

Abstract
Alternative splicing is a pervasive process in eukaryotic organisms. More than 90% of human genes have alternatively spliced products, and aberrant splicing has been shown to be associated with many diseases. Current methods employed in the detection of splice variants include prediction by clustering of expressed sequence tags, exon microarray, and mRNA sequencing, all methods focusing on RNA-level information. There is a lack of tools for analyzing splice variants at the protein level. Here, we present SpliceVista, a tool for splice variant identification and visualization based on mass spectrometry proteomics data. SpliceVista retrieves gene structure and translated sequences from alternative splicing databases and maps MS-identified peptides to splice variants. The visualization module plots the exon composition of each splice variant and aligns identified peptides with transcript positions. If quantitative mass spectrometry data are used, SpliceVista plots the quantitative patterns for each peptide and provides users with the option to cluster peptides based on their quantitative patterns. SpliceVista can identify splice-variant-specific peptides, providing the possibility for variant-specific analysis. The tool was tested on two experimental datasets (PXD000065 and PXD000134). In A431 cells treated with gefitinib, 2983 splice-variant-specific peptides corresponding to 939 splice variants were identified. Through comparison of splice-variant-centric, protein-centric, and gene-centric quantification, several genes (e.g. EIF4H) were found to have differentially regulated splice variants after gefitinib treatment. The same discrepancy between protein-centric and splice-centric quantification was detected in the other dataset, in which induced pluripotent stem cells were compared with parental fibroblast and human embryotic stem cells. In addition, SpliceVista can be used to visualize novel splice variants inferred from peptide-level evidence. In summary, SpliceVista enables visualization, detection, and differential quantification of protein splice variants that are often missed in current proteomics pipelines.
Eukaryotic genes are composed of exonic (protein-coding) and intronic (non-coding) regions. Alternative splicing is a process in which pre-mRNA is cut at junction sites and the resulting exonic sequences are reconnected in different ways to form different versions of mature mRNA. It has been shown that 92% to 94% of human genes can undergo alternative splicing (1, 2). This process plays an essential role in increasing the proteome diversity in eukaryotic organisms. For multiexon mRNAs, different splicing patterns can occur, such as exon skipping (an exon is either included or excluded from the mature mRNAs), alternative 5′ or 3′ splicing (exons are spliced in different lengths), or mutually exclusive splicing (exons are selectively spliced to be exclusively present in different splice forms). Alternative splicing is carried out by the spliceosome, which consists of five small nuclear ribonucleoprotein particles, U1, U2, U4, U5, and U6, and more than 150 other proteins (3). Mutations in splicing sites or in the main components of the splicing machinery will affect the genes' splicing patterns and potentially give rise to alternative protein products that might have different conformations, functions, or subcellular locations. Disruption of the splicing machinery has been shown to be associated with many human diseases such as cystic fibrosis, Alzheimer disease, and cancer (4–6).
Large efforts have been put into the identification of gene products generated by alternative splicing. This is a challenging task, as alternative splice forms are often temporal, tissue specific, and low abundant (7). So far most work has been done starting at the mRNA level, making use of the vast amount of public-domain expressed sequence tag data as well as RNA sequencing data (8–11). Expressed sequence tags or RNA sequencing reads that belong to one gene are clustered together and then aligned with the genomic sequence in order to identify alternative splicing events. These efforts have resulted in many publically available alternative splicing databases. Most of them are generated by mining data from GenBank, UniGene and Swiss-Prot. The Evidence Viewer Database (EVDB)1 is one of the relational databases that support splice variant searches by sequence and gene symbol query (12). This database uses high-quality transcripts from NCBI GenBank and RefSeq, which are then aligned to the chromosomal sequence so that their exon structures can be determined. EVDB contains 81,142 non-redundant human splice variants in the most recent build (completed in June 2010). The ECgene database is an alternative splicing database constructed according to genome-based expressed sequence tag clustering. Splice variants in the database are assigned different evidence levels based on the minimum number of clones used to cover the transcripts (13).
Mass spectrometry (MS)-based proteomics enables large-scale identification and quantification of proteins. The most commonly used workflow for MS-based proteomics is the so-called bottom-up approach or shotgun proteomics, in which proteins are digested into peptides to facilitate efficient MS analysis. Bioinformatic methods are then used to infer the protein-level events (14). A challenge in shotgun proteomics is the protein inference problem, which refers to the task of determining which proteins the identified peptides are derived from. The difficulty arises because some peptides are shared by several proteins. Once the protein mixture is digested by a protease, peptides from all proteins are mixed together, and the protein context of each and every peptide is lost. This leads to ambiguities in the identification of proteins present in the sample. The existence of several protein isoforms (e.g. alternative splicing forms) further complicates the identification process, as protein isoforms usually have very similar sequences. After tryptic digestion, it is impossible to distinguish between different protein isoforms with absolute certainty if no splice-variant-specific peptides (SVSPs) are identified. Nevertheless, MS-based proteomics has been used to identify known and novel splice variants by including the sequences of known and predicted protein variants in the search database (15–17).
In quantitative proteomics, the protein inference problem also affects the accuracy of protein quantification, as protein quantity measurements can be compromised by peptides that are wrongly assigned to a particular protein or protein variant. To address this problem, we recently developed a tool, PQPQ (protein quantification by peptide quality control) (18), to detect protein variants in MS-based shotgun proteomics data. This method is based on the assumption that peptides derived from a given protein variant will have a correlated quantitative pattern over samples. PQPQ takes all high-confidence peptide spectrum matches (PSMs) and clusters them based on their quantitative pattern over samples. PSMs derived from different protein isoforms that are differentially expressed or regulated will have different quantitative patterns and will consequently be grouped in different clusters by PQPQ. PQPQ may thus detect protein variants based on quantitative patterns, even in cases where the database search of the MS/MS data has failed to detect those protein variants.
