

Abstract
Simultaneous electrophysiological and fluorescent imaging recording methods were used to study the role of changes of membrane potential or current in regulating the intracellular calcium concentration. Changing environmental conditions, such as the light-dark cycle, can modify neuronal and neural network activity and the expression of a family of circadian clock genes within the suprachiasmatic nucleus (SCN), the location of the master circadian clock in the mammalian brain. Excitatory synaptic transmission leads to an increase in the postsynaptic Ca2+ concentration that is believed to activate the signaling pathways that shifts the rhythmic expression of circadian clock genes. Hypothalamic slices containing the SCN were patch clamped using microelectrodes filled with an internal solution containing the calcium indicator bis-fura-2. After a seal was formed between the microelectrode and the SCN neuronal membrane, the membrane was ruptured using gentle suction and the calcium probe diffused into the neuron filling both the soma and dendrites. Quantitative ratiometric measurements of the intracellular calcium concentration were recorded simultaneously with membrane potential or current. Using these methods it is possible to study the role of changes of the intracellular calcium concentration produced by synaptic activity and action potential firing of individual neurons. In this presentation we demonstrate the methods to simultaneously record electrophysiological activity along with intracellular calcium from individual SCN neurons maintained in brain slices.
Keywords: Neuroscience, Issue 82, Synaptic Transmission, Action Potentials, Circadian Rhythm, Excitatory Postsynaptic Potentials, Life Sciences (General), circadian rhythm, suprachiasmatic nucleus, membrane potential, patch clamp recording, fluorescent probe, intracellular calcium
Introduction
Changes in gene expression are known to occur in neurons as a consequence of synaptic signaling. Signaling by the excitatory neurotransmitter glutamate can depolarize the neuronal membrane potential eventually leading to gene transcription and translation1,2. Activation of ionotropic receptors by glutamate allows extracellular calcium ions to enter the cell, which is thought to play a critical role as a second messenger in activating gene transcription. Evaluating the relationship between membrane electrical activity, such as action potential firing frequency, and changes of intracellular calcium concentration requires the combination of two methods - whole cell patch clamping and quantitative imaging of fluorescent calcium probes3-5, allowing the relationship to be studied in individual neurons. The single cell recording technique allows the recording of the activity of individual neurons in identifiable portions of the brain. The whole cell recording technique allows the membrane voltage or current to be controlled allowing for experimental manipulation of specific ion channel currents. Using micropipettes filled with fluorescent calcium probes also ensures that the neuron is well filled with calcium probe. This technique has a clear advantage when working with brain slice preparations from adult slice preparations, since these neurons are particularly difficult to load using the more common cell permeant probes and reduces potential background fluorescence issues6-8.
Light is the principal way mammals adjust their circadian clock, which is located in the hypothalamic suprachiasmatic nucleus (SCN). Light information transduced in the retina is transmitted 9-11 via the retinohypothalamic tract (RHT) where glutamate is released in the SCN12,13. Glutamate opens NMDA and AMPA ionotropic receptors located on SCN neurons producing an influx of calcium and sodium, and initiating an intracellular signaling cascade that ultimately leads to altering the expression of a family of clock genes14-17 and shifts in phase of the circadian clock18-20. However, calcium can enter neurons either directly through ionotropic glutamate receptors or through membrane depolarization and activation of voltage-dependent calcium channels (VDCC)21. We therefore developed an experimental protocol to investigate the relationship between the intracellular calcium concentration and membrane electrical activity in SCN neurons, such as occurs with action potential firing and from synaptic input22.
Protocol
1. Preparation of Hypothalamic Brain Slices
Obtain, in advance, Institutional Animal Care and Use Committee (IACUC) approval for any procedure involving animals. The animal procedures described here are consistent with the AVMA Guidelines for the Euthanasia of Animals 2013 and were approved in advance by the Oregon Health & Science University IACUC.
Prepare 500 ml of the slicing buffer without the MgCl2 and CaCl2 (Table 1). Bubble the solution with 95% O2 and 5% CO2 at room temperature then add the MgCl2 and CaCl2 while continuing to aerate. Adjust the osmolarity to 300 mOsm with sucrose. The osmolarity is measured with a vapor pressure osmometer.
