

Abstract
For the first time, nickel-catalyzed silyl-Heck reactions are reported. Using simple phosphine-supported nickel catalysts, direct activation of silyl triflates has been achieved. These results contrast earlier palladium-catalyzed systems, which require iodide additives to activate silyl-triflates. These nickel-based catalysts exhibit good functional group tolerance in the preparation of vinyl silanes, and unlike earlier systems, allows for the incorporation of trialkylsilanes larger than Me3Si.
1. INTRODUCTION
Unsaturated organosilanes are potent nucleophiles and highly useful intermediates in organic synthesis.1 Recently, we have established the silyl-Heck reaction as a novel route to access both allyl and vinyl silanes.2,3 This general method allows for the direct silylation of terminal alkenes using silyl halides and transition metal catalysts, in a reaction that we believe is analogous to classical Heck arylation (Figure 1).4
Figure 1.
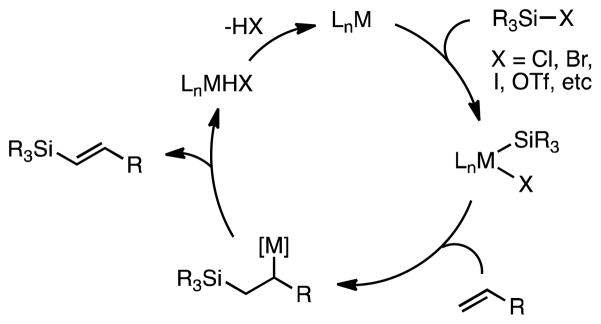
Proposed Mechanism for Silyl-Heck Reaction.
Our previous work has focused exclusively on the use of palladium-based catalysts in this transformation.2a, b In these processes, we have found that the use of iodosilanes is required. These can either be used directly or prepared in situ from silyl chlorides, bromides, or triflates and simple iodide salts.5 Significantly, even the most reactive silyl triflate, trimethylsilyl triflate, fails to undergo reaction under palladium-catalyzed conditions without added iodide. Silicon-oxygen bonds are known to be very strong, we attribute this to the reluctance of palladium to insert into the Si-OTf bond.6
An active interest in our group is developing a catalyst capable of engaging silyl halides other than iodosilanes in silyl-Heck type reactions. This interest is fueled by the recognition that iodosilanes are potent Lewis acids, and thus have attenuated functional group compatibility. In addition, access to silyl iodides is limited, with only Me3SiI being commercially available. In contrast, a much wider variety of silyl chlorides and triflates can be purchased or readily prepared, making methods that can directly utilize these reagents attractive to develop.7
In cross-coupling chemistry of carbon electrophiles, nickel catalysts have proven adept at the activation of strong carbon-heteroatom bonds (such as aryl ethers and carboxylates), particularly in comparison to palladium catalysts.8 Despite the fact that silyl bromides and iodides have been shown to oxidatively add to a variety of late transition metal complexes, to our knowledge such reactions involving nickel compounds have not been described.3a–e, 9 Based upon the precedent with strong C–X bonds, we decided to investigate silyl-Heck type reactions with nickel-based catalysts.10
Herein, we report the first examples of nickel-catalyzed silyl-Heck type reactions. We show that, unlike in palladium-catalyzed reactions, these nickel-catalyzed reactions are able to utilize silyl triflate electrophiles without the need for iodide additives. Using this system, a variety of styrene derivatives and related terminal alkenes lacking allylic hydrogen atoms can be successfully transformed into E-vinyl silanes. As significantly, for the first time, this nickel-based catalyst system allows for the direct preparation of vinyl trialkyl silanes from trialkylsilyl electrophiles larger than trimethylsilane. We believe that this new catalytic system not only provides promise for developing general base-metal catalysts for this class of reaction, but also greatly expands the types of unsaturated organosilanes that can be accessed using the silyl-Heck reaction.
2. RESULTS AND DISCUSSION
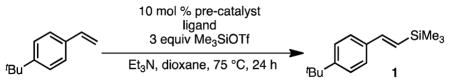
To begin our investigation of nickel-catalyzed silyl-Heck reactions, we studied the reaction of 4-tert-butyl styrene with trimethylsilyltriflate (Me3SiOTf) without iodide additives (Table 1). Consistent with our previous observations, palladium-based catalysts provided only trace yield of desired vinyl silane 1 (entry 1). In our hands, this outcome is not improved by variation of either palladium pre-catalyst, nature of phosphine ligand, metal:ligand ratio, solvent, or temperature (not shown). In contrast, a modest screen of catalysts derived from Ni(COD)2 and phosphine ligands revealed a significantly different outcome. Whereas catalysts employing triaryl phosphines (entries 2 and 3) or mixed arylalkyl phosphines (entries 4 and 5) were ineffective, moderately bulky trialkyl phosphines provided highly active catalysts (entries 6–10). Interestingly, however, there seems to be a steric limit with regard to ligand size; the very bulky tBu3P was ineffective (entry 11). Further optimization revealed that a highly effective catalyst was obtained using tBuPCy2 and Ni(COD)2 when a 1.5:1 ligand:metal ratio was employed (entry 13).
Table 1.
