

Abstract
In mice with temporally-induced cardiac-specific deficiency of acyl-CoA synthetase-1 (Acsl1H−/−), the heart is unable to oxidize long-chain fatty acids and relies primarily on glucose for energy. These metabolic changes result in the development of both a spontaneous cardiac hypertrophy and increased phosphorylated S6 kinase (S6K), a substrate of the mechanistic target of rapamycin, mTOR. Doppler echocardiography revealed evidence of significant diastolic dysfunction, indicated by a reduced E/A ratio and increased mean performance index, although the deceleration time and the expression of sarco/endoplasmic reticulum calcium ATPase and phospholambin showed no difference between genotypes. To determine the role of mTOR in the development of cardiac hypertrophy, we treated Acsl1H−/− mice with rapamycin. Six to eight week old Acsl1H−/− mice and their littermate controls were given i.p. tamoxifen to eliminate cardiac Acsl1, then concomitantly treated for 10 weeks with i.p. rapamycin or vehicle alone. Rapamycin completely blocked the enhanced ventricular S6K phosphorylation and cardiac hypertrophy and attenuated the expression of hypertrophy-associated fetal genes, including α-skeletal actin and B-type natriuretic peptide. mTOR activation of the related Acsl3 gene, usually associated with pathologic hypertrophy, was also attenuated in the Acsl1H−/− hearts, indicating that alternative pathways of fatty acid activation did not compensate for the loss of Acsl1. Compared to controls, Acsl1H−/− hearts exhibited an 8-fold higher uptake of 2-deoxy[1-14C]glucose and a 35% lower uptake of the fatty acid analog 2-bromo[1-14C]palmitate. These data indicate that Acsl1-deficiency causes diastolic dysfunction and that mTOR activation is linked to the development of cardiac hypertrophy in Acsl1H−/− mice.
Keywords: fatty acid oxidation, mTOR, insulin resistance, gene expression, ventricular function, substrate switching
Introduction
It has been a recurring question as to whether altered cardiomyocyte metabolism activates pathways that lead to hypertrophy. Hypertrophied and stressed hearts increase their reliance on glucose as a major fuel, and it has been hypothesized that this alteration in glucose homeostasis underlies the heart’s conversion, during pathologic hypertrophy, to a fetal pattern of gene expression [1]. However, it is unclear whether hypertrophy leads to increased glucose use or vice versa, or whether cardiac hypertrophy is an essential adaptation that is required to maintain normal heart function.
Long-chain acyl-CoA synthetase-1 (Acsl1), one of 5 independent Acsl isoforms, catalyzes the activation of long-chain fatty acids to acyl-CoAs that are directed towards mitochondrial β-oxidation [2, 3]. The Acsl1 isoform is specifically required for fatty acid oxidation in highly oxidative tissues like heart [4], skeletal muscle [4], and brown adipose [5]. Normally, β-oxidation provides 60–90% of the ATP required for myocardial contraction [6–8], but in hearts from temporally-induced, multi-tissue Acsl1 deficient mice (Acsl1T−/−) or from mice with a cardiomyocyte-specific Acsl1 deficiency (Acsl1H−/−), fatty acid oxidation in ventricles is 90% lower than in littermate controls, and the uptake of [14C]glucose into the heart increases 8-fold [4]. Although hypertension is not present, both Acsl1T−/− and Acsl1H−/− cardiomyocytes become hypertrophied within 10 weeks of the temporal initiation of the knockout [4]. In these mouse models, the altered fuel use is primary, rather than the consequence of hypertension or hypertrophy.
Left ventricular hypertrophy (LVH) results from a variety of stresses, including ischemia, infarction, valvular insufficiency, diabetes, and hypertension [9]. As a response to systemic hypertension, LVH is considered to be maladaptive and pathologic because it is a stronger predictor of morbidity and mortality than is hypertension itself [10], and hypertrophy may ultimately result in heart failure and sudden death [9]. Pathologic hypertrophy is believed to result from events that involve biomechanical deformation signals, activation of G-protein-coupled receptors and their intracellular signaling pathways, and activation of the PI3K-Akt pathway initiated by insulin and other growth signals [9]. The growth-promoting mechanistic target of rapamycin (mTOR) kinase may be involved, because when rapamycin is used to inhibit mTOR in several mouse models, myocardial dysfunction improves [11–13], and the hypertrophy associated with excess thyroid hormone, constitutively active Akt, pressure overload, or over-expressed focal adhesion kinase, diminishes [12, 14–16]. In contrast, in a mouse model with cardiac-specific deficiency of mTOR’s partner Raptor (Regulatory-associated protein of mTOR), a dilated cardiomyopathy ensues, with death occurring within 6 weeks [17]. When cardiac- specific Raptor−/− hearts are challenged with pressure overload-induced hypertrophy, dilated cardiomyopathy ensues within 1 week, adaptive cardiomyocyte growth does not occur, and apoptosis and autophagy are enhanced [17]. Compared to isolated working wildtype hearts, palmitate oxidation by the Raptor knockout hearts decreased 51% and glucose oxidation increased 24%. These changes in fuel use were attributed to declines in PPARα and PGC1α expression, paralleling decreases in their target genes, including CPT1β, MCD-1, and SCOT, and a concomitant 2.2-fold higher expression of GLUT1 [17].
We previously showed in hearts lacking Acsl1 that mTOR complex-1 (mTORC1) is activated, as indicated by a 5-fold increase in phosphorylation of the mTORC1 target, p70 S6 kinase (S6K) [4], and we hypothesized that the inability to activate long-chain fatty acids for mitochondrial oxidation initiates metabolic shifts that activate mTOR. In Acsl1-deficient hearts, these shifts result in multiple inputs that signal the enhanced availability of glucose and other nutrients, demonstrated by the 8-fold increase in glucose flux and increased glucose metabolism and the higher ventricle content of amino acids. Although both Acsl1-deficient models have activated mTORC1, only the multi-tissue Acsl1T−/− mice have diminished phosphorylation of AMP-activated kinase (AMPK) [4], an inhibitor of mTORC1 signaling [18]. Cardiac hypertrophy was judged to be pathologic in both Acsl1T−/− and Acsl1H−/− hearts because of the upregulation of the fetal genes, α-skeletal actin, brain natriuretic peptide, and Acsl3. We hypothesized that the cardiomyocyte hypertrophy that develops in Acsl1-deficient hearts results from the promotion of cardiac remodeling and hypertrophy by mTORC1. We treated Acsl1H−/− mice and their littermate controls with rapamycin to determine whether hypertrophy in this metabolic model resulted from mTOR activation, and, if so, whether the hypertrophy was required to maintain normal systolic function when Acsl1 deficiency impaired the use of fatty acids for energy production.
