

Abstract
Sex differences in neuronal susceptibility to ischemic injury and neurodegenerative disease have long been observed, but the signaling mechanisms responsible for those differences remain unclear. Primary disassociated embryonic neuronal culture provides a simplified experimental model with which to investigate the neuronal cell signaling involved in cell death as a result of ischemia or disease; however, most neuronal cultures used in research today are mixed sex. Researchers can and do test the effects of sex steroid treatment in mixed sex neuronal cultures in models of neuronal injury and disease, but accumulating evidence suggests that the female brain responds to androgens, estrogens, and progesterone differently than the male brain. Furthermore, neonate male and female rodents respond differently to ischemic injury, with males experiencing greater injury following cerebral ischemia than females. Thus, mixed sex neuronal cultures might obscure and confound the experimental results; important information might be missed. For this reason, the Herson Lab at the University of Colorado School of Medicine routinely prepares sex-stratified primary disassociated embryonic neuronal cultures from both hippocampus and cortex. Embryos are sexed before harvesting of brain tissue and male and female tissue are disassociated separately, plated separately, and maintained separately. Using this method, the Herson Lab has demonstrated a male-specific role for the ion channel TRPM2 in ischemic cell death. In this manuscript, we share and discuss our protocol for sexing embryonic mice and preparing sex-stratified hippocampal primary disassociated neuron cultures. This method can be adapted to prepare sex-stratified cortical cultures and the method for embryo sexing can be used in conjunction with other protocols for any study in which sex is thought to be an important determinant of outcome.
Keywords: Neuroscience, Issue 82, male, female, sex, neuronal culture, ischemia, cell death, neuroprotection
Introduction
Mammals are sexually dimorphic with males and females exhibiting different traits and characteristics. Sex differences extend even to the brain and are evident in neuronal susceptibility to injury and disease (reviewed in1-5). For example, the male brain is more susceptible to ischemic neuronal injury from stroke or cardiac arrest followed by resuscitation (reviewed in6). Parkinson's disease (reviewed in7) and schizophrenia (reviewed in8) are more common in human males than in females, while the female human brain appears more susceptible to Alzheimer's disease (reviewed in9), and mood disorders such as depression (reviewed in10). Despite observable differences between sexes in susceptibility to neuronal injury and disease, most current treatment paradigms fail to consider sex and are similar in both men and women. Greater success might be achieved if sex differences were accounted for and understood.
In vivo animal models of neuronal injury and disease demonstrate sexual dimorphism similar to humans and are critical to enhancing our understanding of sex differences and how those differences contribute to neuronal survival and function. Evidence has demonstrated, for example, that male rodents are more susceptible than females to ischemic neuronal injury11-15. This difference is thought to be at least partially dependent upon the molecular signaling of testosterone, the primary male sex steroid hormone, and estrogen, the primary female sex steroid hormone. Studies with gonectomized male and female rodents reveals that testosterone and the testosterone metabolite, dihydrotestosterone (DHT) exacerbate and estrogen decreases neuronal injury following cerebral ischemia12-14,16-18. However, sex differences in cerebral ischemia are present in both neonate and adult animals19,20. Neonates experience surges in estrogen or testosterone in utero during development, but have relatively low levels of hormone after birth until puberty (reviewed in21). Thus, the presence or absence of primary sex hormone is not the only determinant of sex-influenced neuronal susceptibility to injury. Indeed, it is likely that sexual dimorphisms are established during development that result in differences in cell signaling and response to ischemia even when hormone levels are low.
Regardless of whether sex differences are dependent on the presence of sex steroid hormone or whether they are due to dimorphisms established during development, it is becoming clear with further study that there are differences in cell signaling between male and females in the brain. In the Herson Lab at the University of Colorado School of Medicine, we have identified one such difference. We have found that the calcium, sodium, and potassium permeable ion channel, TRPM2 (reviewed in22,23), induces neuronal cell death following ischemic insult in vivo in the male but not the female mouse24. We demonstrate that pharmacological inhibition or genetic knockdown of TRPM2 is protective in the male but not the female mouse in our in vivo middle carotid arterial occlusion (MCAO) model of stroke24.
Despite their usefulness, in vivo animal models of ischemic neuronal injury may, at times, be limited for molecular studies of neuronal death, due to the difficulty or inability to specifically target neurons for investigation. For this reason, our lab uses primary disassociated cortical or hippocampal neurons in vitro to complement our in vivo work. Primary disassociated neuron cultures present a useful tool to investigate the cell signaling pathways involved in neuronal injury and disease. Neurons in culture can be maintained in a tightly controlled environment, with or without glial input. In the absence of glia, any responses observed in culture are neuronal. Pharmacological agents can be administered to culture without concerns about absorption across the blood brain barrier, allowing for the inhibition or activation of specific components of a given signal transduction pathway. Molecular biology can be used to overexpress or knockdown various proteins of interest, and electrophysiological measurements can be conducted on individual cells, something that would not be possible in an in vivo model system.