Identification of Nickel-Based Catalyst.
| |||
|---|---|---|---|
| entry | pre-catalyst | ligand (mol %) | yield 1 |
| 1 | (COD)Pd(CH2TMS)2 | tBuPPh2 (30) | 0% |
| 2 | Ni(COD)2 | PPh3 (30) | 0% |
| 3 | Ni(COD)2 | P(o-tol)3 (30) | 0% |
| 4 | Ni(COD)2 | tBuPPh2 (30) | 12% |
| 5 | Ni(COD)2 | Cy2PPh (30) | 11% |
| 6 | Ni(COD)2 | nBu3P (30) | 69% |
| 7 | Ni(COD)2 | PCy3 (30) | 57% |
| 8 | Ni(COD)2 | PCyp3 (30) | 65% |
| 9 | Ni(COD)2 | tBuPCy2(30) | 71% |
| 10 | Ni(COD)2 | tBu2PCy (30) | 55% |
| 11 | Ni(COD)2 | tBu3P (30) | 7% |
| 12 | Ni(COD)2 | tBuPCy2 (20) | 85% |
| 13 | Ni(COD)2 | tBuPCy2 (15) | 90% |
Using these optimized conditions, we studied the scope of the nickel-catalyzed silyl-Heck reaction (Table 2). A variety of styrenyl alkenes participate in the reaction. On preparative scale (1 mmol), vinyl silane 1 was isolated in 82% yield. Likewise, unsubstituted styrene could be silylated in 89% isolated yield under these conditions (2). A variety of ethereal substrates were also tolerated, including those with both electron-donating para-methoxy (3) and electron-withdrawing meta-methoxy groups (4) in good yield (71% and 76%, respectively). Silyl ethers (5) and dioxoles (6) were also well tolerated. Aromatic fluorides proved amiable to the reaction conditions; fluorinated vinyl styrene 7 was isolated in 66% yield. Unfortunately, larger aromatic halogens were not compatible with the silylation conditions. For example, the use of 4-chlorostyrene as substrate led to a complex mixture of products without detectable formation of desired vinyl styrene 8. Also problematic were highly electron-deficient or electron-rich styrenes. For example, the formation of ester 9 was not observed, and dimethylamino product 10 was formed in low yield. Strained rings, such as benzocyclobutane (11), and steric bulk on the aromatic group ortho to the alkene (12), however, were well tolerated. Some heterocyclic substrates could also be silylated using this protocol. For example, silylation of N-vinyl carbazole led to vinyl silane 13 in high yield.11 However, in other cases, such as in the formation of benzofuran 14, yields proved to be suboptimal. Finally, more complex vinyl silanes, such as pinacol borane 15 and estradiol-derived 16, could also be accessed using the nickel-catalyzed protocol. In the case of 15, tricyclopentyl phosphine (Cyp3P) proved to be a slightly more effective ligand than tBuPCy2, demonstrating that some ligand optimization might prove necessary in order to maximize vinyl silane yield.12 Overall, while these yields are slightly lower and scope is somewhat more limited than our previously reported palladium-catalyzed silyl-Heck protocol involving Me3SiI, we believe that this reaction enjoys sufficient substrate scope to make it a synthetically viable alternative, particularly given the advantages of using a nonprecious metal, nickel-based catalyst and a silyl triflate as the silylating reagent.
Table 2.
Substrate Scope with Respect to Styrene.

|
Isolated yields. All reactions run at 0.5 M concentration.
30 mol % Cyp3P used in place of tBuPCy2.
As mentioned above, silyl triflates are much more abundant than silyl iodides. We therefore wanted to investigate the scope of the transformation with respect to the silyl triflate. We deemed this to be a significant goal as vinyl silanes bearing groups other than trimethylsilyl exhibit improved reactivity in a variety of transformations. In particular, vinyl benzyl silanes are highly effective in Hiyama cross-coupling,13 and those bearing aromatic functionality on silicon are faster in both acylation and oxidation reactions.14 The expansion of the silyl-Heck reaction to include these silanes would allow direct preparation of these products from alkenes.
Initial investigations using tBuPCy2 and the above-optimized conditions revealed that silyl triflates larger than trimethylsilyl triflate do participate in the reaction. However, we rapidly identified the use of Cy3P with a ligand:metal ratio of 3:1 as an alternative catalyst that provided generally higher yields with larger silanes.
Scope studies using 4-tert-butyl styrene and this latter catalyst system are outlined in Table 3. Dimethylsilyl triflates containing one primary alkyl group, such as nBuMe2SiOTf or BnMe2SiOTf participate well under these conditions (17 and 18), providing similar yields to Me3SiOTf. One secondary substituent, such as in iPrMe2SiOTf, can also be tolerated without loss of yield (19). However, a tertiary silyl substituent proved to be beyond the steric limit under these conditions; using Cy3P as ligand, none of desired vinyl silane 20 was observed using tBuMe2SiOTf (TBSOTf). However, switching to the smaller ligand nBu3P and using elevated temperatures did allow for the formation of 20. Despite the modest yield, this transformation is remarkable as it presumably involves oxidative addition at a silicon center that bears a fully substituted adjacent center (akin to a neopentylic carbon center). Silyl triflates bearing aromatic groups are also good substrates for the nickel-catalyzed silyl-Heck reaction. Both phenyldimethyl and diphenylmethyl vinyl silanes can be prepared in good yield in this way (21 and 22). Finally, triethylsilyl triflate also participates in the reaction; 23 was prepared in 65% yield. However, triisopropylsilyl triflate appears to be too large (even under forcing conditions). As the previously developed palladium-catalyzed reaction only tolerates Me3SiI (used directly or generated in situ), these results greatly expand the types of electrophilic trialkylsilanes that can participate in the silyl-Heck reaction.
Table 3.
Substrate Scope with Respect to Silyl Triflate.

Isolated yields. Unless otherwise noted, all reactions run at 0.5 M concentration.
40% nBu3P in place of PCy3,105 °C, 1.0 M concentration.