Materials and Methods
Animal Treatment
Mouse protocols were approved by the University of North Carolina Institutional Animal Care and Use Committee and conformed to the NIH Guide for the Care and Use of Laboratory Animals. Mice were housed in a pathogen-free barrier facility (12 h light/dark cycle) with free access to water and food (Prolab RMH 3000 SP76 chow). Mice with Loxp sequences inserted on either side of exon 2 in the Acsl1 gene were backcrossed to the C57Bl/6J strain six times and then interbred with mice in which Cre expression is driven by an α-myosin heavy chain promoter that is induced by tamoxifen (B6.Cg-Tg(Myh6-cre/Esr1)1Jmk/J, Jackson Labs) to generate tamoxifen-inducible, heart-specific Acsl1H−/− knockout mice. Six to 8 week old Acsl1H−/− and littermate Acsl1flox/flox control male mice were injected i.p. with tamoxifen (Sigma, St. Louis, MO) (75 mg/kg BW), dissolved in corn oil (20 mg/ml) for 4 consecutive days (3 mg/40 g body weight). Starting on the next day, subgroups of mice were injected daily i.p. for either 2 weeks or 10 weeks with rapamycin (1 mg/kg in PBS/8% ethanol, 10% Tween 20/10% PEG-400) or with the vehicle alone. Ten weeks after tamoxifen induction, the mice were anesthetized with avertin and heart ventricles were removed and snap frozen in liquid nitrogen. To isolate total membranes, hearts were homogenized with 10 up-and-down strokes, using a motor-driven Teflon pestle and glass mortar in ice-cold buffer (10 mM Tris [pH 7.4], 1mM EDTA, 0.25 M sucrose, 1 mM dithiothreitol). Homogenates were centrifuged at 100,000 x g for 1 h at 4°C. The membrane pellet was then resuspended in buffer. Protein content was determined by the BCA assay (Pierce, Rockford, IL) with BSA as the standard. Plasma was collected from mice in 10% 0.5 M EDTA. Glucose tolerance tests were performed by i.p. injection with D-glucose (2.5 g/kg body wt), and tail blood glucose was measured at baseline, 15, 30, 60, and 120 min using a One Touch Ultra glucometer (Lifescan, Inc., Milpitas, CA).
Echocardiography and Doppler Analysis
Cardiac echocardiography was performed (blinded to mouse type and treatment) on conscious mice at the indicated time points using a VisualSonics Vevo 770 or Vevo 2100 ultrasound biomicroscopy system (VisualSonics, Inc., Toronto, Ontario, Canada). A model 707B (30 MHz) or model MS-550D (22–55 MHz) scan head was used on the Vevo 770 and Vevo 2100, respectively, as previously described [19]. Two dimensional guided M-mode echocardiography was performed in the parasternal long-axis view at the level of the papillary muscle on loosely restrained conscious mice. Wall thickness was then determined by measurements of epicardial to endocardial leading edges. Doppler analysis of the mitral valve to determine the mitral inflow velocity was performed on lightly anesthetized mice (2% (vol/vol) isoflurane/100% oxygen) as previously described [4, 20, 21]. Mitral valve flow Doppler was acquired by positioning the transducer angled cranially in a supine mouse at 45 degrees in an epigastric position to achieve an apical four-chamber view. Peak E and A heights were determined on mitral valve sequential waveforms in at least 5 waveforms. The mean performance index (ICT+IRT/ET) is calculated as the isovolumetric contraction time (ICT) plus the isovolumetric relaxation time (IRT) divided by the ejection time [22, 23]. E-wave deceleration time (DT) was also measured. These measures were determined by Doppler waveform analyses of the mitral valve (as described above) and aortic valve along with EKG measurements to ensure the correct waveform times. The mean performance index (MPI; (ICT + ET+IRT)−ET/ET) can determine if either systolic or diastolic dysfunction is present [24, 25] and has been successfully used in mice [26]. M-mode and Doppler measurement data represent 3–6 averaged cardiac cycles from at least 2 scans per mouse.
Acsl assay
Acsl initial rates were measured with 50 μM [1-14C]palmitic acid (Perkin Elmer, Waltham, MA), 10 mM ATP, and 0.25 mM CoA in total membrane fractions from ventricles (2–6 μg protein) [27]. This assay measures the total ATP-mediated activation of long-chain fatty acids by long-chain Acsl isoforms plus very-long-chain Acsl isoforms (ACSVL, also called FATP). No Acsl activity was measurable in the soluble (cytosolic) fraction.
RT-PCR
Total RNA was isolated from heart ventricles (RNeasy Fibrous Tissues Kit, Qiagen, Alameda, CA). cDNA was amplified by real-time PCR using SYBR Green (Applied Biosystems, Foster City, CA) detection with specific primers for the gene of interest and normalized to Gapdh or Tubulin and expressed as arbitrary units of 2−ΔCT relative to the control group (Table 2).
Table 2.
Quantitative Real Time PCR Primers
| Gene | Forward | Reverse |
|---|---|---|
| Acsl1 | GGTACCACTGATGGTATTCG | CATGGTTCTGACATCGTCGT |
| Acsl3 | AACCTGTCAGTTCCAAACCG | GCCAATTATAGTGCCCCAGA |
| Acsl4 | TGCCAAAAGTGACCAGTCCTATG | TGTTACCAAACCAGTCTCGGGG |
| Acsl5 | TCCCAGCCCACCTCTGATGATGTG | ACAAACTGTCCCGCCGAATG |
| Acsl6 | AAAGGCTAAGAGACCGGAGC | GTCAGGAAAGGCTCAGTTGC |
| Anf | CCATATTGGAGCAAATCCTGTG | CGGCATCTTCTCCTCCAGGT |
| α-Ska | TTGACGTGTACATAGATTGACTCGTTT | TGGCTGGCTTTAATGCTTCA |
| Asns | GCGGCCTCCAACCGGTCTTGTC | GCTACAGGCGGACTGCAGGCA |
| Bnp | GGGAGAACACGGCATCATTG | ACAGCACCTTCAGGAGATCCA |
| Glut1 | CAATGAAGTTTGAGGTCCAGTTGG | TGTGGTGTCGCTGTTTGTTGTAG |
| Glut4 | AGCTCGTTCTACTAAGAGCAC | TCGTCATTGGCATTCTGGTTG |
| Gapdh | GGTGCTGAGTATGTCGTGGA | ACTGTGGTCATGAGCCCTTC |
| PLN | ATGACGACGATTCAAATCTCTTGG | CATGGGTTTGCAAAGTTAGGCATAA |
| SERCA1/2 | GACGAGTTTGGGGAGCAGCT | CAGGTGGTGATGACAGCAGG |
| Tubulin | ACCGAAGCTGAGAGCAACAT | CACCATTTACCCCCAATGAG |
Abbreviations: Acsl, acyl-CoA synthetase; Anf, atrial natriuretic factor; α-Ska, α-skeletal actin; Asns, asparagine synthetase; Bnp, B-type natriuretic peptide; Gapdh, glyceraldehyde 3-phosphate dehydrogenase; Glut, glucose transporter; PLN, phospholambin; SERCA, sarco/endoplasmic reticulum calcium ATPase
Immunoblots
Total protein lysates were isolated in lysis buffer (20 mM Tris base, 1% Triton-X-100, 50 mM NaCl, 250 mM sucrose, 50 mM NaF, 5 mM Na2P2O7, plus protease inhibitor (#11836153 Roche, Florence, SC). Equal amounts of protein (40–60 μg) were loaded and resolved on 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes. Blots were probed with antibodies against P-p70 S6K (Thr389) and were then stripped and reprobed with p70 S6K.
