue using an anti-C8orf55 antibody. Left panel: lower magnification. Right panel: higher magnification. The arrowhead shows areas of stained tumor cells and the arrow shows normal colon epithelial glands. Bar, 100 μm.
Examination of C8orf55 Expression in Various Cancer Tissues using Tissue Microarrays
The expression of the tumor markers used in clinical practice, such as CEA and CA19–9, was shown to be higher in multiple cancer types. Therefore, we investigated whether C8orf55 was expressed in various cancer tissues using tissue microarrays (TMA), which contained 1150 cores from 14 common cancer tissues and 280 cores from corresponding normal tissues (supplemental Fig. S6). TMA revealed that the expression of C8orf55 was high in many of the cores prepared from colon cancer tissue, but was negligible in those from normal colon tissues (Fig. 5). TMA also showed that that the expression of C8orf55 was significantly higher in colon cancer tissue than in normal tissue. Immunostaining for C8orf55 was stronger in cancer tissues such as those form the stomach and breast than in normal tissues (Fig. 5). These results demonstrated that C8orf55 may be a potential biomarker for colorectal, stomach, and breast cancer.
Fig. 5.
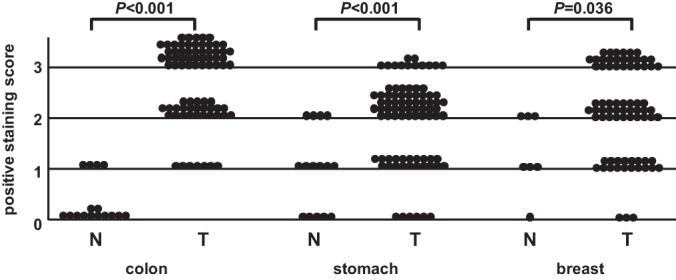
Tissue microarray of C8orf55. C8orf55 immunohistochemistry score (staining intensity) between normal and cancer tissues. Statistical analyses were performed using the Wilcoxon test.
DISCUSSION
A number of large-scale proteomic analyses of cancer tissues for biomarker discovery have been reported to date (30–32); however, few studies have validated the candidate proteins identified because of the absence of an appropriate validation method. SRM/MRM was recently shown to be an efficient validation method (3–5) and several studies, including our own, reported the identification of biomarker candidates by quantitative shotgun proteomics using the iTRAQ labeling method and verification by SRM/MRM (19, 21, 33). In the present study, we performed a proteomic analysis of membrane fractions prepared from colorectal cancer tissue to identify novel biomarker candidates for diagnosis and/or therapeutic targets. We identified membrane proteins, the expression levels of which were altered with the development and progression of colorectal cancer, using comprehensive quantitative analysis with iTRAQ. The most significant achievement of this study was the SRM/MRM-based confirmation and simultaneous large-scale verification using an independent set of tissue samples. Of the 105 biomarker candidate proteins identified by iTRAQ, changes in the expression of 69 proteins were confirmed by SRM/MRM, with significant differences being verified in 44 proteins between groups. This discovery-confirmation-verification workflow should be able to identify more reliable biomarkers for the clinical diagnosis of colon cancer. To the best of our knowledge, we have performed the largest verification of biomarker candidate membrane proteins to date. This verification process using SRM/MRM enabled us to select more potential candidates and prioritize the subsequent validation, and may represent a rapid and effective method to identify novel biomarkers.
We were able to identify 5566 proteins in the membrane fraction in the present study, 3087 (58.4%) of which were predicted to be membrane proteins. This number was markedly higher than that previously reported (34–38); however, non-membrane proteins were also identified in addition to membrane proteins, and this was attributed to the preparation of crude membrane fractions using a simple method. One of the reasons for the increased rate of membrane protein identification was the PTS method-based isolation of membrane proteins (12, 13). The PTS method enables the efficient isolation of membrane proteins and allows the use of a high detergent concentration to achieve the efficient solubilization of very hydrophobic membrane proteins in the cleavage procedure of membrane proteins. Thus, this method may provide deeper proteome coverage for the identification of tissue membrane proteins.
We focused on membrane proteins in this study because membrane proteins are not only involved in the regulation of cell signaling and cell-cell interactions, but are also suitable therapeutic targets for cancers (39). One of the greatest advances in the treatment of cancer in recent years has been the discovery of molecular-targeted drugs, which has resulted in the development of many antibody drugs. Membrane proteins are clearly the best targets for antibody drugs. In this study, we identified a number of previously unreported membrane proteins, the expression of which changed with the development and progression of colorectal cancer. These membrane proteins may be novel therapeutic targets for antibody drug discovery.