In the case of reactions using larger silyl triflates (described in Table 3), the major byproduct is alkene 25 (Figure 2). We hypothesize that this styrene dimer arises via a metal hydride-mediated Heck-type pathway.15 Minor amounts of similar dimers are also observed as byproducts in reactions using Me3SiOTf (Table 1); however, formation of these dimers is less significant. These results suggest that the dimerization pathway becomes more competitive with increasing steric bulk of the silyl triflate, likely due to the difficulty of oxidative addition.
Figure 2.
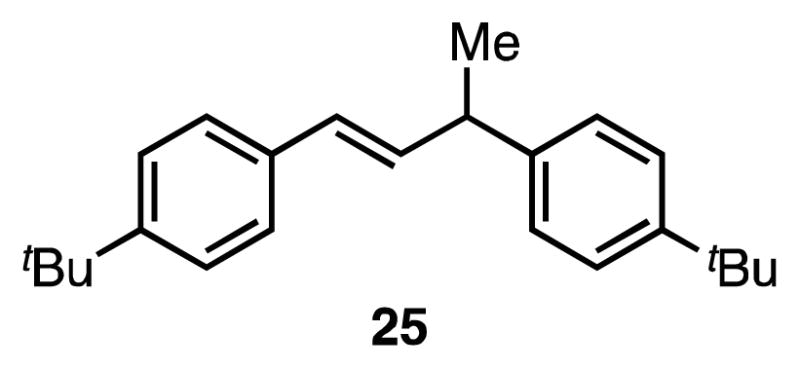
Major byproduct from silyl-Heck reactions of larger silyl triflates.
3. CONCLUSIONS
For the first time, we have demonstrated a nickel-catalyzed silyl-Heck reaction, the first demonstration of a first-row transition metal catalyst in this type of reaction. We have shown that simple phosphine-supported nickel-based catalysts are not only capable of silylating styrene derivatives, but are also capable of promoting the reaction with silyl triflate reagents without the need for in situ generation of silyl iodides. Moreover, good substrate scope with respect to the alkene has been observed. More importantly, for the first time electrophilic trialkylsilanes bearing alkyl groups larger than methyl have been shown to participate in Heck-like reactions. These results provide promising leads for the further development of silyl-Heck reactions using inexpensive catalysts and silylating reagents.
4. EXPERIMENTAL SECTION
4.1 General Experimental Details
Dioxane, tetrahydrofuran, and dichloromethane were dried on alumina according to published procedures.16 Triethylamine was distilled from CaH2 and then sparged with nitrogen. 2-Dimethylaminoethanol was distilled under vacuum from anhydrous potassium carbonate and sparged with nitrogen. Trifluoromethanesulfonic acid (TfOH) was distilled under vacuum and stored under nitrogen in a teflon-sealed vessel. Trimethylsilyl-, triethylsilyl-, (Oakwood Chemical), tert-butyldimethylsilyl- (Combi-Blocks) and tri-iso-propylsilyl- (Gelest) trifluoromethanesulfonate were distilled under vacuum and degassed prior to use. All hot glassware was oven dried for a minimum of four hours or flame-dried under vacuum prior to use. All other substrates and reagents were purchased in highest analytical purity from commercial suppliers. Liquid substrates were sparged with nitrogen before use, and all others were used as received. Column chromatography was performed with 5–20 μm or 40–63 μm silica gel (Silicycle) with the eluent reported in parentheses. Analytical thin-layer chromatography (TLC) was performed on precoated glass plates and visualized by UV or by staining with KMnO4.
4.2 Instrumentation
NMR spectra were obtained on a Bruker AV400 MHz FT-NMR spectrometer equipped with a Bruker CryoPlatform (400 MHz 1H, 101 MHz 13C, and 376 MHz 19F) or on a Bruker AVIII 600 MHz FT-NMR spectrometer (600 MHz 1H, 151 MHz 13C), in the indicated deutero-solvent and were recorded at ambient temperatures. Chemical shifts are reported in ppm. 1H NMR were calibrated using the residual protio-solvent as a standard. 13C NMR spectra are calibrated using the deutero-solvent as a standard and were recorded using the attached proton test.17,19F spectra are referenced to an external FCCl3 sample. IR spectra were recorded on a Nicolet Magna 560 FTIR spectrometer as thin films. GCMS data was collected using an Agilent 6850 series GC and 5973 MS detector. High resolution MS was attained on a Waters GCT Premier spectrometer using electron impact ionization (EI).
4.3 General Procedures
General Procedure A – Reactions of Alkenes with Trimethylsilyl trifluoromethanesulfonate: In a glovebox (N2 atmosphere), dicyclohexyl-tert-butylphosphine (15 mol %) and Ni(COD)2 (10 mol %) were added to a 2 dram vial equipped with a stirbar. Solid alkenes were also added at this time. Dioxane and triethylamine (5 equiv) were then added sequentially, followed by liquid alkene (1 equiv) if applicable. The vial was sealed with a Teflon-lined septum cap and removed from the glovebox. The reaction mixture was stirred at room temperature until homogeneous. Trimethylsilyl trifluoromethanesulfonate (3 equiv) was then added via syringe at room temperature with stirring. The vessel was then heated in an oil bath at 75 °C with stirring for 24 h. The reaction was removed from the oil bath and cooled to room temperature. The reaction vessel was then opened to air, and brine and diethyl ether or hexanes were added. The brine layer was removed, and the organic layer was washed twice with brine. The combined aqueous layers were back-extracted twice with diethyl ether or hexanes. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The product was purified using flash silica chromatography, eluting with the indicated solvent noted in parenthesis.
