

Abstract
Phospho-Ser129 α-synuclein is the modified form of α-synuclein that occurs most frequently within Parkinson's disease pathological inclusions. Here we demonstrate that the antidiabetic drug metformin significantly reduces levels of phospho-Ser129 α-synuclein and the ratio of phospho-Ser129 α-synuclein to total α-synuclein. This effect was documented in vitro in SH-SY5Y and HeLa cells as well as in primary cultures of hippocampal neurons. In vitro work also elucidated the mechanisms underlying metformin's action. Following metformin exposure, decreased phospho-Ser129 α-synuclein was not strictly dependent on induction of AMP-activated protein kinase, a primary target of the drug. On the other hand, metformin-induced phospho-Ser129 α-synuclein reduction was consistently associated with inhibition of mammalian target of rapamycin (mTOR) and activation of protein phosphatase 2A (PP2A). Evidence supporting a key role of mTOR/PP2A signaling included the finding that, similar to metformin, the canonical mTOR inhibitor rapamycin was capable of lowering the ratio of phospho-Ser129 α-synuclein to total α-synuclein. Furthermore, no decrease in phosphorylated α-synuclein occurred with either metformin or rapamycin when phosphatase activity was inhibited, supporting a direct relationship between mTOR inhibition, PP2A activation and protein dephosphorylation. A final set of experiments confirmed the effectiveness of metformin in vivo in wild-type C57BL/6 mice. Addition of the drug to food or drinking water lowered levels of phospho-Ser129 α-synuclein in the brain of treated animals. These data reveal a new mechanism leading to α-synuclein dephosphorylation that could be targeted for therapeutic intervention by drugs like metformin and rapamycin.
Keywords: mice, mTOR, Parkinson, phosphorylation, PP2A, rapamycin
Post-translational modifications modulate the toxic properties of proteins involved in human neurodegenerative diseases and, for this reason, could be targeted for therapeutic intervention. Both tau and α-synuclein (α-syn) are substrates for post-translational phosphorylation in Alzheimer's and Parkinson's disease (PD), respectively.1, 2 Hyperphosphorylation disrupts the normal function of tau and results in disassembly and depolymerization of microtubules.1, 3 Lewy bodies (LBs) and Lewy neurites (LNs) are post-mortem pathological hallmarks found within neuronal perikarya and axonal processes throughout the CNS of all patients who died with idiopathic PD.4 Fibrillar α-syn is a major component of these intraneuronal inclusions, and phosphorylation of the serine residue at position 129 (phospho-Ser129) is the most abundant modification of α-syn in LBs and LNs.2, 5 Formation and accumulation of phospho-Ser129 α-syn is therefore likely to play an important role in PD pathogenetic processes.6
Kinase inhibitors could be used to reduce protein phosphorylation. A potential limitation of this drug approach, however, is the fact that several kinases are capable of phosphorylating tau or α-syn. For example, casein kinases, G-protein-coupled receptor kinases and Polo-like kinases are all able to convert α-syn into phospho-Ser129 α-syn.7, 8, 9 Thus, significant production of phospho-Ser129 α-syn would likely occur even after pharmacologic inhibition of one of these kinases. A different strategy for decreasing α-syn phosphorylation has been proposed in a recent study and involves an enhancement of protein dephosphorylation.10 The results of this study demonstrated that protein phosphatase 2A (PP2A) is the primary enzyme involved in phospho-Ser129 α-syn dephosphorylation. They also revealed that treatment with eicosanoyl-5-hydroxytryptamide (EHT), an enhancer of PP2A activity, was effective in reducing levels of phospho-Ser129 α-syn in the mouse brain.
Additional evidence in support of enhanced dephosphorylation as a promising therapeutic strategy in neurodegenerative diseases comes from investigations assessing the effects of the antidiabetic drug metformin on tau. Metformin was found to lower tau phosphorylation both in vitro and in vivo.11 Interestingly, this effect was due to activation of PP2A and increased dephosphorylation of PP2A-dependent tau epitopes at Ser202, Ser356 and Ser262. PP2A also catalyzes α-syn dephosphorylation.10 The present study was therefore designed to determine whether treatment with metformin lowers phospho-Ser129 α-syn levels and, if so, to elucidate the biochemical pathways responsible for this effect. We report that metformin inhibits mammalian target of rapamycin (mTOR) and enhances PP2A activity. By doing so, it significantly decreases the ratio of phospho-Ser129 α-syn to total α-syn in cell culture systems as well as in the mouse brain.
Results
Metformin reduces levels of phospho-Ser129 α-syn in SH-SY5Y cell cultures
The effect of metformin on phospho-Ser129 α-syn levels was assessed in SH-SY5Y cells. Cells were first treated with retinoic acid to induce differentiation from a neuroblastic toward a more neuronal phenotype.12 The latter was morphologically characterized by an extension of neuritic processes (Figures 1a and b). Differentiated SH-SY5Y cells expressed relatively low amounts of α-syn and, consequently, levels of phospho-Ser129 α-syn were barely detectable by immunocytochemistry (Figure 1c) or western blot analysis (Figure 1f). To circumvent this limitation, we used an inducible system and triggered overexpression of human wild-type α-syn (hα-syn) 72 h before incubations with metformin. Under these conditions, SH-SY5Y cells displayed moderate immunoreactivity for phospho-Ser129 hα-syn as detected by immunocytochemistry (Figure 1d). Western blot analysis confirmed enhanced expression of soluble (predominantly monomeric) hα-syn and presence of phospho-Ser129 hα-syn in the supernatant fraction of homogenates from induced cells (Figures 1e and f). Following induction, monomeric hα-syn was also present in urea-extracted pellets in which levels of phospho-Ser129 hα-syn were barely detectable, however (Figures 1e and f).
Figure 1.