The Herson Lab expands the usefulness of primary disassociated neuronal cultures by first sexing the embryos used to prepare the cultures. Male and female sex-stratified neuron cultures demonstrate clearly that male neurons are more susceptible than female neurons to neuronal cell death following oxygen and glucose deprivation25. Oxygen and glucose deprivation is an accepted in vitro model of neuronal ischemia as ischemia reduces oxygen and glucose availability to the brain. Furthermore, we have demonstrated with electrophysiological experiments in disassociated, sex-stratified neurons that TRPM2 is activated in male but not female neurons24,26. We are currently investigating the molecular sex-specific regulators of TRPM2 activity24,26. Here, we share and discuss our protocol for establishing sex-stratified primary embryonic disassociated hippocampal neuron cultures and present representative data using this method that suggests that the DNA repair enzyme, PARP-1, contributes to ischemic cell death in a sex specific manner similar to TRPM2. PARP-1 is activated by DNA breakage due to oxidative stress (reviewed in27,28). When DNA damage is minimal, PARP-1 activity enhances cell survival, but when damage is excessive, increased PARP-1 activity exacerbates cell death. PARP-1 produces poly-(ADP)ribose, a known activator of the TRPM2 channel28-31, and some evidence suggests that PARP-1 is activated preferentially in the male but not female brain in response to oxidative stress32,33. Thus, we hypothesize that PARP-1 activates TRPM2 in the male but not female, inducing neuronal cell death in response to oxygen and glucose deprivation.
Protocol
All procedures in this protocol were conducted according to the National Institute of Health guidelines for the ethical treatment and care of animals, and according to an approved IACUC animal use protocol.
1. Preparation of Tissue Culture Dishes, Culture Media, and Dissection Instruments to be Performed at Least One Day Before Dissection
Prepare 1 L borate buffer: 3.1 g/L boric acid and 4.75 g/L Borax in double distilled H2O. Adjust pH to 8.4. Filter-sterilize and store at 4 °C until use.
- Dilute poly-D-lysine to 0.1 mg/ml.
- Under a laminar-flow tissue culture hood, place enough diluted poly-D-lysine to generously cover the bottom of each well of a multi-well, culture-treated, sterile tissue culture plate. (If desired, precleaned, sterile, glass-cover slips can be added to the bottom of each well before the addition of poly-D-lysine. Cover slips can float after the addition of poly-D-lysine, so they should be pushed down with sterile forceps if necessary.)
- Incubate the plates overnight in the incubator at 37 °C.
Prepare 1 L of dissection buffer: 350 mg/L NaHCO3, 2.38 g/L HEPES, 1.44 g/L MgSO4, and 300 mg/L BSA in 1x Hanks Balanced Salt Solution without Ca, Mg, sodium pyruvate, or phenol red. Adjust pH to 7.3. Filter-sterilize and store at 4 °C until needed.
Prepare 500 ml plating medium: 10% Fetal Bovine Serum, 100 units/ml each of penicillin and streptomycin, and 2.5 mM glutamine in MEM without phenol red. Filter-sterilize. Store at 4 °C until needed. Discard any unused medium after 1 month.
Prepare 500 ml feeding medium: 2% B-27 supplement, 100 units/ml each of penicillin and streptomycin, and 2.0 mM GlutaMAX (not glutamine) in Neurobasal without phenol red. Filter-sterilize. Store at 4 °C until needed. Discard any remaining feeding medium after 1 month.
Prepare 10 ml of 100 mM CaCl2, 500 mM EDTA, and 1 M NaOH as needed according to standard methods.
Autoclave the surgical/dissection instruments and store under sterile conditions until needed. This procedure requires two sharp extra-fine tipped angled forceps, a fine tipped curved forceps, a serrated curved forceps, a small rat-toothed forceps, one pair of micro-surgical scissors, one pair of fine, narrow tipped surgical scissors, one pair of medium tipped scissors, and a blunt-ended dissection probe. Additional sterile instruments can be added according to personal preference.
2. Preparation of Tissue Culture Dishes, Culture Media, Dissection Instruments, and Dissection Area to be Performed on the Day of Dissection
- Wash the poly-D-lysine coated tissue culture plates. Under the laminar flow biosafety cabinet, aspirate the poly-D-lysine with a vacuum line or pipette, add water to each well, and incubate at room temperature for ~5 min. Aspirate the water and replace it with clean, sterile water. Repeat 3x.
- Allow the plates to fully dry at room temperature.
- Prepare the dissection area by wiping down the counter and dissection scope with 70% ethanol.
- Saturate a large Kimwipe with 70% ethanol, and lay out the autoclaved dissection tools. Cover with another Kimwipe.
- Fill a large, flat dish with ice. Label and fill an appropriate number of 60 mm culture dishes with cold dissection solution. Prepare enough dishes to keep the male and female heads, brains, and hippocampi separate (if applicable).
- Fill two larger 100 mm culture dishes with cold dissection buffer.
- Set aside a separate 100 mm dish without buffer (dry).
- Establish an area for biohazard waste and another for animal tissue (to be disposed of appropriately after the dissection, according to institutional standards).
Warm 30 ml plating media in a 37 °C water bath. This warm solution will be used in the cell disassociation phase of the protocol.
Place 1 ml of plating media into each well of 24-well, poly-D-lysine coated, tissue culture plate. Use an appropriate smaller volume if smaller plates are used. Place the covered plate into a 37 °C incubator at 5% CO2 to warm and equilibrate to the CO2 during the dissection.
- Prepare the papain solution.
- Label two 15 ml sterile conical tubes, one as "male", and the other as "female".
- Add the following to each tube: 10 ml of dissection buffer, 150 μl 100 mM CaCl2, 10 μl 500 mM EDTA, 10μl 1 M NaOH, and 150 U of papain.
- Gently mix by inverting the tube three times, then warm in a 37 °C water bath. Note: Each vial of papain will have a different unit of activity per mg of papain, thus the volume of papain added to solution will differ from vial to vial. The papain will be cloudy initially, but the solution will clear as the papain dissolves completely.
