

Abstract
Bone morphogenetic protein 2 (BMP2) is known to activate unfolded protein response (UPR) signaling molecules, such as BiP (IgH chain-binding protein), PERK (PKR-like ER-resistant kinase), and IRE1α. Inositol-requiring enzyme-1a (IRE1a), as one of three unfolded protein sensors in UPR signaling pathways, can be activated during ER stress. Granulin-epithelin precursor (GEP) is an autocrine growth factor that has been implicated in embryonic development, tissue repair, tumorigenesis, and inflammation. However, the influence on IRE1a in BMP2-induced osteoblast differentiation has not yet been elucidated. Herein we demonstrate that overexpression of IRE1a inhibits osteoblast differentiation, as revealed by reduced activity of alkaline phosphatase (ALP) and osteocalcin; however, knockdown of IRE1a via the RNAi approach stimulates osteoblastogenesis. Mechanistic studies revealed that the expression of IRE1a during osteoblast was a consequence of JunB transcription factor binding to several AP1 sequence (TGAG/CTCA) in the 5′-flanking regulatory region of the IRE1a gene, followed by transcription. In addition, GEP induces IRE1a expressions and this induction of IRE1a by GEP depends on JunB. Furthermore, IRE1a inhibition of GEP-induced osteoblastogenesis relies on JunB. Besides, GEP is required for IRE1a inhibition of BMP2-induced bone formation. Collectively, these findings demonstrate that IRE1a negatively regulates BMP2-induced osteoblast differentiation and this IRE1a inhibition effect depends on GEP growth factor. Thus, IRE1a, BMP2, GEP growth factor, and JunB transcription factor form a regulatory loop and act in concert in the course of osteoblastogenesis.
Keywords: inositol-requiring enzyme-1a, osteoblastogenesis, GEP, BMP2, unfolded protein response
It is known that the differentiation of uncommitted mesenchymal cells to osteoblasts is a fundamental process in embryonic development and the repair of bone defect. The bone morphogenetic proteins (BMPs) are important regulators of this process and have a key role in the BMP2-induced osteogenic differentiation and bone formation.1, 2, 3 Recently, another BMP2 signaling pathway in osteoblasts, mediated by the unfolded protein response (UPR), was introduced by Murakami et al.4 The expression levels of the ER stress markers, BiP (IgH chain-binding protein), CHOP (C/EBP homologous protein) and EDEM (ER degradation- enhancing α-mannosidase-like protein), were upregulated by BMP2 stimulation. Jang et al.5 reported that BMP2 stimulated ATF6 (activating transcription factor 6) transcription by enhancing the direct binding of RUNX2 to the OSE2 (osteoblast-specific cis-acting element 2) motif of the ATF6 promoter region.
Inositol-requiring enzyme-1a (IRE1a), as one of three unfolded protein sensors in UPR signaling pathways, senses ER unfolded proteins through an ER lumenal domain that becomes oligomerized during ER stress.6, 7 On activation in response to ER stress, IRE1 has at least two different actions in mammalian cells. First, the endoribonuclease activity of IRE1 cleaves XBP1U mRNA, converting it into a potent transcriptional activator XBP1S (X-box binding protein1 spliced). This cleavage initiates an unconventional splicing reaction, which leads to the production of an active transcription factor and induction of various adaptive genes.8, 9 Second, recent studies have shown that IRE1 links ER stress to the activation of JNK signaling pathways. IRE1α has a central role in the ER stress response.10, 11
Granulin-epithelin precursor (GEP), also known as progranulin, is a 593-amino acid-secreted glycoprotein with an apparent molecular weight of 80 kDa.12, 13, 14 It has been isolated as a differentially expressed gene from mesothelial differentiation,15 macrophage development,16 sexual differentiation of the brain,17 synovium in rheumatoid arthritis, and osteoarthritis.18 Although GEP functions mainly as a secreted growth factor, it was also found to be localized inside cells and to directly modulate intracellular activities.19, 20, 21, 22 The role of GEP in the stimulation of cellular proliferation has been well characterized. In addition, more evidence implicated that GEP is involved in the regulation of differentiation, development, and pathological processes. GEP was also shown to be a crucial mediator of wound response and tissue repair.23, 24, 25 In our previous study, we reported that GEP induced chondrocyte differentiation, endochondral bone formation, and cartilage repair.26 Furthermore, GEP was reported to promote bone regeneration and formation.27 GEP also regulates myogenic differentiation by reducing MyoD, an important transcription factor for myogenesis.28
It was known that IRE1 is involved in the switch between the prosurvival UPR, differentiation, and initiation of cell death pathways during ER stress. In mammalian cells, the termination of IRE1 activity is an important factor in controlling cell fate after UPR activation.29, 30 We previously found that BMP2 induces ER stress during chondrocyte differentiation and activates the IRE1α-XBP1 pathway.31 However, little is known about the modulation and physiological significance of IRE1a in osteoblast differentiation. Specifically, whether IRE1a influences GEP-mediated osteoblastogenesis have not been delineated; the molecular mechanism on how to regulate GEP-mediated osteoblastogenesis by IRE1a also remains unknown. In this study, we report that (1) IRE1a inhibits the BMP2-mediated osteogenic differentiation; (2) JunB upregulates endogenous IRE1a expression and enhance this inhibition; (3) GEP induces IRE1a expressions and this induction depends on JunB; (4) GEP is required for IRE1a inhibition of BMP2-induced bone formation; (5) IRE1a inhibition of GEP-induced osteoblastogenesis is, at least partially, mediated by the transcription factor JunB. Thus, IRE1a, BMP2, GEP, and JunB constitute a regulatory feedback loop during osteoblast differentiation and bone formation.