Here we present a novel tool, SpliceVista, which enables and facilitates splice-variant-centered interrogation of shotgun proteomics data. SpliceVista retrieves gene structure and translated sequences from two alternative splicing databases, EVDB and ECgene, and maps identified peptides to splice variants. The visualization module plots the exon composition of each splice variant and aligns identified peptides with its transcript positions. If quantitative MS data are used, SpliceVista plots the quantitative patterns for each peptide. In addition, a simplified version of the PQPQ algorithm is included in the package to provide users the option to cluster peptides based on their quantitative patterns. Given that splice variants affect gene function and aberrant splicing forms have been shown to be related to many human diseases such as cancer (4–6), we envision that SpliceVista will be an important tool in splice-variant-associated biomarker discovery and biological research on variant-specific proteome changes.
MATERIALS AND METHODS
Algorithm Availability and Requirements
SpliceVista was written in Python 2.7.2. It consists of five modules: converter.py, mergepsm.py, download.py, mapping.py, and visualization.py. It also includes a simplified version of PQPQ (named clusterpeptide.py) that mainly does the peptide clustering. A detailed manual for the program can be found in supplemental file 1. The program is free to use and instructions for downloading the program is in the manual.
Preprocessing of MS Data
The following information needs to be extracted from the MS output from database searching: protein accession I.D., peptide sequence, and quantitative data (if available). The gene symbol is then assigned to each protein by the Python script converter.py and is used to retrieve known protein splice variants from the EVDB.
SpliceVista Workflow
SpliceVista is designed to identify and visualize splice variants based on MS-identified peptides. There are four main parts of the program (Fig. 1):
Data preprocess. In this step, all PSMs are assigned a gene symbol from the protein I.D. and grouped into peptides.
Download. SpliceVista uses the gene symbol to retrieve all known splice variants of a gene from the EVDB. The identifiers of splice variants used in EVDB are consistent with those in GenBank. Nucleotide sequences are extracted from GenBank according to their I.D.s and then translated into amino acid sequences.
Mapping. In this step, all identified peptides are grouped by gene, and genes are analyzed one by one. For each gene, all the identified peptides are mapped to the gene's splice variants from EVDB or the ECgene database. The genomic and transcript positions of each peptide are reported in the output file.
Visualization. The data from previous steps are used for visualizing the exon structures of each splice variant lined up with the identified peptides. The splice variants in EVDB are visualized with only exons scaled to size. The predicted splice variants derived from the ECgene database (including both known, defined as present in Ensembl 72, and unknown, only in the ECgene database) are visualized with introns and exons scaled to the corresponding size. In addition, if PQPQ is used, the peptide clusters based on quantitative patterns are visualized, allowing connections between specific peptides and detected quantitative peptide clusters.
Fig. 1.
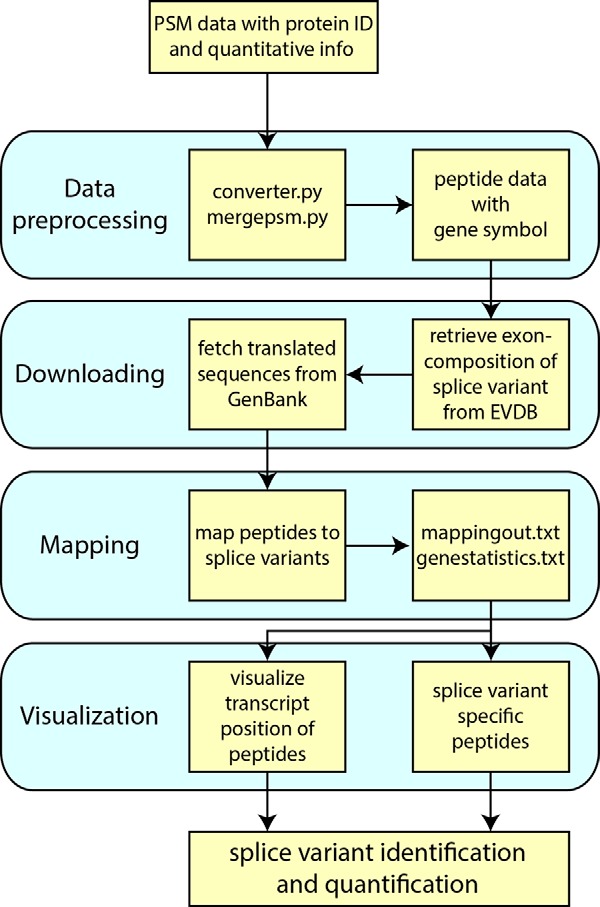
Workflow of SpliceVista. The blue boxes explain the four main steps of SpliceVista. The yellow boxes depict the detailed workflow of SpliceVista. Given the peptide data, converter.py assigns each protein I.D. a gene symbol, which is used to retrieve known splice variants of this gene and its exon structure in EVDB. Peptide sequences are mapped to the translated sequence of splice variants retrieved from GenBank. Genomic coordinates of the peptides and their transcript positions are reported in the output. Known splice variants are identified by splice-variant-specific peptides that map uniquely to the splice variants. Quantification of splice variants can then be done via quantification of splice-variant-specific peptides.
Output Files from SpliceVista
There are two important output files from SpliceVista: mappingout.txt and genestatistic.txt (see Table S1 and S2 in supplemental file 2). The genomic and transcript positions, quantitative data, and PQPQ clustering results for each peptide can be found in the mappingout.txt file. Other files, subexons.txt, splicingvar.txt, and varseq.fa, which are retrieved from databases (EVDB and GenBank), are necessary for mapping peptides in the visualization module. See the user manual (supplemental file 1) for detailed information.