3. Harvesting, Sexing, and Dissection of E18 Embryos
Anesthetize the pregnant female mouse with isoflurane or other appropriate anesthesia. Euthanize the mouse under deep anesthesia by decapitation or cervical dislocation.
Working quickly, clean the female's abdomen with 70% ethanol. If desired, the abdomen can be shaved before being sprayed with ethanol to reduce risks of contamination. Lift the abdominal skin with the rat-toothed forceps. Make a large incision in the abdomen with the medium-tipped scissors, cutting through skin and abdominal muscles, exposing the internal organs.
Carefully remove the embryos, lifting each entire uterine horn clear of the abdomen, being careful not to let it contact any external tissue if possible (to avoid contamination). Place the uterine horns containing the embryos into a dish with cold dissection saline and transfer to ice.
- Carefully remove each pup from the uterine horns and the embryonic sac. The pups are fragile, so this is best done by cutting the uterine tissue between each embryo to expose the embryonic sac, and then carefully slicing the sac on the surface with the edge of the small, narrow scissors. The embryonic fluid and the pup can then be gently pushed through the opening without damage to the embryo.
- If the incision in the embryonic sac is not large enough for the fluid and pup to come out easily, then enlarge the incision carefully until the pup slides out with minimal force.
- Do not squeeze the sac too forcefully to force the embryo out, as this too could damage the embryos.
Remove the head from a single embryo. Leave the head in a dish of dissection saline on ice. Place the body of the embryo on its back on the dry 100 mM dish under the dissection scope. Using the curved medium-tipped forceps in one hand and the serrated curved forceps in the other, gently flatten the pup out, pushing the back legs down and ventral, the front legs down and dorsal.
Remove the tail with scissors if necessary; the tail tends to curl up over the abdomen and can get in the way of the dissection.
Release the back legs and pinch the abdomen at the root of the umbilical cord, called the umbilicus. Lift up gently. Release the front legs and with the other hand and the micro-scissors, make a small inverted "v" in the abdomen just above the umbilicus with the point of the "v" pointing cranial.
Exchange the scissors for the curved forceps again. Turn the forceps over so that the point is up and away from the animal. With gentle force, push down and dorsal on the rib-cage with the closed, rounded edge of the curved forceps, while pulling down and ventral with the other forceps, spreading the pup out ventral to dorsal. This spreading motion will open the abdominal cavity, exposing the intestines, liver, and other internal organs.
- Using the curved forceps, very gently push the intestines and liver cranially using repeated sweeping motions. This will gradually expose the kidneys, bladder, and most importantly, the internal sex organs.
- Male pups are identified by the presence of striated testes found just below the kidneys on each side. The testes are approximately ⅓-½ the size of the kidneys and occasionally will have dropped to sit just above the bladder.
- Females are most readily identified by a lack of testes, but can also be identified by the small uterus that sits above the bladder, and fallopian tubes that extend up toward the kidneys. Under high-power on the dissection scope, the ovaries can also be identified as small, dark organs at the end of the fallopian tubes immediately below and sometimes underneath the kidneys bilaterally.
Once the sex is determined, discard the body and place the head in an appropriately labeled dish (male or female). Keep the heads on ice in dissection solution.
Repeat steps 3.5-3.10 until each pup has been sexed. For best results, wipe the dish clean with a Kimwipe between each dissection; fluid makes the pups slippery and difficult to grasp for the dissection.
Once all embryos have been sexed and the male and female heads have been separated, proceed with the rest of the dissection taking especial care to keep all male and female-derived tissue separate.
Transfer one brain at a time to a 100 mM dish filled with cold dissection buffer. Under the dissection scope, use the curved forceps to pierce the eyes of the embryo to anchor it for dissection. Using the microsurgical scissors, make a shallow midline incision, splitting the skin and skull down the midline, from base of skull to top of the head just above the nasal passage.
- Gently push aside the skin and skull bilaterally and very gently extract brain by carefully scooping it from the forebrain towards the brainstem with the base of the micro-surgical scissors or with a closed, angled forceps.
- Carefully use the forceps to transfer the extracted brain to a clean, labeled dish filled with cold dissection buffer. Keep the dish on ice.
- Repeat this process until all brains have been harvested.
- This step should yield two plates of whole brains in dissection buffer, one for male and one for female tissue.
Place a clean dish beneath the dissection scope and fill with dissection buffer. Pour just enough solution to keep the brains moist throughout the dissection of the hippocampus; too much will cause the brain to float and make handling difficult. If desired, precoat the dissection dish with sterile wax. The wax provides nonslip surface upon which to work and protects the tips of delicate dissection instruments; scratching the bottom of a noncoated dish can bend or dull the sharp edges of the tools.
Change the dissection scope to a high-power setting, and adjust the focus accordingly. Carefully transfer one brain at a time to dissection dish. Using the extra-fine tipped forceps, tease apart each brain hemisphere and pinch them off of the brainstem.
- Very carefully, remove the meninges from each hemisphere. (The meninges are a pinkish membrane, rich with blood vessels, and are a common source of glial contamination in a neuron culture.)
- To remove the meninges, delicately pinch, lift, and pull at the membrane, being careful not to pierce the brain tissue beneath.
- Pull the meninges away from the brain, allowing the brain to roll a bit beneath the forceps but keeping it in place so that the membrane can be pulled away.