General Procedure B – Reactions of Larger Silyl Triflates: In a glovebox (N2 atmosphere), tricyclohexylphosphine (30 mol %) and Ni(COD)2 (10 mol %) were added to a 2 dram vial equipped with a stirbar. Dioxane, triethylamine (5 equiv), and 1-tert-butyl-4-vinylbenzene (1 equiv) were then added, sequentially. The vial was sealed with a Teflon-lined septum cap and removed from the glovebox. The reaction mixture was stirred at room temperature until homogeneous. The appropriate silyl trifluoromethanesulfonate reagent (3 equiv) was then added via syringe at room temperature with stirring. The vessel was then heated in an oil bath at 75 °C with stirring for 24 h, after which time N,N-dimethyl ethanolamine (3 equiv) was added via syringe with stirring at 75 °C. The vessel was stirred at 75 °C for approximately 1 minute before stirring at room temperature for approximately 15 minutes. The reaction vessel was then opened to air, and hexanes and HCl (1M aqueous) were added. The HCl layer was removed, and the organic layer was washed twice with HCl (1M aqueous). The combined aqueous layers were back-extracted with hexanes. The combined organic layers were dried over MgSO4 and concentrated in vacuo. The product was purified using flash silica chromatography, eluting with the indicated solvent noted in parenthesis.
4.4 Characterization data
4.4.1 (E)-(4-(tert-butyl)styryl)trimethylsilane (1)
Following general procedure A: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 190 mg of 1 (82%) of a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 8.7 Hz, 2H), 7.35 (d, J = 8.7 Hz, 2H), 6.85 (d, J = 19.1 Hz, 1H), 6.42 (d, J = 19.1 Hz, 1H), 1.31 (s, 9H), 0.14 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 151.2, 143.4, 135.8, 128.6, 126.2, 125.6, 34.7, 31.4, −1.0; FTIR (cm−1): 2957, 1248, 986, 868, 838. HRMS (EI) m/z, calcd for [C15H24Si]: 232.1647; found: 232.1668.
4.4.2 (E)-trimethyl(styryl)silane (2)
Following general procedure A: styrene (115 μL, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 158 mg of 2 (89%) of a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.44 (d, J = 7.0 Hz, 2H), 7.33 (t, J = 7.4 Hz, 2H), 7.25 (t, J = 7.2 Hz, 1H), 6.88 (d, J = 19.2 Hz, 1H), 6.48 (d, J = 19.1 Hz, 4H), 0.16 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 143.7, 138.5, 129.7, 128.7, 128.1, 126.5, −1.1; FTIR (cm−1) 2955, 1247, 988, 866, 843. HRMS (EI) m/z, calcd for [C11H16Si]: 176.1021; found: 176.1048.
4.4.3 (E)-(4-methoxystyryl)trimethylsilane (3)
Following general procedure A: 4-vinyl-anisole (134 μL, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 146 mg of 3 (71%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 7.38 (d, J = 8.8 Hz, 2H), 6.91 – 6.75 (m, 3H), 6.31 (d, J = 19.1 Hz, 1H), 3.81 (s, 3H), 0.14 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 159.6, 143.1, 131.5, 127.7, 126.8, 114.0, 55.5, −1.0; FTIR (cm−1) 2958, 1608, 1510, 1251, 1033, 993, 835, 798. HRMS (EI) m/z, calcd for [C12H18OSi]: 206.1127; found: 206.1140.
4.4.4 (E)-(3-methoxystyryl)trimethylsilane (4)
Following general procedure A: 3-vinyl-anisole (139 μL, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (5% Et2O:hexanes) and concentrated in vacuo to yield 159 mg of 4 (77%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.27 (t, J = 7.8 Hz, 1H), 7.06 (d, J = 7.7 Hz, 1H), 7.01 (t, J = 2.0 Hz, 1H), 6.87 (d, J = 19.2 Hz, 1H), 6.83 (dd, J = 2.7, 0.8 Hz, 1H), 6.50 (d, J = 19.1 Hz, 1H), 3.86 (s, 3H), 0.18 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 159.9, 143.5, 139.9, 130.0, 129.6, 119.2, 114.0, 111.3, 55.5, −1.1; FTIR (cm−1) 2954, 1263, 865, 838. HRMS (EI) m/z, calcd for [C12H18OSi]: 206.1127; found: 206.1148.
4.4.5 (E)-tert-butyldimethyl(3-(2-(trimethylsilyl)vinyl)phenoxy)silane (5)
Following general procedure A: tert-butyldimethyl(3-vinylphenoxy)silane18 (234 mg, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 236 mg of 5 (77%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.18 (t, J = 7.8 Hz, 1H), 7.04 (d, J = 7.7 Hz, 1H), 6.91 (t, J = 2.1 Hz, 6H), 6.80 (d, J = 19.1 Hz, 1H), 6.73 (dd, J = 8.0, 2.3 Hz, 1H), 6.43 (d, J = 19.1 Hz, 1H), 0.99 (s, 9H), 0.20 (s, 6H), 0.15 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 156.0, 143.5, 140.0, 129.7, 129.5, 119.9, 119.8, 118.0, 25.9, 18.4, −1.1, −4.2; FTIR (cm−1) 2956, 2859, 1575, 1280, 985, 838. HRMS (EI) m/z, calcd for [C17H30OSi2]: 306.1835; found: 306.1819.