Results
Overexpression of IRE1a inhibits the BMP2-mediated osteogenic differentiation
BMP2 potently induces osteoblast differentiation in a variety of cell types and controls the expression of genes that are activated during osteoblast differentiation, including ALP and osteocalcin (OCL).32, 33 It is reported that IRE1α is associated with the regulation of osteoblast differentiation and bone formation,34, 35 but the molecular mechanism by which IRE1a regulates osteogensis remains unknown. To address this issue, we sought to determine whether IRE1a affect the BMP2 activity by using the pluripotent mesenchymal C2C12 cells, a well-established cell model for studying osteogenesis in vitro.36, 37
For this purpose, we generated a recombinant adenovirus encoding IRE1a and detected the influence on BMP2 expression; next we determined the effect of IRE1a on the BMP2-mediated activity of ALP and production of OCL, two marker genes widely used for osteoblasts. As shown in Figures 1a and b, 300 ng/ml BMP2 led to a high ALP activity and a robust production of OCL. This BMP2-dependent induction of ALP and OCL was dramatically inhibited by infection with Ad-IRE1a in a dose-dependent manner. More significantly, similar results were also observed with primary bone marrow stromal (BMSC) cells. In addition, Ad-IRE1a inhibits the expression of BMP2 in the course of osteogenesis in a dose-dependent manner (Figures 1c and e). Figures 1d and f are the semi-quantification of relative levels of BMP2 and IRE1α in the C2C12 and BMSC. These data suggest IRE1a is a potent inhibitor of BMP2-mediated gene activation in the course of osteogenesis.
Figure 1.

IRE1a inhibits the BMP2-induced osteogenesis assayed by ALP and OCL. (a) IRE1a inhibits the BMP2-dependent ALP activity in a dose-dependent manner. C2C12 cell lines and BMSCs were infected either Ad-GFP (MOI=50, serves as a control) or BMP2 (300 ng/ml) with or without IRE1a (at different MOI) for 4 days and the cell lysates were used for determining the ALP activity. (b) IRE1a inhibits the BMP2-dependent OCL production in a dose-dependent manner. C2C12 cell lines and BMSCs were infected as described in a and the cell culture media were used for determining the OCL level. (c and e) Expression of IRE1a and BMP2 in C2C12 and BMSCs infected with either Ad-GFP (MOI=50, serves as a control) or BMP2 (300 ng/ml) with or without IRE1a (at different MOI=10, 20, 50) for 4 days. Cell lysates were prepared from C2C12 (c) and BMSCs (e) infected with various adenoviruses, as indicated, and detected by western blotting with anti-IRE1a, anti-BMP2, and anti-tubulin (internal control) antibodies. (d and f) Semi-quantification of protein relative levels of BMP2 and IRE1α in the C2C12 (d) and BMSCs (f) infected with various adenoviruses, as indicated. Levels were normalized against those of tubulin by MJ Opticon Monitor Analysis Software (Bio-Rad); data were expressed as means±S.D. (n=3). Every treatment group compared with control groups, respectively, *P<0.05 or **P<0.01
Knockdown of IRE1a stimulates BMP2-induced osteoblast differentiation
Next, we determined how to influence BMP2-induced osteoblastogenesis by knocking down IRE1a using the small interfering RNA (siRNA) approach. A real-time PCR was performed to verify the RNA level of IRE1a after infected siIRE1a adenovirus. As shown in Figure 2a, infection with siIRE1a1 adenovirus resulted in 78% reduction and in 70% reduction with siIRE1a2 adenovirus. C2C12 cells infected with siIRE1a1, siIRE1a2 adenovirus, or control adenovirus (CTR) were treated with BMP2. As shown in Figure 2, knockdown of IRE1a clearly increased the expression of BMP2 in C2C12 cells (Figure 2b) and BMSC cells (Figure 2c). Figure 2d is the semi-quantification of protein relative levels of BMP2 in the C2C12 and BMSC infected with siIRE1a1 and siIRE1a2 adenovirus. Next, both cultures infected with siIRE1a1, siIRE1a2 adenovirus, or control adenovirus were cultured in the presence of BMP2 (300 ng/ml) and the effect of siIRE1a on the BMP2-mediated activity of ALP and production of OCL were detected. As shown in Figures 2e and f, prominently enhanced the BMP2-induced ALP activity (Figure 2e) and a robust production of OCL (Figure 2f) in siIRE1a-infected cells were observed compared with those in control cell lines, suggesting that IRE1a is a negative mediator for osteoblast differentiation. BMP2-induced ALP activity and OCL expression in osteoblastogenesis were largely improved by knockdown of IRE1a. These findings clearly indicated that endogenous IRE1a can inhibit BMP2-mediated osteoblast differentiation.
Figure 2.

siIRE1a enhances the BMP2-mediated osteogenesis assayed by ALP and OCL. (a) siRNA against IRE1a mRNA efficiently inhibited expression of endogenous IRE1a in C2C12 cells. C2C12 cells were infected with either siIRE1a adenovirus or control adenovirus (CTR), and total RNA was collected for real-time PCR analysis. Expression of IRE1a was normalized against the GAPDH endogenous control. The normalized values were then calibrated against the control value, here set as 1. *P<0.05. (b and c) Expression of IRE1a and BMP2 in C2C12 and BMSCs infected with indicated adenoviruses. Cell lysates were prepared from C2C12 (b) and BMSCs (c) infected with siIRE1a1, siIRE1a2, and siIRE1a1+siIRE1a2 adenoviruses, as indicated, and detected by western blotting with anti-IRE1a, anti-BMP2, and anti-tubulin (internal control) antibodies. (d) Semi-quantification of protein relative levels of BMP2 and IRE1α in the C2C12 (b) and BMSC(c) infected with various adenoviruses, as indicated in b and c. Levels were normalized against those of tubulin by MJ Opticon Monitor Analysis Software (Bio-Rad); data were expressed as means±S.D. (n=3). Every treatment group compared with control groups, respectively, *P<0.05 or **P<0.01. (e) siIRE1a increases the BMP2-dependent ALP activity. C2C12 cell lines and BMSCs were infected either Ad-GFP (MOI=50, serves as a control) or BMP2 (300 ng/ml) with or without siIRE1a1, siIRE1a2. The cell lysates were used for determining the ALP activity. (f) siIRE1a enhances the BMP2-dependent OCL production. C2C12 cell lines and BMSCs were infected as described in e, and the cell culture media were used for determining the OCL level
JunB binds to the AP1 sequence (GTGAG/CTCAG) in the IRE1a 5′-flanking region
Having determined that IRE1a inhibits BMP2-induced osteoblast differentiation, we next sought to elucidate the transcriptional control mechanism of the IRE1a gene. In our initial sequence analysis of the human IRE1a promoter, we found there are at least four AP1 sequences (TGAG/CTCA) in human IRE1a gene 5′-flanking regulatory region (Figure 3a).38 We then used electrophoretic mobility shift assays (EMSAs) to determine whether JunB can actively bind to these sequences. The JunB-binding sequence (5′-CGCTGCTGACTCAGTTCTGCGTC-3′) within the human IRE1a 5′-flanking region was used as a probe. The EMSA assay revealed that JunB does bind to the sequence (Figure 3b, lane3). This binding was not affected by the addition of unrelated oligonucleotides, but was prevented by unlabeled oligodeoxynucleotide probes (Figure 3b, lanes 1 and 2). As expected, the addition of antibodies to JunB resulted in a ‘super-shift' of the complex (Figure 3b, lane 4). Once the interaction between JunB and the AP1 sequence became clear, we sought to identify the JunB binding element (AP1) in the IRE1a promoter by performing EMSA assay using wild-type (WT) and two mutant AP1 probes. All probes were incubated with nuclear extracts prepared from AdJunB-infected C2C12 cells; the binding of JunB to various probes is summarized and shown in Figure 3c. Mutants 1 and 2, in which the first four nucleotides (TGAC) and the last four nucleotides (CTCA) were altered, respectively, did not bind to JunB (Figure 3c, lanes 2 and 3), demonstrating that these AP1 sequence (TGAG/CTCA) are essential for binding to JunB.