Dose- and time-dependent effect of metformin on phospho-Ser129 α-syn levels in SH-SY5Y cells. SH-SY5Y cells with tetracycline-inducible expression of hα-syn were immunostained with an anti-β-III tubulin antibody, and representative images were taken before (a) and after (b) differentiation treatment with retinoic acid. Differentiated cultures were costained with Dapi (blue) and an anti-phospho-Ser129 α-syn antibody (green); representative images show cells before (c) and after (d) treatment with tetracycline. Scale bars=50 μm. Cells were differentiated with retinoic acid and incubated without or with tetracycline. After cell homogenization, levels of hα-syn (e) and phospho-Ser129 hα-syn (f) were assayed by western blotting in the supernatant and pellet fractions. Cells pretreated with retinoic acid and tetracycline were then incubated for 16 h in the absence or presence of metformin at different concentrations. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by immunoblotting in the supernatant fraction (g). Semiquantitative analysis of band intensities is expressed as hα-syn/tubulin ratio (h) or phospho-Ser129 hα-syn/hα-syn ratio (i). In separate experiments, differentiated cells pretreated with tetracycline were incubated in the presence of 2.5 mM metformin. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by western blotting at 0 h and after 16 and 24 h (j). Analysis of band intensities is expressed as phospho-Ser129 hα-syn / hα-syn ratio (k). Data in (h, i and k) are the means±S.E.M. of at least three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 versus the respective control group (black bar). †††P<0.001 versus the group treated with 0.5 mM metformin (i). †P<0.05 versus the group treated with metformin for 16 h (k)
In all subsequent experiments, potential changes in phospho-Ser129 hα-syn were evaluated in supernatant fractions. To determine the effect of metformin on hα-syn phosphorylation, cells were incubated with no drug or different concentrations of the drug (0.5, 1.0 and 2.5 mM) for 16 or 24 h. Metformin did not affect the amount of total hα-syn (Figures 1g, h and j). On the other hand, the ratio of phospho-Ser129 hα-syn over total hα-syn was significantly reduced by metformin in both a concentration-dependent (Figures 1g and i) and time-dependent (Figures 1j and k) manner. No overt toxicity was observed morphologically (e.g., shape and density of the cells) and biochemically (e.g., release of lactate dehydrogenase) at the beginning as well as the end of metformin treatment (data not shown).
Treatment with metformin enhances phospho-Ser129 α-syn dephosphorylation
Metformin is capable of inducing PP2A activity.11 To determine the role that enhanced dephosphorylation may have in the reduction of phospho-Ser129 hα-syn caused by metformin, experiments were carried out using the phosphatase inhibitor okadaic acid (OKA). OKA is a potent blocker of PP2A activity although, at higher concentrations, it also inhibits other serine/threonine protein phosphatases.13 For these experiments, OKA was added at concentrations in the 10−7–10−8 M (shorter and longer incubations, respectively) range; these concentrations have previously been used for specific PP2A inhibition in cell cultures.14, 15 SH-SY5Y cells were treated with OKA alone or incubated with both OKA and metformin. Addition of OKA alone significantly enhanced phospho-Ser129 hα-syn (Figures 2a and b), underscoring the importance of enzymatic dephosphorylation in the regulation of phospho-Ser129 hα-syn levels. Measurements were then compared in cultures treated with OKA alone versus cells exposed to both OKA and metformin. Levels of total hα-syn and phospho-Ser129 hα-syn were found to be unchanged in cells incubated with OKA versus OKA plus metformin (Figures 2a and b), indicating that the ability of metformin to decrease phospho-Ser129 hα-syn is counteracted by OKA and is therefore dependent on protein phosphatase activity.
Figure 2.

OKA, a PP2A inhibitor, prevents metformin-induced reduction of phospho-Ser129 α-syn. SH-SY5Y cells were pretreated with retinoic acid and tetracycline. Cells were then incubated for 16 h with no addition or in the presence of OKA alone (15 nM), metformin alone (2.5 mM) or both OKA and metformin. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by immunoblotting (a). Semiquantitative analysis of band intensities is expressed as phospho-Ser129 hα-syn/hα-syn ratio (b). In separate experiments, cells were treated with no addition or with 300 nM OKA for 4 h, and levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by western blotting (c, first two lanes). In cultures preincubated with OKA, the medium was removed, cells were washed with fresh medium and incubations were continued for 16 h in the absence or presence of 2.5 mM metformin. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by western blotting (c, third and fourth lanes). Analysis of band intensities is expressed as phospho-Ser129 hα-syn/hα-syn ratio (d). Data in (b and d) are the means±S.E.M. of at least three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 versus the respective control group (black bar). †P<0.05 versus the group treated with OKA alone (b). ##P<0.01 versus the group treated with both OKA and metformin (b). †††P<0.001 versus the group pretreated with OKA for 4 h (d). #P<0.05 versus the group pretreated with OKA and then incubated in the absence of metformin (d)
In separate experiments, SH-SY5Y cells were treated with or without OKA for only a limited period of time, that is, 4 h. Then, the medium was removed, cells were washed with fresh medium and incubations were continued in the absence or presence of metformin. Pretreatment with OKA resulted in enhanced phospho-Ser129 hα-syn (Figure 2c). Levels of phosphorylated hα-syn declined, however, during further incubation in OKA-free medium (Figure 2c), consistent with reversible enzyme inhibition and progressive restoration of phosphatase activity.16 It is noteworthy that this reduction of phospho-Ser129 hα-syn after OKA removal was significantly more pronounced in cells treated with metformin (Figures 2c and d). Thus, metformin regained its effectiveness in parallel to the restoration of phosphatase activity, further demonstrating a direct role of dephosphorylation in metformin-induced reduction of phospho-Ser129 hα-syn.