4.4.6 (E)-(2-(benzo[d][1,3]dioxol-5-yl)vinyl)trimethylsilane (6)
Following general procedure A: 5-vinylbenzo[d][1,3]dioxole18 (148 mg, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (hexanes) and concentrated in vacuo to yield 127 mg of 6 (57%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.00 (d, J = 1.8 Hz, 1H), 6.86 (dd, J = 8.0, 1.7 Hz, 1H), 6.80 – 6.74 (m, 2H), 6.27 (d, J = 19.1 Hz, 1H), 5.95 (s, 2H), 0.14 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 148.2, 147.6, 143.1, 133.4, 127.4, 121.5, 108.3, 105.6, 101.2, −1.0; FTIR (cm−1) 2954, 2895, 1489, 1248, 866, 839. HRMS (EI) m/z, calcd for [C12H16O2Si]: 220.0920; found: 220.0933.
4.4.7 (E)-(4-fluorostyryl)trimethylsilane (7)
Following general procedure A: 4-fluorostyrene (119 μL, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (hexanes) and concentrated in vacuo to yield 128 mg of 8 (66%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.40 (ddt, J = 8.3, 5.4, 2.5 Hz, 2H), 7.01 (app tt, J = 8.5, 1.9 Hz, 2H), 6.82 (d, J = 19.1 Hz, 1H), 6.38 (d, J = 19.1 Hz, 1H), 0.15 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 162.7 (d, J = 247.3 Hz), 142.4 (s), 134.7 (s), 129.4 (d, J = 2.2 Hz), 128.0 (d, J = 8.0 Hz), 115.5 (d, J = 21.5 Hz), −1.1 (s); 19F NMR (376 MHz, CDCl3) δ −114.2; FTIR (cm−1) 2956, 1507, 1248, 836. HRMS (EI) m/z, calcd for [C11H15FSi]: 194.0927; found: 194.0945.
4.4.8 (E)-(2-(bicyclo[4.2.0]octa-1(6),2,4-trien-3-yl)vinyl)trimethylsilane (11)
Following general procedure A: 4-vinylbenzocyclobutene (130 mg, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 157 mg of 11 (78%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 10.1 Hz, 1H), 7.18 (s, 1H), 7.00 (d, J = 7.5 Hz, 1H), 6.85 (d, J = 19.1 Hz, 1H), 6.38 (d, J = 19.1 Hz, 1H), 3.16 (s, 4H), 0.14 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 146.2, 146.1, 144.7, 137.5, 127.8, 126.1, 122.7, 120.0, 29.6, 29.4, −1.0; FTIR (cm−1) 2955, 2930, 1247, 985, 866, 837. HRMS (EI) m/z, calcd for [C13H18Si]: 202.1178; found: 202.1194.
4.4.9 (E)-(2,4-dimethylstyryl)trimethylsilane (12)
Following general procedure A: 2,4-dimethystyrene (146 μL, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (hexanes) and concentrated in vacuo to yield 126 mg of 12 (62%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.43 (d, J = 7.9 Hz, 1H), 7.10 (d, J = 19.0 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.96 (s, 1H), 6.33 (d, J = 19.0 Hz, 1H), 2.35 (s, 3H), 2.31 (s, 3H), 0.16 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 141.2, 137.6, 135.3, 134.9, 131.2, 130.2, 127.0, 125.3, 21.3, 19.7, −1.0; FTIR (cm−1) 2954, 1247, 987, 868, 842. HRMS (EI) m/z, calcd for [C13H20Si]: 204.1334; found: 204.1350.
4.4.10 (E)-9-(2-(trimethylsilyl)vinyl)-9H-carbazole (13)
Following general procedure A: N-Vinyl carbazole (193 mg, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 248 mg of 13 (93%) as a white solid. 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.8 Hz, 2H), 7.72 (d, J = 8.3 Hz, 2H), 7.48 (ddd, J = 8.3, 7.2, 1.3 Hz, 2H), 7.38 – 7.27 (m, 3H), 6.04 (d, J = 17.2 Hz, 1H), 0.27 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 139.4, 133.6, 126.3, 124.2, 120.8, 120.4, 113.5, 110.9, 77.2, −0.6; FTIR (cm−1) 2953, 1610, 1447, 834, 751, 721. HRMS (EI) m/z, calcd for [C17H19NSi]: 265.1287; found: 265.1262.
4.4.11 (E)-trimethyl(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)styryl)silane (15)
Following a modification to general procedure A: 4-pinacolatoboryl styrene19 (230 mg, 1 mmol), tricyclopentylphosphine (72 mg, 0.3 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (3–15% CH2Cl2:hexanes) and concentrated in vacuo to yield 123 mg of 15 (41%) as a white solid. 1H NMR (600 MHz, CDCl3) δ 7.76 (d, J = 8.0 Hz, 2H), 7.43 (d, J = 8.0 Hz, 2H), 6.88 (d, J = 19.1 Hz, 1H), 6.55 (d, J = 19.1 Hz, 1H), 1.35 (s, 12H), 0.16 (s, 9H); 13C NMR (151 MHz, CDCl3) δ 143.7, 141.1, 135.2, 131.1, 125.8, 83.9, 25.0, −1.1 (the carbon attached to boron was not observed); FTIR (cm−1) 2953, 1607, 1358, 1141, 1090, 869. HRMS (EI) m/z, calcd for [C17H27BO2Si]: 302.1873; found: 302.1893.