At this point, you may further dissect the brain to use the tissue of choice. For example, dissect the hippocampus from each hemisphere. Place one hemisphere with the lateral side down. Observe the large ventricle cavity, which creates a flap covering the hippocampus. Gently tease this flap back until the crescent-shaped hippocampus can be observed. The hippocampus is slightly raised and sits between the mid-brain and cortex.
- Using two pair of sharp, extra-fine forceps, hold the brain hemisphere open with one hand and cut the hippocampus away from the cortex and mid-brain with the other.
- Place the hippocampus in a correctly labeled dish (male or female) filled with dissection buffer and kept on ice.
- Dissect the hippocampus from the remaining hemisphere and repeat this process with each brain.
- Transfer the hippocampi into appropriately labeled 15 ml conical tubes.
- Gently wash twice with cold dissection buffer. Use vacuum suction very carefully at this stage. The hippocampi are small and can be irrecoverably lost to the vacuum. Prevent this by allowing the tissue to settle to the bottom of the tube, remove as much liquid as possible from the tube with a pipette, discard the solution, and replace with fresh, clean dissection buffer.
- Repeat step 3.20.1.
4. Neuronal Dissociation
Transfer the hippocampi to appropriately labeled, prewarmed papain solution. Place in a 37 °C water bath and incubate for 8-10 min, gently inverting the tubes to mix every 2-3 min.
Remove the papain mix and tissue from the water-bath. Allow the tissue to settle, and then carefully remove as much of the papain solution as possible with a pipette, leaving the tissue at the bottom of the tube. Wash the tissue 3x with plating media. The serum in the plating media quenches the papain enzymatic digestion. Trypsin inhibitor is needed if serum is not used in the plating media.
- Remove the plating media.
- Add 2 ml of fresh neurobasal medium and gently dissociate tissue using a 5 ml serological pipette set on the slowest speed.
- Dissociation is performed by gently pipetting up and down 8x. Be careful not to introduce bubbles into solution. For best results, pick the solution and tissue up in the pipette and then release the solution onto the side of the conical tube, allowing the tissue to slide down the side and back to the bottom of the tube.
- After the 8th trituration, allow the tissue to settle and collect the cloudy media on top. This media contains disassociated cells.
- Transfer it to a new, labeled tube.
- Add an additional 2 ml to the undisassociated tissue.
- Triturate again another 8x.
- Harvest the cloudy media.
- Repeat this process 3-5x. Some undissociated tissue will remain. While it is possible to triturate until all tissue is dissociated, excessive trituration results in poor neuronal health and is not recommended.
Add extra medium to the conical tubes as necessary to equalize the volumes and weights between male and female tissue. Centrifuge the cells in a spinning bucket centrifuge at 228 x g for 5 min. Carefully remove the supernatant.
Gently resuspend the cells in 2 ml fresh plating media and filter the solution through a mesh cell strainer into a clean, labeled, 50 ml conical tube.
Add 50 μl cell suspension to 450 μl of 0.4% Trypan Blue. Incubate the cells for 2 min in Trypan Blue, mix gently, and load 10 μl onto a hemocytometer. Count the cells excluding those that have taken up the Trypan Blue dye; dark blue cells are dead or dying.
5. Plating and Culture Maintenance
Calculate the correct volume of cell suspension to add to each well of the tissue culture plate to yield the desired cell density. Neuron cultures are very sensitive to cell density and do best at medium to high density unless cocultured with glia or fed with glial conditioned media. In this protocol, cells are plated at 150,000-200,000 cells/cm2.
- Remove the tissue culture plate prefilled with plating media from the incubator (prepared in step 2 above).
- Gently swirl the cell suspension in the 50 ml conical tube, then pipette the desired volume of cell suspension (calculated above in step 5.1) to the center of each well.
- Repeat, swirling the cell suspension each time before pipetting to ensure equal cell density in each well. Cells settle quickly and cell density can change from well to well if the swirling step is skipped.
Once cells have been added to each well, gently swirl the plate to disperse the cells, and then immediately replace the dish in the incubator. Leave the cells undisturbed for at least 24 hr.
At least 24 hr after plating the cells, warm complete neurobasal feeding media to 37 °C. Aspirate the plating media from each well and replace it with feeding media. Aspirate and replace from one well at a time to minimize neuronal stress and improve cell survival.
Neuron cultures should be fed every 4-7 days by carefully removing half of the medium and adding back an equal volume of fresh medium. Under these conditions, cultures can be maintained for two weeks. With primary neuronal cultures, some cell death is expected. Thus, as the cultures mature, the density will decrease. In this protocol, cell density decreases by approximately 30% by day 10 in vitro.
Representative Results
Proper sexing of embryonic mice is a critical step in the preparation of sex-stratified primary neuron cultures. To ensure the male and female hippocampi were cultured separately from correctly sexed pups, embryos were separated by sex and their cortex and hippocampus were harvested. Genomic DNA was isolated from the harvested tissue of each embryo according to standard procedures, being careful to keep sex separate. Polymerase Chain Reaction (PCR) was performed using primers for the male-specific gene, SRY and the universal marker myogenin (Myog) (Figure 1). SRY, or sex-determination region on the Y chromosome, is responsible for testes formation and is commonly used as a sex marker. We observed that Myog was present in each male and female brain tissue tested and the SRY gene was present only in embryo tissue identified as "male" by sexing, but it was not present in tissue identified as "female".