Visualization Module of SpliceVista
Given a gene symbol for a protein, SpliceVista (visualization.py) can generate an image that contains three panels (Fig. 2). The top panel displays the exon structure of all known splice variants. The middle panel displays the transcript positions of identified peptides. If PQPQ is applied, each peptide is assigned to a cluster in which all peptides show a correlated quantitative pattern. In the bottom panel, the quantitative patterns of the different clusters are drawn in the same order as in the middle panel. The bars represent the mean intensity ratio of all PSMs for each unique peptide, with the standard deviation indicated by vertical lines (error bars).
Fig. 2.

SpliceVista visualization overview. The figure shows the SpliceVista output picture of the BAX gene detected in the A431 whole cell fraction. In the top panel, the exon composition of the gene is depicted, and the gene symbol is written in the upper left corner. The white boxes are all sub-exons of the gene present in the database. The beige boxes show the exon compositions of splice variants, and their accession numbers are marked on the left. The shadow on the transcript indicates the location of identified peptides. Below the transcript variant, in the middle panel, the colored lines represent identified peptides that are aligned to corresponding positions on transcripts. The number underneath the lines is a numbering given to the peptide (the numbering is sorted based on genomic coordinates of peptides). If PQPQ-based grouping (18) of the peptides in quantitative clusters has been performed, each peptide is filled with the same color as the cluster that it was assigned to by PQPQ. On the left, the boxes in different colors represent different clusters. After the box is the name assigned to the cluster (for example, “cluster1”) and then the number of unique peptides that belong to this cluster (in brackets). The histogram in the bottom panel is the relative quantitative pattern of each cluster. Each group of bars represents one peptide, and the number of bars is equal to the sample size. The height of one bar is the mean of the relative intensity ratio of all PSMs from one peptide in this cluster (in this dataset, iTRAQ 8-plex was used for relative quantification). The black vertical lines indicate the standard deviation of the intensity ratio of the PSMs connected to the peptide (the number in the bracket after each peptide is the number of PSMs). For those without black lines, there is only one PSM for the peptide in that cluster. The picture generated by SpliceVista is high resolution and more detailed, and a clear view can be achieved by zooming in.
In Silico Analysis of All Human Protein Isoforms
A list of 21,494 protein-coding genes was downloaded from Ensembl 63. Of those, 18,372 genes had splice variants in EVDB (i.e. 76,827 splice variants in total). 3072 of them belong to genes with only one known splice variant in the database. In silico trypsin digestion of the human proteome (Ensembl 63) was performed, resulting in 832,421 unique peptides (6 amino acids ≤ peptide length ≤ 40 amino acids) that were later mapped to all splice variants. To compare with trypsin, in silico lysC digestion of human proteome, which generates on average longer peptides, was also performed, yielding 447,880 unique peptides (6 amino acids ≤ peptide length ≤ 40 amino acids). In the simulated protease digestion, one missed cleavage was allowed only in cases of consecutive cleavage sites (KK, KR, RK, or RR for trypsin, and KK for lysC), and no cutting was done if the cleavage site was followed by proline.
A431 Human Cell Line Proteomics Data
Sample Preparation
In order to exemplify the key functions of SpliceVista, we used it to analyze A431 human cell line (epidermoid carcinoma cell line, cell line number ACC 91) proteomics data. The sample preparation and mass spectrometry experiment are described in Ref. 19. Briefly, 24 h after seeding, A431 cell cultures (in duplicate) were treated with gefitinib and harvested 2 h, 6 h, and 24 h after treatment. Controls were left untreated (duplicates at 0 h). Protein samples from A431 whole cell extraction and three subcellular fractions were then digested by trypsin (see experimental setup and subcellular fractionation procedures in supplemental file 2 Fig. S1). The resulting peptide mixtures were arranged into four sets (whole, light, medium, and heavy), and peptides at different time points in each set were labeled with 8-plex iTRAQ (AB Sciex, Framingham, MA). Peptide mixtures (200 μg) in each set were separated by means of isoelectric focusing (20) using five different immobilized pH gradient gel strips (provided by GE Healthcare Bio-Sciences AB, Uppsala, Sweden; pH ranges of the five strips were 3.7–4.9, 3.70–4.05, 4.00–4.25, 4.20–4.45, and 4.39–4.99, and all of them were 24 cm long). After completion of the isoelectric focusing, each immobilized pH gradient strip was divided into 72 fractions, and peptides from each fraction were transferred into a 96-well micro-titer plate by liquid handling robotics (GE Healthcare prototype) and dried in a SpeedVac. Five MS-based experiments (corresponding peptides collected from five immobilized pH gradient gel strips) were performed using a hybrid LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific) for each of the four peptide-mixture sets (whole cell, light, medium, and heavy fractions). Detailed mass spectrometry analysis can be found in Ref. 19.
Mass Spectrometry Data Processing
Searching Ensembl 63 Human Protein Database
All MS/MS spectra were searched by Sequest/Percolator using the software platform Proteome Discoverer (v1.3.0.339, Thermo Scientific) and a target-decoy strategy. The reference database used was the human protein subset of Ensembl 63 (76,501 protein entries). A precursor mass tolerance of 10 ppm and product mass tolerances of 0.02 Da for HCD Fourier transform MS and 0.8 Da for collision-induced dissociation IT MS were used. Additional settings were trypsin with 1 missed cleavage; carbamidomethylation on cysteine and iTRAQ 8-plex on lysine and N-terminal as fixed modifications; and oxidation of methionine and phosphorylation on serine, threonine, or tyrosine as variable modifications. Quantification of iTRAQ 8-plex reporter ions was done using an integration window tolerance of 20 ppm. PSMs found at a 1% false discovery rate (FDR) were exported.