Prepare 1 L of the recording media without the MgCl2 and CaCl2 (Table 2). Bubble the solution with 95% O2 and 5% CO2 then add the MgCl2 and CaCl2 while continuing to aerate. Adjust the pH to 7.2-7.4 with NaOH (10 N) and the osmolarity to 300 mOsm with sucrose. The addition of HEPES buffer is optional and was used to further stabilize pH.
The slicing and recording solutions may be stored in the refrigerator for several days prior to use.
Prepare 3% agarose and pour into a sterile Petri dish (6 mm deep) and refrigerate.
Cut three cubes of agarose about 6 mm x 6 mm x 6 mm.
Deeply anesthetize a four to eight week old Sprague-Dawley rat with isoflurane. Test for the depth of anesthesia. The depth of anesthesia is deemed appropriate when there is no withdrawal reflex of the hind limb following pinching of the footpad. Once appropriately anesthetized, the rat is euthanized by rapid decapitation.
Rapidly remove the brain being careful to cut the optic nerves first, and place in ice-cold slicing solution saturated with 5% CO2 and 95% O2 and allow to cool for 2-3 min.
Trim the brain. Place the brain using forceps on a moistened (with slicing solution) circular filter paper set on a glass Petri dish resting on ice.
Cut the brain coronally to remove the remaining cerebellum and brain stem, then the rostral cortex and the sides, to leave a block of brain containing the hypothalamus carefully preserving the side containing SCN, optic nerves, and optic chiasm.
Spread a thin layer of cyanoacrylic glue large enough to attach the brain and agarose blocks in an ice-cold but dry microtome-slicing chamber.
Transfer the ice-cold brain block caudal side down onto the glue with the side containing the SCN positioned for cutting first with the optic nerves forming a "V" pointing up.
Place the agarose blocks on the other 3 sides. Fill the chamber with ice-cold slicing buffer continuously bubbled with 5% CO2 and 95% O2. The cyanoacrylic glue will solidify with moisture holding the brain and agarose blocks in place.
Cut coronal hypothalamic slices (220-250 µm thick) containing the SCN with the vibrating blade microtome. Typically a slice is chosen where the optic chiasm is about 2-4 mm across, appearing like a white band with SCN just dorsal appearing as two small “indentations” in the optic chiasm on either side of the 3rd ventricle (Figure 1A).
Place the slices in the recording chamber (36 °C) mounted on the stage of a Leica DMLFS microscope. The chamber is filled with continuously flowing recording solution (2 ml/min) aerated with 95% O2 and 5% CO2 and preheated with an inline heater.
Perform whole-cell patch clamp and calcium imaging recordings in SCN neurons 0.5-8 hr after slice preparation.
2. Patch Clamp Recording
Prepare the electrode solution (intracellular) solution in half the total final volume of water.
Adjust the solution to pH 7.3 with KOH (1 N).
Add the remaining water. The final osmolarity of the solution should be in the range of 280-290 mOsm.
Pass the electrode solution through a 0.2 µm filter, aliquot (0.5 ml) into microfuge tubes, and store at -20 °C.
Prepare the calcium probe bis-fura-2 hexapotassium salt stock solution (10 mM) by mixing 1 mg bis-fura-2 hexapotassium salt with 99.29 µl of 0.2 µm filtered water.
Aliquot into separate tubes (2-5 µl each) and freeze (-20 °C).
Thaw an aliquot of internal solution and the Ca2+ probe shortly before use. Add 2 µl of the bis-fura-2 stock solution to 400 µl of the internal solution for a final 50 µM calcium probe concentration. Briefly triturate to mix the solutions. The calcium probe concentration (10-250 µM) varies depending on experimental design and imaging system. Note that bis-fura-2 is comprised of two linked fura-2 molecules.
Pull whole-cell patch electrodes, in two stages to an outside tip diameter of approximately 1 µm and resistances of 7-10 MΩ when filled with the intracellular solution.