Membrane proteins are also suitable biomarkers for the screening and diagnosis of various cancers. Diagnostic biomarkers are ideally detected and quantified in biological fluids such as the plasma and/or urine; however, soluble proteins derived from tissue leakage are often very difficult to detect because there are very few and they are unstable. In contrast, membrane proteins and extracellular proteins are potentially shed and secreted from cells into the circulation; some are actively secreted as microvesicles, such as exosomes, which are very stable and may be potential biomarkers. Several previous studies reported the potential for diagnosing malignant tumors, such as colorectal cancer, melanoma, and glioblastoma, by analyzing exosomal proteins (40–42). Thus, the membrane proteins identified in this study may be promising biomarker candidates for the diagnosis of colorectal cancer.
We observed variations in the quantitative results obtained from iTRAQ and SRM. The samples used for iTRAQ were fractionated with a SCX column, while those for SRM were not. Therefore, variations may have occurred in the quantitative results obtained from iTRAQ and SRM because of differences in the complexities of the samples analyzed. Splicing isoforms or post-translational modifications may also have been involved in these variations because iTRAQ ratios were calculated as the average of all contributing peptide iTRAQ measurements and SRM ratios were obtained by measuring a target peptide.
We investigated differences in the expression levels of proteins between polyps and cancer tissues without metastasis in the present study using proteomic analysis to identify characteristic expression profiles in cancer. Although a number of previous biomarker studies identified hundreds of candidate proteins by comparing cancer tissues with matched normal tissues, many proteins unrelated to malignant properties may also have been included because cancer is generally not directly derived from normal tissues. Thus, the best negative control would be benign tumors, ideally premalignant lesions. In this regard, colorectal polyps are considered to be the best control for colorectal cancer. Moreover, a comparison between different stages of cancer tissues, including benign tumors, is the optimal procedure to identify more useful biomarker candidates.
In our study, C8orf55 was confirmed by SRM/MRM and Western blotting, the findings of which were further verified by multiple cancer tissue microarrays (TMA1150). TMA1150 had 1150 cores from 50 or 100 cases of 14 cancer types and was previously shown to be useful for evaluating changes in protein expression in multiple cancers (25). TMA1150 can also be used to examine the expression of target proteins in various cancer tissues as well as in dozens of cases of colorectal cancer. The extensive validation of the expression of identified candidates in various types of cancer tissues is important in order to determine their usefulness as biomarkers for diverse cancers. In this regard, multi-cancer TMA is a very effective method that can be used to rapidly and simply evaluate the expression patterns of various cancers. TMA1150 revealed that the expression of C8orf55 was higher not only in colon cancer tissue, but also in other cancer tissues, which suggested that these proteins have the potential to be biomarkers for stomach and breast cancer as well as colon cancer.
In conclusion, we successfully performed a SRM/MRM-based large-scale verification of biomarker candidate membrane proteins for colorectal cancer tissues. The methods described here can be readily applied to any type of cancer tissue and can contribute to the identification of novel biomarkers for the diagnosis and therapeutic targets of diseases.
Supplementary Material
Acknowledgments
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository (43) with the dataset identifier PXD000851.
Footnotes
Author contributions: H.K. and T.T. designed research; H.K., S.M., T.K., R.N., and J.F. performed research; R.N., S.W., M.K., Y.K., K.M., J.F., T.M., Y.I., H.M., and F.N. contributed new reagents or analytic tools; H.K., T.K., J.A., S.W., M.K., J.F., and T.T. analyzed data; H.K. and T.T. wrote the paper.
* This work was supported by a Grant-in-Aid, Research on Biological Markers for New Drug Development H20–0005, to T.T. from the Ministry of Health, Labor and Welfare of Japan. It was also supported by Grants-in-Aid 21390354 to T.T and 22590545 to H.K. from the Ministry of Education, Science, Sports and Culture of Japan.
 This article contains supplemental Figs. S1 to S6 and Tables S1 to S9.
This article contains supplemental Figs. S1 to S6 and Tables S1 to S9.
1 The abbreviations used are:
- iTRAQ
- isobaric tag for relative and absolute quantitation
- SRM
- selected reaction monitoring
- MRM
- multiple reaction monitoring
- PTS
- phase-transfer surfactant
- SI-peptide
- stable isotope-labeled peptide
- CID
- collision-induced dissociation
- HCD
- higher energy collision-induced dissociation
- IHC
- immunohistochemistry
- LC-MS/MS
- liquid chromatography tandem mass spectrometry
- CE
- collision energy
- LTQ
- linear ion trap
- FDR
- false discovery rate
- TMA
- tissue microarray.
