

Abstract
Inhibition of distinct ubiquitin E3 ligases might represent a powerful therapeutic tool. ITCH is a HECT domain-containing E3 ligase that promotes the ubiquitylation and degradation of several proteins, including p73, p63, c-Jun, JunB, Notch and c-FLIP, thus affecting cell fate. Accordingly, ITCH depletion potentiates the effect of chemotherapeutic drugs, revealing ITCH as a potential pharmacological target in cancer therapy. Using high throughput screening of ITCH auto-ubiquitylation, we identified several putative ITCH inhibitors, one of which is clomipramine—a clinically useful antidepressant drug. Previously, we have shown that clomipramine inhibits autophagy by blocking autophagolysosomal fluxes and thus could potentiate chemotherapy in vitro. Here, we found that clomipramine specifically blocks ITCH auto-ubiquitylation, as well as p73 ubiquitylation. By screening structural homologs of clomipramine, we identified several ITCH inhibitors and putative molecular moieties that are essential for ITCH inhibition. Treating a panel of breast, prostate and bladder cancer cell lines with clomipramine, or its homologs, we found that they reduce cancer cell growth, and synergize with gemcitabine or mitomycin in killing cancer cells by blocking autophagy. We also discuss a potential mechanism of inhibition. Together, our study (i) demonstrates the feasibility of using high throughput screening to identify E3 ligase inhibitors and (ii) provides insight into how clomipramine and its structural homologs might interfere with ITCH and other HECT E3 ligase catalytic activity in (iii) potentiating chemotherapy by regulating autophagic fluxes. These results may have direct clinical applications.
Keywords: HECT, ubiquitin, E3 ligase, inhibitors, autophagy, antidepressant
Ubiquitylation regulates cell physiology and pathology by complex posttranslational protein modifications.1, 2, 3 Protein ubiquitylation consists of three discrete steps carried out by ubiquitin (Ub)-activating enzymes (E1), Ub-conjugating enzymes (E2) and Ub ligases (E3).4 E3 ligases are classified into three subfamilies based on the sequence homology of their E2 binding domains: RING (Really Interesting New Gene), HECT (Homologous to E6AP Carboxyl Terminus) and U-boxes.5 The HECT E3 ligases are unique among the E3s, because they possess intrinsic catalytic activity.6 The basic structural motif of the HECT E3 ligases is a large C-terminal module of circa 350 residues. The reaction requires several steps, (i) binding to E2, (ii) Ub loading, forming a Ub–thioester intermediate with the catalytic cysteine located at the C-terminus of the HECT domain, and (iii) Ub transfer to the substrate protein with a conformational transition and rotation of the catalytic domain onto an hinge region.7
The HECT E3s gain their substrate specificity by selective protein–protein interaction domains, accounting for their classification into three subfamilies: HERC E3s containing RCC1-like domains (RLDs), C2-WW-HECT E3s possessing WW domains, and SI(ngle)-HECT E3s lacking either RLDs or WW domains.6 The C2-WW-HECT E3s are the best-characterized subgroup of HECT ligases. They contain an N-terminal protein kinase C (PKC)-related C2 domain and two to four tryptophan-containing WW domains that regulate E3-substrate interaction.6 ITCH belongs to this sub-family.8
ITCH was originally identified because radiation-induced disruption of the Itch gene induced a lethal autoimmune inflammatory condition.9 ITCH was later found to be crucial in the control of the proteosomal degradation of several important substrates involved in the regulation of the programmed cell death pathway and/or the inflammatory response, including the p53 family members p73 and p63,10, 11, 12, 13, 14, 15, 16, 17 the cellular FLICE-inhibitory protein,18 the small redox proteins Thioredoxin,19 the membrane-targeted death ligand tBid,20 the tumor suppressor RASSF5/NORE1 RASSF5,21 the large tumor suppressor 1 LATS1,22 the lysosomal-associated protein multispanning transmembrane 5 LAPTM5,23 the epithelial kinase receptor ErbB4,24 members of the activator protein 1 c-Jun and JunB,25 the phospholipase C-γ1 and protein kinase θ PLC-γ1/PKC-θ,26, 27 the Ub-editing enzyme A20,28 Notch1,29 proteins involved in the TGFβR signaling Smad and TIEG130, 31 and the chemokine receptor 4 CXCR4.32
Since ITCH is relevant in regulating immunological responses as well as cancer progression,33, 34, 35, 36 and because HECT E3s are the most druggable of the entire family, we decided to investigate the possibility of identifying ITCH inhibitors using a high throughput screen (HTS). To identify such inhibitors we developed an ELISA-based HTS using purified recombinant proteins and auto-ubiquitylation as readout for ITCH activity. Here, we characterized one of the inhibitor compounds, clomipramine, able to block p73 ubiquitylation by binding to ITCH in an irreversible manner and inhibiting its charging with Ub. We found that, in addition to their ITCH inhibitory activity, clomipramine and its structural homolog norclomipramine, induce autophagosome accumulation and synergize with chemotherapeutics in killing cancer cell lines in vitro.
Results
HTS assay to detect ITCH inhibitors
To identify low molecular weight compounds that are potential ITCH inhibitors, we established an ELISA-based HTS assay. To reduce the complexity of the assay we decided to use ITCH auto-ubiquitylation as the readout for ITCH activity (Figure 1a).8, 37 Our results demonstrate that the signal detected in our experimental setting was strictly dependent on ITCH Ub ligase activity, as a parallel experiment using mutant enzymatically inactive ITCH (E3m) did not generate any significant signal (Figure 1b). We then measured the dynamic range of our in vitro ubiquitylation assay using increasing concentrations of immobilized wild type ITCH and observed a dose-dependent change in signal intensity with an EC50 of ∼5 ng per well (Figure 1c). As predicted, mutant ITCH tested in parallel gave only a baseline signal at all concentrations tested, further confirming that the signal detected in our experimental conditions was dependent on the Ub ligase activity of ITCH.
Figure 1.

High throughput screening (HTS) for ITCH inhibitors. (a) Schematic representation of the layout of the ELISA assay used for the HTS. Recombinant GST-ITCH or ubiquitin ligase dead mutant GST-ITCH C830A were immobilized to glutathione-coated ELISA plates and incubated with either complete ubiquitylation reaction mixtures containing E1, E2 (UbcH7) and FLAG-ubiquitin or partial mixtures as indicated in (b). Reactions were performed for 1 h at RT and stopped by washing with PBST before development with HRP-conjugated anti-FLAG antibody for 1 h at RT. After final washes, the wells were incubated with TMB substrate for 15 min at RT, and then stopped with acid and OD450 nm measurements were made with a plate reader. (b) Different combinations of the ubiquitin reaction components were used to evaluate the specificity of the ubiquitylation reaction (E3, GST-ITCH; E3m, E3 ligase dead mutant GST-ITCH C830A). (c) Complete reactions were performed as in (b) using a range of E3 or E3m concentrations immobilized to the ELISA plate as shown. (d) Normalized screening data for 1040 compounds from the NINDS library. Immobilized GST-ITCH was incubated with test compounds (10 μM) and E1/E2/Ub mix. After incubation at RT for 2 h, plates were developed with HRP-conjugated anti-FLAG and TMB substrate as above. Percentage residual activity (%RA) was calculated using the formula %RA=(OD450 sample—mean OD450 low controls)/(mean high controls—mean OD450 low controls) × 100. Z prime values were calculated with a threshold of >0.5 set for acceptable data. 70 and 50% thresholds are indicated with lines. (e) Confirmation of six positive hits using does response and the ELISA system as in Figure 1c
HTS identification of ITCH inhibitors
Using the assay described in the previous paragraph we screened 1040 compounds from the NINDS library and ∼20 000 diverse compounds from various commercial suppliers at a final concentration of 10 μM each. The automated assay was robust according to an average recorded Z′ of 0.7 (range 0.5–0.8). The normalized single shot screening data for the NINDS set is shown in Figure 1d. Potential ITCH inhibitors (‘hits') were identified from test wells yielding normalized residual activity results <3 S.D. from the mean of the high controls. Hits were further ranked more stringently according to 70 and 50% residual activity thresholds. The overall hit rate at the 50% cutoff was low (0.23%), suggesting that the ITCH inhibition with small molecules is relatively rare. Of 46 compounds showing <50% residual activity that were initially identified as hits, 20 were confirmed at single point and 6 of these showed dose-dependent inhibition of ITCH (Figure 1e and Table 1).