Reporter-ion-based quantification of proteins was done following Proteome Discoverer's default settings: only PSMs from unique peptides and with precursor interference < 50% were used for quantification; the quantitative ratios of each PSM were normalized to have the same protein median ratios between iTRAQ channels. Protein tables including all identified proteins (at a 1% FDR) and their quantitative data can be found in supplemental file 3. The raw data, pep.XML, and Proteome Discoverer MSF files associated with this paper are available in the ProteomeXchange repository (dataset I.D.: PXD000065).
Searching ECgene Database Combined with Ensembl 72 Human Protein Database
The ECgene splice variant database (at high and low evidence levels) was downloaded (see peptide overlap of ECgene databases and Ensembl 72 in supplemental file 2 Fig. S2). MS/MS spectra from A431 whole cell samples (five MS experiments; peptides separated on immobilized pH gradient gel strips with pH ranges 3.7–4.9, 3.70–4.05, 4.00–4.25, 4.20–4.45, and 4.39–4.99) were searched against two different databases: the ECgene database (high evidence level) concatenated with the Ensembl 72 database, and the ECgene database (low evidence level) concatenated with the Ensembl 72 database. The same software and parameters were used as described above, except that the only variable modification used was oxidation of methionine. Peptide and protein quantification were not included in this workflow.
RESULTS
Human Protein Isoforms with Unique Sequences and Isoform-specific Peptides
To evaluate the potential and limitations of bottom-up MS-based proteomics for splice-variant-specific analysis, we performed a theoretical analysis. In the simulated trypsin digestion, 146,818 (18%) tryptic peptides were uniquely mapped to a specific splice variant (i.e. they were SVSPs) (Fig. 3). 65,916 of them mapped to a gene with only one known splice variant. Conversely, 16,935 (22%) splice variants were shown to have SVSPs. Because lysC generates longer peptides than trypsin (see the peptide-length distribution for trypsin and lysC digestion in supplemental file 2 Fig. S3), one could expect better coverage of splice junction sites with lysC digestion. However, the proportion of SVSPs produced by lysC digestion (17% SVSPs corresponding to 13,593 splice variants) was not greater than that of those produced by trypsin. This result can in part be explained by the fact that many lysC peptides will be too long (>40 amino acids) to be detected via LC-MS analysis.
Fig. 3.

Theoretical analysis of human splice-variant-specific peptides. Pie chart of the number of theoretical splice-variant-specific peptides (SVSPs). 146,818 trypsin-digested peptides and 74,798 LysC-digested peptides were splice variant specific, corresponding to 16,935 and 13,593 splice variants, respectively. 3072 splice variants in the database were from single-isoform genes, and these generated approximately half of the SVSPs. When all SVSPs are combined, 17,562 splice variants in the EVDB can be identified via MS, assuming the presence of all splice variants and 100% sequence coverage.
Combining peptides generated by both trypsin and lysC digestion resulted in a modest increase in the number of isoforms with SVSPs (23%) relative to those obtained with trypsin alone. These results indicate that three out of four protein isoforms cannot be uniquely identified (Fig. 3) using these two enzymes.
Identification of Splice Variants in A431 Cell Line Data
As described previously, a splice variant is identified if one or more peptides are uniquely mapped to its sequence. To test the applicability of the method to proteomics data generated via MS-based shotgun proteomics, we used SpliceVista to analyze human cancer cell line data (A431). In the whole cell lysate, 607 unambiguous splice variants and 1680 SVSPs were identified (Table I). All SVSPs reported in the A431 dataset are derived from genes with multiple splice variants (see these SVSPs and their mapping output from SpliceVista in supplemental file 4). SVSPs from single isoform genes were not counted. With subcellular fractionation, the number of unique peptides identified increased, as did the number of SVSPs and corresponding splice variants (Fig. 4). Expectedly, these data demonstrate that by using subcellular fractionation, we can increase SVSPs and splice variant identifications due to increased peptide coverage (see sequence coverage with and without subcellular fractionation in supplemental file 2 Fig. S4).
Table I. Overview of unique peptide and splice variant identifications in each subcellular fraction and whole cell.
| Subcellular fraction | Heavy fraction | Medium fraction | Light fraction | Whole cell |
|---|---|---|---|---|
| Total number of identified genes | 8015 | 8062 | 6562 | 7762 |
| Total number of genes found in EVDB and analyzed by SpliceVista | 7260 | 7244 | 5900 | 6945 |
| Number of unique peptides identified | 40,689 | 40,760 | 32,435 | 37,099 |
| Number of unique peptides that can be mapped to EVDB and have quantitative data | 36,077 | 36,417 | 28,726 | 32,969 |
| Number of splice-variant-specific peptides (SVSPs) | 1963 | 2027 | 1309 | 1680 |
| Number of splice variants with SVSPs mapped by SpliceVista | 672 | 681 | 512 | 607 |
| Number of genes with splice variants identified | 669 | 678 | 509 | 606 |
Fig. 4.
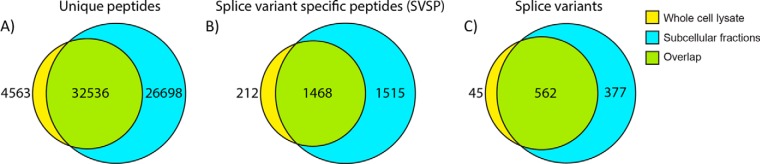
Identified splice-variant-specific peptides in the A431 cell line dataset. Venn diagram comparing the output of whole cell lysate analysis with the analysis of the combined subcellular fractions. From left to right are illustrated the number of (A) unique peptides, (B) splice-variant-specific peptides, and (C) splice variants.