REFERENCES
- 1. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents, Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 2. Ong S. E., Blagoev B., Kratchmarova I., Kristensen D. B., Steen H., Pandey A., Mann M. (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386 [DOI] [PubMed] [Google Scholar]
- 3. Whiteaker J. R., Zhang H., Zhao L., Wang P., Kelly-Spratt K. S., Ivey R. G., Piening B. D., Feng L. C., Kasarda E., Gurley K. E., Eng J. K., Chodosh L. A., Kemp C. J., McIntosh M. W., Paulovich A. G. (2007) Integrated pipeline for mass spectrometry-based discovery and confirmation of biomarkers demonstrated in a mouse model of breast cancer. J. Proteome Res. 6, 3962–3975 [DOI] [PubMed] [Google Scholar]
- 4. Keshishian H., Addona T., Burgess M., Mani D. R., Shi X., Kuhn E., Sabatine M. S., Gerszten R. E., Carr S. A. (2009) Quantification of cardiovascular biomarkers in patient plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics 8, 2339–2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Whiteaker J. R., Lin C., Kennedy J., Hou L., Trute M., Sokal I., Yan P., Schoenherr R. M., Zhao L., Voytovich U. J., Kelly-Spratt K. S., Krasnoselsky A., Gafken P. R., Hogan J. M., Jones L. A., Wang P., Amon L., Chodosh L. A., Nelson P. S., McIntosh M. W., Kemp C. J., Paulovich A. G. (2011) A targeted proteomics-based pipeline for verification of biomarkers in plasma. Nat. Biotechnol. 29, 625–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ahram M., Litou Z. I., Fang R., Al-Tawallbeh G. (2006) Estimation of membrane proteins in the human proteome. In Silico Biol. 6, 379–386 [PubMed] [Google Scholar]
- 7. Hopkins A. L., Groom C. R. (2002) The druggable genome. Nat. Rev. Drug Discovery 1, 727–730 [DOI] [PubMed] [Google Scholar]
- 8. Chen E. I., Cociorva D., Norris J. L., Yates J. R., 3rd. (2007) Optimization of mass spectrometry-compatible surfactants for shotgun proteomics. J. Proteome Res. 6, 2529–2538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mitra S. K., Gantt J. A., Ruby J. F., Clouse S. D., Goshe M. B. (2007) Membrane proteomic analysis of Arabidopsis thaliana using alternative solubilization techniques. J. Proteome Res. 6, 1933–1950 [DOI] [PubMed] [Google Scholar]
- 10. Zhang N., Chen R., Young N., Wishart D., Winter P., Weiner J. H., Li L. (2007) Comparison of SDS- and methanol-assisted protein solubilization and digestion methods for Escherichia coli membrane proteome analysis by 2-D LC-MS/MS. Proteomics 7, 484–493 [DOI] [PubMed] [Google Scholar]
- 11. Zhou J., Zhou T., Cao R., Liu Z., Shen J., Chen P., Wang X., Liang S. (2006) Evaluation of the application of sodium deoxycholate to proteomic analysis of rat hippocampal plasma membrane. J. Proteome Res. 5, 2547–2553 [DOI] [PubMed] [Google Scholar]
- 12. Masuda T., Tomita M., Ishihama Y. (2008) Phase transfer surfactant-aided trypsin digestion for membrane proteome analysis. J. Proteome Res. 7, 731–740 [DOI] [PubMed] [Google Scholar]
- 13. Masuda T., Saito N., Tomita M., Ishihama Y. (2009) Unbiased quantitation of Escherichia coli membrane proteome using phase transfer surfactants. Mol. Cell. Proteomics 8, 2770–2777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomonaga T., Matsushita K., Yamaguchi S., Oh-Ishi M., Kodera Y., Maeda T., Shimada H., Ochiai T., Nomura F. (2004) Identification of altered protein expression and post-translational modifications in primary colorectal cancer by using agarose two-dimensional gel electrophoresis. Clin. Cancer Res. 10, 2007–2014 [DOI] [PubMed] [Google Scholar]
- 15. Nishimori T., Tomonaga T., Matsushita K., Oh-Ishi M., Kodera Y., Maeda T., Nomura F., Matsubara H., Shimada H., Ochiai T. (2006) Proteomic analysis of primary esophageal squamous cell carcinoma reveals downregulation of a cell adhesion protein, periplakin. Proteomics 6, 1011–1018 [DOI] [PubMed] [Google Scholar]