Table 1. Validation of compounds identified in the high throughput screen.
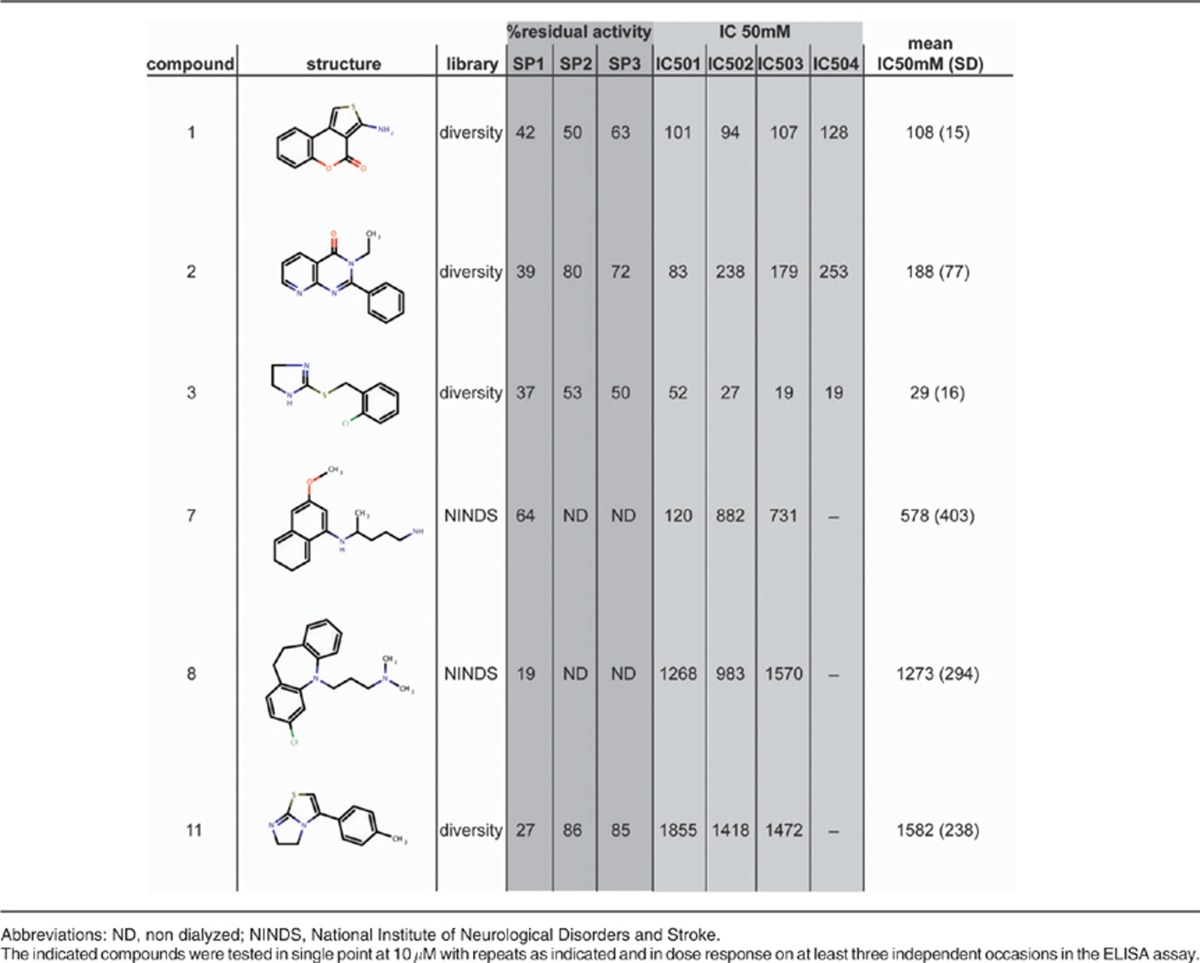
Validation of putative ITCH inhibitors
To validate the results obtained from the HTS we used a more direct assay to visualize the formation of Ub conjugates. For this purpose, recombinant GST-ITCH was incubated with E1 and E2 in the presence of candidate inhibitors or vehicle and auto-ubiquitylation was determined by Western blotting using anti-GST antibodies (Figure 2a). Interestingly, using this assay we found that only clomipramine (compound 8, MRT0003986) significantly inhibited ITCH auto-ubiquitylation, as demonstrated by the disappearance of high molecular weight ITCH species present in the positive control (Figure 2a; lane 8 versus 2).
Figure 2.

Validation of the identified putative ITCH inhibitors. (a) GST-ITCH (ITCH WT) or E3 ligase dead mutant GST-ITCH (ITCH MUT) were incubated in ubiquitylation assay buffer with DMSO or the putative ITCH inhibitors (1 mM). The reaction products were subjected to Western blot analysis. (b) 35S labeled p73 was reacted with ITCH or E3 ligase dead mutant ITCH in the ubiquitylation assay buffer in the presence of DMSO or putative ITCH inhibitors (1 mM) as indicated. The reaction was subjected to SDS-PAGE and resolved by autoradiogram. (c) p73 ubiquitylation assay performed as in (b) in the presence of the indicated concentrations of clomipramine (compound 8). As control for ubiquitylation reaction E3 ligase dead mutant ITCH (lane 1) substituted the WT ITCH. (d) The indicated substrates labeled with 35S were incubated with the indicated E3 ligases in the presence or absence of clomipramine as indicated. The reaction was resolved by SDS-PAGE and radiogram. Control reactions were performed without the E2 as indicated
In accordance with the auto-ubiquitylation experiment, we found that clomipramine significantly inhibited ITCH-dependent ubiquitylation of p73, a well characterized ITCH substrate, as demonstrated by the disappearance of high molecular weight p73 species present in the positive control (Figure 2b; lane 8 versus 2). As expected, incubation of p73 with the ligase dead ITCH mutant did not produce any detectable high molecular weight p73 Ub conjugates (Figure 2b; lane 1). Inhibition of ITCH-dependent p73 ubiquitylation by clomipramine was dose-dependent achieving complete inhibition at 0.8 mM (Figure 2c; lane 6). These results are consistent with the findings that clomipramine inhibited ITCH auto-ubiquitylation activity and support the conclusion that it is an ITCH E3 ligase inhibitor.