2-Deoxyglucose and 2-bromopalmitate uptake
Anesthetized mice were injected retro-orbitally with 5 μCi 2-deoxy[1-14C]glucose in saline or 1.5 μCi 2-bromo-[1-14C]palmitate complexed to BSA in PBS (Moravek Biochemicals, Brea, CA) Tissues were harvested and flash frozen in liquid N2 30 min after injection. Radioactivity was measured in tissue homogenates by scintillation and normalized to the number of DPM present in 10 μl of serum obtained 5 min after injection ((dpm/mg tissue)/dpm in 10 μl serum).
Statistics
Data are presented as mean ± SE for each treatment group. Differences between treatment groups were evaluated by one-way ANOVA with Tukey multiple-comparison posttests. All statistical analyses were performed using GraphPad Prism (version 5.0; GraphPad Software, San Diego CA). Differences between means with p <0.05 were considered statistically significant.
Results
Rapamycin inhibited Acsl1H−/− -induced spontaneous cardiac hypertrophy
Compared to vehicle-treated controls, 10 weeks after tamoxifen induction, Acsl1H−/− deficient mice developed a spontaneous cardiac hypertrophy (Table 1) [4]. Hypertrophy was confirmed by echocardiographic analysis in which both anterior and posterior wall thickness indices (IVS, LVPW) and the LV mass were significantly increased compared to sibling controls (Table 1). Because our previous studies identified activated mTOR and its downstream target, phosphorylated p70 S6 kinase, we treated mice with rapamycin to inhibit mTOR in order to determine directly whether mTOR links Acsl1 deficiency with the observed cardiac hypertrophy.
Table 1.
Conscious echocardiographic analysis of Acsl1H−/− hearts 10 weeks after tamoxifen (or control) induction.
| Control n=4 |
Acsl1H−/− n=8 |
Control n=5 |
Acsl1H−/− n=9 |
|
|---|---|---|---|---|
| Vehicle | Rapamycin | |||
| Heart Rate (bpm) | 660.0 ± 33.8 | 650.1 ± 18.2 | 675.8 ± 28.9 | 686.1 ± 22.7 |
| IVS,d (mm) | 1.13 ± 0.05 | 1.32 ± 0.08* | 1.00 ± 0.05 | 1.11 ± 0.05 |
| LVID,d (mm) | 2.69 ± 0.13 | 2.70 ± 0.13 | 2.83 ± 0.11 | 2.60 ± 0.06 |
| LVPW,d (mm) | 1.04 ± 0.05 | 1.18 ± 0.06* | 0.94 ± 0.05 | 1.04 ± 0.05 |
| IVS,s (mm) | 1.71 ± 0.07 | 1.94 ± 0.06* | 1.62 ± 0.13 | 1.70 ± 0.05 |
| LVID,s (mm) | 1.37 ± 0.15 | 1.27 ± 0.11 | 1.32 ± 0.12 | 1.16 ± 0.07 |
| LVPW,s (mm) | 1.50 ± 0.06 | 1.85 ± 0.07* | 1.50 ± 0.09 | 1.56 ± 0.04 |
| LV Vol;d (μl) | 27.1 ± 3.4 | 27.8 ± 3.2 | 30.6 ± 2.7 | 24.9 ± 1.4 |
| LV Vol;s (μl) | 5.2 ± 1.4 | 4.4 ± 1.0 | 4.6 ±1.1 | 3.3 ± 0.5 |
| FS (%) | 49.1 ± 4.4 | 53.1 ± 2.9 | 53.6 ± 3.0 | 55.4 ± 1.7 |
| EF (%) | 81.4 ± 4.0 | 84.8 ± 2.6 | 85.4 ± 2.8 | 87.2 ± 1.3 |
| LV Mass (mg) | 99.9 ± 0.8 | 128.3 ± 7.5* | 90.6 ± 5.9 | 95.2 ± 6.2 |
Differences between treatment groups were evaluated by one-way ANOVA, followed by an all pairwise Holm-Sidak multiple comparison procedure; *p<≤0.05 versus all other groups.
IVS, interventricular septum; LVID, left ventricular inner diameter; LVPW, left ventricular posterior wall; LV Vol, left ventricular volume; % EF, ejection fraction; fractional shortening; LV, left ventricular; d, diastole; s, systole. Data represent mean ± SEM;
p<0.05.
When Acsl1H−/− deficient mice induced with tamoxifen were treated with rapamycin, the spontaneous hypertrophy observed in Acsl1H−/− deficient mice was inhibited, as evidenced by wall thickness indices and LV mass measurements that did not differ from littermate controls (Table 1), as well as the inhibition of increases in heart weight (Fig. 1). Although Acsl1H−/− deficient mice did not exhibit systolic dysfunction (Table 1), we asked whether they had evidence of diastolic dysfunction in vivo. Doppler analysis of blood flow through the mitral and aortic valves was performed in control and Acsl1H−/− mice with and without treatment with rapamycin, to determine the E/A ratio and the mean performance index as indicators of diastolic and diastolic/systolic dysfunction, respectively (Fig. 1, Table 3). At 2 weeks after tamoxifen treatment, when total Acsl activity has decreased by 50% [4], the diastolic function was normal in both vehicle-treated Acsl1H−/− mice. However, because rapamycin itself caused mild diastolic dysfunction in both control and knockout animals at 2 weeks (data not shown), these latter results cannot be interpreted. By 10 weeks, however, vehicle-treated Acsl1H−/− mice exhibited a nearly 50% decline in diastolic function and a parallel increase of >50% in mean performance index (Fig. 2A–C), consistent with a defect in diastolic function. The mitral deceleration time did not change (Table 3), and the mRNA expression of SERCA (sarco/endoplasmic reticulum calcium ATPase) and its phospholambin regulator (PLN), which are frequently, but not invariably [28, 29], diminished with diastolic dysfunction, were not altered in Acsl1H−/− mice before or after rapamycin treatment (SERCA1/2: vehicle control, 1.11±0.24; rapamycin control, 1.41±0.35; vehicle Acsl1H−/−, 0.83±0.09; rapamycin Acsl1H−/−, 1.16±0.15; PLN: vehicle control, 1.11±0.0.21; rapamycin control, 1.33±0.25; vehicle Acsl1H−/−, 0.85±0.09; rapamycin Acsl1H−/−, 1.23±0.14; n=5–6). Because the 10 week rapamycin treatment itself induced diastolic dysfunction in both control and Acsl1 deficient mice, as previously described [30–32], it was not possible to interpret the effects of Acsl1-related mTOR activity on diastolic function.