Detection of Differentially Regulated Splice Variants in A431 Data
The A431 dataset contained quantitative information generated via iTRAQ 8-plex labeling of samples from a gefitinib treatment time course study. Duplicate samples were taken at 2, 6, and 24 h after gefitinib treatment and compared with duplicate untreated controls, and results were reported as ratios using the average of the controls as the denominator. In the current study, we performed three different quantitative analyses on the genes with splice variants identified from this dataset (Table I): gene-centric analysis, protein-centric analysis, and splice-variant-centric analysis (Fig. 5). In gene-centric analysis, the relative expression level of a gene is calculated as the mean ratio of all PSMs identified for that gene. In protein-centric analysis, the conventional approach in proteomics, the relative expression level is calculated as the mean ratio of all PSMs identified by the search engine for a protein. In splice-variant-centric analysis, the relative expression level of a certain splice variant of a gene is calculated by taking the mean ratio of PSMs specific to that splice variant. The difference relative to the conventional protein-centric analysis is that only PSMs that uniquely map to a splice variant are used to quantify it. Because more than 90% of genes can undergo alternative splicing (1, 2), there is a potential risk of averaging out the differences of differentially regulated splice variants when doing gene- or protein-centric quantitative analysis if the gene or protein contains peptides shared among splice variants.
Fig. 5.
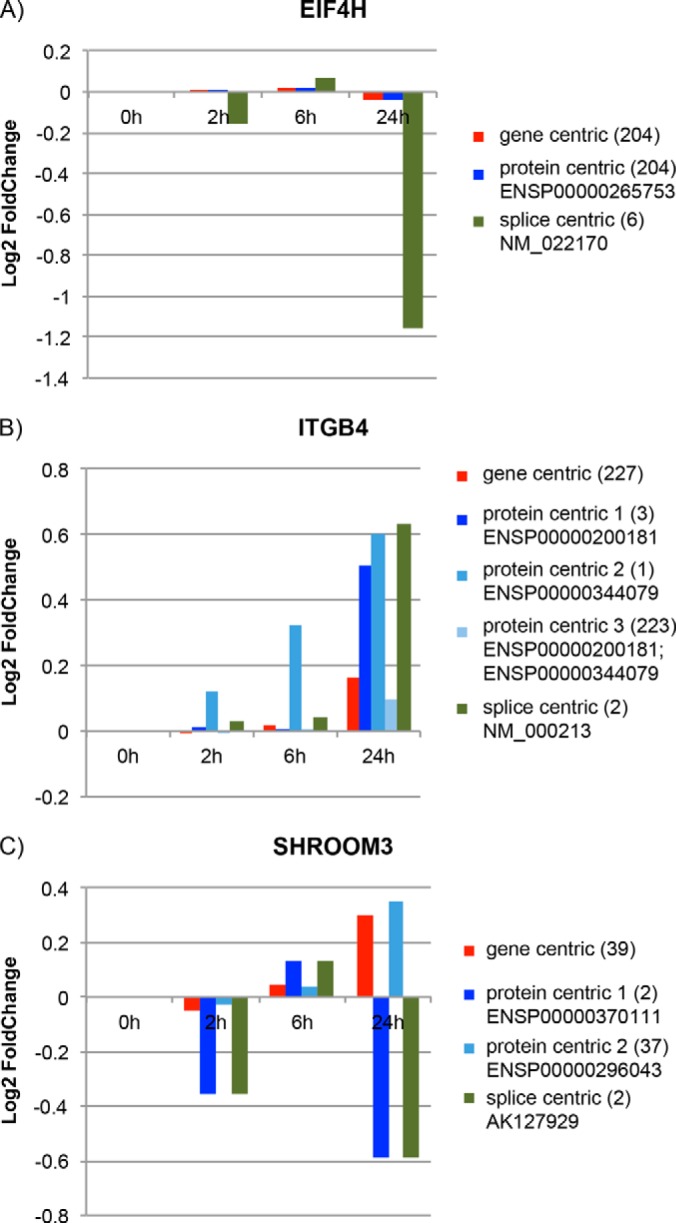
Comparison of gene-centric, protein-centric, and splice-variant-centric quantitative analysis. The figure presents three examples (EIF4H, ITGB4, and SHROOM3) from the A431 dataset in which discrepancies were observed among gene-centric, protein-centric, and splice-variant-centric analyses. Fold changes are reported for different time points after treatment with gefitinib. The numbers in the parentheses are the number of PSMs used for quantification in each analysis. In gene-centric analysis, all PSMs mapping to that gene were used to calculate the fold change. In protein-centric analysis, PSM grouping was done by the search engine. In splice-variant-centric analysis, only PSMs from SVSPs are used for fold-change calculation. For further details, see the “Results” section.