To evaluate whether clomipramine is a general inhibitor of E3 ligases or specific for ITCH, we tested whether clomipramine can inhibit other E3 ligases. To answer this question we assessed the effect of clomipramine on the auto-ubiquitylation of Ring1B, a RING domain E3 ligase, the ubiquitylation of Ring1B by the HECT E3 ligase E6AP38 and the ubiquitylation of Dronc by the RING E3 ligase DIAP39 (Figure 2d). The specificity of the ubiquitylation reaction was confirmed by excluding the E2 in the control lanes (Figure 2d; lanes 1, 4, 7 and 10). As expected, clomipramine inhibited p73 ubiquitylation by ITCH (Figure 2d; lane 2 versus 3). Interestingly, the RING E3 ligases Ring1B and DIAP were resistant to clomipramine inhibition and retained their ubiquitylation activity (Figure 2d; lanes 5 versus 6 and 11 versus 12), while the HECT E3 ligase E6AP was blocked by clomipramine (Figure 2d; lanes 8 versus 9). Together these results suggest that clomipramine is an efficient inhibitor of ITCH with some general degree of specificity for HECT E3 ligases; it does not inhibit any RING E3 ligase tested so far. Moreover as E6AP and ITCH show no significant sequence homology except in their HECT domain it is reasonable to speculate that clomipramine primarily acts directly on the catalytic domain of these two enzymes.
Clomipramine inhibits Ub transthiolation of ITCH
Because ubiquitylation requires the concerted action of E1, E2 and E3, we asked whether the clomipramine inhibitory effect in the Ub conjugation assays was a result of direct inhibition of ITCH E3 ligase activity or an impairment of E1 and/or E2 activity. To answer this question we incubated recombinant E2 (UbcH7) with E1 in the presence or absence of clomipramine and analyzed the E2 Ub transthiolation by Coomassie staining (Figure 3a). Our results show that clomipramine does not inhibit UbcH7 Ub transthiolation, as demonstrated by the appearance of a shift in the UbcH7 band comparable with the samples incubated with vehicle solution (Figure 3a; lanes 2 versus 5). As a control of E2 Ub transthiolation inhibition we used β-mercaptoethanol that efficiently prevented thioester bond formation between Ub and UbcH7 (Figure 3b; lanes 3, 6). In addition, we also repeated similar experiments using only E1 and obtained comparable results (data not shown), indicating that both E1 and E2 were completely resistant to the inhibitory effect of clomipramine. These results are in line with the finding that clomipramine does not block the activity of RING1B or DIAP1 (Figure 2d). We then tested whether clomipramine was acting on ITCH Ub transthiolation or on the ITCH Ub chain elongation activity. To this end, we performed similar experiments as described in Figure 3a using recombinant GST-ITCH and Western blot analysis using anti-GST antibodies (Figure 3b). In contrast to the results obtained in the E2 Ub transthiolation experiments, we observed a strong inhibitory effect of clomipramine on ITCH Ub transthiolation (Figure 3b; lane 5).
Figure 3.

Clomipramine mechanism of inhibition. (a) HIS-UbcH7 was incubated with E1 either with DMSO or clomipramine in the presence or absence of β-mercaptoethanol as indicated along with ubiquitylation assay buffer. Reaction was subjected to SDS-PAGE and the gel was stained with Coomassie blue. (b) GST-ITCH or E3 ligase dead mutant GST-ITCH were incubated either with DMSO or clomipramine in the presence or absence of β-mercaptoethanol and ubiquitylation reaction buffer. The reaction was resolved by Western blot using anti-GST antibodies. (c) GST-ITCH was incubated with DMSO or clomipramine as indicated and then dialyzed. The dialyzed GST-ITCH was subjected to ubiquitylation reaction. The reaction was resoled using Western blot and anti-GST antibodies. As control, untreated GST-ITCH was added to the reaction as indicated, N.D., non dialyzed. (d) 35S radio-labeled p73 was reacted with GST-ITCH prepared as in c
Next we asked whether the inhibitory effect of clomipramine on ITCH catalytic activity is reversible or irreversible. To answer this question we incubated GST-ITCH with clomipramine or vehicle control (DMSO), subjected it to dialysis to dilute low affinity and unbound clomipramine and subsequently incubated it with E1 and E2 in Ub assay buffer. Auto-ubiquitylation activity was clearly evident in the DMSO pretreated ITCH samples but totally absent in the clomipramine pretreated samples (Figure 3c; lanes 1 versus 4). As an internal reaction control we added untreated GST-ITCH to the dialyzed samples and found adequate auto-ubiquitylation activity, even in the samples that were preincubated with clomipramine before the dialysis (Figure 3c; lane 5 and 6). In agreement with these results, we observed that clomipramine pretreated and dialyzed ITCH failed to promote the formation of p73 Ub conjugate species (Figure 3d; lanes 4 versus 8).
Together these data suggest that clomipramine is a specific ITCH inhibitor that interferes with the HECT Ub transthiolation activity of this enzyme in an irreversible manner.
Activity of structural analogs of clomipramine
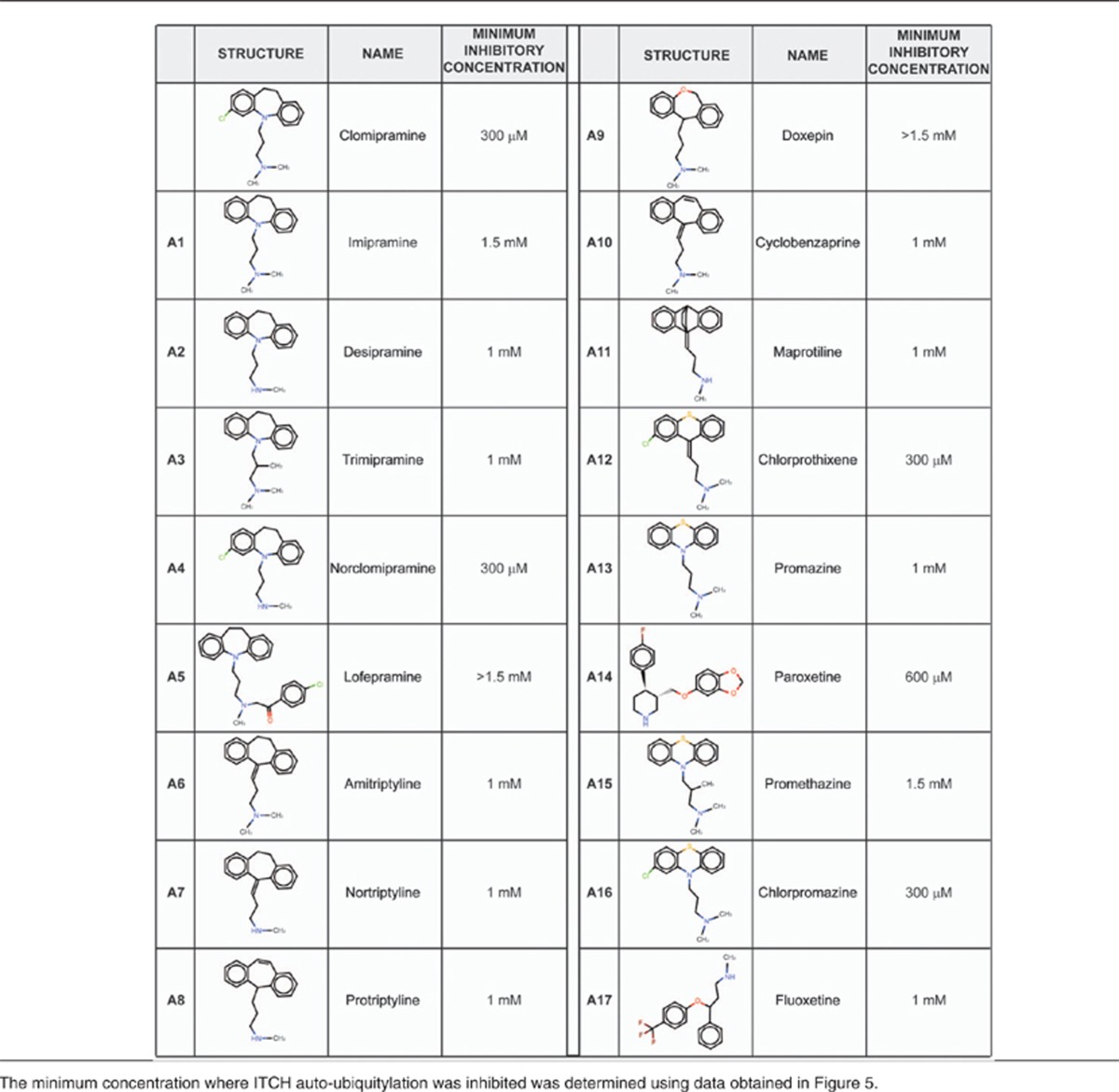
To obtain a qualitative insight into the putative molecular moieties that are essential for ITCH inhibition by clomipramine we tested 17 clomipramine structural analogs for their ability to block ITCH Ub ligase activity. To this end, similar experiments as described in Figure 2a were performed using increasing doses of the different compounds tested (Figure 4). The structures of the analogs and minimum tested concentrations achieving ITCH auto-ubiquitylation inhibition are shown in Table 2. The results obtained show that all the compounds that were found to inhibit ITCH activity at a concentration close to 300 μM contained a Cl group at the aromatic moiety. In addition, we found that the extent of methylation of the final amine group on the side chain decreases the inhibitory effect of the compounds (Figure 4f). Interestingly, norclomipramine, the active metabolite of clomipramine, was found to be the most efficient ITCH inhibitor tested.