Figure 3.

JunB binds to the AP1 sequence of the IRE1a promoter in vitro (EMSA) and in vivo (ChIP). (a) DNA sequence of the 50-bp AP1 (−122 to −72 bp) of the IRE1a promoter. Putative JunB-binding elements (AP1) are underlined. (b) JunB binds to the AP1 of the IRE1a promoter in vitro (EMSA). Ten micrograms of nuclear extracts (NE) prepared from C2C12 cells transfected with pcDNA3.1(−)-JunB was incubated with Dig-labeled JunB-binding site (ER stress response element) probe in reaction buffer (20 μl). For competition experiments, a 100-fold excess of WT oligodeoxynucleotide was added. After 15 min of incubation, the Dig-labeled AP1 probe was added, and the reaction mixture was incubated for an additional 15 min and analyzed by gel electrophoresis. Arrows indicate free DNA probe (bottom) and DNA/protein complex (up). (c) Mutations of AP1 sequence abolish the binding of JunB to the IRE1a gene promoter (DNA). EMSA was performed using the same protein fractions as in b. Probes are as indicated above the lanes. The specific protein–DNA band is indicated by an arrow. Mutant nucleotides are lowercase and underlined. The binding intensity of JunB to these probes is indicated as follows:+, binding; −, no binding. (d) Myc-tagged JunB binds to the transfected IRE1a-specific reporter construct −2065IRE1aluc. C2C12 cells transfected with −2065IRE1aluc and the expression plasmid pcDNA3.1(−)-JunB were cross-linked by formaldehyde treatment and lysed. Cell lysates were subjected to immunoprecipitation with control IgG (lane 1) or with anti-myc (lane 2) or anti-JunB (lane 3) antibody. Purified DNA from the cell lysate (Input DNA; upper panel) and DNA recovered from immunoprecipitation (IP; lower panel) were amplified by PCR. (e) Endogenous JunB binds to the AP1 sequence of the IRE1a gene. ChIP assays were performed in C2C12 cells treated with BMP2 for 3 days. C2C12 cells treated with formaldehyde were lysed, and DNA was sheared by sonication. Cell lysates were subjected to immunoprecipitation with either control IgG (IgG; lane 1) or anti-JunB antibodies (lane 3). DNA recovered from the immunoprecipitation was amplified by PCR. Input DNA (lane 2) was used as a positive control
We then performed a chromatin immunoprecipitation (ChIP) assay to further confirm that JunB binds to the AP1 sequence in the regulatory region of the IRE1a gene. ChIP analysis was carried out using C2C12 cells infected with Ad-JunB and transfected with −2065IRE1a-luc for 2 days; after reaching confluency, DNA-binding proteins were cross-linked to the DNA using formaldehyde. The cells were lysed, the chromatin was sheared by sonication, and protein–DNA complexes were immunocoprecipitated using IgG (negative control), anti-myc and anti-JunB antibodies. The DNA recovered from the immunoprecipitation was then amplified by PCR using primers that span the JunB-binding site. As shown in Figure 3d, immunoprecipitated complexes were formed using both anti-myc (lane 2 in Figure 3d) and anti-JunB (lane 3 in Figure 3d) antibodies, but not with control IgG (lane 1 in Figure 3d), indicating that myc-tagged JunB binds to the AP1 sequence of the IRE1a promoter in transfected living cells. The PCR results demonstrate that DNA bearing the JunB-binding site was successfully immunoprecipitated by anti-JunB antibodies (Figure 3d, lane 3), but not by control IgG (Figure 3d, lane 1). To determine the role of JunB during osteoblastogenesis, ChIP assays were also performed in C2C12 cells treated with BMP2 (300 ng/ml) for 3 days (Figure 3e). Endogenous protein–DNA complexes were then covalently cross-linked with formaldehyde, and following cell lysis the chromatin was sheared by sonication. Endogenous JunB–DNA complexes were immunoprecipitated with either IgG (negative control; Figure 3e, lane 1) or anti-JunB antibody (Figure 3e, lane 3), and the DNA purified from these co-precipitations and input DNA (positive control; Figure 3e, lane 2) were analyzed by PCR. We observed amplified products of the IRE1a promoter from input DNA and DNA isolated from anti-JunB antibody-precipitated complexes, but not from pre-immune serum. These results demonstrate that endogenous JunB binds to the native AP1 sequence in the IRE1a gene promoter during osteoblast differentiation.
JunB transactivates IRE1a-specific reporter genes
We next sought to determine whether the binding of JunB to AP1 sequences results in the induction of IRE1a expression. For this purpose, four deletion reporter gene plasmids (−2065IRE1a-luc, −959 IRE1a-luc, −614IRE1a-luc, and −426IRE1aluc) were generated in which segments containing AP1 sequences from the 5′-flanking region of IRE1a (−2065 to +252, −959 to +252, −614 to +252, and −426 to +252) were placed upstream of a gene encoding luciferase in the pGL3 vector (Figure 4a). Transfection of the reporter plasmids into C2C12 cells resulted in luciferase expression; constructs containing longer lengths of the IRE1a 5′-flanking region segment demonstrated higher expression levels (Figure 4b). Co-transfection of C2C12 cells with the reporter plasmids and a JunB expression plasmid strongly increased reporter gene expression in a dose-dependent manner (Figure 4b). To dissect the importance of the JunB-binding site in the 5′-flanking region of the IRE1a gene, several point mutation reporter constructs of −2065 IRE1a-luc were generated (Figure 4c) and their transactivation by JunB tested. Mutations in the first JunB-binding site (closer to transcriptional initiation site) and the second binding sites resulted in clear reduction in reporter gene activities, whereas mutations in the third or fourth JunB-binding site gave rise to negligible effects on the JunB-mediated transactivation of the IRE1a-specific reporter gene (Figure 4d). To further verify the role of JunB-binding sites in driving IRE1a expression, the AP1 sequence of the −426IRE1a-luc plasmid was altered (Figure 4e). The alteration of three nucleotides (TCA to GTC) severely reduced the transactivation of the reporter gene by JunB (Figure 4f). This confirms that the AP1 sequences are responsible for driving JunB-mediated IRE1a expression.