Figure 1.
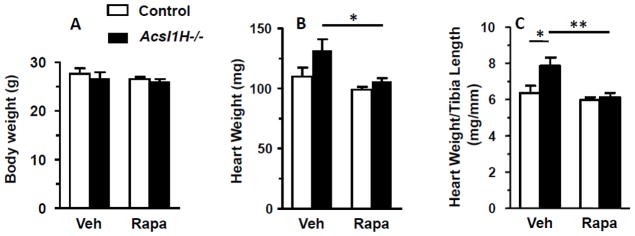
Rapamycin treatment diminished cardiac hypertrophy. A) Body weight of control and Acsl1H−/− mice. B) Absolute heart weight and C) relative heart weight (heart weight/tibia length) in control and Acsl1H−/− mice treated with vehicle (Veh) or rapamycin (Rapa). The values are means plus SEMs (error bars). *, p < 0.05 compared with littermate controls. n = 6–10 per group.
Table 3.
Doppler echocardiographic analysis of Acsl1H−/− hearts 10 weeks after tamoxifen or vehicle (control) induction.
| Control n=7 |
Acsl1H−/− n=7 (n=6 for DT) |
|
|---|---|---|
| Vehicle | ||
| ET (ms) | 51.0 ± 2.9 | 45.1 ± 1.8 |
| IRT (ms) | 16.1 ± 1.8 | 16.1 ± 1.8 |
| ICT+ET+IRT (ms) | 60.4 ± 2.4 | 65.8 ± 5.4 |
| MPI | 0.20 ± 0.06 | 0.48 ± 0.10* |
| E (mm/s) | 683.3 ± 22.2 | 564.6 ± 41.6* |
| A (mm/s) | 374.3 ± 35.7 | 553.0 ± 37.0* |
| E/A Ratio | 1.92 ± 0.18 | 1.04 ± 0.08* |
| DT (ms) | 19.2 ± 3.7 | 18.8 ± 4.0 |
E (early) and A (atrial) ventricular filling velocity; ET, ejection time; IRT, isovolumetric relaxation time; ICT, isovolumetric contraction time; MPI, mean performance index =(ICT+ET+IRT)−ET/ET); DT, mitral deceleration time. Data represent mean ± SEM;
p<0.05 (T-test).
Figure 2.
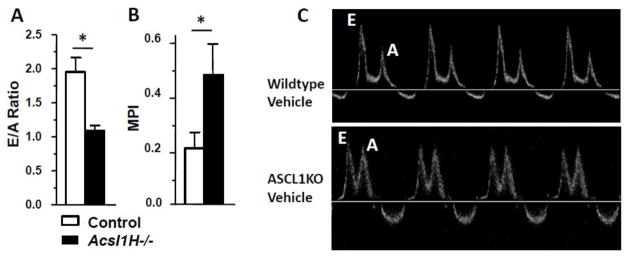
Acsl1H−/− mice exhibited diastolic dysfunction. A) E/A ratio (ratio between early (E) and late (atrial - A) ventricular filling velocity) (n = 4–6 per group); and B) MPI (mean performance index) (n = 4–6 per group) in control and Acsl1H−/− mice. C) Representative mitral valve Doppler analysis (n = 8–9 per group). The values are means plus SEMs (error bars). *, p < 0.05 compared with littermate controls.
Pathologic cardiac hypertrophy is usually accompanied by upregulation of genes that are normally expressed in fetal hearts [33], and we previously reported increases in the mRNAs for α-Ska and Bnp in heart from the multi-tissue knockout of Acsl1 [4]. Similar to its effect in Acsl1H−/− ventricles on the fetally-expressed gene Acsl3, the rapamycin-mediated block of hypertrophy also diminished the expression of α-Ska and Bnp, and it normalized the lowered expression of the ventricle pathogenic marker, atrial natriuretic factor (Anf) (Fig. 3).
Figure 3.
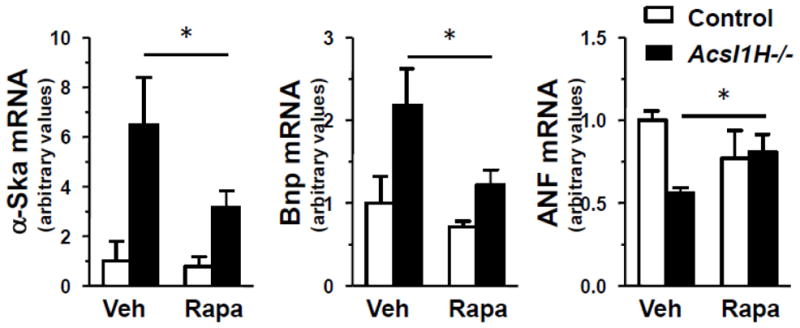
Rapamycin treatment of Acsl1H−/− mice attenuated the expression of pathologic hypertrophy markers. mRNA abundance of fetal gene markers, α-skeletal actin (α-Ska), B-type natriuretic peptide (Bnp), and atrial natriuretic factor (ANF) in ventricles from control and Acsl1H−/− mice treated with vehicle (Veh) or rapamycin (Rapa). The values are means plus SEMs (error bars). Values that are significantly different (p <0.05) from the values for the control are indicated by an asterisk. n = 3 per group.
Rapamycin reversed the Acsl1 deficiency-induced phosphorylation of p70 S6 kinase in Acsl1H−/− mice
Compared to their littermate controls, the absence of Acsl1 resulted in the activation of mTOR in cardiac ventricles, confirmed by the markedly enhanced phosphorylation of p70 S6 kinase (Fig. 4A). Treatment with rapamycin for 10 wk completely blocked this enhanced S6K phosphorylation. By 10 weeks after inducing the MHC-mediated cre recombinase with tamoxifen, Acsl1 mRNA was virtually absent in both vehicle-treated and rapamycin-treated ventricles (Fig. 4B). Other Acsl isoforms contributed only 10% of total Acsl activity in the heart (Fig. 5A) and did not compensate functionally for the loss of Acsl1. The expression of Acsl4, 5, and 6 did not change (Fig. 4B), but Acsl3 mRNA, the major isoform expressed in fetal heart [34], increased 2.5-fold, probably because pathologic hypertrophy causes fetal gene expression to increase [1, 35]. Treatment with rapamycin normalized the increase in Acsl3 expression, but had no effect on other Acsl isoforms and did not alter the amount of residual Acsl activity (Fig. 5A).