Figure 4.

ITCH inhibitions by structural analogs of Clomipramine. (a–e) GST-ITCH was incubated with the indicated concentrations of the indicated structural analogs (A1 to A17) of clomipramine (CL) in ubiquitylation assay buffer. The reactions were resolved by SDS-PAGE and stained with Coomassie. As a control, E2 was excluded from the reaction. The intensity of nonubiquitylated GST-ITCH band was quantified. (f) Clomipramine molecular moieties important for ITCH inhibition. The presence of a Chloride (circle in red) on the Benzene ring increases the inhibitory activity of the compounds tested by roughly fivefold or more. The length of the side chain is also critical. Shorter or longer chains dramatically decrease the inhibitor effect. The Amine number 1 can be substituted by a C=C double bond, without affecting the inhibition capacity of the compound. The extent of methylation of amine number 2 affects the inhibitor effect of the compounds tested. A dimethylamine group in this position decreases the inhibitory potency by a factor of two compared with a monomethylamine
Table 2. Structural analogs of Clomipramine and their ITCH inhibition activity.
Clomipramine and Norclomipramine inhibit autophagy and synergize with chemotherapeutics in killing cancer cells
Autophagy is a major cellular process involved in detoxification, and autophagy inhibitors have been show to promote the activity of anti-cancer drugs40 and are therefore under investigation as potential therapeutic agents in treating cancer.41 Clomipramine has been shown to synergize with doxorubicin in killing HeLa and MEF cells by blocking autophagic flux.40 Therefore, we tested whether clomipramine has activity on other cancer cell lines, can synergize with other chemotherapeutics and whether these activities are present in clomipramine's structural analog norclomipramine.
First, we measured autophagosome accumulation in RT112 or HT-1376 cells treated with the ITCH inhibitors or chemotherapeutics separately or in combination using monodansylcadaverine fluorescence as readout for autophagosomes.42 As expected, clomipramine or norclomipramine, alone increased autophagosome accumulation (Supplementary Figure S1a–d). In addition, while gemcitabine or mitomycin alone increased autophagosome accumulation (Supplementary Figure S1a–d), the combination of an ITCH inhibitor with these chemotherapeutic agents resulted in further increased autophagosome accumulation as compared with the individual compounds alone (Supplementary Figure S1a–d). Together these results support the premise that the two putative ITCH inhibitors block autophagic flux in RT112 and HT-1376 cells and are in line with our preliminary previously published data.
Next we asked whether the identified putative ITCH inhibitors could synergize with chemotherapeutics. To this end we treated bladder cancer cells HT-1376 with clomipramine alone or in combination with the chemotherapeutic agent gemcitabine and measured cell growth and induction of cell death. Indeed, the combination of clomipramine and gemcitabine was shown to be synergistic (Figures 5a and c and Supplementary Figure S2). Similar results were obtained using RT112 cells treated with norclomipramine alone or in combination with mitomycin (Figures 5b and d and Supplementary Figure S3). The data was analyzed using CalcuSyn software (Biosoft, Cambridge, UK)43 to determine synergism between the compounds (Supplementary Table 1).
Figure 5.

Synergistic effect of putative ITCH inhibitors with anti-cancer agents on human bladder cancer cells. Clomipramine/GEM and norclomipramine/MIT synergistic effect on HT-1376 (a) and RT112 (b) cell growth inhibition, respectively. We have evaluated the growth inhibition induced by different concentrations of clomipramine and GEM on HT-1376, and of norclomipramine and MIT on RT112 cells. We have performed these experiments with MTT assay and the resulting data were elaborated with the dedicated software CalcuSyn.43 In the figure it is also shown the isobologram analysis of the effects on growth inhibition of clomipramine/GEM and norclomipramine/MIT combinations, used at equitoxic (50 : 50) concentrations for all cell lines and also in sequential administration, GEM 24 h after clomipramine for HT-1376 (c), and MIT 24 h after norclomipramine for RT112 (d). CI, combination index. Each point is the mean of at least four different replicate experiments
To test whether ITCH inhibitors can inhibit cell growth, we challenged a panel of breast, prostate and bladder cancer cell lines (Supplementary Figures 4–6) with different doses of the clomipramine and norclomipramine, for 48 or 72 h (Supplementary Figures 4–6) and measured cell growth using a MTT assay. Both clomipramine and norclomipramine exhibited IC50 values at low micromolar concentrations for all of the tested cell lines (Supplementary Table 2–4). Because in vitro clomipramine and norclomipramine inhibited ITCH at high micromolar concentrations (300 μM; Supplementary Table 1) but inhibited cancer cell growth at a low micromolar concentrations (6–30 μM; Supplementary Table 2–4) these data suggest that the putative ITCH inhibitors block cell growth through an ITCH independent mechanism and support the conclusion that clomipramine blocks cell growth by blocking autophagic flux.
Discussion
HTS reveals clomipramine as an ITCH inhibitor regulating autophagy
We undertook the challenge of performing a HTS for inhibitors of E3 ligases and successfully identified small molecule ITCH inhibitors, providing a proof of concept and a screening strategy for identification of HECT E3 ligase inhibitors. This provides a proof of concept for a screening strategy for the identification of other HECT E3 ligase inhibitors in the future. Our first unexpected result was the identification of the antidepressant drug clomipramine as an E3 inhibitor. Although E3s were not known as a chemical entity at the time when the molecular mechanism of these antidepressant drugs was identified, this suggests the possibility either of a novel mechanism of action, or that a HECT-containing E3 acts on that pathway, for example, regulating the degradation of the serotonin receptor or its recycling mechanism. Alternatively, the effect of clomipramine on HECT E3s could be an off-target effect, although very powerful. A second surprise was the discrepancy in concentration for the biochemical and the cellular effects of clomipramine. On this point, we have to stress that the biochemical machinery of proteolysis is extremely complicated, unlike the kinases or phosphatases, and therefore the concentration for its biochemical visualization could differ using an in vivo cellular readout. A third unexpected result, already partially published by our group40 was the ability of clomipramine to inhibit autophagy. Is this related to the molecular mechanism of action as an antidepressant? do all antidepressant drugs, including those chemically unrelated to clomipramine, share this autophagic regulation? clearly this is an interesting point for future investigations. Here, we expanded our previous observation40 showing an evident synergism for chemotherapy sensitivity. Indeed, we found that clomipramine shares some structural commonalities with chloroquine, a well-established inhibitor of autophagic flux and that clomipramine and its structural homolog blocked autophagic flux and synergized with chemotherapeutics in inhibiting the growth and viability of a panel of different cancer cells. This could form a prelude to an ad hoc clinical trial.