Figure 4.

Upregulation of IRE1a-specific reporter constructs and regulation of mutant IRE1a promoter transcriptional activity by JunB. (a) Schematic representation of four IRE1a-specific reporter gene constructs. The indicated segments from the 5′-flanking region of the IRE1a gene were linked to a simian virus 40 5′-flanking region (SV) and a DNA segment encoding luciferase (Luc). AP1 sequences are indicated by ovals. The numbers indicate the distance, in nucleotides, from the first nucleotide of intron 1. (b) JunB can drive the expression of IRE1a-specific reporter gene in C2C12 cells. The indicated reporter constructs were transfected into C2C12 cells together with various amounts of a pcDNA3.1(−)-JunB expression plasmid, as indicated, along with a pSVgal internal control plasmid. Forty eight hours after transfection, the cultures were collected and lysed, and β-galactosidase and luciferase activities were determined. Luciferase activity was normalized to β-galactosidase activity. The relative luciferase activity of the leftmost bar was set to 1. Compared with the cells untransfected with pcDNA3.1(−)-JunB plasmid (0) in each group of indicated reporter constructs. *P<0.05. (c) Schematic of four mutants of −2065IRE1a-luc reporter gene. The AP1 sequence in the −2065IRE1a-luc reporter construct was altered, as indicated. The mutant JunB-binding sites (AP1) are indicated by stars; mutant nucleotides are underlined. (d) Mutations of the JunB-binding sites results in the reduction in JunB-activated reporter gene expression. The indicated reporter construct and the pSVgal internal control plasmid were transfected into C2C12 cells together with 3 mg of pcDNA3.1(−) (control) or the pcDNA3.1(−)-JunB expression plasmid. The cultures were processed and the reporter gene activities analyzed as described in b. Compared with WT reporter construct. *P<0.05. (e) Schematic of mutated JunB-binding site (AP1) in −426IRE1aluc reporter construct. The JunB-binding site (AP1) in −426IRE1a-luc reporter construct was altered, as indicated. The mutant JunB binding site is indicated by a star; mutant nucleotides are indicated by arrows. Compared with WT reporter construct. ** P<0.01. (f) Alteration of the JunB-binding site abolishes JunB-induced reporter gene expression. C2C12 cells were transfected, processed, and the reporter gene activities analyzed as described in b
JunB upregulates endogenous IRE1a expression
To confirm whether exogenous overexpression of JunB results in the increased expression of endogenous IRE1a, we infected C2C12 cells with Ad-JunB and performed real-time PCR (Figure 5a) and immunoblotting (Figure 5b) to determine endogenous IRE1a mRNA and protein levels. As shown in Figure 5a, IRE1a mRNA levels were significantly upregulated in C2C12 cells that were overexpressing JunB, when compared with untreated cells and cells infected with the control Ad-GFP. These results were confirmed by immunoblotting (Figures 5b and c). Taken together, these results demonstrate that JunB upregulates endogenous IRE1a expression in the infected cells. In order to establish whether IRE1a transcription is regulated by JunB physiologically, we then examined the effect of knockdown of JunB on IRE1a mRNA levels in the course of osteoblastogenesis. First, we detected the siJunB adenovirus can significantly suppress JunB expression in C2C12 cells (Figure 5d). Next, C2C12 cells infected with siJunB adenovirus or control adenovirus (CTR) were treated with BMP2 for various time points. As shown in Figure 5e, the knockdown of JunB dramatically inhibited IRE1a expression during osteoblast differentiation.
Figure 5.

JunB upregulates endogenous IRE1a gene expression. (a) Real-time PCR assay of IRE1a mRNA level in C2C12 cell lines infected with Ad-JunB expression plasmid or control Ad-GFP. It is found that IRE1a mRNA was upregulated in C2C12 cell lines infected with Ad-JunB (MOI=50) adenovirus when compared with control C2C12 cells. The normalized values were then calibrated against the control value. The units are arbitrary and the left bar indicates a relative level of IRE1a mRNA of 1; *P<0.05. (b) Exogenous expression of JunB increased the IRE1a gene expression in C2C12 cells. Cell lysates from untreated C2C12 cells (CTR) or from C2C12 cells infected with Ad-JunB (MOI=50) or Ad-GFP were subjected to SDS-PAGE and detected by anti-IRE1a antibody, or anti-JunB, or anti-tubulin, respectively. Tubulin is served as an internal control. (c) Semi-quantification of protein relative levels of JunB and IRE1α in the C2C12 infected with various adenoviruses, as indicated in b. Levels were normalized against those of tubulin by MJ Opticon Monitor Analysis Software (Bio-Rad); data were expressed as means±S.D. (n=3). Every treatment group was compared with control groups, respectively, *P<0.05. (d) siRNA against JunB mRNA efficiently inhibited the expression of endogenous JunB in C2C12 cells. Cells were infected with either siJunB or control siRNA (CTR), and total RNA was collected for real-time PCR. Expression of JunB was normalized against the GAPDH endogenous control. The normalized values were then calibrated against the control value, here set as 1. *P<0.05. (e) Repression of JunB largely abolished IRE1a expression in BMP2-induced C2C12 cells. C2C12 cells were treated with BMP2 (300 ng/ml), then infected with either control siRNA (CTR) or JunB siRNA (siJunB) adenovirus. Total RNA was extracted from the cells and the mRNA levels of IRE1a and GAPDH were measured by real-time PCR at various time points. The units are arbitrary and the relative level of IRE1a mRNA on day 0 was set to 1. All experiments were repeated three times. Every siJunB treatment group was compared with control groups, respectively, *P<0.05
GEP induces IRE1a expressions in vitro and the induction of IRE1a by GEP depends on JunB
We next used C2C12 cells and BMSC cells to examine whether IRE1a is the downstream target of GEP. C2C12 cells and BMSC cells pretreated with 300 ng/ml of BMP2 for 1 week were cultured with or without GEP for various time points and the level of IRE1a mRNA was measured using real-time PCR. As revealed in Figure 6a, IRE1a mRNA was increased to 2.2-fold at day 1 and to 2.3-fold by day 3 in the GEP-untreated control C2C12 cells. GEP enhanced the level of IRE1a mRNA to 4.7-fold at day 1 and to 6.1-fold by day 3 in the GEP-treated C2C12 cells. In the case of BMSC cells, IRE1a mRNA was slightly increased to 1.8-fold at day 1 and to 2.3-fold by day 3 in the GEP-untreated control BMSC cells; GEP significantly induced IRE1a mRNA to 3.2-fold by day 1 and to 4.3-fold by day 3 in the GEP-treated BMSC cells. Collectively, these findings demonstrate that IRE1a is a GEP-inducible gene in osteoblastogenesis. Furthermore, induction of IRE1a by GEP was also visualized via western blotting (Figures 6b and c). To further examine whether the induction of IRE1a by GEP depends on JunB in osteoblast differentiation, first, C2C12 cells were pretreated with 300 ng/ml of BMP2 for 1 week, and then were cultured without (control) or with recombinant 300 ng/ml of GEP and different dosage of siJunB adenvious. The result showed that siJunB abolished GEP-induced IRE1a expression in a dose-dependent manner in the course of BMP2-stimulated osteogenesis. GEP-mediated induction of IRE1a was partially lost when JunB was knocked down by siRNA (Figure 6d). These results indicate that the expression of IRE1a induced by GEP is dependent on JunB in osteogenesis.