Backfill the electrode with a few µl of the internal solution containing the calcium probe filtered with a 4 mm syringe filter (0.2 µm) for low volume samples via a Microfil tube.
Apply a small amount of positive pressure to the electrode as it is guided into the recording chamber under red light illumination while being viewed on a video monitor.
Place the microelectrode over the SCN using a low power objective (4X). The SCN is identified as the two translucent areas on either side of the third ventricle and dorsal to the optic chiasm (Figure 1A).
Switch the microscope to a high power magnification (40X or 63X). Subregions of the SCN and individual SCN neurons can be targeted for recording at the higher magnification.
Apply gentle positive pressure to the microelectrode as it is advanced onto the surface of a neuron.
Apply gentle negative pressure to the microelectrode to form a seal with resistances of 3-10 GΩ.
Adjust the membrane voltage to -60 mV with the amplifier controlled with acquisition software.
Apply additional negative pressure to the electrode to rupture the cell membrane to whole cell mode.
Upon entering whole cell mode the SCN neuron soma and dendrites fill with fluorescent probe.
Membrane voltage is measured in current-clamp mode or current in voltage-clamp mode while the calcium probe image data is being monitored and stored (described below). View acquired images along with a graph of 340 nm/380 nm ratio fluorescence data during the experiment.
3. Measurement of Intracellular Ca2+
Quantitative Ca2+ measurements are obtained by recording a pair of images by rapidly exciting the tissue with UV light at 340 nm and 380 nm via a monochronometer with a 10 nm bandwidth and passed through a UG11 optical filter to restrict harmonic wavelengths above 400 nm, a 400 nm DCLP dichroic and emitted light through a 510±40 nm emission filter.
The images are acquired using a cooled CCD camera with acquisition time and binning adjusted to minimize photobleaching and maximize recording speed.
Quantitative imaging software is used to acquire and display the images.
Select regions of interest from the initial image to be converted to relative fluorescence intensity unit data (i.e. soma, dendrite, etc.). Also select a region for measurement of the background fluorescence (Figure 1B).
Raw images and quantified optical data are continually saved during the experiment. After the experiment the saved images are generally reanalyzed when more time can be taken to select more accurately the regions of interest to be quantified.
The data can be presented as the estimated intracellular calcium concentration (Est[Ca2+]i). The ratio (R) of emitted light following excitation and background subtraction at 340 nm divided by 380 nm along with the maximum and minimum fluorescence at 340 nm and 380 nm is used to calculate the calcium concentration4. Background subtraction and other calculations are performed using data analysis software.
An in vitro determination of maximum and minimum bis-fura-2 fluorescence can be performed using 10 mM CaCl2 for Ca2+ bound and 10 mM EGTA for Ca2+ free conditions. The Ca2+ disassociation constant, Kd of the probe is taken to be 370 nM for bis-fura-2 (Invitrogen).
Representative Results
Using hypothalamic brain slices, we simultaneously recorded changes in intracellular calcium in whole cell mode under both voltage and current clamp conditions. The microelectrode shown in Figure 1A is lowered into position under high magnification (40X or 63X UV objectives) with a small amount of applied positive pressure. After touching the neuron, a gigaohm seal is formed with gentle suction. In voltage-clamp mode after setting the cell membrane potential to -60 mV, additional suction breaks the membrane creating the whole cell configuration. The neuron will rapidly fill with probe when viewed on the monitor. Dendritic processes in a similar focal plane as the soma can be visualized (Figure 1A inset). Alternatively, the focus could be adjusted to measure calcium changes in distal dendrites. Recording in voltage clamp mode at -60 mV we simulated action potential firing using brief (2 msec) steps to +20 mV at 10 Hz, which is in the physiological range for the daytime action potential firing rate of many SCN neurons (Figures 1B and 1C). Different regions of interest (ROI) of the neuron including the proximal dendrite were quantified and the background at each wavelength was subtracted to determine the ratio of emitted fluorescence from excitation at 340 nm and 380 nm of light (Figure 1B, inset). The bis-fura-2 filled microelectrode can be seen but is mainly out of the focal plane. Pseudocolor applied to the image with software can sometimes facilitate visualization. Differences in the rate of intracellular calcium influx can be observed between different regions of the neuron and the magnitude of the response increased with depolarizing current steps. Combining location data from images taken with 63X and 4X objectives (Figure 1A) we can assess the anatomical location within the SCN from each neuron recorded.