Figure 4.

Rapamycin reversed the upregulated phosphorylation of p70 S6 kinase in Acsl1H−/− mice and diminished the expression of Acsl3 mRNA. A) Representative immunoblot from male mice were treated with vehicle (Veh) or rapamycin (1 mg/kg) daily i.p. (Rapa) from 1 wk after tamoxifen treatment until sacrifice 10 wk after the tamoxifen treatment. Total (p70 S6K) and phosphorylated p70 S6K (P-p70 S6K) at Thr389 in ventricles from control and Acsl1H−/− mice. n=4. B) Acsl isoenzyme mRNA abundance in ventricles from control and Acsl1H−/− mice treated with vehicle or rapamycin. n = 3–6. The values are means plus SEMs (error bars). *, p<.05.
Figure 5.

Rapamycin treatment had no effect on Acsl activity or the altered fuel uptake, but caused glucose intolerance. A) Acsl specific activity in ventricles from control and Acsl1H−/− mice treated with vehicle (Veh) or rapamycin (Rapa). B) Uptake of 2-bromo[1-14C]palmitate into the ventricles of control and Acsl1H−/− mice. C) 2-Deoxy[1-14C]glucose uptake into the ventricles of control and Acsl1H−/− mice treated with vehicle or rapamycin. n = 3–5 per group. D) mRNA abundance of Glut1 and Glut4 in ventricles from control and Acsl1H−/− mice treated with vehicle (Veh) or rapamycin (Rapa). The values show means plus SEMs (error bars); *, p<.005 compared with littermate controls and #, p<.03 compared with vehicle control. n = 5–6 per group. E) Plasma glucose at intervals after i.p. glucose (2.5 g/kg body wt) in control and Acsl1H−/− mice treated with vehicle or rapamycin. n = 5 per group.
Rapamycin treatment had no effect on Acsl activity or the altered fuel uptake
In the heart, activation of long-chain fatty acids by Acsl1 is required for their subsequent metabolism by mitochondrial β-oxidation [4]. Compared to controls, Acsl specific activity with the long-chain fatty acid palmitate was 90% lower in both vehicle- and rapamycin-treated ventricles (Fig. 5A), and the uptake of the non-metabolizable fatty acid analog 2-bromo-[14C]palmitate was markedly lower (Fig. 5B). Consistent with the diminished ability of the Acsl1H−/− ventricles to use fatty acid as its primary fuel, ventricle uptake of the non-metabolizable glucose analog 2-deoxy-[14C]glucose was 7.9-fold higher in the Acsl1H−/− ventricles; this enhanced uptake was unchanged by rapamycin treatment (Fig. 5C). Surprisingly, despite the marked increase in the uptake of 2-deoxy-[14C]glucose, the expression of both Glut1 and Glut4 mRNA was lower in Acsl1H−/− mice than in controls, showing a discrepancy between glucose transport and transporter mRNAs (Fig. 5D). Rapamycin treatment had no effect on mouse weight (Fig. 1A), but the drug abrogated the hypertrophy that develops in hearts that lack Acsl1 [4] (Fig. 1B, C), even though the Acsl1H−/− hearts continued to remain highly dependent on glucose. In the control Acsl1flox/flox hearts, however, rapamycin treatment increased the uptake of 2-deoxy[14C]glucose (Fig. 5C), likely because long-term treatment with rapamycin inhibits mTORC2-mediated phosphorylation of Akt, thereby inducing glucose intolerance, as shown by glucose tolerance tests performed in vehicle- and rapamycin-treated mice (Fig. 5E) [36].
Discussion
The 90% decrease in total Acsl activity in both Acsl1T−/− and Acsl1H−/− heart ventricles [4] shows that Acsl1 is the major Acsl isoform in heart. Acsl1, a 78 kDa protein with a predicted N-terminal transmembrane domain, is located on several intracellular organelles, including the outer mitochondrial membrane where, in heart and adipose tissue, it appears to direct long-chain fatty acids into the mitochondria for β-oxidation. When Acsl1 is missing in white or brown adipose tissue [5] or in cardiomyocytes [4], the oxidation of palmitate (and of oleate or linoleate; data not shown) is reduced by more than 80%. Consequently, the deficient tissues must rely on glycolysis and the oxidation of glucose to provide fuel for normal function. In beating hearts, the inability to use fatty acids results in a compensatory 8-fold increase in glucose uptake, and the subsequent enhancement in glucose flux and metabolism upregulates mTORC1 [4], a complex in which mTOR kinase associates with several partners including Raptor and Pras40, to phosphorylate p70 S6K, thereby facilitating the translational control of protein synthesis [37]. The upregulation of mTOR in Acsl1H−/− hearts that depend on glucose as their major energy substrate is reminiscent of the activation of mTOR in isolated rat hearts stimulated to contract in the presence of a high glucose load [38], in the contractile impairment caused by cardiac over-expression of GLUT1 that increases glucose uptake in the context of a high fat diet [39], and in CD36-null mouse hearts which have diastolic dysfunction [40]. Pathologic hypertrophy is generally accompanied by the upregulation in ventricles of fetal genes like α-Ska and Bnp [1, 41], as observed in Acsl1T−/− [4] and Acsl1H−/− ventricles.
mTORC1 is activated by nutrients that signal the availability of sufficient substrate and energy for growth. Thus, mTOR activity is increased by elevated plasma insulin [42] and by tissue branched chain amino acids, particularly leucine [43]. mTOR is also activated by hypertension [44]. None of these features were present in Acsl1-deficient mice [4], but, other positive nutrient signals were prominent, including an increase in general amino acid availability and enhanced glycolytic flux [45]. Insulin-dependent activation of mTOR requires glucose phosphorylation [45], and mTOR is active when activated cellular AMPK is low, but unlike the 3-fold higher ventricle content of glucose-6-phosphate and the low AMPK activity in the multi-tissue knockout of Acsl1 (Acsl1T−/−) [4], the Acsl1H−/− heart-specific model had unaltered glucose-6-phosphate content and a phosphoAMPK/total AMPK ratio that was increased relative to that of controls (data not shown). Despite the increased uptake of 2-deoxy-[14C]glucose, mRNA expression of Glut1 and Glut4 were actually lower in Acsl1H−/− hearts. Glut1 and Glut4 are among the most abundant Glut transcripts in heart, but their expression varies in mouse cardiac dysfunction models [46], and both isoforms are downregulated 80% in failing human hearts [47]. These decreases are thought to be a manifestation of the heart reversion to a fetal metabolic profile, which is also supported by the increases in α-Ska and Bnp expression in the Acsl1H−/− hearts.
mTORC1 is highly sensitive to rapamycin, and chronic treatment with rapamycin also disturbs mTORC2 signaling by blocking the association of mTOR with its partner Rictor (Raptor-independent companion of mTOR) [48]. Although upregulating mTORC1 diminishes insulin-induced phosphorylation of IRS-1(Ser636/Ser639) and treatment with rapamycin acutely reverses this phosphorylation [49], chronic rapamycin treatment disrupts mTORC2 and suppresses its ability to phosphorylate Akt at S473, thereby lessening insulin signaling [36]. In addition to disrupting insulin signaling in peripheral tissues, rapamycin is toxic for β-cells and diminishes insulin secretion [50]. As previously reported in mice and humans [51], treatment with rapamycin for 10 weeks caused marked glucose intolerance to develop in the Acsl1H−/− mice. Thus, in the present study, glucose intolerance rapidly resulted from inhibition of both mTORC1 and mTORC2.