Figure 6.

IRE1a expression induced by GEP depends on JunB. (a) GEP induces the expression of IRE1a mRNA, assayed by real-time PCR. C2C12 cells and BMSC cells were pretreated with 300 ng/ml of BMP2 for 1 week, then were cultured without (control) or with recombinant 300 ng/ml of GEP for various time periods, as indicated. The normalized values against GAPDH mRNA were calibrated against controls (day 0), given the value of 1. (b) GEP increase the level of IRE1a protein, assayed by western blotting. C2C12 cells were pretreated with 300 ng/ml of BMP2 for 1 week, then treated with or without 300 ng/ml of GEP for various time periods were stained with IRE1a antibody. Tubulin protein served as an internal control. (c) Semi-quantification of protein relative levels of IRE1α in the C2C12 treated with 300 ng/ml of GEP, as indicated in b. Levels were normalized against those of tubulin by MJ Opticon Monitor Analysis Software (Bio-Rad); data were expressed as means±S.D. (n=3). Every GEP treatment group was compared with control groups, respectively, *P<0.05. (d) siJunB abolished GEP-induced IRE1a expression, assayed by real-time PCR. C2C12 cells were pretreated with 300 ng/ml of BMP2 for 1 week, then were cultured without (control) or with recombinant 300 ng/ml of GEP and different dosage siJunB adenovirus (MOI=10, 20, 50). The normalized values against GAPDH mRNA were calibrated against controls, given the value of 1. The values are the mean±S.D. of three independent experiments. Every GEP+siJunB treatment group was compared with GEP treatment group, respectively, *P<0.05; **P<0.01
GEP is required for IRE1a expression and IRE1a inhibition of GEP-induced osteoblastogenesis relies on JunB
We next determined whether IRE1a expression depends on GEP signaling in vivo via performing immunohistochemistry with the sections of long bone from 18.5-day-old WT (GEP+/+) and GEP-null (GEP−/−) mouse embryos. As expected, IRE1a demonstrates a prominent expression in the proliferating and prehypertrophic chondrocytes in the embryonic growth plate of control mice (Figure 7a, left). However, in GEP knockout (GEP−/−) embryonic growth plates, we found that IRE1a is dramatically reduced (Figure 7a, right), indicating that in vivo expression of IRE1a is tightly controlled by GEP signaling.
Figure 7.

IRE1a expression depends on GEP signaling and inhibits GEP-mediated osteogenesis assayed by ALP and OCL. (a) IRE1a is markedly reduced in the growth plate chondrocytes of GEP-null embryos, revealed by immunohistochemistry. Sections of long bone from 18.5-day-old GEP-null (GEP−/−) and WT (GEP+/+) mouse embryos were stained with anti-IRE1a antibody (brown, indicated with arrows) and counterstained with Methyl green (green). Bar=100 mm. (b) IRE1a reduces the expression of GEP mRNA in the course of BMP2-stimulated osteogenesis, assayed by real-time PCR. C2C12 cells and BMSC cells were pretreated with 300 ng/ml of BMP2 for 1 week, then were cultured with Ad-GFP control (CTR) or with Ad-IRE1a (MOI=50) infected for various time periods, as indicated. The normalized values against GAPDH mRNA were calibrated against controls (day 0), given the value of 1. Every Ad-IRE1a treatment group was compared with control groups, respectively. *P<0.05. (c) IRE1a inhibits the level of IRE1a protein in the course of BMP2-stimulated osteogenesis, assayed by western blotting. C2C12 cells were pretreated with 300 ng/ml of BMP2 for 1 week, then infected with Ad-GFP control (CTR) or without Ad-IRE1a (MOI=50) for various time periods and were stained with GEP antibody. Tubulin protein served as an internal control. (d) IRE1a inhibits the GEP-induced ALP activity in a dose-dependent manner. C2C12 cell lines and BMSCs were infected either Ad-GFP (MOI=50, serves as a control) or 300 ng/ml GEP protein with or without Ad-IRE1a (at different MOI) for 4 days, and the cell lysates were used for determining the ALP activity. (e) IRE1a inhibits the GEP-induced OCL production in a dose-dependent manner. C2C12 cell lines and BMSCs were infected as described in d, and the cell culture media were used for determining OCL level. (f) The IRE1a inhibition GEP-induced ALP activity is required for JunB. C2C12 cell lines and BMSCs were infected either Ad-GFP (MOI=50, serves as a control) or 300 ng/ml GEP protein with or without Ad-IRE1a, Ad-JunB and siJunB for 4 days, then the cell lysates were used for determining the ALP activity. (g) The IRE1a inhibition of GEP-induced OCL production is required for JunB. C2C12 cell lines and BMSCs were infected as described in f and the cell culture media were used for determining OCL level