- 16. Seimiya M., Tomonaga T., Matsushita K., Sunaga M., Oh-Ishi M., Kodera Y., Maeda T., Takano S., Togawa A., Yoshitomi H., Otsuka M., Yamamoto M., Nakano M., Miyazaki M., Nomura F. (2008) Identification of novel immunohistochemical tumor markers for primary hepatocellular carcinoma; clathrin heavy chain and formiminotransferase cyclodeaminase. Hepatology 48, 519–530 [DOI] [PubMed] [Google Scholar]
- 17. Katada K., Tomonaga T., Satoh M., Matsushita K., Tonoike Y., Kodera Y., Hanazawa T., Nomura F., Okamoto Y. (2012) Plectin promotes migration and invasion of cancer cells and is a novel prognostic marker for head and neck squamous cell carcinoma. J. Proteomics 75, 1803–1815 [DOI] [PubMed] [Google Scholar]
- 18. Lazebnik Y. (2010) What are the hallmarks of cancer? Nat. Rev. Cancer 10, 232–233 [DOI] [PubMed] [Google Scholar]
- 19. Muraoka S., Kume H., Watanabe S., Adachi J., Kuwano M., Sato M., Kawasaki N., Kodera Y., Ishitobi M., Inaji H., Miyamoto Y., Kato K., Tomonaga T. (2012) Strategy for SRM-based verification of biomarker candidates discovered by iTRAQ method in limited breast cancer tissue samples. J. Proteome Res. 11, 4201–4210 [DOI] [PubMed] [Google Scholar]
- 20. Muraoka S., Kume H., Adachi J., Shiromizu T., Watanabe S., Masuda T., Ishihama Y., Tomonaga T. (2013) In-depth membrane proteomic study of breast cancer tissues for the generation of a chromosome-based protein list. J. Proteome Res. 12, 208–213 [DOI] [PubMed] [Google Scholar]
- 21. Narumi R., Murakami T., Kuga T., Adachi J., Shiromizu T., Muraoka S., Kume H., Kodera Y., Matsumoto M., Nakayama K., Miyamoto Y., Ishitobi M., Inaji H., Kato K., Tomonaga T. (2012) A strategy for large-scale phosphoproteomics and SRM-based validation of human breast cancer tissue samples. J. Proteome Res. 11, 5311–5322 [DOI] [PubMed] [Google Scholar]
- 22. Shiromizu T., Adachi J., Watanabe S., Murakami T., Kuga T., Muraoka S., Tomonaga T. (2013) Identification of missing proteins in the nextprot database and unregistered phosphopeptides in the phosphositeplus database as part of the chromosome-centric human proteome project. J. Proteome Res. DOI 10.1021/pr300825v [DOI] [PubMed] [Google Scholar]
- 23. Rappsilber J., Ishihama Y., Mann M. (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 75, 663–670 [DOI] [PubMed] [Google Scholar]
- 24. Selevsek N., Matondo M., Sanchez Carbayo M., Aebersold R., Domon B. Systematic quantification of peptides/proteins in urine using selected reaction monitoring. Proteomics 11, 1135–1147 [DOI] [PubMed] [Google Scholar]
- 25. Kitano H., Kageyama S., Hewitt S. M., Hayashi R., Doki Y., Ozaki Y., Fujino S., Takikita M., Kubo H., Fukuoka J. (2010) Podoplanin expression in cancerous stroma induces lymphangiogenesis and predicts lymphatic spread and patient survival. Arch. Pathol. Lab Med. 134, 1520–1527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yang B., Gao J., Rao Z., Shen Q. (2013) Clinicopathological and Prognostic Significance of alpha5beta1-integrin and MMP-14 Expressions in Colorectal Cancer. Neoplasma DOI 10.4149/neo_2013_034 [DOI] [PubMed] [Google Scholar]
- 27. Zougman A., Hutchins G. G., Cairns D. A., Verghese E., Perry S. L., Jayne D. G., Selby P. J., Banks R. E. (2013) Retinoic acid-induced protein 3: identification and characterisation of a novel prognostic colon cancer biomarker. Eur. J. Cancer 49, 531–539 [DOI] [PubMed] [Google Scholar]
- 28. Sillars-Hardebol A. H., Carvalho B., de Wit M., Postma C., Delis-van Diemen P. M., Mongera S., Ylstra B., van de Wiel M. A., Meijer G. A., Fijneman R. J. (2010) Identification of key genes for carcinogenic pathways associated with colorectal adenoma-to-carcinoma progression. Tumour Biol. 31, 89–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prutki M., Poljak-Blazi M., Jakopovic M., Tomas D., Stipancic I., Zarkovic N. (2006) Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer Lett. 238, 188–196 [DOI] [PubMed] [Google Scholar]