mTOR-dependent reduction of phospho-Ser129 α-syn
The mechanism of action of metformin involves several pathways and molecular interactions. A primary effect of the drug is to enhance the phosphorylation of AMP-activated protein kinase (AMPK).17 Phosphorylation/activation of AMPK has a regulatory effect on mTOR, resulting in inhibition of mTOR signaling.18, 19 mTOR inhibition could in turn enhance PP2A activity and thus account for a decrease in phospho-Ser129 hα-syn.20 To test whether metformin-induced reduction of phospho-Ser129 hα-syn in SH-SY5Y cells was associated with AMPK activation and inhibition of mTOR, levels of phospho-Ser79 acetyl-CoA carboxylase (p-ACC) and phospho-Ser240/244 S6 ribosomal protein (p-RPS6) were assayed. p-ACC is a specific product of AMPK phosphorylation. p-RPS6 is an indicator of mTOR/PP2A signaling, being both a product of mTOR phosphorylation and a target of PP2A.21, 22 Results indicated that p-ACC was significantly increased as a consequence of metformin addition, reflecting activation of AMPK (Figures 3a and b). Data also showed levels of p-RPS6 to be reduced after treatment with the drug, consistent with mTOR inhibition/PP2A activation (Figures 3c and d).
Figure 3.
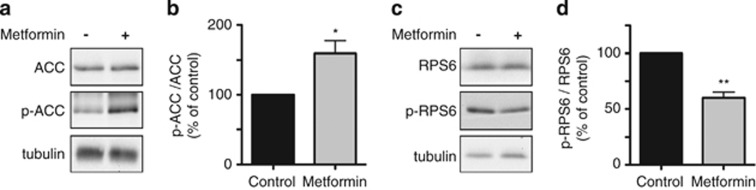
Effects of metformin on AMPK- and mTOR-dependent phosphorylation. SH-SY5Y cells were pretreated with retinoic acid and tetracycline. Cells were then incubated for 16 h with no addition or in the presence of 2.5 mM metformin. Levels of ACC, p-ACC (the product of AMPK-dependent phosphorylation of ACC at Ser79) and tubulin were assayed by immunoblotting (a). Levels of RPS6, p-RPS6 (the product of mTOR-dependent phosphorylation of RPS6 at Ser240/244) and tubulin were measured by western blotting (c). Semiquantitative analysis of band intensities is expressed as p-ACC/ACC (b) or p-RPS6/RPS6 (d) ratios. Data in (b and d) are the means±S.E.M. of at least three independent experiments. *P<0.05 and **P<0.01 versus the respective control group (black bar)
If mTOR inhibition and consequent PP2A activation play a role in the reduction of phospho-Ser129 hα-syn caused by metformin, other mTOR modulators, such as the mTOR inhibitor rapamycin, would also be expected to affect phospho-Ser129 hα-syn levels. To test this hypothesis, measurements of p-RPS6 and phospho-Ser129 hα-syn were compared in SH-SY5Y cells incubated in the absence versus presence of rapamycin. mTOR inhibition by rapamycin was indicated by a marked decrease in p-RPS6 and paralleled by a significant reduction of phospho-Ser129 hα-syn (Figures 4a and c). The decrease in phosphorylated hα-syn could not be attributed to an overall reduction of hα-syn as total levels of the protein were unchanged in untreated cells versus cells exposed to rapamycin (Figures 4a and b).
Figure 4.

Levels of phospho-Ser129 α-syn are decreased by the mTOR inhibitor rapamycin. SH-SY5Y cells were pretreated with retinoic acid and tetracycline. Cells were then incubated for 16 h with no addition or in the presence of 2 μM rapamycin. Levels of RPS6, p-RPS6, hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by immunoblotting (a). Semiquantitative analysis of band intensities is expressed as hα-syn/tubulin ratio (b) or phospho-Ser129 hα-syn/hα-syn ratio (c). In separate experiments, cells were incubated for 16 h with no addition or in the presence of OKA alone (15 nM), rapamycin alone (2 μM) or both OKA and rapamycin. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by immunoblotting (d). Semiquantitative analysis of band intensities is expressed as phospho-Ser129 hα-syn/hα-syn ratio (e). Data in (b, c and e) are the means±S.E.M. of at least three independent experiments. **P<0.01 and ***P<0.001 versus the respective control group (black bar). †††P<0.001 versus the group treated with OKA alone. ###P<0.001 versus the group treated with both OKA and rapamycin
Similarities between the effects of rapamycin and metformin were further indicated by experiments in which cells were cotreated with rapamycin and OKA. As a consequence of this cotreatment, that is, when phosphatase activity was inhibited by OKA, rapamycin failed to cause any reduction of phospho-Ser129 hα-syn (Figures 4d and e). Taken together, results of these experiments support a direct relationship between mTOR inhibition, PP2A activation, enhanced α-syn dephosphorylation and decreased ratio phospho-Ser129 α-syn over total α-syn.
AMPK-independent reduction of phospho-Ser129 α-syn by metformin
To further elucidate the role of AMPK activation in decreasing phospho-Ser129 hα-syn, experiments were carried out in HeLa cells overexpressing hα-syn after transient transfection. AMPK activation requires phosphorylation of AMPK by another upstream kinase, liver kinase B1 (LKB1).23 Because HeLa cells are constitutively deficient of LKB1 (Figure 5a),24, 25 we postulated that treatment with metformin would not result in AMPK activation in these cells. p-ACC was used as a marker of AMPK-dependent phosphorylation; supporting our assumption, its levels were found to be unaffected by addition of metformin to HeLa cells (Figures 5b and c). Phosphorylation of hα-syn was then assessed in this model system and interestingly, despite lack of AMPK activation, metformin was still capable of decreasing phospho-Ser129 hα-syn (Figures 5d and e).
Figure 5.