4.4.12 tert-butyldimethyl(((8R,9S,13S,14S,17S)-13-methyl-3-((E)-2-(trimethylsilyl)vinyl)-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-17-yl)oxy)silane (16)
Following general procedure A: tert-butyldimethyl(((8R,9S,13S,14S,17S)-13-methyl-3-vinyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthren-17-yl)oxy)silane2b (396 mg, 1 mmol), tBuPCy2 (38 mg, 0.15 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and Me3SiOTf (540 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 283 mg of 16 (60%) as a white foam. 1H NMR (400 MHz, CDCl3) δ 7.25 – 7.18 (m, 2H), 7.15 (s, 1H), 6.82 (d, J = 19.1 Hz, 1H), 6.40 (d, J = 19.2 Hz, 1H), 3.64 (t, J = 8.2 Hz, 1H), 2.98 – 2.73 (m, 2H), 2.30 (dt, J = 12.8, 3.0 Hz, 1H), 2.21 (td, J = 11.5, 11.0, 3.9 Hz, 1H), 2.08 – 1.79 (m, 3H), 1.76 – 1.59 (m, 1H), 1.59 – 1.09 (m, 7H), 0.89 (s, 9H), 0.74 (s, 3H), 0.14 (s, 9H), 0.04 (s, 3H), 0.03 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 143.6, 140.7, 137.0, 135.9, 128.5, 127.0, 125.7, 123.7, 81.9, 49.9, 44.7, 43.7, 38.8, 37.3, 31.1, 29.7, 27.4, 26.4, 26.0, 23.4, 18.3, 11.5, −1.0, −4.3, −4.6; FTIR (cm−1) 2926, 1248, 1095, 866, 836. HRMS (EI) m/z, calcd for [C29H48OSi2]: 468.3244; found: 468.3259.
4.4.13 (E)-butyl(4-(tert-butyl)styryl)dimethylsilane (17)
Following general procedure B: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), Cy3P (84 mg, 0.3 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and n-butyldimethylsilyl trifluoromethanesulfonate7b (790 mg, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. N,N-dimethyl ethanolamine (300 μL, 3 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 165 mg of 17 (60%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.38 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 19.2 Hz, 1H), 6.41 (d, J = 19.1 Hz, 1H), 1.35 – 1.28 (m, 13H), 0.88 (t, J = 6.8 Hz, 3H), 0.64 – 0.59 (m, 2H), 0.12 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 151.2, 143.8, 135.9, 127.8, 126.2, 125.6, 34.8, 31.5, 26.7, 26.3, 15.6, 14.0, −2.8; FTIR (cm−1) 2956, 1982, 986, 837. HRMS (EI) m/z, calcd for [C18H30Si]: 274.2117; found: 274.2092.
4.4.14 (E)-benzyl(4-(tert-butyl)styryl)dimethylsilane (18)
Following general procedure B: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), Cy3P (84 mg, 0.3 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and benzyldimethylsilyl trifluoromethanesulfonate7a (895 mg, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. N,N-dimethyl ethanolamine (300 μL, 3 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (0–5% CH2Cl2:hexanes) and concentrated in vacuo to yield 206 mg of 18 (67%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.36 (app s, 4H), 7.22 (t, J = 7.6 Hz, 2H), 7.08 (t, J = 7.4 Hz, 1H), 7.03 (app d, J = 7.3 Hz, 2H), 6.84 (d, J = 19.2 Hz, 1H), 6.38 (d, J = 19.2 Hz, 1H), 2.21 (s, 2H), 1.33 (s, 9H), 0.13 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 151.4, 144.6, 140.1, 135.7, 128.4, 128.3, 126.4, 126.2, 125.6, 124.1, 34.8, 31.4, 26.3, −3.2; FTIR (cm−1) 2961, 1493, 832, 698. HRMS (EI) m/z, calcd for [C21H28Si]: 308.1960; found: 308.1950.
4.4.15 (E)-(4-(tert-butyl)styryl)(isopropyl)dimethylsilane (19)
Following general procedure B: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), Cy3P (84 mg, 0.3 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and isopropyldimethylsilyl trifluoromethanesulfonate7a (750 mg, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. N,N-dimethyl ethanolamine (300 μL, 3 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 157 mg of 19 (60%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 19.2 Hz, 1H), 6.41 (d, J = 19.2 Hz, 1H), 1.32 (s, 9H), 0.98 (d, J = 7.1 Hz, 6H), 0.93 – 0.80 (m, 1H), 0.10 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 151.2, 144.4, 135.9, 126.4, 126.2, 125.6, 34.7, 31.4, 17.8, 14.0, −5.1; FTIR (cm−1) 2955, 2863, 1267, 987, 839. HRMS (EI) m/z, calcd for [C17H28Si]: 260.1960; found: 260.1968.
4.4.16 (E)-tert-butyl(4-(tert-butyl)styryl)dimethylsilane (20)
Following a modification of general procedure B: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), nBu3P (81 mg, 0.4 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and tert-butyldimethylsilyl trifluoromethanesulfonate (690 μL, 3 mmol) were reacted in dioxane (1 mL) at 110 °C for 24 h. N,N-dimethyl ethanolamine (300 μL, 3 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (hexanes) and concentrated in vacuo to yield 86 mg of 20 (31%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.39 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.5 Hz, 2H), 6.87 (d, J = 19.2 Hz, 1H), 6.43 (d, J = 19.1 Hz, 1H), 1.32 (s, 9H), 0.91 (s, 9H), 0.11 (s, 6H); 13C NMR (101 MHz, CDCl3) δ 151.2, 144.7, 135.9, 126.2, 125.8, 125.6, 34.8, 31.4, 26.6, 17.0, −5.9; FTIR (cm−1) 2954, 2856, 1247, 987, 828. HRMS (EI) m/z, calcd for [C18H30Si]: 274.2117; found: 274.2103.