Is the effect of clomipramine dependent of p73? as discussed in the Introduction, ITCH, like any other E3, has a significant number of substrates, and discriminating among different E3 targets may be not easy.44 In turn, p73 can be ubiquitylated by multiple mechanisms, including Cul4A,45 PIR2,46 antizyme,47 FBX045,48 calpain49 as well as de-ubiquitylated.50, 51 Consequently, the effect of an E3 inhibitor could depend on the tissue, on the cellular micro-environment and stimuli, as well as on a distinct combination of multiple E3 substrates present in the cell.
Putative mechanism of action of the ITCH inhibitor clomipramine
Using biochemical assays we found that the identified compound, namely clomipramine, blocks ITCH Ub transthiolation in an irreversible manner. Clomipramine inhibited some other HECT E3 ligases but not RING E3 ligases leading us to hypothesize a putative mechanism of inhibition.
To further increase our understanding of how clomipramine derivatives might obstruct ITCH Ub conjugation activity, modeling studies were performed on the HECT domain of ITCH. Two potential binding sites were found (see Figure 6a), and in those full-flexible docking of clomipramine was performed. The low-energy conformations of the ligand within each binding site are displayed in Figure 6a. If the ligand were to bind between the N- and C-lobes, it could prevent the interaction of Ub with the catalytic C871, and could also preclude the relative rearrangement of the two lobes for Ub transfer from E2, as was theoretically predicted for E6AP7 and observed in NEDD4L.52 If the ligand were to bind within the N-lobe, it might also interfere with protein rearrangement blocking ITCH Ub transthiolation. In both cases, burial of the Cl atom within the binding site could explain the observed sensitivity of the binding affinity to Cl deletion (Table 2).
Figure 6.

HECT domain of ITCH with potential Clomipramine binding sites. (a) Two conformations of the 613–622 loop (missing in the experimental structure 3TUG) are shown. Low-energy conformations of docked clomipramine in the two sites are displayed in yellow and gray carbons. The catalytic C871 is also displayed as reference. Color code: C-lobe, red; N-lobe large subdomain, magenta; N-lobe small subdomain, cyan; flexible connecting loops, yellow. (b) The indicated mechanism shows the direct interaction between the catalytic sulfidrilic group of ITCH HECT domain and the aromatic ring of the ITCH inhibitors. The resonance-stabilized adduct favors the elimination of the halogen group with the formation of a new covalent bond. (c) Comparison between the structures of chloroquine and clomipramine showing the similarity of their scaffolds: highlighted with stars and boxes are the Cl atoms and the alkyl chains, respectively
A possible alternative interpretation of the results is that these compounds interact directly with the thiol group of the catalytic cysteine (Figure 6b). In this case, a potential mechanism for the reaction may consist of two steps; the first is the addition of the nucleophilic sulfur atom, which is rate-limiting, at the aromatic ring. During this process a new carbon–sulfur bond is created as a stable intermediate in the form of a resonance-stabilized carbon anion. The second step is the elimination of the halogen leaving group which restores the aromatic system. This possible model is supported by the findings that clomipramine irreversibly inhibits ITCH and that it can also inhibit other HECT E3 ligases that have a similar catalytic cysteine. Interestingly, we observed that the structures of the ITCH inhibitors are similar to the autophagy inhibitor chloroquine and present a moiety that resembles the chloroquine aromatic scaffold (Figure 6c). These include the heterocyclic structure leading the nitrogen atom, the chloro substituent as the reactive site for the inhibition reaction and the alkyl chain bearing a tertiary amine reported to be important for the activity of autophagy inhibitors that promote the deacidification of lysosomes and reduce tumor growth in vivo.53
Discriminating between these possible mechanisms of action will require further studies.
Conclusion
Growing amount of evidence is accumulating indicating that deregulation of ITCH and other members of the family of Nedd4-like E3 ligases that have a crucial role in the onset and progression of different medical conditions.6, 54 Here, we have (i) successfully performed an HTS for an HECT- containing E3 ligase inhibitors, (ii) identified clomipramine, and its structural analogs, as inhibitors of ITCH, (iii) shown their ability to regulate autophagy and chemosensitivity, leading to immediate potential clinical application. Clearly, these results require significant further work to clarify the open points discussed above. Nonetheless, the results presented here not only provide valuable information for the development of new research tools, that may be extremely useful to study ITCH and other HECT E3 ligases, but also show great translational potential.
Materials and Methods
Plasmids
Expression vectors were described previously: p73, ITCH, ITCH mutant, Ub and UBCH7;11 RINGB and E6AP;38 DIAP and Dronc.39
GST fusion proteins
GST fusion proteins were expressed in E. coli BL21 (DE3) and purified on glutathione–sepharose beads (Amersham Biosciences, Little Chalfont, UK) following standard procedures. Briefly, Glutathione S-transferase (GST)-tagged ITCH protein and inactivated ITCH mutant (C830A) were expressed in bacteria. E. coli BL21 CodonPlus(DE3)-RIL cells (Stratagene, La Jolla, CA, USA) were transformed with either wild type or mutant constructs prepared in the expression vector pGEX-6P1 (Amersham Biosciences). Saturated cultures were prepared by inoculation of LB medium containing ampicillin (LB/amp) with growth overnight at 37 °C. For expression, overnight cultures were diluted 1/100 in LB/amp at 37 °C until they reached an OD of 0.40. At this point the temperature was reduced to 15 °C and IPTG (50 μM final concentration) was added to induce expression for 3–4 h with shaking at 250 r.p.m. Cell lystates were prepared and the GST fusion proteins were purified on glutathione–sepharose beads (Amersham Biosciences) using standard procedures.
E1 and E2
E1 Ub activating enzyme was expressed in the Baculovirus system and purified by Ni-NTA chromatography (Qiagen, Manchester, UK) according to the manufacturer's instructions. Following expression in E. coli BL21 CodonPlus(DE3)-RIL cells (Stratagene), purified E2 was obtained by Ni-NTA affinity chromatography (Qiagen) according to the manufacturer's instructions.
FLAG-tagged Ub
Purified recombinant Ub containing an amino terminal FLAG sequence (DYKDDDDK) was purchased from Sigma Aldrich (Gillingham, UK; cat. No.U5382).
Anti-FLAG M2 peroxidase conjugate
The anti-FLAG M2 antibody conjugated to hydrogen peroxidase was purchased from Sigma Aldrich (cat. No. A8592).
TMB ELISA substrate
The peroxidase substrate 3,3′,5,5′-tetramethylbenzidine (TMB), was prepared by making a 1% w/v TMB stock (Sigma Aldrich T-2885) in DMSO and diluting 1/10 into 0.1 M sodium acetate buffer at pH 4.5 containing 0.01% v/v hydrogen peroxide just before use.
Compound libraries
We screened the NINDS library of 1040 bioactive compounds (ICCB Longwood, Harvard Medical School) and ∼20 000 diverse compounds from a range of commercial suppliers (Maybridge (Trevillett, UK), Interbioscreen (Moscow, Russia), SPECs (Delft, Netherlands), Peakdale (Chapel-en-le-Frith, UK) and Biofocus (Chesterford Pk, UK)). Library compounds were solubilized in DMSO at 1 mM in 384-well plates in preparation for screening.