Effects of metformin in HeLa cells. As compared with SHSY5Y cells, HeLa cells do not express LKB1, as indicated by the lack of LKB1 immunoreactivity by immunoblotting (a). Levels of ACC, p-ACC and tubulin were compared in HeLa cells incubated in the absence or presence of 2.5 mM metformin. Proteins were assayed by western blotting (b). Band intensities were quantified and expressed as p-ACC/ACC ratio (c). HeLa cells were transiently transfected with hα-syn and then incubated for 16 h in the absence or presence of metformin at different concentrations. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by immunoblotting (d). Semiquantitative analysis of band intensities is expressed as phospho-Ser129 hα-syn/hα-syn ratio (e). In separate experiments, hα-syn-expressing HeLa cells were incubated for 16 h with no addition or in the presence of OKA alone (20 nM), metformin alone (2.5 mM) or both OKA and metformin. Levels of hα-syn, phospho-Ser129 hα-syn and tubulin were assayed by immunoblotting (f). Semiquantitative analysis of band intensities is expressed as phospho-Ser129 hα-syn/hα-syn ratio (g). Levels of RPS6, p-RPS6 and tubulin were compared by western blot analysis in HeLa cells incubated in the absence or presence of metformin at different concentrations (h). Band intensities were quantified and expressed as p-RPS6/RPS6 ratio (i). Data in (c, e, g and i) are the means±S.E.M. of at least three independent experiments. *P<0.05, **P<0.01 and ***P<0.001 versus the respective control group (black bar). †P<0.05 versus the group treated with 0.5 mM metformin (e). †P<0.05 versus the group treated with OKA alone, and #P<0.05 versus the group treated with both OKA and metformin (g). †P<0.05 versus the group treated with 0.5 mM metformin, and #P<0.05 versus the group treated with 1 mM metformin (i)
The reduction of phospho-Ser129 hα-syn caused by metformin in HeLa cells required phosphatase activity as no decrease was seen in cells coincubated with metformin and OKA (Figures 5f and g). Furthermore, measurements of p-RPS6 revealed a significant reduction of this mTOR/PP2A product after treatment of HeLa cells with metformin alone (Figures 5h and i). Data are therefore compatible with the interpretation that, in HeLa cells exposed to metformin, mTOR inhibition and PP2A activation are achieved via AMPK-independent pathways. Then, similar to the results obtained in SH-SY5Y cultures, this mTOR/PP2A signaling leads to a reduction of phospho-Ser129 hα-syn.
Metformin decreases phospho-Ser 129 α-syn in primary cultures of hippocampal neurons and in the mouse brain
A final set of in vitro experiments was carried out in primary cultures of hippocampal neurons. This model system was chosen after both immunohistochemical and biochemical evaluations revealed that normal (i.e., nontransfected) neurons isolated from embryonic mouse hippocampus featured robust α-syn expression and, at the same time, contained detectable levels of phospho-Ser129 α-syn (Figures 6a–c and d). Measurements in cultures incubated in the absence or presence of metformin showed a significant difference in the ratio of phosphorylated α-syn over total α-syn; this ratio was decreased because of a reduction of phospho-Ser129 α-syn (Figures 6d–f). It is noteworthy that metformin was effective at concentrations in the 10−6 M range, that is, within the range of concentrations reached in the plasma of patients treated with this antidiabetic drug.26
Figure 6.

Effect of metformin on phospho-Ser129 α-syn levels in primary hippocampal neurons. Representative images of primary hippocampal neurons immunostained with antibodies against α-syn (a) and phospho-Ser129 α-syn (b). Colocalization is shown in the merged panel (c). Scale bar=100 μm. Primary hippocampal neurons were incubated for 24 h in the absence or presence of 5 μM metformin. Levels of α-syn, phospho-Ser129 α-syn and tubulin were assayed by immunoblotting (d). Semiquantitative analysis of band intensities is expressed as α-syn/tubulin ratio (e) or phospho-Ser129 α-syn/α-syn ratio (f). Data in (e and f) are the means±S.E.M. of at least three independent experiments. *P<0.05 versus the control group (black bar)
To determine whether metformin affected brain levels of phospho-Ser129 α-syn in vivo, the drug was administered to mice using two different regimens. Animals were either treated with 5 g/kg metformin in food pellets for 1 month or with 5 g/l metformin in the drinking water for 6 months. Post-mortem brain (forebrain plus brainstem) tissue was homogenized and used for measurements of total α-syn and phospho-Ser129 α-syn. Regardless of the administration regimen, metformin caused a significant reduction of phosphorylated protein; the ratio of phospho-Ser129 α-syn over total α-syn was decreased by ∼25% and 50% in animals treated with metformin for 1 month and 6 months, respectively (Figures 7a–c and d–f).
Figure 7.

Effect of metformin on phospho-Ser129 α-syn levels in the mouse brain. Mice received 5 g/kg metformin in the food pellet for 1 month (a, b and c) or 5 g/l metformin in the drinking water for 6 months (d, e and f). Levels of α-syn, phospho-Ser129 α-syn, tubulin and actin were assayed by immunoblotting of the brain homogenates (a and d). Semiquantitative analysis of band intensities is expressed as α-syn/tubulin ratio (b), α-syn/actin ratio (e) or phospho-Ser129 α-syn/α-syn ratio (c and f). Data in (b and c) are the means±S.E.M. of 20 mice/group. Data in (e and f) are the means±S.E.M. of four mice/group. **P<0.01 and ***P<0.001 versus the respective control group (black bar)
Discussion
Results of this study demonstrate that induction of phosphatase activity represents an effective strategy to lower phospho-Ser129 α-syn levels. First evidence in support of this conclusion was provided by Lee et al.10 who, in an earlier investigation, were able to enhance phosphatase activity via inhibiting PP2A demethylation; reduced demethylation and consequent enzyme stimulation ultimately led to a decrease in phospho-Ser129 α-syn. Here, we describe a different mechanism of PP2A activation and reduction of phospho-Ser129 α-syn that involves the mTOR signaling pathway and is featured by a widely used pharmacologic agent, the antidiabetic drug metformin.