Similarly, recording in current-clamp mode the intracellular calcium response to changes in membrane potential can be measured. A small change of current near the action potential firing threshold can induce action potential firing and a corresponding elevation of calcium (Figure 2). Recorded in the soma, the calcium concentration increase lags behind the changes in membrane voltage but decays after cessation of action potential firing to a new plateau. Using these methods we have previously shown that synaptically-evoked excitatory postsynaptic potentials (EPSPs) do not significantly alter somatic intracellular calcium without subsequently induced action potential firing22, suggesting that postsynaptic action potential firing is required for calcium to affect subsequent transcriptional and translation events which may represent a way these neurons differentiate synaptic input signaling at different times of the day and night.
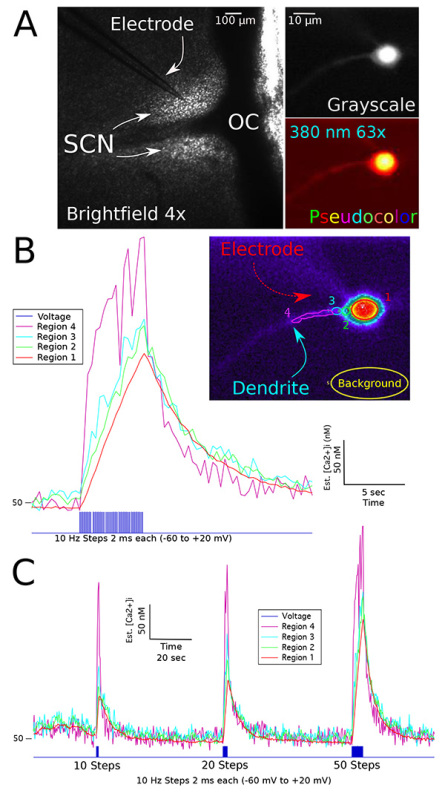 Figure 1.Calcium imaging of a voltage clamped SCN neuron.A: Bright field image of a coronal slice of hypothalamus containing the SCN and optic chiasm (OC). A glass microelectrode filled with a bis-fura-2 containing internal solution is sealed onto a single neuron. Inset: Recorded neuron with bis-fura-2 fluorescence (380 nm excitation) images in grayscale and pseudocolor. B: Calcium responses to a series of 50 voltage steps recorded from a voltage-clamped SCN neuron. Voltage-gated sodium currents were inhibited with TTX (0.5 µM). Inset: Screen capture image of somatic and dendritic regions of interest corresponding to the calcium responses shown in the graph. C: Calcium responses to a variable number of voltage steps. The right recording is the same data shown in B. Click here to view larger image.
Figure 1.Calcium imaging of a voltage clamped SCN neuron.A: Bright field image of a coronal slice of hypothalamus containing the SCN and optic chiasm (OC). A glass microelectrode filled with a bis-fura-2 containing internal solution is sealed onto a single neuron. Inset: Recorded neuron with bis-fura-2 fluorescence (380 nm excitation) images in grayscale and pseudocolor. B: Calcium responses to a series of 50 voltage steps recorded from a voltage-clamped SCN neuron. Voltage-gated sodium currents were inhibited with TTX (0.5 µM). Inset: Screen capture image of somatic and dendritic regions of interest corresponding to the calcium responses shown in the graph. C: Calcium responses to a variable number of voltage steps. The right recording is the same data shown in B. Click here to view larger image.