Based on Doppler echocardiography, the Acsl1H−/− mice had a significant decrease in diastolic function. However, the normal deceleration time suggests that this represents an early stage of diminished function [52], and the normal expression of SERCA and phospholambin suggest that calcium loading is not likely to be responsible [29]. Interestingly, although rapamycin inhibited the increased cardiac growth in the Acsl1H−/− mice, it did not appear to relieve the diastolic dysfunction. This finding was confounded by the fact that rapamycin treatment of control mice for as little as 2 weeks induced its own mild diastolic dysfunction. However, because Acsl1H−/− hearts were not affected to a greater extent than control mice, these data suggest that the cardiac hypertrophy is driven by the ability of the increased glucose flux to activate mTORC1, independent of the mechanisms by which Acsl1H−/− diastolic dysfunction occurs. Rapamycin analogs are given as immunosuppressants after organ transplantation, but rapamycin-related abnormalities in cardiac diastolic function have not been observed [53–55]. In fact, in humans with age-induced activated mTOR, caloric restriction diminishes diastolic dysfunction [56], and in cardiac transplantation patients, sirolimus treatment is associated with improved diastolic function [57].
The dependence on glucose of Acsl1-deficient hearts allowed us to re-examine the problem of fuel metabolism and hypertrophy. Although the adult heart normally relies on long chain fatty acids for 60–90% of its fuel, it is believed that the heart functions best when it retains metabolic flexibility and can switch readily between the use of fatty acid and glucose substrates [58]. Numerous studies have reported preferential cardiac use of fatty acid during fasting and diabetes [59, 60], whereas use of glucose is higher with hypoxia or failure [61–63]. Despite cardiac hypertrophy in Acsl1-deficient hearts, systolic function remained normal, and when transverse arch banding was performed, control and Acsl1T−/− mice responded similarly to the increased pressure overload; heart weight doubled and fractional shortening decreased to ~40% in hearts from both genotypes [4]. The, elevations in the expression of fetal marker genes normally associated with pathologic hypertrophy, α-Ska, Bnp, and Acsl3, all decreased with rapamycin treatment, suggesting that their increased expression had resulted directly from the activation of mTORC1, and not from an underlying cardiomyocyte problem related to fuel use or energy production. Although Anf expression increases when hypertrophy develops after transverse aortic constriction [64], Anf expression was diminished in Acsl1H−/− ventricles but was normalized by rapamycin treatment.
In summary, this study strongly indicates that the cardiac hypertrophy observed in Acsl1H−/− mice occurs secondary to the mTORC1 activation that results from enhanced glycolytic flux, although mTORC2 might also be playing a role. Systolic function in Acsl1H−/− mice remains normal in older animals [4], and ventricular hypertrophy was not required to support normal systolic function or to prevent worsening of diastolic dysfunction. Although these data suggest the possibility that abrogation of cardiac hypertrophy might be useful, the detrimental effects of rapamycin on glucose metabolism warrant additional investigation.
Highlights.
Mice with heart-specific acyl-CoA synthetase-1 deficiency cannot oxidize fatty acids
Acsl1H−/− hearts depend primarily on glucose for energy
Echocardiography showed that Acsl1H−/− hearts have significant diastolic dysfunction
Rapamycin treatment showed that activated mTORC1 causes cardiac hypertrophy
Cardiac hypertrophy was not required to maintain normal systolic function
Acknowledgments
Grants. This work was supported by grants from the National Institutes of Health [DK59935 and DK59935S1 to RAC, DK056350 for the UNC Nutrition Obesity Research Center, HL104129 to MSW, and T32 HL069768 to TJG], from the American Heart Association [12GRNT12030144 to RAC], and from the Leducq Foundation (to MSW).
Abbreviations
- Acsl
long-chain acyl-CoA synthetase
- FA
fatty acid
- mTOR
mechanistic target of rapamycin
- mTORC1
mTOR complex-1
- MPI
mean performance index
Footnotes
Authors contributions. DSP, TJG, FP, JME, and MSW performed experiments; DSP, TJG, and MSW analyzed data and prepared figures; DSP, MSW, and RAC conceived and designed research and drafted the manuscript.
Conflict of Interest: None declared
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
David S. Paul, Email: david_paul@med.unc.edu.
Trisha J. Grevengoed, Email: tgrevengoed@gmail.com.
Florencia Pascual, Email: fpascual@live.unc.edu.
Jessica M. Ellis, Email: jessie.ellis@gmail.com.
Monte S. Willis, Email: monte_willis@med.unc.edu.