The property of metformin to decrease phospho-Ser129 α-syn was documented in three in vitro systems. In SH-SY5Y and HeLa cells, the drug was effective at concentrations in the 10−3 M range, whereas a reduction of phospho-Ser129 α-syn was achieved in primary cultures of hippocampal neurons at relatively lower concentrations (in the 10−6 M range). Most likely, these differences in drug efficacy reflect variations in metformin's ability to gain access into cells. Because metformin is actively carried across cell membranes via organic cation transporters (OCTs),27 its intracellular action would depend upon the presence and level of expression of these transporters. A need for high drug concentrations in our experiments with SH-SY5Y and HeLa cells is therefore consistent with the reported deficiency of OCT expression in these cells.28, 29 On the other hand, the presence of OCTs in brain regions such as the hippocampus and cerebral cortex likely underlies drug effectiveness in primary hippocampal cultures as well as in vivo after administration to mice.30
In both SH-SY5Y and HeLa cells treated with metformin, lowering of phospho-Ser129 α-syn was paralleled by a decrease p-RPS6, supporting a relationship between reduced α-syn phosphorylation and mTOR/PP2A signaling. Direct evidence in support of this relationship came from experiments in the presence of the canonical mTOR inhibitor, rapamycin. Data revealed that, similar to metformin, rapamycin induced a marked decrease in phospho-Ser129 α-syn. At least two mechanisms could explain these findings. mTOR inhibition is known to induce autophagic pathways of protein degradation.31 It is possible, therefore, that lower levels of phosphorylated protein may be a consequence of enhanced clearance of unmodified α-syn and/or phospho-Ser129 α-syn. The second mechanism linking mTOR to decreased phospho-Ser129 α-syn relates to mTOR–PP2A interactions by which mTOR suppression can lead to but also result from PP2A activation.20, 32 For example, mTOR-dependent phosphorylation of α4 (mammalian homolog of yeast Tap42) promotes the formation of an α4–PP2A complex that maintains the enzyme in a suppressed state. When mTOR signaling is suppressed, dissociation of this complex would favor the assembly of all three PP2A subunits (A, B and C) and, ultimately, lead to phosphatase activation.20
Experiments were designed to discriminate between these two mechanisms, that is, induction of autophagy or enhanced phosphatase activity via mTOR/PP2A signaling. Data indicated that levels of total α-syn remained unchanged in the presence of either metformin or rapamycin, making it unlikely that reduced phosphorylation would result from enhanced degradation of the unmodified protein. A significant role of autophagy in decreasing phospho-Ser129 α-syn was also ruled out by experiments in which cells were treated with either metformin or rapamycin in the presence of the protein phosphatase blocker OKA. Under these conditions, no reduction of phosphorylated protein was observed, consistent with the interpretation that the ability of metformin and rapamycin to lower phospho-Ser129 α-syn requires phosphatase activity. Taken together, data support a sequence of drug-induced events involving mTOR inhibition, PP2A activation and enhanced dephosphorylation of phospho-Ser129 α-syn.
Following treatment of SH-SY5Y cells with metformin, increased levels of p-ACC indicated activation of AMPK. AMPK is known to down-regulate mTOR.18, 19 Therefore, drug-induced AMPK stimulation could be an upstream signal leading to mTOR-dependent phospho-Ser129 α-syn reduction in SH-SY5Y cells. It is noteworthy, however, that this AMPK pathway is not the only mechanism that could mediate the effect of metformin on phosphorylated α-syn. This conclusion is supported by our findings in HeLa cells. HeLa cells lack LKB1, a kinase required for metformin-induced AMPK activation.23, 24 Nonetheless, a significant decrease in phospho-Ser129 α-syn was observed in these cells after incubation with the drug. Findings in HeLa cells also revealed that (1) metformin exposure lowered p-RPS6 levels, and (2) OKA counteracted metformin-induced reduction of phospho-Ser129 α-syn. Thus, results indicate that mTOR inhibition, PP2A activation and consequent α-syn dephosphorylation can still be triggered by metformin via AMPK-independent pathways. One of these pathways may involve metformin-induced inhibition of Rag proteins, a family of GTPases capable of stimulating mTOR signaling.33 Another pathway could relate to the ability of metformin to increase the expression of REDD1 (regulated in development and DNA damage responses 1), a negative regulator of mTOR.34 Finally, AMPK-independent α-syn dephosphorylation may result from a direct effect of metformin on the binding between the ubiquitin ligase MID1 and PP2A. Dissociation of this MID1/PP2A complex by metformin would result in reduced PP2A degradation, enhanced phosphatase activity and mTOR downregulation.11, 35
In summary, the results of this study elucidate a new mechanism leading to α-syn dephosphorylation and identify two drugs, metformin and rapamycin, capable of decreasing α-syn phosphorylation. The precise role of Ser129 phosphorylation in α-syn pathology has yet to be fully understood.5, 6 Nevertheless, several lines of evidence indicate that preventing hyperphosphorylation is likely to attenuate α-syn toxic potential and may represent a valuable therapeutic strategy.10, 36, 37, 38 Particularly relevant in this respect are the results of a previous study using the agent EHT that, similar to metformin and rapamycin, reduces phospho-Ser129 α-syn levels by enhancing PP2A activity.10 A diet supplemented with EHT was found to ameliorate neuropathological features and behavioral deficits in transgenic mice overexpressing hα-syn, thus supporting a relationship between decreased α-syn phosphorylation and neuroprotection.10
A final important note concerns the double effect of metformin on α-syn and tau; metformin-induced PP2A activation not only reduces phospho-Ser129 α-syn but was also found to counteract the accumulation of hyperphosphorylated tau.11 Tau and α-syn aggregates are pathological hallmarks of Alzheimer's and Parkinson's disease, respectively, but can also frequently coexist in the brain of patients with typical or atypical (e.g., PD with dementia) forms of these diseases.39, 40 Copathology is associated with more pronounced clinical manifestations and less benign prognosis.39, 40 Moreover, synergistic tau-α-syn interactions of pathologic relevance are indicated by experimental findings both in vitro and in animal models, including evidence of mutually enhanced protein fibrillation.36, 41, 42 Mixed-aggregate pathology and synergistic injury underscore the relevance of therapeutics capable of targeting both α-syn and tau. Metformin features this double action and, for this reason, its potential use against human neurodegenerative diseases warrants consideration.