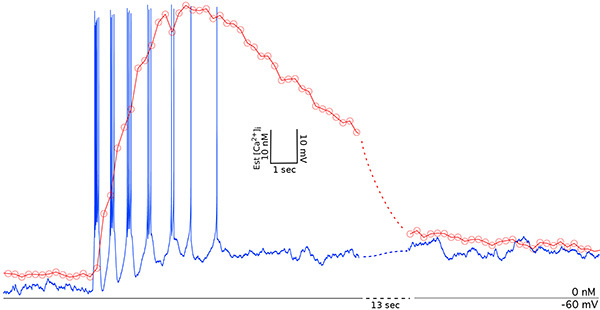 Figure 2. Simultaneous recording of membrane voltage and intracellular calcium in an SCN neuron. The current-clamp experiment was performed in the presence of picrotoxin (50 µM). A small depolarization of the membrane potential triggers several bouts of action potential firing with a corresponding rise and subsequent fall of intracellular calcium. Click here to view larger image.
Figure 2. Simultaneous recording of membrane voltage and intracellular calcium in an SCN neuron. The current-clamp experiment was performed in the presence of picrotoxin (50 µM). A small depolarization of the membrane potential triggers several bouts of action potential firing with a corresponding rise and subsequent fall of intracellular calcium. Click here to view larger image.
Discussion
The methods described above provide a powerful tool to simultaneously record the link between neuron membrane electrical activity and the intracellular calcium concentration. The method has a number of strengths in that it combines two very well characterized methods - whole cell patch clamp recording and measurement of intracellular calcium using fluorescent dyes. Our approach is similar to those described by other investigators6,23.
A number of items must be taken into consideration when using the method. First is the use of the whole cell patch configuration, which leads to the dialyzation of the cellular contents with the intracellular microelectrode solution. The dialyzation can lead to the rundown of voltage-gated ion channels and the loss of second messenger mediated signals24-26. Second, cell injury or damage to the gigaohm seal can result in increasing levels of calcium that may be associated with decreasing cellular input resistance24. Third, the intercellular solution containing the probe should be filtered just before filling the microelectrode to avoid plugging the microelectrode tip. By minimizing these factors, intracellular calcium and membrane electrical activity may be simultaneously recorded in patch clamped neurons for more than 30 min. We have had some success using the beta-Escin perforated patch method27 and bis-fura-2 to slow dialyzation and increase recording time. Fourth, calcium and a chelator EGTA or BAPTA are common constituents of intercellular solutions used to stabilize the intracellular calcium concentration. However, we have not included these under our recording conditions and dialyzing the neuron may alter calcium homeostasis and lower the basal calcium levels. We also did not add magnesium to the internal solution since it can also bind to fura-2. However, since fura-2 binds Mg2+ with a much lower affinity than calcium (Kd 5.6 mM)28 this may be optional. Fifth, the affinity of bis-fura-2 for calcium can be altered by a number of intracellular factors within a cell including pH and cell viscosity4,29. Finally, bis-fura-2 can buffer the intracellular calcium concentration and this should be considered when deciding on the probe concentration in the internal solution5. Thus the constituents of the internal solution may be modified based on neuronal type and conditions, experimental questions being addressed, and the equipment available for the study.
It is important to note that quantitative calcium imaging using bis-fura-2 typically uses the ultraviolet light wavelengths at 340 nm and 380 nm. Protective equipment must be used to avoid injury to eyes (UV eye protection) and skin (lab coats, etc.). In addition, UV producing bulbs may produce ozone requiring adequate ventilation especially important since such equipment is often located in small rooms where ambient light can be controlled.
The affinity of the calcium probe for calcium is an important consideration. If a high affinity probe is chosen then large calcium currents may saturate the probe leading to an under estimation of the magnitude of the calcium signal5. Whereas a low affinity probe may be more useful in regions where high calcium concentrations occur such as in distal dendritic processes and spines. Similarly, low affinity probes may miss changes at low calcium concentrations. In general, somatic changes of calcium can be measured using a quantitative ratiometric calcium probe such as bis-fura-2 or fura-2.
Data may be presented as a simple ratio quantified from a region of interest from a pair of images following background (BG) subtraction at each wavelength. While not as quantitative, ratiometric probes may also be excited at a single wavelength as one would use other single excitation wavelength probes for experiments requiring very fast recording time frames or if a monochronometer or other wavelength changing (e.g. filter wheel) device is not available.