Pharmacological inhibition of PARP1 with PJ34 is neuroprotective against oxygen and glucose deprivation (OGD) in primary hippocampal neuron cultures derived from male embryos, but PARP1 inhibition has no protective effect in cultures prepared from female embryos (Figure 2). Male neuron cultures prepared from PARP1 knockout mice were also protected against OGD induced cell death. Treatment of wild type male neurons with the TRPM2 channel inhibitor 2-APB in addition to PJ34 had no further neuroprotective effect against OGD. 2-APB alone is neuroprotective in OGD in wild-type males (previously published data), but 2-APB is not protective in male or female PARP1 knockout mice. These data support the hypothesis that PARP1 activity exacerbates neuron cell death in male but not female neurons through the activation of the TRPM2 channel.
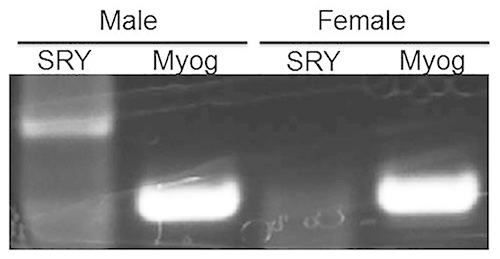 Figure 1.Sex stratification of neuronal cultures is confirmed by the presence of SRY gene product in male but not female embryonic tissue. PCR amplification of the male-specific SRY gene was observed only in embryonic tissue identified as ‘male' using the sexing methods described in this protocol. SRY was not observed in female tissue. The universal marker, myogenin (Myog), was observed in both male and female tissue, and serves as a positive control for the PCR reaction. Click here to view larger image.
Figure 1.Sex stratification of neuronal cultures is confirmed by the presence of SRY gene product in male but not female embryonic tissue. PCR amplification of the male-specific SRY gene was observed only in embryonic tissue identified as ‘male' using the sexing methods described in this protocol. SRY was not observed in female tissue. The universal marker, myogenin (Myog), was observed in both male and female tissue, and serves as a positive control for the PCR reaction. Click here to view larger image.
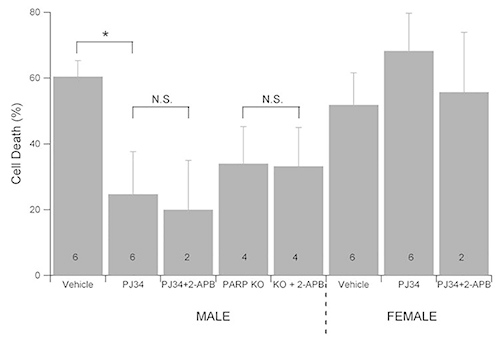 Figure 2.PARP1 inhibition or knockout is neuroprotective in the male but not female neuron culture. 10 day in vitro sex stratified neuron cultures from wild-type or PARP1 knockout mouse embryos were treated with PARP1 inhibitor (10 μM PJ34) and/or TRPM2 inhibitor (100 μM 2-APB) and subjected to oxygen and glucose deprivation (OGD) in an anoxic, temperature controlled chamber with 5% CO2. 24 hr after OGD, cell survival was assessed by MTT assay. PARP1 inhibition or knockdown was neuroprotective in neuron cultures derived from male embryos, but had no effect in neuron cultures from female neurons. 2-APB provided no further protection. Click here to view larger image.
Figure 2.PARP1 inhibition or knockout is neuroprotective in the male but not female neuron culture. 10 day in vitro sex stratified neuron cultures from wild-type or PARP1 knockout mouse embryos were treated with PARP1 inhibitor (10 μM PJ34) and/or TRPM2 inhibitor (100 μM 2-APB) and subjected to oxygen and glucose deprivation (OGD) in an anoxic, temperature controlled chamber with 5% CO2. 24 hr after OGD, cell survival was assessed by MTT assay. PARP1 inhibition or knockdown was neuroprotective in neuron cultures derived from male embryos, but had no effect in neuron cultures from female neurons. 2-APB provided no further protection. Click here to view larger image.
Discussion
Sex stratified primary disassociated hippocampal neuronal cultures in which male and female hippocampi are dissected, disassociated, and cultured separately present a useful tool for investigating sex-specific cell signaling in neuron cell death. While neuroscientists commonly use primary neuronal cultures, protocols vary between labs depending on their specific experimental aims and interests. Many researchers choose to coculture neurons with glia, either in mixed culture or by plating the neurons on a glass cover slip suspended on a preestablished astrocyte layer. Astrocytes are thought to secrete growth hormones that are beneficial to neuron survival in culture. However, in order to simplify the experimental system, the protocol described in this study results in neuronal cultures without substantial glial influence (less than 10% glia contamination).
Protocols also vary between labs in regards to the age of the embryo from which the hippocampus is harvested, and, for that matter, protocols vary with regards to which region of the brain is harvested. This protocol uses E18 embryos for dissection. Other common ages include E15-16. E18 embryos are larger and easier to sex and dissect than younger embryos. Furthermore, E18 embryos have experienced a second surge of sex steroid hormone (around E16-17), which is believed to masculinize or feminize the male and female brain. Sex-specific differences in response to ischemia may be more robust in cultures prepared from older embryos, though this remains to be tested.
Determining the sex of the embryos presents a challenge in this protocol because the embryos are very small and fragile. Care must be taken so as not to penetrate the gut or destroy the sex organs while attempting to move the intestines and liver out of the way. A light, steady, gentle hand is critical during the dissection, but once the technique is mastered, sexing is relatively fast and easy.