Three genes identified in the A431 dataset were selected to exemplify three typical situations in which different results are observed depending on whether gene-centric, protein-centric, or splice-variant-centric analysis is used (Fig. 5). (i) Down-regulation of gene EIF4H was only seen in splice-centric analysis, with no obvious regulation observed in either gene-centric or protein-centric analysis (Fig. 5A). The protein reported (ENSP00000265753) in the conventional protein-centric analysis contains the SVSPs used for splice-variant-centric analysis. The distinct quantitative pattern detected in the splice-variant-centric analysis implies that at least one additional unreported splice variant is present. Moreover, the unreported variant is highly abundant relative to the variant identified by SVSPs. When doing gene-centric or protein-centric analysis, the unreported but dominant splice variant averages out the down-regulation signal of the identified splice variant. (ii) Gene-centric, protein-centric, and splice-centric analyses all showed different results for the ITGB4 gene (Fig. 5B). Here, three different PSM populations were found via protein-centric analysis, corresponding to two different variants (ENSP00000200181 and ENSP00000344079) and a protein group containing shared PSMs between these two variants. In addition, the quantitative pattern of that protein group indicates that at least one additional variant could have been present and dominant. (iii) For the SHROOM3 gene, two variants with different quantitative patterns were found in the protein-centric analysis. One of these variants contains an SVSP (two PSMs) mapping to the variant reported in the splice-centric analysis (Fig. 5C). However, the other variant (37 PSMs) is the highly abundant one, and thus makes the major contribution to the gene-centric signal. In all three cases, the gene-centric quantification result was an averaged outcome of all identified PSMs mapped to the gene and was dominated by the protein variant that contained the most PSMs. With SpliceVista, we are able to quantify splice variants specifically and in some cases infer hidden variants by comparing splice-centric analysis to gene-centric and protein-centric analysis (see the comparison for all genes from the heavy fraction with SVSPs identified in supplemental file 2 Fig. S6).
PQPQ-based quantitative clustering combined with information about peptides' transcript position was investigated to see whether peptides uniquely mapped to gene EIF4H's splice variant (NM_022170) had a correlated quantitative pattern. As shown in Fig. 6, three peptides (4, 5, and 6) were clustered together and showed down-regulation at 24 h relative to the other peptides. As shown by the transcript positions of the peptides, peptide 4 (DDFNSGFR) and peptide 5 (DDFNSGFRDDFLGGR) were unique to the splice variant NM_022170. Although peptide 6 (DDFLGGR) was not uniquely mapped to NM_022170, it could be derived from this splice variant based on its quantitative pattern. The other splice variant shared peptides showed no significant regulation, implying that splice variant NM_022170 of EIF4H is differentially regulated. Because all peptides are assigned with genomic coordinates by SpliceVista, it is also possible to compare protein-level data with RNA-level data. As shown in supplemental file 2 Fig. S5, the splice-variant-specific change of gene EIF4H identified at the protein level was compared with RNA-sequencing data.
Fig. 6.

SpliceVista visualization of EIF4H gene. The figure shows the SpliceVista output picture for the EIF4H gene detected in A431 cell line whole cell samples. Gene EIF4H has six exons and four known splice variants (exon 6 was cut out to enable better resolution). Nine unique peptides were identified for EIF4H and grouped in three clusters. Cluster 1 (blue), which included peptides 4, 5, and 6, had a unique pattern that showed clear down-regulation in the last two samples (replicates at 24 h after drug treatment). Peptide 4 (DDFNSGFR) and peptide 5 (DDFNSGFRDDFLGGR) were uniquely mapped to splice variant NM_022170. Peptide 6 (DDFLGGR) was not unique to splice variant NM_022170, but it is very likely that this peptide was derived from NM_022170 based on its quantitative pattern, which is similar to those of peptides 4 and 5. In the middle panel, the number in the bracket after each cluster is the number of unique peptides grouped in this cluster. In the bottom panel, the number in the bracket after each peptide is number of PSMs.
Discovery and Visualization of Novel Splice Variant Peptides in A431 Data
SpliceVista can also be used to visualize novel protein isoforms not yet reported in the EVDB. To exemplify this feature, we searched the A431 data against the ECgene database, which contains predicted splice variants based on expressed sequence tag data. In total, 31,985 and 31,023 unique peptides were identified at a 1% FDR in the ECgene high (high evidence level) concatenated with Ensembl 72 database and ECgene low (low evidence level) concatenated with Ensembl 72 database, respectively. Of these, 30,708 peptides were identified in both searches. As more or less the same number of peptides was identified in both searches, we focused on peptides identified from the high-evidence-level database. In that search, 223 unique peptides were exclusively identified (with Xcorr > 2) in the ECgene high and not in the Ensembl 72 database. SpliceVista was used to map the 223 peptides to their genomic positions (a list of these 223 peptides and their genomic coordinates is provided in supplemental file 5). We then searched these 223 peptides in the NCBI human non-redundant protein sequence database using BLASTP (21). Of these, 157 peptides had at least one mismatch to the sequences in the database and were therefore considered novel. One of the peptides mapping to a novel splice variant of gene PLCB2 is shown in Fig. 7.
Fig. 7.

SpliceVista visualization of a novel PLCB2 variant. The figure shows a novel peptide (GSAAQNSSFMPVSLQRHQR) identified from previously unknown splice variant H15C2281.2 of gene PLCB2 in the ECgene database. In the figure, introns and exons (beige boxes) are scaled to its size, and green and red lines indicate start codons and stop codons, respectively. The blue line with the number 1 below it indicates the peptide's genomic position; it is also marked as a blue line on the splice variant the peptide is derived from. Discovery of this novel peptide also complies with RNA sequencing data (not shown here). Four splice variants are shown with their complete structures; parts of other splice variants are cut out.