4.4.17 (E)-(4-(tert-butyl)styryl)(phenyl)dimethylsilane (21)
Following general procedure B: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), Cy3P (84 mg, 0.3 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and phenyldimethylsilyl trifluoromethanesulfonate7c (850 mg, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. N,N-dimethyl ethanolamine (300 μL, 3 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 205 mg of 21 (70%) as a colorless oil. 1H NMR (600 MHz, CDCl3) δ 7.58 – 7.55 (m, 2H), 7.39 (d, J = 8.4 Hz, 4H), 7.37 – 7.33 (m, 5H), 6.93 (d, J = 19.1 Hz, 1H), 6.54 (d, J = 19.2 Hz, 1H), 1.32 (s, 9H), 0.42 (s, 6H); 13C NMR (151 MHz, CDCl3) δ 151.5, 145.3, 138.9, 135.6, 134.1, 129.1, 127.9, 126.4, 126.2, 125.6, 34.8, 31.4, −2.3; FTIR (cm−1) 2960, 1247, 1112, 841, 821, 729, 698. HRMS (EI) m/z, calcd for [C20H26Si]: 294.1804; found: 294.1788.
4.4.18 (E)-(4-(tert-butyl)styryl)(methyl)diphenylsilane (22)
Following general procedure B: 1-tert-butyl-4-vinylbenzene (137 μL, 0.75 mmol), Cy3P (63 mg, 0.225 mmol), Ni(COD)2 (20.6 mg, 0.075 mmol), Et3N (529 μL, 3.75 mmol), and diphenylmethylsilyl trifluoromethanesulfonate7c (780 mg, 2.25 mmol) were reacted in dioxane (1.5 mL) at 75 °C for 24 h. N,N-dimethyl ethanolamine (225 μL, 2.25 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (2%–10% CH2Cl2:hexanes) and concentrated in vacuo to yield 198 mg of 22 (74%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.59 (dd, J = 7.5, 1.8 Hz, 4H), 7.47 – 7.33 (m, 10H), 6.97 (d, J = 19.1 Hz, 1H), 6.73 (d, J = 19.0 Hz, 1H), 1.33 (s, 9H), 0.72 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 151.7, 147.1, 136.7, 135.5, 135.1, 129.4, 128.0, 126.5, 125.6, 123.9, 34.8, 31.4, −3.5; FTIR (cm−1) 2961, 1427, 1111, 800, 699. HRMS (EI) m/z, calcd for [C25H28Si]: 356.1960; found: 356.1955.
4.4.19 (E)-(4-(tert-butyl)styryl)triethylsilane (23)
Following general procedure B: 1-tert-butyl-4-vinylbenzene (183 μL, 1 mmol), Cy3P (84 mg, 0.3 mmol), Ni(COD)2 (27.5 mg, 0.1 mmol), Et3N (700 μL, 5 mmol), and triethylsilyl trifluoromethanesulfonate (680 μL, 3 mmol) were reacted in dioxane (2 mL) at 75 °C for 24 h. N,N-dimethyl ethanolamine (300 μL, 3 mmol) was added after reaction. The product was purified by flash chromatography on silica gel (petroleum ether) and concentrated in vacuo to yield 178 mg of 23 (65%) as a colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.40 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 8.6 Hz, 2H), 6.88 (d, J = 19.3 Hz, 1H), 6.38 (d, J = 19.3 Hz, 1H), 1.32 (s, 9H), 0.98 (t, J = 7.9 Hz, 9H), 0.65 (q, J = 7.9 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 151.2, 144.7, 135.9, 126.1, 125.6, 125.0, 34.7, 31.4, 7.6, 3.7; FTIR (cm−1) 2954, 2874, 987, 788, 731. HRMS (EI) m/z, calcd for [C18H30Si]: 274.2117; found: 274.2093.
Supplementary Material
Acknowledgments
The University of Delaware (UD), the NSF (CAREER CHE1254360), and the Research Corporation (Cottrell Scholars Program) are gratefully acknowledged for funding and additional support. SESM acknowledges graduate fellowship support from NIH/NIGMS CBI Training Grant 5T32 GM 08550-16. NMR and other data were acquired at UD on instruments obtained with the assistance of NSF and NIH funding (NSF MRI CHE0421224 and CHE1229234, NSF CRIF MU CHE0840401, NIH P20 GM103541).
Footnotes
Supplementary data associated with this article (NMR spectra of products) can be found in the online version, at http:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.(a) Hosomi A, Endo M, Sakurai H. Chem Lett. 1976;5:941–942. [Google Scholar]; (b) Masse CE, Panek JS. Chem Rev. 1995;95:1293–1316. [Google Scholar]; (c) Fleming I, Barbero A, Walter D. Chem Rev. 1997;97:2063–2192. doi: 10.1021/cr941074u. [DOI] [PubMed] [Google Scholar]; (d) Brook MA. Silicon in Organic, Organometallic, and Polymer Chemistry. Wiley; Chichester: 2000. [Google Scholar]; (e) Denmark SE, Fu J. Chem Rev. 2003;103:2763–2793. doi: 10.1021/cr020050h. [DOI] [PubMed] [Google Scholar]; (f) Fleming I, Dunoguès J, Smithers R. Organic Reactions. John Wiley & Sons, Inc; New York: 2004. The Electrophilic Substitution of Allylsilanes and Vinylsilanes; pp. 57–575. [Google Scholar]; (g) Denmark SE, Liu JHC. Angew Chem Int Ed. 2010;49:2978–2986. doi: 10.1002/anie.200905657. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Nakao Y, Hiyama T. Chem Soc Rev. 2011;40:4893–4901. doi: 10.1039/c1cs15122c. [DOI] [PubMed] [Google Scholar]
- 2.(a) McAtee JR, Martin SES, Ahneman DT, Johnson KA, Watson DA. Angew Chem Int Ed. 2012;51:3663–3667. doi: 10.1002/anie.201200060. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Martin SES, Watson DA. J Am Chem Soc. 2013;135:13330–13333. doi: 10.1021/ja407748z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Martin SES, Watson DA. Synlett. 2013;24:2177–2182. doi: 10.1055/s-0033-1339795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For early studies related to the silyl-Heck reaction see: Yamashita H, Hayashi T, Kobayashi T, Tanaka M, Goto M. J Am Chem Soc. 1988;110:4417–4418.Chatani N, Amishiro N, Murai S. J Am Chem Soc. 1991;113:7778–7780.Yamashita H, Kobayashi T, Hayashi T, Tanaka M. Chem Lett. 1991;20:761–762.Chatani N, Amishiro N, Morii T, Yamashita T, Murai S. J Org Chem. 1995;60:1834–1840.Yamashita H, Tanaka M, Goto M. Organometallics. 1997;16:4696–4704.Terao J, Jin Y, Torii K, Kambe N. Tetrahedron. 2004;60:1301–1308.Terao J, Torii K, Saito K, Kambe N, Baba A, Sonoda N. Angew Chem Int Ed. 1998;37:2653–2656. doi: 10.1002/(SICI)1521-3773(19981016)37:19<2653::AID-ANIE2653>3.0.CO;2-3.