- 30. Luo Y., Wang L., Wang J. (2013) Developing proteomics-based biomarkers for colorectal neoplasms for clinical practice: Opportunities and challenges. Proteomics Clin. Appl. 7, 30–41 [DOI] [PubMed] [Google Scholar]
- 31. Lehtio J., De Petris L. (2010) Lung cancer proteomics, clinical and technological considerations. J. Proteomics 73, 1851–1863 [DOI] [PubMed] [Google Scholar]
- 32. Kalinina J., Peng J., Ritchie J. C., Van Meir E. G. (2011) Proteomics of gliomas: initial biomarker discovery and evolution of technology Neuro Oncol. 13, 926–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Thingholm T. E., Bak S., Beck-Nielsen H., Jensen O. N., Gaster M. (2011) Characterization of human myotubes from type 2 diabetic and nondiabetic subjects using complementary quantitative mass spectrometric methods. Mol. Cell. Proteomics 10, DOI 10.1074/mcp.M110.006650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Josic D., Clifton J. G. (2007) Mammalian plasma membrane proteomics. Proteomics 7, 3010–3029 [DOI] [PubMed] [Google Scholar]
- 35. Chen J. S., Chen K. T., Fan C. W., Han C. L., Chen Y. J., Yu J. S., Chang Y. S., Chien C. W., Wu C. P., Hung R. P., Chan E. C. (2010) Comparison of membrane fraction proteomic profiles of normal and cancerous human colorectal tissues with gel-assisted digestion and iTRAQ labeling mass spectrometry. FEBS J. 277, 3028–3038 [DOI] [PubMed] [Google Scholar]
- 36. Han C. L., Chen J. S., Chan E. C., Wu C. P., Yu K. H., Chen K. T., Tsou C. C., Tsai C. F., Chien C. W., Kuo Y. B., Lin P. Y., Yu J. S., Hsueh C., Chen M. C., Chan C. C., Chang Y. S., Chen Y. J. (2011) An informatics-assisted label-free approach for personalized tissue membrane proteomics: case study on colorectal cancer. Mol. Cell. Proteomics 10, DOI 10.1074/mcp.M110.003087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Besson D., Pavageau A. H., Valo I., Bourreau A., Belanger A., Eymerit-Morin C., Mouliere A., Chassevent A., Boisdron-Celle M., Morel A., Solassol J., Campone M., Gamelin E., Barre B., Coqueret O., Guette C. (2011) A quantitative proteomic approach of the different stages of colorectal cancer establishes OLFM4 as a new nonmetastatic tumor marker. Mol. Cell. Proteomics 10, DOI 10.1074/mcp.M111.009712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Polisetty R. V., Gautam P., Sharma R., Harsha H. C., Nair S. C., Gupta M. K., Uppin M. S., Challa S., Puligopu A. K., Ankathi P., Purohit A. K., Chandak G. R., Pandey A., Sirdeshmukh R. (2012) LC-MS/MS analysis of differentially expressed glioblastoma membrane proteome reveals altered calcium signaling and other protein groups of regulatory functions. Mol. Cell. Proteomics 11, DOI 10.1074/mcp.M111.013565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rucevic M., Hixson D., Josic D. (2011) Mammalian plasma membrane proteins as potential biomarkers and drug targets. Electrophoresis 32, 1549–1564 [DOI] [PubMed] [Google Scholar]
- 40. Choi D. S., Park J. O., Jang S. C., Yoon Y. J., Jung J. W., Choi D. Y., Kim J. W., Kang J. S., Park J., Hwang D., Lee K. H., Park S. H., Kim Y. K., Desiderio D. M., Kim K. P., Gho Y. S. (2011) Proteomic analysis of microvesicles derived from human colorectal cancer ascites. Proteomics 11, 2745–2751 [DOI] [PubMed] [Google Scholar]
- 41. Peinado H., Aleckovic M., Lavotshkin S., Matei I., Costa-Silva B., Moreno-Bueno G., Hergueta-Redondo M., Williams C., Garcia-Santos G., Ghajar C., Nitadori-Hoshino A., Hoffman C., Badal K., Garcia B. A., Callahan M. K., Yuan J., Martins V. R., Skog J., Kaplan R. N., Brady M. S., Wolchok J. D., Chapman P. B., Kang Y., Bromberg J., Lyden D. (2012) Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shao H., Chung J., Balaj L., Charest A., Bigner D. D., Carter B. S., Hochberg F. H., Breakefield X. O., Weissleder R., Lee H. (2012) Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med. 18, 1835–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.