Assay for ITCH auto-ubiquitylation and HTS
ITCH auto-ubiquitylation was assayed by ELISA. GST-ITCH was coated onto clear glutathione-coated 384-well ELISA plates (Pierce, Rockford, IL, USA) at 7 ng/20 μl per well for 1 h at room temperature (RT). Low control wells (n=24 per plate) were coated with GST-tagged ITCH E3 ligase dead mutant. E2 (UbcH7) was preincubated with E1 and FLAG-Ub (Sigma) off-plate in a bulk mix (E1/E2/Ub mix). Briefly, a mixture was prepared containing 51 μg/ml UbcH7, 468 ng/ml E1, 1.25 mM ATP in ubiquitylation buffer (25 mM Tris pH 8.0 containing 100 mM NaCl, 4 mM MgCl2. and 50 μM DTT) and incubated at RT for 45 min. After three washes with PBST (phosphate-buffered saline pH 7.2 containing 0.1% tween 20), 10 μl of test compounds pre-diluted to 20 uM in ubiquitylation buffer were added in single point to assay wells using automated robotics (Beckman Fx, High Wycombe, UK), followed by 10 μl of E1/E2/Ub mix. High control wells (n=24) were incubated with 10 μl ubiquitylation buffer containing 2%v/v DMSO followed by 10 μl E1/E2/Ub mix. The plates were sealed and incubated for 2 h at RT, after which the reactions were terminated by washing three times with PBST. Bound FLAG peptide corresponding to incorporation into the E3 was detected by addition of 20 μl anti-FLAG M2 monoclonal antibody conjugated to horseradish peroxidise (Sigma) diluted 1/10,000 in PBST and incubated for 1 h at RT, followed by three washes with PBST and addition of 20 μl 0.1% w/v 3,3′′,5,5′′-TMB (Sigma) in 0.1 M sodium acetate buffer pH 4.5 containing 0.01% v/v hydrogen peroxide for 15 min. The reactions were stopped by mixing with 5 μl 1 M HCl and reading absorbance at 450 nm using a Safire 2 plate reader (Tecan, Reading, UK). Percentage residual activity (%RA) was calculated using the formula %RA=(OD450 sample—mean OD450 low controls)/(mean high controls—mean OD450 low controls) × 100. Z prime values were calculated with a threshold of >0.5 set for acceptable data. Hits were identified using a cutoff of <50% residual activity. Dose response experiments were performed in the same way, except that concentrated compound stocks were used (10 μM in DMSO) that were serially diluted in 100% DMSO in half log series before addition to the assay plate. The final DMSO concentration present in the compound test wells was matched in the high and low controls. Curve fitting was performed using a 4 parameter logistic model (Graphpad Prism, La Jolla, CA, USA) with baseline constrained to zero.
Ubiquitylation assays
In vitro assays were performed as described previously.55 In vitro transcription/translation of proteins was performed using the wheat germ lysate system TNT kit (Promega, Madison, WI, USA), in the presence of [35S]Met (Amersham Biosciences), according to the manufacturer's protocol. The ubiquitylation reaction mixture contained: 1 μl of E1 and 1 μg of a E2 (UbcH7 for ITCH and E6AP; Ubch5c for Ring1B; UbcD1 for DIAP), 1 μg of purified E3 enzyme for GST-ITCH, GST-ITCH MUT, E6AP, DIAP1 and 0.3 μl for Ring1B, 40 mM Tris–HCl (pH 7.6), 2 mM dithiothreitol, 2.5 mM ATPγS, 2 mM MgCl2 and 5 μg of Ub. After incubation for 90 min at 37 °C, the reactions were terminated by adding 5 × Laemmli buffer, resolved by SDS-PAGE, followed by autoradiography.
Western blotting
Proteins were separated on SDS-PAGE and blotted onto nitrocellulose membranes. Filters were blocked with TBST 5% nonfat dry milk and incubated with primary antibodies for 2 h at RT. Filters were incubated for 1 h at RT using the appropriate horseradish peroxidase-conjugated secondary antibody (rabbit and mouse BioRad). Detection was performed with the enhanced chemiluminescence Supersignal West Pico (Pierce). Anti-GST is a monoclonal antibody (Abcam, Cambridge, UK).
Computerized homology structural modeling
For modeling purposes, the unbound structure of HECT domain of ITCH (PDB code 3TUG) was used. The 613–622 loop, missing in the PDB structure, was built in silico and subjected to a stochastic global energy optimization using the ICM program (Molsoft L.L.C., San Diego, CA, USA).56 As the missing loop is assumed to be flexible, two low-energy conformations of the loop were retained. Clomipramine was seeded in the two predicted binding sites (Figure 5), and the ligand-receptor complex structure was optimized using a flexible ligand–flexible receptor docking protocol,57, 58, 59 where low-energy conformations were collected in a conformational stack. The structure of clomipramine was downloaded from the GPCR Ligand Library 60 (http://cavasotto-lab.net/Databases/GDD/).
Acknowledgments
This work has been supported by the Medical Research Council and MRCT; grants from AIRC (2008-2010_33-08) (#5471) (2011-IG11955), AIRC 5xmille (#9979), Telethon Grant GGPO9133, Ministry of Education and Science of the Russian Federation (11.G34.31.0069), to GM. This work was also supported by the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (PICT-2011-2778 to CNC and PICT-2011-1231 and PICT-2011-2783 to MR) and FOCEM-Mercosur (COF 03/11). C.N.C. thanks Molsoft LLC for providing an academic license for the ICM program. Research in the laboratory of AC is supported by grants from the Dr Miriam and Sheldon G. Adelson Medical Research Foundation (AMRF), the Israel Science Foundation (ISF), the I-CORE Program of the Planning and Budgeting Committee and the ISF (Grant 1775/12), the EU TreatPolyQ Network and the Deutsch-Israelische Projektkooperation (DIP). AC is an Israel Cancer Research Fund (ICRF) USA Professor.