Splice-variant-specific Analysis on Human 4Skin hiPS Cells, Parental Fibroblast Cell Lines, and Human ES Cells
To demonstrate SpliceVista's compatibility with other proteomics datasets, the stem cell dataset created by Munoz et al. (22) was downloaded from ProteomeXchange (PXD000134). MSF files for two experiments in which 4Skin hiPS cells were compared with the parental fibroblast cell line and human ES cells were downloaded and opened in Proteome Discoverer (version 1.3, Thermo Electron). PSMs with a 1% FDR cutoff were extracted for each experiment. Then the peptides were mapped to splice variants in EVDB (see statistics of SVSP identifications and comparison to A431 cell line dataset in supplemental file 2 Table S3). The numbers of splice variants reported in the two mass spectrometry experiments were 390 and 397, respectively. On average, we found SVSPs for 7% of the identified genes in this dataset. This is comparable to the A431 cell line dataset, in which we found SVSPs for 9% of the identified genes.
The number of overlapping proteins with SVSPs identified in both MS experiments from the study by Munoz et al. was 296. Protein-centric analysis of these proteins was compared with splice-variant-specific analysis. As shown in Fig. 8A, for most proteins, protein-centric analysis of the Fibro/hiPS ratio was consistent with splice-variant-specific results. However, some proteins marked as red showed large differences in their Log2(Fibro/hiPS) value between protein-centric and splice-centric analysis, indicating that peptides assigned to the protein might be derived from differentially regulated splice variants. The top 10 proteins ranked by Fibro/hiPS ratio differences between splice-variant- and protein-centric analysis are shown in Fig. 8B.
Fig. 8.
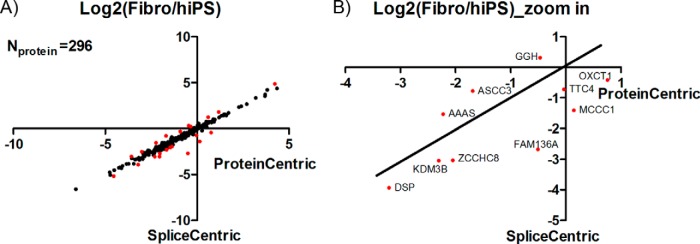
Comparison of protein-centric and splice-centric quantitative analysis in the stem cell dataset. A, comparison of Fibro/hiPS protein ratios determined via protein-centric analysis and splice-variant-centric analysis on proteins with splice variants identified. 296 proteins with splice variants identified in both MS experiments (biological replicates) were included. The Fibro/hiPS ratio in the figure was calculated as the mean of the two replicates. The red dots indicate proteins that showed differences in log2(Fibro/hiPS) values greater than 0.5 (1.41-fold) between protein-centric and splice-variant-centric analyses. B, zoomed view of the top 10 proteins ranked according to differences in log2(Fibro/hiPS) between protein-centric and splice-variant-centric quantification.
DISCUSSION
Shotgun proteomics is being widely applied in large-scale characterizations of proteins because of its intrinsic ability to systematically profile the entire protein complement in the sample, both qualitatively and quantitatively. However, the lack of tools for shotgun proteomics data analysis limits the exploration of the large amount of data generated, leading to many missed protein isoforms that are relevant to specific biological events.
The tool presented herein, SpliceVista, simplifies the detailed analysis of known and predicted splice variants by enabling their visualization and by mapping peptide evidence to these variants. Splice-variant-specific peptides (SVSPs) are the key for both identification and quantification of splice variants by SpliceVista. Hence, splice-variant-specific analysis is limited to the genes with identified SVSPs. Increased protein sequence coverage obtained via pre-fractionation methods at the subcellular, protein, or peptide level will increase the likelihood of identifying more SVSPs. However, we acknowledge the limitation of using shotgun proteomics to identify splice variants, as at most one out of four splice variants can be uniquely identified even assuming 100% sequence coverage in the MS analysis.
In MS-based proteomics, the unique peptides for a gene can come from several splice variants that usually possess high sequence similarity. Unless the gene has only one splice variant, it should be noted that in gene-centric analysis a gene's quantitative pattern is a mixed outcome of all present splice variants. Consequently, the quantitative pattern can be dominated by one or a few highly abundant splice variants, as most copies of the peptides identified come from those abundant protein variants. As shown in this study, the same problem can occur in protein-centric analysis as a result of ambiguous assignment of peptides to protein variants. SVSPs mapped by SpliceVista provide the possibility of splice-variant-centric analysis at the protein level. The quantification of a splice variant is then done through quantification of its SVSPs. In practice, we most often have unique peptides from only one splice variant of a gene. Nevertheless, differentially regulated hidden splice variants can still be detected indirectly by comparing gene-centric or protein-centric to splice-variant-centric analyses as illustrated here.
SpliceVista also enables remapping of peptide data to a splice variant database, such as EVDB, and thus splice-variant-specific analysis can be performed on already generated peptide data. This makes SpliceVista compatible for analysis and re-analysis of most MS-based proteomics datasets. Additionally, SpliceVista can be used to map and visualize novel splice variant peptides identified by searching MS data against the ECgene database. When including predicted splice variants in the peptide search space, it is important to be aware of the potential problems: the increased search space tends to increase false discoveries, and the expected low occurrence of findings in the novel (predicted) part of the database can lead to an increased FDR. It is therefore important to validate these novel splice variant identifications with independent experimental evidence.
In summary, the presented program, SpliceVista, can assist users in the identification and quantification of splice variants in MS-based proteomics. First, the program reports the number of known splice variants of the gene and aligns identified peptides with their transcript positions. Given this information, users can easily screen out the peptides unique to splice variants. Second, the given genomic coordinates of each peptide make it possible to compare data with the results from RNA-level experiments such as RT-PCR and RNA sequencing. Third, the visualization feature of SpliceVista can help users interpret MS-based proteomics data for a specific gene associated with splice variant information. If peptide clustering by PQPQ is applied, SpliceVista will also present clustering results and histograms of the different quantitative patterns detected. This information, combined and visualized by SpliceVista, enables users to identify and evaluate splice-variant-specific quantitative patterns and infer alternative splicing regulation. With these features, SpliceVista will serve as a tool for the exploration of splice-variant-specific information from high-throughput proteomics data and the generation of hypotheses associated with alternative splicing.