To confirm whether IRE1a inhibits GEP expression in the course of BMP2-stimulated osteogenesis, C2C12 cells and BMSC cells were pretreated with 300 ng/ml of BMP2 for 1 week, then were cultured without (control) or with Ad-IRE1a (MOI=50) infected for various time points, and the level of GEP mRNA and protein were measured using real-time PCR (Figure 7b) and immunoblotting analysis (Figure 7c), respectively. As shown in Figure 7b, IRE1a reduced the level of GEP mRNA by day 1 and day 3 in comparison with the control group in C2C12 cells and BMSC cells. Furthermore, inhibition of GEP by IRE1a was also visualized via western blotting (Figure 7c). Collectively, these findings demonstrate that IRE1a inhibits GEP expression in osteoblastogenesis. We next investigated whether endogenous IRE1a inhibits GEP-mediated osteoblast differentiation and whether this inhibition relies on JunB. For this purpose, we generated three kinds of recombinant adenoviruses encoding IRE1a, JunB, and siJunB, respectively. It is known that osteoblast differentiation was monitored by examining the expression of ALP and OCL, two marker genes widely used for osteoblasts.32, 33 As revealed in Figures 7d and e, infection of C2C12 cells and BMSC cells with adenovirus encoding IRE1a (Ad-IRE1a), but not control Ad-GFP, decreased the GEP-induced ALP expression and the OCL production. The expression of ALP and the production of OCL were strongly inhibited by infection with Ad-IRE1a, but not control Ad-GFP, in a dose-dependent manner. These results indicated that IRE1a can inhibit GEP-mediated osteoblast differentiation. In addition, the IRE1a inhibition of GEP-induced osteoblastogenesis is dependent on JunB. As revealed in Figures 7f and g, overexpression of JunB can augment this IRE1a inhibition, whereas knocking down JunB using the siRNA approach dramatically abolishes this IRE1a inhibition. These results indicated that JunB is required for, at least in part, the IRE1a inhibition of GEP-triggered osteoblast differentiation.
GEP is required for IRE1a inhibition of BMP2-induced bone formation
We next determined whether GEP was involved in IRE1a inhibition of BMP2-induced osteoblastogenesis. First, we infected C2C12 cells with Ad-IRE1a at different dosage and performed immunoblotting (Figure 8a) and real-time PCR (Figures 8b and c) to determine endogenous BMP2 mRNA and protein levels. As shown in Figures 8b and c, BMP2 mRNA levels were significantly downregulated in C2C12 cells and BMSC cells when compared with untreated cells and cells infected with the control Ad-GFP. These results were confirmed by immunoblotting (Figure 8a). BMP2 protein levels were clearly reduced after infected Ad-IRE1a in C2C12 cells in a dose-dependent manner. Taken together, these findings demonstrate that IRE1a reduces endogenous BMP2 expression in the infected cells.
Figure 8.

GEP was required for BMP2-induced osteoblastogenesis in vitro. (a) IRE1a decreases the level of BMP2 protein, assayed by western blotting. C2C12 cells treated with or without Ad-IRE1a (MOI=10, 20, 50) were stained with IRE1a antibody. Tubulin protein served as an internal control. (b and c) IRE1a reduces the expression of BMP2 mRNA, assayed by real-time PCR. C2C12 (b) and BMSC (c) cells were cultured without (control) or with AdIRE1a, as indicated. The normalized values against GAPDH mRNA were calibrated against controls (day 0), given the value of 1. (d) Deficiency of GEP impaired BMP2-induced OCL activity and IRE1a inhibition effect, detected by OCL activity assay. BMSCs from WT and GEP−/− mice were cultured in the absence or presence of 300 ng/ml BMP2 for 4 days, and OCL activity assay was performed. (e) Deficiency of GEP impaired BMP2-induced ALP activity and IRE1a inhibition effect, detected by ALP activity assay. BMSCs from WT and GEP−/− mice were cultured in the absence or presence of 300 ng/ml BMP2 for 4 days, and ALP activity assay was performed. (f) BMP2-induced OCL activity and IRE1a inhibition were restored when GEP was re-expressed, detected by OCL activity assay. BMSCs from GEP−/− mice were cultured in the absence or presence of 300 ng/ml GEP for 4 days, and OCL activity assay was performed. (g) BMP2-induced ALP activity and IRE1a inhibition were restored when GEP was re-expressed, detected by ALP activity assay. BMSCs from GEP−/− mice were cultured in the absence or presence of 300 ng/ml GEP for 4 days, and ALP activity assay was performed. The values are the mean±S.D. of three independent experiments. *P<0.05, **P<0.01 versus control group
Second, we detected whether GEP is required for BMP2-induced osteoblastogenesis inhibited by IRE1a. For this purpose, we collected primary BMSCs from bone marrow of WT and GEP−/− mice, and cultured them in the presence of 300 ng/ml BMP2. Next, ALP activity and OCL production were assayed in the cell lysates 4 days after infection. As shown in Figures 8d and e, deficiency of GEP resulted in significantly lower ALP activity and OCL production compared with WT group following BMP2 stimulation (**P<0.01). Besides, there was no significant difference of ALP activity and OCL production in GEP-deficient BMSCs with or without Ad-IRE1a. These results indicated that GEP has an important role in BMP2-induced osteogenic activity. Deficiency of GEP impaired BMP2-induced osteoblastogenesis and IRE1a inhibition effect. As revealed in Figures 8f and g, BMP2-induced ALP activity and OCL production were restored in GEP-deficient BMSCs after treating with GEP (300 ng/ml; **P<0.01), and that IRE1a inhibits BMP2-induced ALP activity and OCL production were also recovered after treating with GEP (300 ng/ml; *P<0.05). Taken together, BMP2-induced osteogenic activity and IRE1a inhibition were restored when GEP was re-expressed. Collectivelly, IRE1a inhibits BMP2 and GEP is required for IRE1a inhibition of BMP2-induced osteoblastogenesis.