We chose to use an in vitro calibration to obtain the parameters needed to estimate the intracellular calcium concentration along with a published Kd. However, more detailed techniques are available for in situ calibrations and to measure the Kd of the probe under specific experimental conditions. We used the excitation wavelengths of 340 nm and 380 nm for bis-fura-2 and estimated the intracellular Ca2+ concentration using the formula: Est[Ca2+]i = (R-Rmin)/(Rmax-R) x (Ff380/Fb380) x Kd, where F is fluorescence intensity at 380 nm of the probe in it's free (f) and bound (b) states, R is the emission ratio following excitation at 340 nm divided by the emission following excitation at 380 nm (i.e. F340/F380), Rmin is the minimum (Ff340/Ff380) and Rmax the maximum (Fb340/Fb380) ratio obtained in the free and bound states respectively, and Kd is the Ca2+ disassociation constant of the indicator4. Note that both Kd and (Ff380/Fb380) are scalar terms while Rmin and Rmax can affect the magnitude of the change in calcium. Care must be taken when estimating the concentration values for calcium. There are a number of unknowns that may be altered by the intracellular chemical environment that may affect the values in the equation.
Table 1. Slicing solution.
| Chemicals | Final Conc (mM) | FW (g/M) | Final (g/L) |
| NaCl | 120 | 58.44 | 7.013 |
| KCl | 2.5 | 74.55 | 0.186 |
| MgCl2 | 5 | 203.31 | 1.017 |
| CaCl2•2H2O | 0.5 | 147.02 | 0.074 |
| NaH2PO4•H2O | 1.2 | 137.99 | 0.166 |
| NaHCO3 | 26 | 84.01 | 2.184 |
| D-Glucose | 10 | 180.16 | 1.802 |
Table 2. Recording solution.
| Chemicals | Final Conc (mM) | FW (g/M) | Final (g/L) |
| NaCl | 120 | 58.44 | 7.013 |
| KCl | 2.5 | 74.55 | 0.186 |
| MgCl2•6H2O | 1.2 | 203.31 | 0.244 |
| CaCl2•2H2O | 2.4 | 147.02 | 0.353 |
| NaH2PO4•H2O | 1.2 | 137.99 | 0.166 |
| NaHCO3 | 26 | 84.01 | 2.184 |
| D-Glucose | 10 | 180.16 | 1.802 |
| HEPES (optional) | 10 | 238.3 | 2.383 |
Table 3. Intracellular (microelectrode) solution.
| Chemicals | Final Conc (mM) | FW (g/M) | Final (mg/25 ml) |
| KCl | 5 | 74.55 | 9.32 |
| Potassium D-gluconate | 140 | 234.2 | 819.70 |
| HEPES | 10 | 238.3 | 59.58 |
| Adenosine 5'-triphosphate dipotassium salt | 4 | 583.4 | 58.34 |
| Guanosine 5'-triphosphate tris salt | 0.4 | 523.2 | 5.23 |
Table of Specific Reagents and Equipment:
| Name of Reagent | Company | Catalog Number | Comments |
| NaCl | Fisher Scientific Co. | S271-3 | |
| KCl | Fisher Scientific Co. | P217-500 | |
| NaH2PO4•H2O | Sigma Chemical Co. | S-9638 | |
| MgCl2•6H2O | Fisher Scientific Co. | M33-500 | |
| CaCl2•2H2O | Fisher Scientific Co. | C79-500 | |
| D-glucose | Fisher Scientific Co. | D16-500 | |
| NaHCO3 | Fisher Scientific Co. | S233-500 | |
| Sucrose | Fisher Scientific Co. | S5-500 | |
| Potassium D-gluconate | Sigma-Aldrich | G4500 | |
| HEPES | Sigma-Aldrich | H4034 | |
| Adenosine 5′-triphosphate dipotassium salt dihydrate | Sigma-Aldrich | A8937 | |
| Guanosine 5′-triphosphate tris salt | Sigma-Aldrich | G9002 | |
| Agarose (ultrapure) | Life Technologies | 15510-027 | Gel pored into sterile Petri dish 6 mm thick layer |
| KOH | Sigma-Aldrich | P-6310 | |
| NaOH | Sigma-Aldrich | S-5881 | |
| bis-fura-2, hexapotassium salt | Invitrogen | B6810 | cell impermeant |
| Tetrodotoxin | Alomone Labs | T-550 | |
| Picrotoxin | Sigma-Aldrich | P-1675 | |
| Name of Equipment | Company | Catalog Number | Comments |
| Microtome | Leica | VT1000S | Tissue slicing |