Dissecting the brain to isolate the hippocampus can be difficult and requires significant practice for the uninitiated. Embryonic brain is soft. The brain tissue must be held gently, with little pressure. Unfortunately, the health and longevity of the disassociated neurons appears to depend on minimizing the time between when the embryo is removed from the sacrificed dame and when the neurons are plated and placed in the incubator. The individual doing the dissection must work as carefully and quickly as possible. For someone who does not have experience with neuron culture, this may mean processing only one or two brains per sex until the necessary speed is developed. This will yield a plate or partial plate of neurons, but is more likely to yield healthy neurons than if several hours are allowed to pass while all brains are dissected. The goal of these authors is to have the neurons in plating media an hour after the embryos have been removed from the dame.
Neurons are stained with Trypan Blue when counted in the hemocytometer so that neuron viability can be assessed. This protocol should yield at least 80-95% viability. Less than that is cause for concern and likely indicates that either the dissection is taking too long, that the neurons were digested in too much papain or for too long, or that the dissector was too rough during the trituration step. The dissection should be done as quickly as possible, as mentioned above. Digestion with papain should be just long enough to make the tissue soft and ‘gummy' around the edges. Too much ‘gumminess' indicates that the tissue is over-digested. The trituration step should be gentle as well, being careful not to introduce air bubbles into the solution. When viability issues are observed during the cell count, the gentleness of the trituration step is usually the first process to adjust. It seems to be the most important step during the dissection.
Once the neurons are disassociated, they are added to the plating media. In this protocol, the plating media contains glutamine instead of the more commonly used GlutaMAX. Extensive trial-and-error has demonstrated an improved survivability in dissociated mouse neurons with glutamine instead of GlutaMAX at the plating stage, though the reason for this improvement is not known. Additionally, this protocol uses serum in the plating media. Serum does contain sex steroid hormones and many choose to plate in the absence of serum (substituting B-27 for serum) when assessing the role of sex or sex-steroid hormones in neuronal function so that the concentration of hormones can be more carefully controlled. However, serum also substantially improves survivability of the culture, likely because serum contains additional growth factors and nutrients not found in serum-free media. We choose to use serum in our plating media because the improved neuronal survival seems to outweigh the disadvantage of having unknown quantities of sex steroid hormones present in the first 24 hr of culture. We conduct our oxygen and glucose deprivation experiments on the 10th day in vitro in each culture, with the assumption that the serum was removed or greatly diluted with cell feeding.
Phenol red is excluded from the plating media and feeding media in this protocol because of evidence linking phenol red to estrogen-mediated cell signaling34-40. While there is evidence also suggesting that the observed estrogen signaling is due to contaminants and not phenol red41, phenol red is not necessary for the survival and health of neuron cultures and is therefore excluded as a precaution. It is possible to supplement with charcoal-stripped FBS if serum is necessary for the health of the cultures.
24 hr after plating, the neurons should be sprouting small neurite outgrowth, and at this point, the plating media is replaced with feeding media. If the neurons do not settle down and establish outgrowths in 24 hr, they will not attach to the culture surface even with additional time. The likely cause of unattached neurons is a poor coating with poly-D-lysine. Several molecular weights of poly-D-lysine are available. 70,000-150,000 kD size is recommended for greatest success. If plating yields poor cell attachment, ensure that the molecular weight of poly-D-lysine is correct. Occasionally, neurons will attach to the surface of the dish in grape-like clusters and will not disperse. This effect is also likely due to poor tissue culture dish preparation. Care should be taken to coat the dishes a full 24 hr in poly-D-lysine solution. Fresh solution should be prepared for the coating if necessary. Poly-D-lysine can be prepared in water, but pH adjusted borate buffer recommended to produce a more dependable coating. It is equally important to use tissue culture treated dishes for plating the neurons. Untreated dishes will not allow adherence of the neurons. If difficulties with neuronal attachment persist, culture dishes can be coated in laminin (0.1 mg/ml) in addition to poly-D-lysine. Coat the plates in poly-D-lysine as described above, wash the plates, and then incubate in laminin at 37 °C for 2 hr. Wash plates in distilled water 3x as described above, and store in plating media until needed.
24-48 hr after the cultures have been established, antimitotic agents can be added if desired to control glial contamination. These agents are common and indeed necessary in postnatal neuronal cultures; without mitotic inhibitors, glial cells will proliferated and overrun the cultures. However, antimitotic agents are excluded from this protocol because glial burden in embryonic neurons is quite low, less than 10% of the culture even at 10 days in vitro. Furthermore, and perhaps more importantly, antimitotic agents appear to negatively affect embryonic neuron survival.
Once the cultures have been plated and established, they should remain relatively healthy for 12-14 days. Neuronal health does decline with each day in vitro. By the 12th day in vitro, this decrease in neuronal viability is very apparent with approximately 30% cell loss. If however, the neurons begin to die after only a few days in culture, then the components of the feeding media or the treatment of the cells postplating may need to be adjusted. Change only one component of media at a time until the problem is resolved. Some common culprits for unhealthy neurons are the B-27 in the feeding media, media pH, incubator CO2, or excessive handling of neurons after plating. Fresh B-27 is best. While this protocol indicates that media with B-27 can be used for 1 month, if cell survival is poor, it is recommended that fresh B-27 be added with each feeding. Additionally, the quality of B-27 can vary from lot to lot, so it is recommended that the lot number be recorded to identify any sudden changes in neuron health related to lot. Correct pH is also critical to the health of neuron cultures; check both the pH of the media and the concentration of CO2 in the incubator. Furthermore, the Herson Lab has found that glutamine is superior to GlutaMAX during neuron plating; thus, glutamine or GlutaMAX concentrations may need alteration if problems in cell viability arise. Use caution while adjusting glutamine concentrations. Glutamine supports cell survival during the early stages of culture, but glutamine can also be excitotoxic, especially in older neuron cultures (>7 DIV). Cell density is also critical for embryonic cultures; density may be increased to improve cell survival. However, it is important to keep cell density the same from experiment to experiment as density will impact experimental outcome. Primary cultured neurons can be very fickle. It is crucial to handle them as little as possible. Remove them from the incubator only for feedings and for as little time as possible. Feed the cells gently, with adequately warm medium, and without jarring the plates.