Discussion
Bone formation occurs through two developmental processes – intramembranous and endochondral ossification. Osteoblast differentiation is a multi-step process where mesenchymal cells differentiate into osteoblast lineage cells, including osteocytes.33, 34, 35 BMP2 has several important roles in a variety of cellular functions ranging from embryogenesis, cell growth, and differentiation to bone development and the repair of bone fractures.39, 40, 41, 42 It is known that BMP2 can activate UPR signaling molecules, such as BiP, IRE1α, and PERK (PKR-like ER-resistant kinase). Murakami et al.4 reported that another BMP2 signaling pathway in osteoblasts was mediated by the UPR of ER stress and the expression levels of the ER stress markers were upregulated by BMP2 stimulation. The IRE1α-XBP1 pathway is involved in osteoblast differentiation through promoting Osterix transcription by XBP1.34
Saito et al.43, 44 reported that treatment of WT primary osteoblasts with BMP2 induced ER stress, leading to an increase in ATF4 protein expression levels. IRE1α is a ubiquitously expressed ER type I transmembrane protein containing both a serine/threonine kinase module and an endoribonuclease domain in its cytosolic region.45, 46 We previously reported that ER stress is induced during chondrocyte differentiation and activates the IRE1a-XBP1 pathway. XBP1S in turn enhances BMP2-induced chondrocyte differentiation as a cofactor of Runx2 for hypertrophic chondrocyte formation.31 However, the role of IRE1a in osteoblastogenesis is poorly understood. Therefore, to detect the impact of IRE1a on how to influence osteoblast differentiation, the current study focused on the role of IRE1a in osteoblastogenesis as well as the molecular events involved.
Here we established that overexpression of IRE1a inhibits, whereas knockdown of IRE1a via siRNA approach enhances, BMP2-induced osteoblast differentiation, as assayed by the activity of ALP and the osteoblast protein OCL production in osteoblastogenesis (Figures 1 and 2). Further, we sought to clarify the molecular mechanism of how to regulate osteoblast differentiation by IRE1a. We found that JunB can bind AP1 sequence of the IRE1a gene promoter and may also act through this cis-acting element to regulate promoter activity (Figure 3). Our data indicate that upregulated IRE1a levels during osteoblastogenesis are mediated by the binding of JunB to AP1 sites within the IRE1a 5′-flanking region. In our luciferase reporter-based assays, we found that one particular AP1 sequence is sufficient to stimulate reporter gene expression (Figure 4). Interestingly, the inclusion of additional binding sites added only modest amounts of reporter activity. Mutation of this site led to the near-complete repression of reporter gene expression, further highlighting the importance of this particular site for JunB-mediated IRE1a expression. Our data also demonstrate that JunB upregulates endogenous IRE1a expression in C2C12 cells. Next, we induced C2C12 cells with BMP2 for induction of osteoblast differentiation, the result showed that knockdown of JunB dramatically inhibited IRE1a expression in BMP2-induced osteoblastogenesis (Figure 5).
As noted, GEP is a multi-functional protein involved in the control of cell proliferation and the differentiation of numerous types of cells and tissues, including skeletal muscle myotubes, cardiac myocytes, macrophages, chondrocytes, and osteoblasts. Multiple signaling pathways are involved in endochondral ossification in epiphyseal growth plate.47, 48 GEP was shown to have a key role in augment chondrogenesis, endochondral bone formation, and cartilage repair;26 promoting bone healing and bone repair;27 and regulating skeletal muscle differentiation by reducing MyoD.28 In this study, we present evidence showing that IRE1a is a GEP-inducible gene in osteoblastogenesis (Figure 6) and the expression of IRE1a induced by GEP is dependent on JunB. JunB is a key transcriptional factor that drives the cell away from myogenic differentiation and toward osteoblast differentiation.49, 50 Furthermore, our data indicate that IRE1a inhibition of GEP-induced osteoblastogenesis also relies on JunB (Figure 7).
Recent studies indicated that GEP acts as the downstream factor of BMP2 in the course of chondrogenesis. This is unsurprising given that GEP is a growth factor with pro-proliferative and tumorigenic effects.51, 52, 53 We previously reported that GEP was a downstream molecule of BMP2 and mediated BMP2-induced skeleton formation; knockdown of GEP suppressed BMP2 function in bone development.26 We next determined whether GEP was involved in IRE1a inhibition of BMP2-induced osteoblastogenesis. For this purpose, we collected primary BMSCs from bone marrow of WT and GEP−/− mice and cultured them in the presence of 300 ng/ml BMP2. Next, ALP activity and OCL production were assayed. The result showed that BMSCs from GEP−/− mice displayed impaired stimulation of osteoblastic marker genes following BMP2 treatment (Figure 8), suggesting that GEP directly participates in BMP2-induced osteoblastogenesis. In addition, deficiency of GEP impaired the IRE1a inhibition of BMP2-induced osteoblastogenesis and IRE1a inhibition effect were restored when GEP was re-expressed. Collectivelly, GEP was required for the IRE1a inhibition of BMP2-induced osteoblast differentiation.
In conclusion, we have identified that IRE1a acts as a novel regulator of osteogenic differentiation. IRE1a is able to inhibit osteoblast differentiation and has modulatory effects on bone development. It was previously reported that BMP2, GEP, and JunB form a negative feedback regulatory loop in the course of myotube formation.28 Our findings that IRE1a, BMP2, GEP, and JunB also constitute a regulatory loop during osteoblast differentiation extend the interplay networks among these molecules. As summarized in Figure 9, we propose a model for the role of IRE1a in osteogenic differentiation. IRE1a functions in concert with BMP2, GEP, and JunB to participate in a regulatory feedback loop in which the transactivation of IRE1a expression by JunB leads to a BMP2-mediated suppression of osteoblastogenesis, a process that involves the expression of JunB, and IRE1a inhibition of BMP2-induced bone formation is dependent on GEP. Our results also suggest that this interplay network might regulate several biological and pathological processes. The elucidation of IRE1a's role and molecular events involved in osteogenic differentiation will better our understanding of normal bone development and the pathogenesis of many orthopedic diseases.
Figure 9.