Materials and Methods
Chemicals
Metformin, retinoic acid and OKA were purchased from Sigma Aldrich (St. Louis, MO, USA). Rapamycin was obtained from Invitrogen (Carlsbad, CA, USA).
Cell cultures
SH-SY5Y cells with tetracycline-inducible expression of wild-type hα-syn were generated as described by Hasegawa et al.43 They were cultured in DMEM/Ham F12 (1 : 1) medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin (pen/strp), 5 mg/ml blasticidin and 300 mg/ml zeocin (all from Invitrogen). Cells were differentiated with 50 μM retinoic acid for 10 days. Then, hα-syn expression was induced with addition of 1 mg/ml tetracycline (Invitrogen) for 24 h. Treatment with metformin began 72 h after induction.
HeLa T-Rex cells were obtained from Invitrogen. They were cultured in DMEM GlutaMAX supplemented with 10% FBS, 1% pen/strp and 5 mg/ml blasticidin. Cells were transiently transfected with pcDNA3.1 (−) wild-type hα-syn using lipofectamine 2000 (Invitrogen). Treatment with metformin began 24 h after transfection.
For primary cultures, hippocampal tissue was isolated from E16 embryonic mouse brains and placed in buffer (pH 7.4) containing 120 mM NaCl, 5 mM KCl, 25 mM HEPES and 9.1 mM glucose. Tissues were dissociated by incubation with trypsin-EDTA (Invitrogen) at 37°C for 10 min. Cells were then resuspended in Neurobasal Medium (Invitrogen) containing 5% FBS, 1% pen/strp, 0.5 mM glutamine and 2% B27 supplement (Invitrogen) and seeded at a density of 1 × 106 onto 3.5 mm dishes coated with poly-L-lysine (Sigma Aldrich). On the next day, the volume of incubation medium was diluted (1 : 1) with the addition of medium devoid of FBS. Then, half of the medium volume was replaced every 48–72 h. Glial cells were killed by addition of AraC (cytosine β-D-arabinofuranoside; Sigma Aldrich) to the culture media at days 2–3. Treatment with metformin began 7 days after cell seeding.
Animals
Female C57BL/6 mice of 10 weeks of age were purchased from Charles River (Sulzfeld, Germany). Animals were housed in a room with a 12/12-h light/dark cycle and free access to food and water. Experimental protocols were approved by the ethical committee of the State Agency for Nature, Environment and Consumer Protection in North Rhine Westphalia. A set of mice was treated with 5 g/kg metformin in their food for 1 month. The drug was mixed with powdered diet and pressed into pellets (ssniff Spezialdiäten GmbH, Soest, Germany). Control animals received food pellets devoid of metformin obtained from the same vendor. A second set of mice was treated with 5 g/l metformin in the drinking water for 6 months. Weight of the animals was checked throughout and at the end of the experiments; no difference was found between the control and treated groups. Mice were killed by cervical dislocation, and their brains (forebrains plus brainstems) were removed and processed immediately for immunoblotting.
Immunoblotting
Brains were homogenized in ice-cold RIPA buffer (pH 8.0) containing protease (cOmplete; Roche Diagnostics, Mannheim, Germany) and phosphatase (PhosStop; Roche) inhibitors; samples were then incubated at 4°C in a rotating carousel for 1 h. Similarly, HeLa cells, SH-SY5Y cells and primary hippocampal neurons were harvested in phosphate buffer saline (PBS), resuspended in RIPA buffer containing protease and phosphatase inhibitors and kept on ice for 20 min. Samples were centrifuged at 12 000 × g for 30 min and, for all experiments with one exception (see below), measurements were carried out in supernatants. In one set of experiments (data are reported in Figures 1e and f), levels of hα-syn and phospho-Ser129 hα-syn were also assayed in the pellet fraction. For these studies, pellets were resuspended and sonicated in PBS containing 8 M urea and protease and phosphatase inhibitors. Proteins were separated by either 15% (tubulin, actin, RPS6, p-RPS6, α-syn and phospho-Ser129 α-syn) or 7% (ACC, p-ACC and LKB1) SDS-PAGE. Samples were transferred to nitrocellulose membrane, blocked with 5% fat-free dry milk and incubated with the following primary antibodies against: α-syn (1 : 1000; BD Transduction Laboratories, San Jose, CA, USA), phospho-Ser129 α-syn (1 : 1000; Wako Pure Chemical Industries, Osaka, Japan), RPS6 (1 : 1000; Cell Signalling Technology, Boston, MA, USA), p-RPS6 (1 : 1000; Cell Signalling Technology), ACC (1 : 1000; Cell Signalling Technology), p-ACC (1 : 1000; Cell Signalling Technology), LKB1 (1 : 1000; Cell Signalling), α-tubulin (1 : 30 000; Sigma Aldrich) and actin (1 : 2000; Sigma Aldrich). Finally, blots were incubated with anti-mouse (1 : 10 000) or anti-rabbit (1 : 10 000) horseradish peroxidase-conjugated secondary antibodies, or with anti-mouse (1 : 5000) or anti-rabbit AP (1 : 5000) alkaline peroxidase-conjugated secondary antibodies (all from Promega Corporation, Madison, WI, USA). Because levels of phospho-Ser129 α-syn are relatively low both in vitro and in vivo, detection of this phosphorylated protein was enhanced by (1) loading greater amounts of cell/tissue proteins onto gels (e.g., 50 and 25 μg protein for phospho-Ser129 α-syn and total α-syn, respectively), (2) using a more sensitive developing reagent (Tropix CDP Star; Applied Biosystems, Foster City, CA, USA) and (3) prolonging exposure time.