The methods and tips presented here should enable one to establish sex-stratified primary disassociated hippocampal neuron cultures within the lab. The methods can likely be adapted for E15-16 embryos and for rat neuron culture. The methods might also work for culturing other brain regions. While the process of growing neurons can be frustrating, the reward of consistently healthy cultures is scientifically important and can be very gratifying.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Work supported in part by the Walter S. and Lucienne Driskill Foundation and NIH R01NS058792.
References
- Andreano JM, Cahill L. Sex influences on the neurobiology of learning and memory. Learn Mem. 2009;16:248–266. doi: 10.1101/lm.918309. [DOI] [PubMed] [Google Scholar]
- Arnold AP, Burgoyne PS. Are XX and XY brain cells intrinsically different. Trends Endocrinol. Metab. 2004;15:6–11. doi: 10.1016/j.tem.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Cahill L. Why sex matters for neuroscience. Nat. Rev. Neurosci. 2006;7:477–484. doi: 10.1038/nrn1909. [DOI] [PubMed] [Google Scholar]
- Jazin E, Cahill L. Sex differences in molecular neuroscience: from fruit flies to humans. Nat. Rev. Neurosci. 2010;11:9–17. doi: 10.1038/nrn2754. [DOI] [PubMed] [Google Scholar]
- Morris JA, Jordan CL, Breedlove SM. Sexual differentiation of the vertebrate nervous system. Nat. Neurosci. 2004;7:1034–1039. doi: 10.1038/nn1325. [DOI] [PubMed] [Google Scholar]
- Lang JT, McCullough LD. Pathways to ischemic neuronal cell death: are sex differences relevant. J. Transl. Med. 2008;6:33. doi: 10.1186/1479-5876-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Den Eeden SK, et al. Incidence of Parkinson's disease: variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003;157:1015–1022. doi: 10.1093/aje/kwg068. [DOI] [PubMed] [Google Scholar]
- Abel KM, Drake R, Goldstein JM. Sex differences in schizophrenia. Int. Rev. Psychiatry. 2010;22:417–428. doi: 10.3109/09540261.2010.515205. [DOI] [PubMed] [Google Scholar]
- Vest RS, Pike CJ. Gender, sex steroid hormones, and Alzheimer's disease. Horm. Behav. 2013;63:301–307. doi: 10.1016/j.yhbeh.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Guasti A, Fiedler JL, Herrera L, Handa RJ. Sex, stress, and mood disorders: at the intersection of adrenal and gonadal hormones. Horm. Metab. Res. 2012;44:607–618. doi: 10.1055/s-0032-1312592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Pazara KE, Linseman KL. Sex differences in postischemic neuronal necrosis in gerbils. J. Cereb. Blood Flow Metab. 1991;11:292–298. doi: 10.1038/jcbfm.1991.61. [DOI] [PubMed] [Google Scholar]
- Hawk T, Zhang YQ, Rajakumar G, Day AL, Simpkins JW. Testosterone increases and estradiol decreases middle cerebral artery occlusion lesion size in male rats. Brain Res. 1998;796:296–298. doi: 10.1016/s0006-8993(98)00327-8. [DOI] [PubMed] [Google Scholar]
- Nakano T, Hurn PD, Herson PS, Traystman RJ. Testosterone exacerbates neuronal damage following cardiac arrest and cardiopulmonary resuscitation in mouse. Brain Res. 2010;1357:124–130. doi: 10.1016/j.brainres.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, et al. Effect of testosterone on functional recovery in a castrate male rat stroke model. Brain Res. 2005;1043:195–204. doi: 10.1016/j.brainres.2005.02.078. [DOI] [PubMed] [Google Scholar]
- Siegel C, Turtzo C, McCullough LD. Sex differences in cerebral ischemia: possible molecular mechanisms. J. Neurosci. Res. 2010;88:2765–2774. doi: 10.1002/jnr.22406. [DOI] [PubMed] [Google Scholar]
- Uchida M, et al. Dose-dependent effects of androgens on outcome after focal cerebral ischemia in adult male mice. J. Cereb. Blood Flow Metab. 2009;29:1454–1462. doi: 10.1038/jcbfm.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, et al. Testosterone increases neurotoxicity of glutamate in vitro and ischemia-reperfusion injury in an animal model. J. Appl. Physiol. 2002;92:195–201. doi: 10.1152/jappl.2002.92.1.195. [DOI] [PubMed] [Google Scholar]
- Cheng J, Alkayed NJ, Hurn PD. Deleterious effects of dihydrotestosterone on cerebral ischemic injury. J. Cereb. Blood Flow Metab. 2007;27:1553–1562. doi: 10.1038/sj.jcbfm.9600457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunez J. Sex and steroid hormones in early brain injury. Rev. Endocr. Metab. Disord. 2012;13:173–186. doi: 10.1007/s11154-012-9219-3. [DOI] [PubMed] [Google Scholar]
- Renolleau S, Fau S, Charriaut-Marlangue C. Gender-related differences in apoptotic pathways after neonatal cerebral ischemia. Neuroscientist. 2008;14:46–52. doi: 10.1177/1073858407308889. [DOI] [PubMed] [Google Scholar]