Glossary
- Ub
ubiquitin
- GST
glutathione S-transferase
- HIS
6 Histidine
- TMB
3,3,5,5-Tetramethylbenzidine
- RING
Really Interesting New Gene
- DMSO
dimethyl sulfoxide
- MDC
monodansylcadaverine
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Disease website (http://www.nature.com/cddis)
Edited by A Stephanou
Supplementary Material
References
- Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin-proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005;12:1178–1190. doi: 10.1038/sj.cdd.4401692. [DOI] [PubMed] [Google Scholar]
- Hershko A. The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 2005;12:1191–1197. doi: 10.1038/sj.cdd.4401702. [DOI] [PubMed] [Google Scholar]
- Melino G. Discovery of the ubiquitin proteasome system and its involvement in apoptosis. Cell Death Differ. 2005;12:1155–1157. doi: 10.1038/sj.cdd.4401740. [DOI] [PubMed] [Google Scholar]
- Ciechanover A. The ubiquitin-proteasome proteolytic pathway. Cell. 1994;79:13–21. doi: 10.1016/0092-8674(94)90396-4. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Bernassola F, Karin M, Ciechanover A, Melino G. The HECT family of E3 ubiquitin ligases: multiple players in cancer development. Cancer cell. 2008;14:10–21. doi: 10.1016/j.ccr.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Raimondo D, Giorgetti A, Bernassola F, Melino G, Tramontano A. Modeling and molecular dynamics of the interaction between the E3 ubiquitin ligase Itch and the E2 UbcH7. Biochem Pharmacol. 2008;76:1620–1627. doi: 10.1016/j.bcp.2008.08.026. [DOI] [PubMed] [Google Scholar]
- Scialpi F, Malatesta M, Peschiaroli A, Rossi M, Melino G, Bernassola F. Itch self-polyubiquitylation occurs through lysine-63 linkages. Biochem Pharmacol. 2008;76:1515–1521. doi: 10.1016/j.bcp.2008.07.028. [DOI] [PubMed] [Google Scholar]
- Perry WL, Hustad CM, Swing DA, O'Sullivan TN, Jenkins NA, Copeland NG. The itchy locus encodes a novel ubiquitin protein ligase that is disrupted in a18H mice. Nat Genet. 1998;18:143–146. doi: 10.1038/ng0298-143. [DOI] [PubMed] [Google Scholar]
- Maisse C, Guerrieri P, Melino G. p73 and p63 protein stability: the way to regulate function. Biochem Pharmacol. 2003;66:1555–1561. doi: 10.1016/s0006-2952(03)00511-2. [DOI] [PubMed] [Google Scholar]
- Rossi M, De Laurenzi V, Munarriz E, Green DR, Liu YC, Vousden KH, et al. The ubiquitin-protein ligase Itch regulates p73 stability. EMBO J. 2005;24:836–848. doi: 10.1038/sj.emboj.7600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M, Aqeilan RI, Neale M, Candi E, Salomoni P, Knight RA, et al. The E3 ubiquitin ligase Itch controls the protein stability of p63. Proc Natl Acad Sci USA. 2006;103:12753–12758. doi: 10.1073/pnas.0603449103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melino G, Knight RA, Cesareni G. Degradation of p63 by Itch. Cell Cycle. 2006;5:1735–1739. doi: 10.4161/cc.5.16.3260. [DOI] [PubMed] [Google Scholar]
- Bellomaria A, Barbato G, Melino G, Paci M, Melino S. Recognition of p63 by the E3 ligase ITCH: effect of an ectodermal dysplasia mutant. Cell Cycle. 2010;9:3730–3739. [PubMed] [Google Scholar]
- Browne G, Cipollone R, Lena AM, Serra V, Zhou H, van Bokhoven H, et al. Differential altered stability and transcriptional activity of DeltaNp63 mutants in distinct ectodermal dysplasias. J Cell Sci. 2011;124 (Pt 13:2200–2207. doi: 10.1242/jcs.079327. [DOI] [PubMed] [Google Scholar]
- Bellomaria A, Barbato G, Melino G, Paci M, Melino S. Recognition mechanism of p63 by the E3 ligase Itch: novel strategy in the study and inhibition of this interaction. Cell Cycle. 2012;11:3638–3648. doi: 10.4161/cc.21918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salah Z, Melino G, Aqeilan RI. Negative regulation of the Hippo pathway by E3 ubiquitin ligase ITCH is sufficient to promote tumorigenicity. Cancer Res. 2011;71:2010–2020. doi: 10.1158/0008-5472.CAN-10-3516. [DOI] [PubMed] [Google Scholar]
- Chang L, Kamata H, Solinas G, Luo JL, Maeda S, Venuprasad K, et al. The E3 ubiquitin ligase itch couples JNK activation to TNFalpha-induced cell death by inducing c-FLIP(L) turnover. Cell. 2006;124:601–613. doi: 10.1016/j.cell.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Zhang P, Wang C, Gao K, Wang D, Mao J, An J, et al. The ubiquitin ligase itch regulates apoptosis by targeting thioredoxin-interacting protein for ubiquitin-dependent degradation. J Biol Chem. 2010;285:8869–8879. doi: 10.1074/jbc.M109.063321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azakir BA, Desrochers G, Angers A. The ubiquitin ligase Itch mediates the antiapoptotic activity of epidermal growth factor by promoting the ubiquitylation and degradation of the truncated C-terminal portion of Bid. FEBS J. 2010;277:1319–1330. doi: 10.1111/j.1742-4658.2010.07562.x. [DOI] [PubMed] [Google Scholar]
- Suryaraja R, Anitha M, Anbarasu K, Kumari G, Mahalingam S. The E3 ubiquitin ligase Itch regulates tumor suppressor protein RASSF5/NORE1 stability in an acetylation-dependent manner. Cell Death Dis. 2013;4:e565. doi: 10.1038/cddis.2013.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho KC, Zhou Z, She YM, Chun A, Cyr TD, Yang X. Itch E3 ubiquitin ligase regulates large tumor suppressor 1 stability [corrected] Proc Natl Acad Sci USA. 2011;108:4870–4875. doi: 10.1073/pnas.1101273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara T, Inoue J, Kozaki K, Imoto I, Inazawa J. HECT-type ubiquitin ligase ITCH targets lysosomal-associated protein multispanning transmembrane 5 (LAPTM5) and prevents LAPTM5-mediated cell death. J Biol Chem. 2011;286:44086–44094. doi: 10.1074/jbc.M111.251694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundvall M, Korhonen A, Paatero I, Gaudio E, Melino G, Croce CM, et al. Isoform-specific monoubiquitination, endocytosis, and degradation of alternatively spliced ErbB4 isoforms. Proc Natl Acad Sci USA. 2008;105:4162–4167. doi: 10.1073/pnas.0708333105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Elly C, Gao B, Fang N, Altman Y, Joazeiro C, et al. Dysregulation of T lymphocyte function in itchy mice: a role for Itch in TH2 differentiation. Nat Immunol. 2002;3:281–287. doi: 10.1038/ni763. [DOI] [PubMed] [Google Scholar]
- Heissmeyer V, Macian F, Im SH, Varma R, Feske S, Venuprasad K, et al. Calcineurin imposes T cell unresponsiveness through targeted proteolysis of signaling proteins. Nat Immunol. 2004;5:255–265. doi: 10.1038/ni1047. [DOI] [PubMed] [Google Scholar]
- Venuprasad K, Elly C, Gao M, Salek-Ardakani S, Harada Y, Luo JL, et al. Convergence of Itch-induced ubiquitination with MEKK1-JNK signaling in Th2 tolerance and airway inflammation. J Clin Invest. 2006;116:1117–1126. doi: 10.1172/JCI26858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shembade N, Harhaj NS, Parvatiyar K, Copeland NG, Jenkins NA, Matesic LE, et al. The E3 ligase Itch negatively regulates inflammatory signaling pathways by controlling the function of the ubiquitin-editing enzyme A20. Nat Immunol. 2008;9:254–262. doi: 10.1038/ni1563. [DOI] [PubMed] [Google Scholar]
- Qiu L, Joazeiro C, Fang N, Wang HY, Elly C, Altman Y, et al. Recognition and ubiquitination of Notch by Itch, a hect-type E3 ubiquitin ligase. J Biol Chem. 2000;275:35734–35737. doi: 10.1074/jbc.M007300200. [DOI] [PubMed] [Google Scholar]