Supplementary Material
Acknowledgments
We thank Henrik Johansson for performing the MS experiments on the A431 cell line samples and Mikael Huss for his help in the analysis of RNA sequencing data from A431 cell line samples.
Footnotes
Author contributions: Y.Z., L.H., J.F., and J.L. designed research; Y.Z. performed research; Y.Z. analyzed data; Y.Z., L.H., J.F., R.M.B., L.M.O., and J.L. wrote the paper.
* This project was financed with support from Karolinska Institutet (KID-funding), the Swedish Research Council, the Swedish Cancer Society, the Cancer Society in Stockholm, the Stockholm County Council, and FP7 EU project GlycoHit.
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- EVDB
- Evidence Viewer Database
- MS
- mass spectrometry
- IT
- ion trap
- HCD
- higher energy collisional dissociation
- PSM
- peptide spectrum match
- iTRAQ
- isobaric tags for relative and absolute quantification
- PQPQ
- peptide quantification by peptide quality control
- SVSP
- splice-variant-specific peptide
- FDR
- false discovery rate
- hiPS cells
- human induced pluripotent stem cells.
REFERENCES
- 1. Wang E. T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S. F., Schroth G. P., Burge C. B. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pan Q., Shai O., Lee L. J., Frey B. J., Blencowe B. J. (2008) Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat. Genet. 40, 1413–1415 [DOI] [PubMed] [Google Scholar]
- 3. Matlin A. J., Clark F., Smith C. W. (2005) Understanding alternative splicing: towards a cellular code. Nat. Rev. Mol. Cell Biol. 6, 386–398 [DOI] [PubMed] [Google Scholar]
- 4. Blencowe B. J. (2006) Alternative splicing: new insights from global analyses. Cell 126, 37–47 [DOI] [PubMed] [Google Scholar]
- 5. Venables J. P. (2004) Aberrant and alternative splicing in cancer. Cancer Res. 64, 7647–7654 [DOI] [PubMed] [Google Scholar]
- 6. Garcia-Blanco M. A., Baraniak A. P., Lasda E. L. (2004) Alternative splicing in disease and therapy. Nat. Biotechnol. 22, 535–546 [DOI] [PubMed] [Google Scholar]
- 7. Black D. L. (2003) Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336 [DOI] [PubMed] [Google Scholar]
- 8. Eyras E., Caccamo M., Curwen V., Clamp M. (2004) ESTGenes: alternative splicing from ESTs in Ensembl. Genome Res. 14, 976–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kan Z., Rouchka E. C., Gish W. R., States D. J. (2001) Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 11, 889–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ramani A. K., Calarco J. A., Pan Q., Mavandadi S., Wang Y., Nelson A. C., Lee L. J., Morris Q., Blencowe B. J., Zhen M., Fraser A. G. (2011) Genome-wide analysis of alternative splicing in Caenorhabditis elegans. Genome Res. 21, 342–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Trapnell C., Pachter L., Salzberg S. L. (2009) TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kahn A. B., Ryan M. C., Liu H., Zeeberg B. R., Jamison D. C., Weinstein J. N. (2007) SpliceMiner: a high-throughput database implementation of the NCBI Evidence Viewer for microarray splice variant analysis. BMC Bioinformatics 8, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim P., Kim N., Lee Y., Kim B., Shin Y., Lee S. (2005) ECgene: genome annotation for alternative splicing. Nucleic Acids Res. 33, D75–D79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 15. Power K. A., McRedmond J. P., de Stefani A., Gallagher W. M., Gaora P. O. (2009) High-throughput proteomics detection of novel splice isoforms in human platelets. PLoS One 4, e5001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hatakeyama K., Ohshima K., Fukuda Y., Ogura S., Terashima M., Yamaguchi K., Mochizuki T. (2011) Identification of a novel protein isoform derived from cancer-related splicing variants using combined analysis of transcriptome and proteome. Proteomics 11, 2275–2282 [DOI] [PubMed] [Google Scholar]
- 17. Menon R., Omenn G. S. (2010) Proteomic characterization of novel alternative splice variant proteins in human epidermal growth factor receptor 2/neu-induced breast cancers. Cancer Res. 70, 3440–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Forshed J., Johansson H. J., Pernemalm M., Branca R. M., Sandberg A., Lehtio J. (2011) Enhanced information output from shotgun proteomics data by protein quantification and peptide quality control (PQPQ). Mol. Cell. Proteomics 10, M111.010264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Branca R. M., Orre L. M., Johansson H. J., Granholm V., Huss M., Perez-Bercoff A., Forshed J., Kall L., Lehtio J. (2014) HiRIEF LC-MS enables deep proteome coverage and unbiased proteogenomics. Nat. Methods 11, 59–62 [DOI] [PubMed] [Google Scholar]
- 20. Eriksson H., Lengqvist J., Hedlund J., Uhlen K., Orre L. M., Bjellqvist B., Persson B., Lehtio J., Jakobsson P. J. (2008) Quantitative membrane proteomics applying narrow range peptide isoelectric focusing for studies of small cell lung cancer resistance mechanisms. Proteomics 8, 3008–3018 [DOI] [PubMed] [Google Scholar]
- 21. Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Munoz J., Low T. Y., Kok Y. J., Chin A., Frese C. K., Ding V., Choo A., Heck A. J. (2011) The quantitative proteomes of human-induced pluripotent stem cells and embryonic stem cells. Mol. Syst. Biol. 7, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.