Immunocytochemistry
For immunocytochemical staining, SH-SY5Y cells or primary hipocampal neurons were plated onto cover slips coated with poly-L-lysine. Cells were rinsed with PBS, fixed with 4% paraformaldehyde and permeabilized with Tris-HCl buffer (pH 7.4) containing 0.25% Triton for 20 min. Nonspecific epitopes were blocked with 1% BSA. Then, incubations with primary antibodies were carried out in buffer containing 0.1% Triton at 4°C for 16 h. The following antibodies were used: anti-α-syn (1 : 1000; BD Transduction Laboratories), anti-phospho-Ser129 α-syn (1 : 200; ab51253 from Abcam plc, Cambridge, UK) and anti-β-III tubulin (1 : 5000; Sigma Aldrich). Phospho-Ser129 α-syn was detected using biotinylated goat-anti-rabbit (1 : 200; Vector Laboratories, Burlingame, CA, USA) and streptavidin-conjugated Dylight fluorophore 488 (1 : 400; Vector Laboratories). α-Syn and β-III Tubulin were detected directly with Dylight 594 (1 : 400; Vector Laboratories) secondary antibody. In some experiments, nuclear staining was achieved by incubation with 4',6-diamidino-2-phenylindole (Dapi) (Biotium Inc., Hayward, CA, USA). Images were acquired with a Zeiss LSM 710 NLO confocal microscope (Jena, Germany).
Statistical analysis
Differences among means were analyzed using one-way ANOVA or Student's t-test (when comparisons involved only two experimental groups). The post hoc analysis was used when differences were observed in ANOVA testing (P<0.05).
Acknowledgments
This study was supported by grants from the Backus Foundation, the Blanche A Paul Foundation and the Centres of Excellence in Neurodegeneration Research (CoEN). We thank Drs. Raffaella Rusconi, Ruth Musgrove, Ayse Ulusoy and Sarah Jewell for assistance with the experiments and manuscript review.
Glossary
- α-Syn
α-synuclein
- PD
Parkinson's disease
- LBs
Lewy bodies
- LN
Lewy neuritis
- PP2a
protein phosphatase 2a
- EHT
eicosanoyl-5-hydroxytryptamide
- mTOR
mammalian target of rapamycin
- hα-syn
human α-synuclein
- OKA
okadaic acid
- AMPK
AMP-activated protein kinase
- p-ACC
phospho-ser79 acetyl-CoA carboxylase
- p-RPS6
phospho-Ser240/244 S6 ribosomal protein
- LKB1
liver kinase B1
- OCT
organic cation transport
- FBS
fetal bovine serum
- PBS
phosphate buffer saline
- Pen/Strp
penicillin/streptomycin
The authors declare no conflict of interest.
Footnotes
Edited by A Verkhratsky
References
- Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzhemer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS, et al. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol. 2002;4:160–164. doi: 10.1038/ncb748. [DOI] [PubMed] [Google Scholar]
- Alonso AD, Grundke-Iqbal I, Barra HS, Iqbal K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci USA. 1997;94:298–303. doi: 10.1073/pnas.94.1.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goerdert M. α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proc Natl Acad Sci USA. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JP, Walker DE, Goldstein JM, de Laat R, Balducci K, Caccavello RJ, et al. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J Biol Chem. 2006;281:29739–29752. doi: 10.1074/jbc.M600933200. [DOI] [PubMed] [Google Scholar]
- Oueslati A, Fournier M, Lashuel HA. Role of post-translational modifications in modulating the structure, function and toxicity of α-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T, et al. Constitutive phosphorylation of the Parkinson's disease associated α-synuclein. J Biol Chem. 2000;275:390–397. doi: 10.1074/jbc.275.1.390. [DOI] [PubMed] [Google Scholar]
- Arawaka S, Wada M, Goto S, Karube H, Sakamoto M, Ren CH, et al. The role of G-protein-coupled receptor kinase 5 in pathogenesis of sporadic Parkinson's disease. J Neurosci. 2006;26:9227–9238. doi: 10.1523/JNEUROSCI.0341-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M, et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J Biol Chem. 2010;285:2807–2822. doi: 10.1074/jbc.M109.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK, et al. Enhanced phosphatase activity attenuates α-synucleinopathy in a mouse model. J Neurosci. 2011;31:6963–6971. doi: 10.1523/JNEUROSCI.6513-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kickstein E, Krauss S, Thornhill P, Rutschow D, Zeller R, Sharkey J, et al. Biguanide metformin acts on tau phosphorylation via mTOR/protein phosphatase 2A (PP2A) signaling. Proc Natl Acad Sci USA. 2010;107:21830–21835. doi: 10.1073/pnas.0912793107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pahlman S, Ruusala AI, Abrahamsson L, Mattsson ME, Esscher T. Retinoic acid-induced differentiation of cultured human neuroblastoma cells: a comparison with phorbolester-induced differentiation. Cell Differ. 1984;14:135–144. doi: 10.1016/0045-6039(84)90038-1. [DOI] [PubMed] [Google Scholar]
- Swingle M, Ni L, Honkanen RE. Small-molecule inhibitors of ser/thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol Biol. 2007;365:23–38. doi: 10.1385/1-59745-267-X:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau RT, Conrad DM, Hoskin DW. Apoptosis induced by protein phosphatase 2A (PP2A) inhibition in T leukemia cells is negatively regulated by PP2A-associated p38 mitogen-activated protein kinase. Cell Signal. 2007;19:139–151. doi: 10.1016/j.cellsig.2006.05.030. [DOI] [PubMed] [Google Scholar]
- Van Kanegan MJ, Adams DG, Wadzinski BE, Strack S. Distinct protein phosphatase 2A heterotrimers modulate growth factor signaling to extracellular signal-regulated kinases and Akt. J Biol Chem. 2005;280:36029–36036. doi: 10.1074/jbc.M506986200. [DOI] [PubMed] [Google Scholar]