References
- 1.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci. 2010;1188:191–198. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellis JM, Frahm JL, Li LO, Coleman RA. Acyl-coenzyme A synthetases in metabolic control. Current opinion in lipidology. 2010;21:212–217. doi: 10.1097/mol.0b013e32833884bb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li LO, Klett EL, Coleman RA. Acyl-CoA synthesis, lipid metabolism and lipotoxicity. Biochim Biophys Acta. 2010;1801:246–251. doi: 10.1016/j.bbalip.2009.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS, Coleman RA. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs fatty acid oxidation and induces cardiac hypertrophy. Mol Cell Biol. 2011;31:1252–1262. doi: 10.1128/MCB.01085-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, Stevens RD, Watkins SM, Muoio DM, Coleman RA. Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab. 2010;12:53–64. doi: 10.1016/j.cmet.2010.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark H, Carling D, Saggerson D. Covalent activation of heart AMP-activated protein kinase in response to physiological concentrations of long-chain fatty acids. Eur J Biochem. 2004;271:2215–2224. doi: 10.1111/j.1432-1033.2004.04151.x. [DOI] [PubMed] [Google Scholar]
- 7.Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. 2005;85:1093–1129. doi: 10.1152/physrev.00006.2004. [DOI] [PubMed] [Google Scholar]
- 8.van der Vusse GJ, Glatz JF, Stam HC, Reneman RS. Fatty acid homeostasis in the normoxic and ischemic heart. Physiol Rev. 1992;72:881–940. doi: 10.1152/physrev.1992.72.4.881. [DOI] [PubMed] [Google Scholar]
- 9.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7:589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 10.Kurdi M, Booz GW. Three 4-letter words of hypertension-related cardiac hypertrophy: TRPC, mTOR, and HDAC. J Mol Cell Cardiol. 2011;50:964–971. doi: 10.1016/j.yjmcc.2011.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, Colucci WS, Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. 2003;107:1664–1670. doi: 10.1161/01.CIR.0000057979.36322.88. [DOI] [PubMed] [Google Scholar]
- 13.Aoyagi T, Kusakari Y, Xiao CY, Inouye BT, Takahashi M, Scherrer-Crosbie M, Rosenzweig A, Hara K, Matsui T. Cardiac mTOR protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2012;303:H75–85. doi: 10.1152/ajpheart.00241.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shioi T, McMullen JR, Kang PM, Douglas PS, Obata T, Franke TF, Cantley LC, Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kuzman JA, O’Connell TD, Gerdes AM. Rapamycin prevents thyroid hormone-induced cardiac hypertrophy. Endocrinology. 2007;148:3477–3484. doi: 10.1210/en.2007-0099. [DOI] [PubMed] [Google Scholar]
- 16.Clemente CF, Xavier-Neto J, Dalla Costa AP, Consonni SR, Antunes JE, Rocco SA, Pereira MB, Judice CC, Strauss B, Joazeiro PP, Matos-Souza JR, Franchini KG. Focal adhesion kinase governs cardiac concentric hypertrophic growth by activating the AKT and mTOR pathways. J Mol Cell Cardiol. 2012;52:493–501. doi: 10.1016/j.yjmcc.2011.10.015. [DOI] [PubMed] [Google Scholar]
- 17.Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, Krishnan J, Lerch R, Hall MN, Ruegg MA, Pedrazzini T, Brink M. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 18.Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL. TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell. 2006;126:955–968. doi: 10.1016/j.cell.2006.06.055. [DOI] [PubMed] [Google Scholar]
- 19.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits Akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of Forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jearawiriyapaisarn N, Moulton HM, Sazani P, Kole R, Willis MS. Long-term improvement in mdx cardiomyopathy after therapy with peptide-conjugated morpholino oligomers. Cardiovasc Res. 2010;85:444–453. doi: 10.1093/cvr/cvp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhou YQ, Foster FS, Parkes R, Adamson SL. Developmental changes in left and right ventricular diastolic filling patterns in mice. Am J Physiol Heart Circ Physiol. 2003;285:H1563–1575. doi: 10.1152/ajpheart.00384.2003. [DOI] [PubMed] [Google Scholar]
- 22.Tei C, Ling LH, Hodge DO, Bailey KR, Oh JK, Rodeheffer RJ, Tajik AJ, Seward JB. New index of combined systolic and diastolic myocardial performance: a simple and reproducible measure of cardiac function--a study in normals and dilated cardiomyopathy. J Cardiol. 1995;26:357–366. [PubMed] [Google Scholar]
- 23.Klein AL, Burstow DJ, Tajik AJ, Zachariah PK, Bailey KR, Seward JB. Effects of age on left ventricular dimensions and filling dynamics in 117 normal persons. Mayo Clin Proc. 1994;69:212–224. doi: 10.1016/s0025-6196(12)61059-3. [DOI] [PubMed] [Google Scholar]
- 24.Bruch C, Schmermund A, Marin D, Katz M, Bartel T, Schaar J, Erbel R. Tei-index in patients with mild-to-moderate congestive heart failure. Eur Heart J. 2000;21:1888–1895. doi: 10.1053/euhj.2000.2246. [DOI] [PubMed] [Google Scholar]
- 25.Bruch C, Schmermund A, Dagres N, Katz M, Bartel T, Erbel R. Severe aortic valve stenosis with preserved and reduced systolic left ventricular function: diagnostic usefulness of the Tei index. J Am Soc Echocardiogr. 2002;15:869–876. doi: 10.1067/mje.2002.120977. [DOI] [PubMed] [Google Scholar]
- 26.Stypmann J, Engelen MA, Troatz C, Rothenburger M, Eckardt L, Tiemann K. Echocardiographic assessment of global left ventricular function in mice. Lab Anim. 2009;43:127–137. doi: 10.1258/la.2007.06001e. [DOI] [PubMed] [Google Scholar]
- 27.Polokoff MA, Bell RM. Limited palmitoyl-CoA penetration into microsomal vesicles as evidenced by a highly latent ethanol acyltransferase activity. J Biol Chem. 1978;253:7173–7178. [PubMed] [Google Scholar]
- 28.Dally S, Corvazier E, Bredoux R, Bobe R, Enouf J. Multiple and diverse coexpression, location, and regulation of additional SERCA2 and SERCA3 isoforms in nonfailing and failing human heart. J Mol Cell Cardiol. 2010;48:633–644. doi: 10.1016/j.yjmcc.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Charles PY, Nan C, Pinto JR, Wang Y, Liang J, Wu G, Tian J, Feng HZ, Potter JD, Jin JP, Huang X. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49:402–411. doi: 10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long C, Cook LG, Hamilton SL, Wu GY, Mitchell BM. FK506 binding protein 12/12.6 depletion increases endothelial nitric oxide synthase threonine 495 phosphorylation and blood pressure. Hypertension. 2007;49:569–576. doi: 10.1161/01.HYP.0000257914.80918.72. [DOI] [PubMed] [Google Scholar]
- 31.Lindenfeld J, Miller GG, Shakar SF, Zolty R, Lowes BD, Wolfel EE, Mestroni L, Page RL, 2nd, Kobashigawa J. Drug therapy in the heart transplant recipient: part II: immunosuppressive drugs. Circulation. 2004;110:3858–3865. doi: 10.1161/01.CIR.0000150332.42276.69. [DOI] [PubMed] [Google Scholar]
- 32.Lindenfeld J, Page RL, 2nd, Zolty R, Shakar SF, Levi M, Lowes B, Wolfel EE, Miller GG. Drug therapy in the heart transplant recipient: Part III: common medical problems. Circulation. 2005;111:113–117. doi: 10.1161/01.CIR.0000151609.60618.3C. [DOI] [PubMed] [Google Scholar]