- McCarthy MM, Konkle AT. When is a sex difference not a sex difference. Front Neuroendocrinol. 2005;26:85–102. doi: 10.1016/j.yfrne.2005.06.001. [DOI] [PubMed] [Google Scholar]
- Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451:243–249. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- Jiang L-H, Yang W, Zou J, Beech DJ. TRPM2 channel properties, functions and therapeutic potentials. Expert Opin. Ther. Targets. 2010;14:973–988. doi: 10.1517/14728222.2010.510135. [DOI] [PubMed] [Google Scholar]
- Jia J, et al. Sex differences in neuroprotection provided by inhibition of TRPM2 channels following experimental stroke. J. Cereb. Blood Flow Metab. 2011;31:2160–2168. doi: 10.1038/jcbfm.2011.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks SL, et al. Mechanism of the sex difference in neuronal ischemic cell death. Neuroscience. 2012;219:183–191. doi: 10.1016/j.neuroscience.2012.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma S, et al. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci. Lett. 2012;530:41–46. doi: 10.1016/j.neulet.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai P, Canto C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- Blenn C, Wyrsch P, Bader J, Bollhalder M, Althaus FR. Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cell Mol. Life Sci. 2011;68:1455–1466. doi: 10.1007/s00018-010-0533-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buelow B, Song Y, Scharenberg AM. The Poly(ADP-ribose) polymerase PARP-1 is required for oxidative stress-induced TRPM2 activation in lymphocytes. J. Biol. Chem. 2008;283:24571–24583. doi: 10.1074/jbc.M802673200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonfria E, et al. TRPM2 channel opening in response to oxidative stress is dependent on activation of poly(ADP-ribose) polymerase. Br. J. Pharmacol. 2004;143:186–192. doi: 10.1038/sj.bjp.0705914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BA. Inhibition of TRPM2 function by PARP inhibitors protects cells from oxidative stress-induced death. Br. J. Pharmacol. 2004;143:515–516. doi: 10.1038/sj.bjp.0705923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagnerova K, et al. Poly (ADP-ribose) polymerase-1 initiated neuronal cell death pathway--do androgens matter. Neuroscience. 2010. pp. 166–476. [DOI] [PMC free article] [PubMed]
- Yuan M, et al. Sex differences in the response to activation of the poly (ADP-ribose) polymerase pathway after experimental stroke. Exp. Neurol. 2009;217:210–218. doi: 10.1016/j.expneurol.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthois Y, Katzenellenbogen JA, Katzenellenbogen BS. Phenol red in tissue culture media is a weak estrogen: implications concerning the study of estrogen-responsive cells in culture. Proc. Natl. Acad. Sci. U.S.A. 1986;83:2496–2500. doi: 10.1073/pnas.83.8.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic DA, Renk M, Kamel F. Estrogenic effects of phenol red on rat pituitary cell responsiveness to gonadotropin-releasing hormone. Life Sci. 1989;44:397–406. doi: 10.1016/0024-3205(89)90264-6. [DOI] [PubMed] [Google Scholar]
- Ernst M, Schmid C, Froesch ER. Phenol red mimics biological actions of estradiol: enhancement of osteoblast proliferation in vitro and of type I collagen gene expression in bone and uterus of rats in vivo. J. Steroid Biochem. 1989;33:907–914. doi: 10.1016/0022-4731(89)90239-2. [DOI] [PubMed] [Google Scholar]
- Hubert JF, Vincent A, Labrie F. Estrogenic activity of phenol red in rat anterior pituitary cells in culture. Biochem. Biophys. Res. Commun. 1986;141:885–891. doi: 10.1016/s0006-291x(86)80125-5. [DOI] [PubMed] [Google Scholar]
- Ortmann O, Sturm R, Knuppen R, Emons G. Weak estrogenic activity of phenol red in the pituitary gonadotroph: re-evaluation of estrogen and antiestrogen effects. J. Steroid Biochem. 1990;35:17–22. doi: 10.1016/0022-4731(90)90139-j. [DOI] [PubMed] [Google Scholar]
- Rajendran KG, Lopez T, Parikh I. Estrogenic effect of phenol red in MCF-7 cells is achieved through activation of estrogen receptor by interacting with a site distinct from the steroid binding site. Biochem. Biophys. Res. Commun. 1987;142:724–731. doi: 10.1016/0006-291x(87)91474-4. [DOI] [PubMed] [Google Scholar]
- Welshons WV, Wolf MF, Murphy CS, Jordan VC. Estrogenic activity of phenol red. Mol. Cell Endocrinol. 1988;57:169–178. doi: 10.1016/0303-7207(88)90072-x. [DOI] [PubMed] [Google Scholar]
- Moreno-Cuevas JE, Sirbasku DA. Estrogen mitogenic action. III. is phenol red a "red herring"? In Vitro Cell Dev. Biol. Anim. 2000;36:447–464. doi: 10.1290/1071-2690(2000)036<0447:EMAIIP>2.0.CO;2. [DOI] [PubMed] [Google Scholar]