- Bialojan C, Takai A. Inhibitory effect of a marine-sponge toxin, okadaic acid, on protein phosphatases. Specificity and kinetics. Biochem J. 1988;256:283–290. doi: 10.1042/bj2560283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–23980. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]
- Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin-dependent translation initiation in breast cancer cells. Cancer Res. 2007;67:10804–10812. doi: 10.1158/0008-5472.CAN-07-2310. [DOI] [PubMed] [Google Scholar]
- Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- Ha J, Daniel S, Broyles SS, Kim KH. Critical phosphorylation sites for acetyl-CoA carboxylase activity. J Biol Chem. 1994;269:22162–22168. [PubMed] [Google Scholar]
- Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012;441:1–21. doi: 10.1042/BJ20110892. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, et al. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–2008. doi: 10.1016/j.cub.2003.10.031. [DOI] [PubMed] [Google Scholar]
- Tiainen M, Ylikorkala A, Makela TP. Growth suppression by Lkb1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA. 1999;96:9248–9251. doi: 10.1073/pnas.96.16.9248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memmott RM, Dennis PA. LKB1 and mammalian target of rapamycin as predictive factors for the anticancer efficacy of metformin. J Clin Oncol. 2009;27:e226. doi: 10.1200/JCO.2009.25.3963. [DOI] [PubMed] [Google Scholar]
- Lalau JD, Lemaire-Hurtel AS, Lacroix C. Establishment of a database of metformin plasma concentrations and erythrocyte levels in normal and emergency situations. Clin Drug Investig. 2011;31:435–438. doi: 10.2165/11588310-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Choi MK, Song IS. Organic cation transporters and their pharmacokinetic and pharmacodynamic consequences. Drug Metab Pharmacokinet. 2008;23:243–253. doi: 10.2133/dmpk.23.243. [DOI] [PubMed] [Google Scholar]
- Ahlin G, Hilgendorf C, Karlsson J, Szigyarto CA, Uhlen M, Artursson P. Endogenous gene and protein expression of drug-transporting proteins in cell lines routinely used in drug discovery programs. Drug Metab Dispos. 2009;37:2275–2283. doi: 10.1124/dmd.109.028654. [DOI] [PubMed] [Google Scholar]
- Ciarimboli G, Deuster D, Knief A, Sperling M, Holtkamp M, Edemir B, et al. Organic cation transporter 2 mediates cisplatin-induced oto- and nephrotoxicity and is a target for protective interventions. Am J Pathol. 2010;176:1169–1180. doi: 10.2353/ajpath.2010.090610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee G, Dallas S, Hong M, Bendayan R. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev. 2001;53:569–596. [PubMed] [Google Scholar]
- Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu E, Knutzen CA, Krauss S, Schweiger S, Chiang GG. Control of mTORC1 signaling by the Opitz syndrome protein MID1. Proc Natl Acad Sci USA. 2011;108:8680–8685. doi: 10.1073/pnas.1100131108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, et al. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase-dependent manner. Cell Metab. 2010;11:390–401. doi: 10.1016/j.cmet.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand-Brustel Y, Auberger P, et al. Metformin, independent of AMPK, induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer Res. 2011;71:4366–4372. doi: 10.1158/0008-5472.CAN-10-1769. [DOI] [PubMed] [Google Scholar]
- Trockenbacher A, Suckow V, Foerster J, Winter J, Krauss S, Ropers HH, et al. MID1, mutated in Opitz syndrome, encodes an ubiquitin ligase that targets phosphatase 2A for degradation. Nat Genet. 2001;29:287–294. doi: 10.1038/ng762. [DOI] [PubMed] [Google Scholar]
- Khandelwal PJ, Dumanis SB, Feng LR, Maguire-Zeiss K, Rebeck G, Lashuel HA, et al. Parkinson-related parkin reduces α-synuclein phosphorylation in a gene transfer model. Mol Neurodegener. 2010;5:47. doi: 10.1186/1750-1326-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbrano F, Sahin G, Farran A, Soares S, Jensen PH, Kirik D, et al. Ser129D mutant α-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson's disease. Neurobiol Dis. 2013;56:47–58. doi: 10.1016/j.nbd.2013.03.014. [DOI] [PubMed] [Google Scholar]
- Braithwaite SP, Stock JB, Mouradian MM. α-Synuclein phosphorylation as a therapeutic target in Parkinson's disease. Rev Neurosci. 2012;23:191–198. doi: 10.1515/revneuro-2011-0067. [DOI] [PubMed] [Google Scholar]
- Galpern WR, Lang AE. Interface between tauopathies and synucleinopathies: a tale of two proteins. Ann Neurol. 2006;59:449–458. doi: 10.1002/ana.20819. [DOI] [PubMed] [Google Scholar]
- Irwin DJ, Lee VM, Trojanowski JQ. Parkinson's disease dementia: convergence of α-synuclein, tau and amyloid-β pathologies. Nat Rev Neurosci. 2013;14:626–636. doi: 10.1038/nrn3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Forman MS, Higuchi M, Golbe LI, Graves CL, Kotzbauer PT, et al. Initiation and synergistic fibrillization of tau and α-synuclein. Science. 2003;300:636–640. doi: 10.1126/science.1082324. [DOI] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. doi: 10.1016/j.cell.2013.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa T, Konno M, Baba T, Sugeno N, Kikuchi A, Kobayashi M, et al. The AAA-ATPase VPS4 regulates extracellular secretion and lysosomal targeting of α-synuclein. PLoS One. 2011;6:e29460. doi: 10.1371/journal.pone.0029460. [DOI] [PMC free article] [PubMed] [Google Scholar]