- 4.Oestreich M. The Mizoroki-Heck Reaction. John Wiley & Sons; Chichester, U.K: 2008. [Google Scholar]
- 5.For eariler reports of the use of iodide salt to in situ generation of Si-I species, see: Olah GA, Narang SC, Gupta BGB, Malhotra R. J Org Chem. 1979;44:1247–1251.
- 6.Walsh R. Acc Chem Res. 1981;14:246–252. [Google Scholar]
- 7.(a) Aizpurua JM, Palomo C. Tetrahedron Lett. 1985;26:6113–6114. [Google Scholar]; (b) Coppi L, Ricci A, Taddei M. Tetrahedron Lett. 1987;28:965–968. [Google Scholar]; (c) Uhlig W. J Organomet Chem. 1993;452:29–32. [Google Scholar]
- 8.(a) Li BJ, Yu DG, Sun CL, Shi ZJ. Chem Euro J. 2011;17:1728–1759. doi: 10.1002/chem.201002273. [DOI] [PubMed] [Google Scholar]; (b) Rosen BM, Quasdorf KW, Wilson DA, Zhang N, Resmerita AM, Garg NK, Percec V. Chem Rev. 2011;111:1346–1416. doi: 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.For additional examples of silicon halide oxidative additions, see: Kuyper J. Inorg Chem. 1978;17:77–81.Usón R, Oro L, Fernandez M. J Organomet Chem. 1980;193:127–133.Yamashita H, Kobayashi T, Hayashi T, Tanaka M. Chem Lett. 1989;18:471–474.Zlota AA, Frolow F, Milstein D. J Chem Soc, Chem Commun. 1989:1826–1827.Yamashita H, Kawamoto A, Tanaka M, Goto M. Chem Lett. 1990;19:2107–2110.Yamashita H, Kobayashi T, Hayashi T, Tanaka M. Chem Lett. 1990;19:1447–1450.Kirss RU. Inorg Chem. 1992;31:3451–3458.Levy CJ, Puddephatt RJ, Vittal JJ. Organometallics. 1994;13:1559–1560.Levy CJ, Vittal JJ, Puddephatt RJ. Organometallics. 1996;15:2108–2117.Gatard S, Chen CH, Foxman BM, Ozerov OV. Organometallics. 2008;27:6257–6263.Esposito O, Roberts DE, Cloke FGN, Caddick S, Green JC, Hazari N, Hitchcock PB. Euro J Inorg Chem. 2009;2009:1844–1850.Mitton SJ, McDonald R, Turculet L. Organometallics. 2009;28:5122–5136.
- 10.For a recent example using nickel catalyst with alkenes and silyl triflates, see: Ng SS, Ho CY, Jamison TF. J Am Chem Soc. 2006;128:11513–11528. doi: 10.1021/ja062866w.
- 11.In this case, as well as all others reported herein, no more than trace product was observed in reactions conducted without catalyst.
- 12.Silylation of terminal alkenes bearing allylic hydrogen atoms, such as 1-decene, were also investigated. However, with these substrates only trace desired product was observed; alkene isomerization predominated.
- 13.(a) Bennetau B. Benzylsilanes. In: Fleming I, editor. Science of Synthesis. Vol. 4. Thieme; Stuttgart: 2002. pp. 825–836. [Google Scholar]; (b) Sore HF, Galloway WRJD, Spring DR. Chem Soc Rev. 2012;41:1845–1866. doi: 10.1039/c1cs15181a. [DOI] [PubMed] [Google Scholar]
- 14.(a) Yamane M, Uera K, Narasaka K. Bull Chem Soc Jpn. 2005;78:477–486. [Google Scholar]; (b) Jones GR, Landais Y. Tetrahedron. 1996;52:7599–7662. [Google Scholar]
- 15.Choi JH, Kwon JK, RajanBabu TV, Lim HJ. Adv Synth Cat. 2013;355:3633–3638. [Google Scholar]
- 16.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- 17.Patt SL, Shoolery JN. J Mag Res. 1982;46:535–539. [Google Scholar]
- 18.Faler CA, Joullié MM. Org Lett. 2007;9:1987–1990. doi: 10.1021/ol0705907. [DOI] [PubMed] [Google Scholar]
- 19.Cambre JN, Roy D, Gondi SR, Sumerlin BS. J Am Chem Soc. 2007;129:10348–10349. doi: 10.1021/ja074239s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.