| Cyanoacrylic glue (Roti-Coll1) | Carl Roth GmbH+Co | Art-Nr. 0258.1 | |
| Microelectrode puller | Narshige International USA | PP-83 | |
| Microelectrode Capillary Tubes | World Precision Instruments | 1B150F-4 | |
| Microfil 34g | World Precision Instruments | MF34G-5 | |
| Syringe filter | Corning | #431212 | |
| Microscope | Leica | DM LFS | With 4X, 40 UV and 63X UV objectives, and epifluorescence |
| CCD Camera | Hamamatsu | ORCA ER | 12 bit CCD |
| Fura-2 Filter Cube | Chroma | 71500A | Set with UG11 filter |
| Polychrome IV | Till Photonics GmBH | Monochronometer | |
| Ultraviolet blocking safety glasses | Ultra-Violet Products | ||
| EPC-9 amplifier | HEKA Eletronik | ||
| Metafluor | Molecular Devices | Imaging Software | |
| Patchmaster | HEKA Eletronik | Data Acquisition Software | |
| Igor version 6 | Wavemetrics | Electrophysiology & Ca2+ Data Analysis | |
| VAPRO 5520 | Westcor | Vapor pressure osmometer |
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The work was funded by a grant from the National Institute of General Medical Sciences (GM096972).
References
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Ginty DD, Bading H, Greenberg ME. Calcium regulation of gene expression in neuronal cells. J. Neurobiol. 1994;25:294–303. doi: 10.1002/neu.480250309. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Higley MJ, Sabatini BL. Calcium signaling in dendrites and spines: practical and functional considerations. Neuron. 2008;59:902–913. doi: 10.1016/j.neuron.2008.08.020. [DOI] [PubMed] [Google Scholar]
- Colwell CS. Circadian modulation of calcium levels in cells in the suprachiasmatic nucleus. Eur. J. Neurosci. 2000;12:571–576. doi: 10.1046/j.1460-9568.2000.00939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda M, Yoshioka T, Allen CN. Developmental and circadian changes in Ca2+ mobilization mediated by GABAA and NMDA receptors in the suprachiasmatic nucleus. Eur. J. Neurosci. 2003;17:58–70. doi: 10.1046/j.1460-9568.2003.02427.x. [DOI] [PubMed] [Google Scholar]
- Irwin RP, Allen CN. GABAergic signaling induces divergent neuronal Ca2+ responses in the suprachiasmatic nucleus network. Eur. J. Neurosci. 2009;30:1462–1475. doi: 10.1111/j.1460-9568.2009.06944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson DM, Dunn FA, Takao M. Phototransduction by retinal ganglion cells that set the circadian clock. Science. 2002;295:1070–1073. doi: 10.1126/science.1067262. [DOI] [PubMed] [Google Scholar]
- Hattar S, Liao HW, Takao M, Berson DM, Yau KW. Melanopsin-containing retinal ganglion cells: architecture, projections, and intrinsic photosensitivity. Science. 2002;295:1065–1070. doi: 10.1126/science.1069609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwick AT, et al. Light-Evoked Calcium Responses of Isolated Melanopsin-Expressing Retinal Ganglion Cells. J. Neurosci. 2007;27:13468–13480. doi: 10.1523/JNEUROSCI.3626-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castel M, Belenky M, Cohen S, Ottersen OP, Storm-Mathisen J. Glutamate-like immunoreactivity in retinal terminals of the mouse suprachiasmatic nucleus. Eur. J. Neurosci. 1993;5:368–381. doi: 10.1111/j.1460-9568.1993.tb00504.x. [DOI] [PubMed] [Google Scholar]
- Card JP, Moore RY. The organization of visual circuits influencing the circadian activity of the suprachiasmatic nucleus. In: Reppert SM, Klein DC, Moore RY, editors. In Suprachiasmatic Nucleus - The Mind's Clock. Oxford: Oxford University Press; 1991. pp. 51–76. [Google Scholar]