- 33.Ghatpande S, Goswami S, Mascareno E, Siddiqui MA. Signal transduction and transcriptional adaptation in embryonic heart development and during myocardial hypertrophy. Mol Cell Biochem. 1999;196:93–97. [PubMed] [Google Scholar]
- 34.de Jong H, Neal AC, Coleman RA, Lewin TM. Ontogeny of mRNA expression and activity of long-chain acyl-CoA synthetase (ACSL) isoforms in Mus musculus heart. Biochim Biophys Acta. 2007;1771:75–82. doi: 10.1016/j.bbalip.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–262. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- 36.Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science. 2012;335:1638–1643. doi: 10.1126/science.1215135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Balasubramanian S, Johnston RK, Moschella PC, Mani SK, Tuxworth WJ, Jr, Kuppuswamy D. mTOR in growth and protection of hypertrophying myocardium. Cardiovasc Hematol Agents Med Chem. 2009;7:52–63. doi: 10.2174/187152509787047603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, Taegtmeyer H. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2:e004796. doi: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation. 2009;119:2818–2828. doi: 10.1161/CIRCULATIONAHA.108.832915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irie H, Krukenkamp IB, Brinkmann JF, Gaudette GR, Saltman AE, Jou W, Glatz JF, Abumrad NA, Ibrahimi A. Myocardial recovery from ischemia is impaired in CD36-null mice and restored by myocyte CD36 expression or medium-chain fatty acids. Proc Natl Acad Sci USA. 2003;100:6819–6824. doi: 10.1073/pnas.1132094100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vikstrom KL, Bohlmeyer T, Factor SM, Leinwand LA. Hypertrophy, pathology, and molecular markers of cardiac pathogenesis. Circ Res. 1998;82:773–778. doi: 10.1161/01.res.82.7.773. [DOI] [PubMed] [Google Scholar]
- 42.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37:217–222. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–602. doi: 10.1152/ajpendo.90645.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soesanto W, Lin HY, Hu E, Lefler S, Litwin SE, Sena S, Abel ED, Symons JD, Jalili T. Mammalian target of rapamycin is a critical regulator of cardiac hypertrophy in spontaneously hypertensive rats. Hypertension. 2009;54:1321–1327. doi: 10.1161/HYPERTENSIONAHA.109.138818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma S, Guthrie PH, Chan SS, Haq S, Taegtmeyer H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc Res. 2007;76:71–80. doi: 10.1016/j.cardiores.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Aerni-Flessner L, Abi-Jaoude M, Koenig A, Payne M, Hruz PW. GLUT4, GLUT1, and GLUT8 are the dominant GLUT transcripts expressed in the murine left ventricle. Cardiovasc Diabetol. 2012;11:63. doi: 10.1186/1475-2840-11-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H. Metabolic gene expression in fetal and failing human heart. Circulation. 2001;104:2923–2931. doi: 10.1161/hc4901.100526. [DOI] [PubMed] [Google Scholar]
- 48.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 49.Khamzina L, Veilleux A, Bergeron S, Marette A. Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology. 2005;146:1473–1481. doi: 10.1210/en.2004-0921. [DOI] [PubMed] [Google Scholar]
- 50.Barlow AD, Nicholson ML, Herbert TP. Evidence for rapamycin toxicity in pancreatic beta-cells and a review of the underlying molecular mechanisms. Diabetes. 2013;62:2674–2682. doi: 10.2337/db13-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Houde VP, Brule S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59:1338–1348. doi: 10.2337/db09-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oh JK, Park SJ, Nagueh SF. Established and novel clinical applications of diastolic function assessment by echocardiography. Circ Cardiovasc Imaging. 2011;4:444–455. doi: 10.1161/CIRCIMAGING.110.961623. [DOI] [PubMed] [Google Scholar]
- 53.Augoustides JG, Riha H. Recent progress in heart failure treatment and heart transplantation. J Cardiothorac Vasc Anesth. 2009;23:738–748. doi: 10.1053/j.jvca.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Shalev A, Nir A, Granot E. Cardiac function in children post-orthotopic liver transplantation: echocardiographic parameters and biochemical markers of subclinical cardiovascular damage. Pediatr Transplant. 2005;9:718–722. doi: 10.1111/j.1399-3046.2005.00365.x. [DOI] [PubMed] [Google Scholar]
- 55.Mourer JS, Ewe SH, Mallat MJ, Ng AC, Rabelink TJ, Bax JJ, Delgado V, de Fijter JW. Late calcineurin inhibitor withdrawal prevents progressive left ventricular diastolic dysfunction in renal transplant recipients. Transplantation. 2012;94:721–728. doi: 10.1097/TP.0b013e3182603297. [DOI] [PubMed] [Google Scholar]
- 56.Shinmura K, Tamaki K, Sano M, Murata M, Yamakawa H, Ishida H, Fukuda K. Impact of long-term caloric restriction on cardiac senescence: caloric restriction ameliorates cardiac diastolic dysfunction associated with aging. J Mol Cell Cardiol. 2011;50:117–127. doi: 10.1016/j.yjmcc.2010.10.018. [DOI] [PubMed] [Google Scholar]
- 57.Raichlin E, Chandrasekaran K, Kremers WK, Frantz RP, Clavell AL, Pereira NL, Rodeheffer RJ, Daly RC, McGregor CG, Edwards BS, Kushwaha SS. Sirolimus as primary immunosuppressant reduces left ventricular mass and improves diastolic function of the cardiac allograft. Transplantation. 2008;86:1395–1400. doi: 10.1097/TP.0b013e318189049a. [DOI] [PubMed] [Google Scholar]
- 58.Kolwicz SC, Jr, Tian R. Metabolic therapy at the crossroad: how to optimize myocardial substrate utilization? Trends Cardiovasc Med. 2009;19:201–207. doi: 10.1016/j.tcm.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Belke DD, Larsen TS, Gibbs EM, Severson DL. Altered metabolism causes cardiac dysfunction in perfused hearts from diabetic (db/db) mice. Am J Physiol Endocrinol Metab. 2000;279:E1104–1113. doi: 10.1152/ajpendo.2000.279.5.E1104. [DOI] [PubMed] [Google Scholar]
- 60.Mazumder PK, O’Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–2374. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 61.Allard MF, Henning SL, Wambolt RB, Granleese SR, English DR, Lopaschuk GD. Glycogen metabolism in the aerobic hypertrophied rat heart. Circulation. 1997;96:676–682. doi: 10.1161/01.cir.96.2.676. [DOI] [PubMed] [Google Scholar]
- 62.Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. 1999;318:36–42. doi: 10.1097/00000441-199907000-00006. [DOI] [PubMed] [Google Scholar]
- 63.Tian R. Transcriptional regulation of energy substrate metabolism in normal and hypertrophied heart. Curr Hypertens Rep. 2003;5:454–458. doi: 10.1007/s11906-003-0052-7. [DOI] [PubMed] [Google Scholar]
- 64.Song X, Kusakari Y, Xiao CY, Kinsella SD, Rosenberg MA, Scherrer-Crosbie M, Hara K, Rosenzweig A, Matsui T. mTOR attenuates the inflammatory response in cardiomyocytes and prevents cardiac dysfunction in pathological hypertrophy. American journal of physiology Cell physiology. 2010;299:C1256–1266. doi: 10.1152/ajpcell.00338.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]