- Bai Y, Yang C, Hu K, Elly C, Liu YC. Itch E3 ligase-mediated regulation of TGF-beta signaling by modulating smad2 phosphorylation. Mol Cell. 2004;15:825–831. doi: 10.1016/j.molcel.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Venuprasad K, Huang H, Harada Y, Elly C, Subramaniam M, Spelsberg T, et al. The E3 ubiquitin ligase Itch regulates expression of transcription factor Foxp3 and airway inflammation by enhancing the function of transcription factor TIEG1. Nat Immunol. 2008;9:245–253. doi: 10.1038/niXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese A, Raiborg C, Santini F, Keen JH, Stenmark H, Benovic JL. The E3 ubiquitin ligase AIP4 mediates ubiquitination and sorting of the G protein-coupled receptor CXCR4. Dev Cell. 2003;5:709–722. doi: 10.1016/s1534-5807(03)00321-6. [DOI] [PubMed] [Google Scholar]
- Rivetti di Val Cervo P, Tucci P, Majid A, Lena AM, Agostini M, Bernardini S, et al. p73, miR106b, miR34a, and Itch in chronic lymphocytic leukemia Blood 20091136498–6499.author reply 9-500. [DOI] [PubMed] [Google Scholar]
- Rossi M, Inoue S, Walewska R, Knight RA, Dyer MJ, Cohen GM, et al. Caspase cleavage of Itch in chronic lymphocytic leukemia cells. Biochem Biophys Res Commun. 2009;379:659–664. doi: 10.1016/j.bbrc.2008.11.154. [DOI] [PubMed] [Google Scholar]
- Hansen TM, Rossi M, Roperch JP, Ansell K, Simpson K, Taylor D, et al. Itch inhibition regulates chemosensitivity in vitro. Biochem Biophys Res Commun. 2007;361:33–36. doi: 10.1016/j.bbrc.2007.06.104. [DOI] [PubMed] [Google Scholar]
- Ishihara T, Tsuda H, Hotta A, Kozaki K, Yoshida A, Noh JY, et al. ITCH is a putative target for a novel 20q11.22 amplification detected in anaplastic thyroid carcinoma cells by array-based comparative genomic hybridization. Cancer Sci. 2008;99:1940–1949. doi: 10.1111/j.1349-7006.2008.00900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberst A, Malatesta M, Aqeilan RI, Rossi M, Salomoni P, Murillas R, et al. The Nedd4-binding partner 1 (N4BP1) protein is an inhibitor of the E3 ligase Itch. Proc Natl Acad Sci USA. 2007;104:11280–11285. doi: 10.1073/pnas.0701773104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaaroor-Regev D, de Bie P, Scheffner M, Noy T, Shemer R, Heled M, et al. Regulation of the polycomb protein Ring1B by self-ubiquitination or by E6-AP may have implications to the pathogenesis of Angelman syndrome. Proc Natl Acad Sci USA. 2010;107:6788–6793. doi: 10.1073/pnas.1003108107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman-Bachinsky Y, Ryoo HD, Ciechanover A, Gonen H. Regulation of the Drosophila ubiquitin ligase DIAP1 is mediated via several distinct ubiquitin system pathways. Cell Death Differ. 2007;14:861–871. doi: 10.1038/sj.cdd.4402079. [DOI] [PubMed] [Google Scholar]
- Rossi M, Munarriz ER, Bartesaghi S, Milanese M, Dinsdale D, Guerra-Martin MA, et al. Desmethylclomipramine induces the accumulation of autophagy markers by blocking autophagic flux. J Cell Sci. 2009;122 (Pt 18:3330–3339. doi: 10.1242/jcs.048181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munafo DB, Colombo MI. A novel assay to study autophagy: regulation of autophagosome vacuole size by amino acid deprivation. J Cell Sci. 2001;114 (Pt 20:3619–3629. doi: 10.1242/jcs.114.20.3619. [DOI] [PubMed] [Google Scholar]
- Chou T-C, Talalay P. Analysis of combined drug effects: a new look at a very old problem. Trends Pharmacol Sci. 1983;4:450–454. [Google Scholar]
- Inoue S, Hao Z, Elia AJ, Cescon D, Zhou L, Silvester J, et al. Mule/Huwe1/Arf-BP1 suppresses Ras-driven tumorigenesis by preventing c-Myc/Miz1-mediated down-regulation of p21 and p15. Gen Dev. 2013;27:1101–1114. doi: 10.1101/gad.214577.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malatesta M, Peschiaroli A, Memmi EM, Zhang J, Antonov A, Green DR, et al. The Cul4A-DDB1 E3 ubiquitin ligase complex represses p73 transcriptional activity. Oncogene. 2012;32:4721–4726. doi: 10.1038/onc.2012.463. [DOI] [PubMed] [Google Scholar]
- Sayan BS, Yang AL, Conforti F, Tucci P, Piro MC, Browne GJ, et al. Differential control of TAp73 and DeltaNp73 protein stability by the ring finger ubiquitin ligase PIR2. Proc Natl Acad Sci USA. 2010;107:12877–12882. doi: 10.1073/pnas.0911828107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulloo I, Gopalan G, Melino G, Sabapathy K. The antiapoptotic DeltaNp73 is degraded in a c-Jun-dependent manner upon genotoxic stress through the antizyme-mediated pathway. Proc Natl Acad Sci USA. 2010;107:4902–4907. doi: 10.1073/pnas.0906782107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschiaroli A, Scialpi F, Bernassola F, Pagano M, Melino G. The F-box protein FBXO45 promotes the proteasome-dependent degradation of p73. Oncogene. 2009;28:3157–3166. doi: 10.1038/onc.2009.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munarriz E, Bano D, Sayan AE, Rossi M, Melino G, Nicotera P. Calpain cleavage regulates the protein stability of p73. Biochem Biophys Res Commun. 2005;333:954–960. doi: 10.1016/j.bbrc.2005.05.188. [DOI] [PubMed] [Google Scholar]
- Bernassola F, Salomoni P, Oberst A, Di Como CJ, Pagano M, Melino G, et al. Ubiquitin-dependent degradation of p73 is inhibited by PML. J Exp Med. 2004;199:1545–1557. doi: 10.1084/jem.20031943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschiaroli A, Skaar JR, Pagano M, Melino G. The ubiquitin-specific protease USP47 is a novel beta-TRCP interactor regulating cell survival. Oncogene. 2010;29:1384–1393. doi: 10.1038/onc.2009.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamadurai HB, Souphron J, Scott DC, Duda DM, Miller DJ, Stringer D, et al. Insights into ubiquitin transfer cascades from a structure of a UbcH5B approximately ubiquitin-HECT(NEDD4L) complex. Mol Cell. 2009;36:1095–1102. doi: 10.1016/j.molcel.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007;6:304–312. doi: 10.1038/nrd2272. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Staub O. HECT E3s and human disease. BMC Biochem. 2007;8 (Suppl 1:S6. doi: 10.1186/1471-2091-8-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell. 2006;24:701–711. doi: 10.1016/j.molcel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- ICM Version 3.7.2ed.MolSoft, LLC: La Jolla, CA; 2012 [Google Scholar]
- Diaz P, Phatak SS, Xu J, Fronczek FR, Astruc-Diaz F, Thompson CM, et al. 2,3-Dihydro-1-benzofuran derivatives as a series of potent selective cannabinoid receptor 2 agonists: design, synthesis, and binding mode prediction through ligand-steered modeling. Chem Med Chem. 2009;4:1615–1629. doi: 10.1002/cmdc.200900226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti MC, Casapullo A, Cavasotto CN, Tosco A, Dal Piaz F, Ziemys A, et al. The binding mode of petrosaspongiolide M to the human group IIA phospholipase A(2): exploring the role of covalent and noncovalent interactions in the inhibition process. Chem-Eur J. 2009;15:1155–1163. doi: 10.1002/chem.200801512. [DOI] [PubMed] [Google Scholar]
- Phatak SS, Gatica EA, Cavasotto CN. Ligand-steered modeling and docking: A benchmarking study in Class A G-Protein-Coupled Receptors. J Chem Inf Model. 2010;50:2119–2128. doi: 10.1021/ci100285f. [DOI] [PubMed] [Google Scholar]
- Gatica EA, Cavasotto CN. Ligand and Decoy Sets for Docking to G Protein-Coupled Receptors. Journal of chemical information and modeling. 2012;52:1–6. doi: 10.1021/ci200412p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.