- Shigeyoshi Y, et al. Light-induced resetting of a mammalian circadian clock is associated with rapid induction of the mPer1 transcript. Cell. 1997;91:1043–1053. doi: 10.1016/s0092-8674(00)80494-8. [DOI] [PubMed] [Google Scholar]
- Albrecht U, Sun ZS, Eichele G, Lee CC. A differential response of two putative mammalian circadian regulators, mper1 and mper2, to light. Cell. 1997;91:1055–1064. doi: 10.1016/s0092-8674(00)80495-x. [DOI] [PubMed] [Google Scholar]
- Yan L, Silver R. Differential induction and localization of mPer1 and mPer2 during advancing and delaying phase shifts. Eur. J. Neurosci. 2002;16:1531–1540. doi: 10.1046/j.1460-9568.2002.02224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowrey PL, Takahashi JS. Genetics of the mammalian circadian system: Photic entrainment, circadian pacemaker mechanisms, and posttranslational regulation. Annu. Rev. Genet. 2000;34:533–562. doi: 10.1146/annurev.genet.34.1.533. [DOI] [PubMed] [Google Scholar]
- Ding JM, et al. Resetting the biological clock: Mediation of nocturnal circadian shifts by glutamate and NO. Science. 1994;266:1713–1717. doi: 10.1126/science.7527589. [DOI] [PubMed] [Google Scholar]
- Ding JM, Fairman LE, Hurst WJ, Kuriashkina LR, Gillette MU. Resetting the biological clock: mediation of nocturnal CREB phosphorylation via light, glutamate, and nitric oxide. J. Neurosci. 1997;17:667–675. doi: 10.1523/JNEUROSCI.17-02-00667.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding JM, et al. A neuronal ryanodine receptor mediates light-induced phase delays of the circadian clock. Nature. 1998;394:381–384. doi: 10.1038/28639. [DOI] [PubMed] [Google Scholar]
- Bollmann JH, Helmchen F, Borst JG, Sakmann B. Postsynaptic Ca2+ influx mediated by three different pathways during synaptic transmission at a calyx-type synapse. J. Neurosci. 1998;18:10409–10419. doi: 10.1523/JNEUROSCI.18-24-10409.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin RP, Allen CN. Calcium Response to Retinohypothalamic Tract Synaptic Transmission in Suprachiasmatic Nucleus Neurons. J. Neurosci. 2007;27:11748–11757. doi: 10.1523/JNEUROSCI.1840-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smetters D, Majewska A, Yuste R. Detecting action potentials in neuronal populations with calcium imaging. Methods. 1999;18:215–221. doi: 10.1006/meth.1999.0774. [DOI] [PubMed] [Google Scholar]
- Spruston N, Johnston D. Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J. Neurophysiol. 1992;67:508–528. doi: 10.1152/jn.1992.67.3.508. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. J. Gen. Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single-channel recording. Vol. 2. New York: Plenum Press; 1995. pp. 31–52. [Google Scholar]
- Sarantopoulos C, McCallum JB, Kwok WM, Hogan Q. Beta-escin diminishes voltage-gated calcium current rundown in perforated patch-clamp recordings from rat primary afferent neurons. J. Neurosci. Methods. 2004;139:61–68. doi: 10.1016/j.jneumeth.2004.04.015. [DOI] [PubMed] [Google Scholar]
- Hatae J, Fujishiro N, Kawata H. Spectroscopic properties of fluorescence dye fura-2 with various divalent cations. Jap. J. Physiol. 1996;46:423–429. doi: 10.2170/jjphysiol.46.423. [DOI] [PubMed] [Google Scholar]
- Poenie M. Alteration of intracellular Fura-2 fluorescence by viscosity: a simple correction. Cell Calcium. 1990;11:85–91. doi: 10.1016/0143-4160(90)90062-y. [DOI] [PubMed] [Google Scholar]