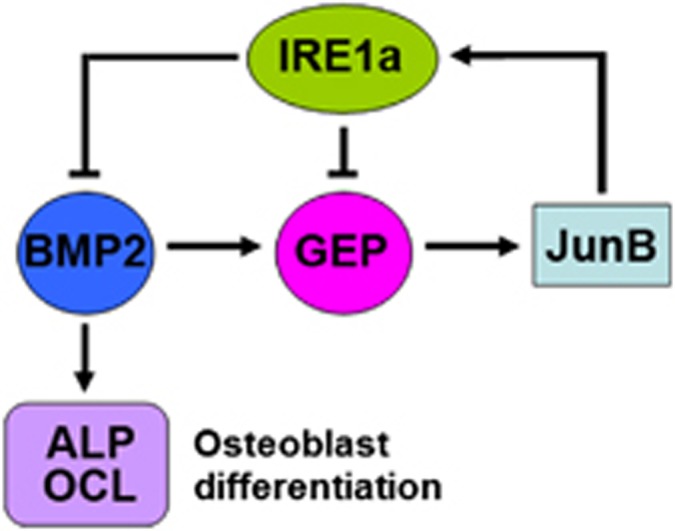
A proposed model for explaining the role and regulation of IRE1a in osteoblast differentiation. GEP induces the expression of IRE1a, whereas IRE1a inhibits BMP2-induced bone formation and GEP-mediated osteoblast differentiation. This inhibition depends on JunB. Thus, IRE1a, BMP2, GEP, and JunB constitute a regulatory feedback loop during bone formation. ‘→' and ‘ ' indicate activation and repression, respectively
' indicate activation and repression, respectively
Materials and Methods
Plasmids and adenoviruses
To generate the pcDNA3.1(−)-IRE1α expression plasmids, the corresponding segments were amplified using PCR with the following primers: IRE1α sense: 5′-GAATTCATGCCGGCCCG GCGGCTGCTGCTG-3′ IRE1α antisense: 5′-AAGCTTGAGGGCGTCTGGAGTCACTGGG GGC-3′ the enzyme sites in the primers are underlined. PCR products were inserted into the pcDNA3.1(−) vector. To generate IRE1α siRNA expression constructs, siRNA corresponding to the coding sequence of the IRE1α gene (5′-ATGCCAATGAACTCTTTCC CTTTT-3′) were cloned into a pSES-HUS vector (an adenoviral shuttle vector expressing siRNA) according to the manufacturer's instructions. Briefly, equimolar amounts of complementary sense and antisense strands were separately mixed, annealed, and slowly cooled to 10 °C in a 50 μl reaction buffer (100 mM NaCl and 50 mM HEPES, pH7.4). The annealed oligonucleotides were inserted into the SfiI sites of pSES-HUS vector. All constructs were verified by nucleic acid sequencing; subsequent analysis was performed using BLAST software (National Institutes of Health). Ad-IRE1α siRNA and adenovirus encoding IRE1α were constructed, respectively, using methods described previously.54, 55, 56 All constructs were verified by nucleic acid sequencing; subsequent analysis was performed using BLAST software (available at http://www.ncbi.nlm.nih.gov/blast/).
Mice
All animal studies were performed in accordance with institutional guidelines and approved by the Institutional Animal Care and Use Committee of Chongqing Medical University. The GEP-knockout (GEP−/−) mice were bought from Jackson Laboratories (Bar Harbor, ME, USA); the generation and genotyping of GEP−/− mice on the basis of Jackson Laboratory's protocol were used for these experiments (http://jaxmice.jax.org/query/).
Isolation and culture of mouse BMSCs
Mouse bone marrow was isolated by flushing the femurs and tibiae of 8–12-week-old female BALB/c mice with 0.6 ml of improved minimal essential medium (Sigma-Aldrich, St. Louis, MO, USA), supplemented with 20% fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin (Invitrogen), and 2 mM glutamine (Invitrogen, Carlsbad, CA, USA), and then it was filtered through a cell strainer (Falcon, BD Biosciences, Northbrook, IL, USA). Cells were centrifuged for 10 min at 260 × g, washed by the addition of fresh medium, centrifuged again, resuspended, and plated out in improved minimal essential medium supplemented with 20% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mM glutamine at a density of ∼2 × 106 cells/cm2 in 25 cm2 plastic culture dishes. The cells were incubated at 37 °C in 5% CO2. After 72 h, non-adherent cells and debris were removed and the adherent cells were cultured continuously. Cells were grown to confluence, washed with phosphate-buffered saline (PBS), and lifted by incubation with 0.25% trypsin, 2 mM EDTA (Invitrogen) for 5 min. Non-detached cells were discarded and the remaining cells were regarded as passage 1 of the BMSC culture. Confluent BMSCs were passaged and plated out at 1 : 2–1 : 3 dilutions. At passage 3, cells were transferred to DMEM (Invitrogen) supplemented with 10% FBS for differentiation studies.
ALP and OCL assays
C2C12 cells or BMSCs transducted with Ad-IRE1a or/and Ad-JunB, were cultured in DMEM containing 10% FBS for 4 days. Cells were then lysed for measuring ALP activity and medium was used for determining OCL production. In brief, the ALP assay mixtures contained 0.1 M 2-amino-2-methyl-1-propanol (Sigma-Aldrich), 1 mM MgCl2, 8 mM P-nitrophenyl phosphate disodium, and cell homogenates. After 30-min incubation at 37 °C, the reaction was stopped with 0.1 N NaOH and the absorbance was read at 405 nm. A standard curve was prepared with P-nitrophenol (Sigma-Aldrich). Each value was normalized to the protein concentration. The amount of OCL secreted into the culture medium was determined by enzyme-linked immunosorbent assay using a mouse OCL assay kit (Biomedical Technologies, Stoughton, MA, USA) per the manufacturer's protocol.
Electrophoretic mobility shift assays
The C2C12 cells were seeded at 5 × 106 cells/well of a six-well plate in medium containing 10% FBS. The cells were cultured overnight and transfected the following day with pcDNA3.1(−)-JunB (3 μg) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. After 48 h, the cells were scraped from the plate by cold PBS, transferred to 10-ml tubes, and centrifuged for 10 min at 4000 rpm. The cell pellet was resuspended in 400 μl of cold buffer (10 mM HEPES (pH 7.9), 1 mM DTT, 10 mM KCl, 0.5 mM PMSF, 0.1 mM EDTA, 0.1 mM EGTA, and 19 protease inhibitors (Roche Applied Science, Penzberg, Upper Bavaria, Germany) by gentle pipetting and placed on ice for 15 min. We then added 25 μl of 10% Nonidet P-40, vortexed vigorously for 10 s, centrifuged at 14 000 rpm for 1 min, aspirated the supernatant, and kept the pellet. We resuspended the nuclear pellet in a 50-ml ice-cold buffer (20 mM HEPES (pH 7.9), 1 mM DTT, 0.4 mM NaCl, 1 mM PMSF, 1 mM EDTA, 1 mM EGTA, and 19 protease inhibitors (Roche Applied Science) and rocked the tube for 30 min at 4 °C. It was then centrifuged for 15 min at 14 000 rpm at 4 °C. Double-stranded oligonucleotides containing a specific JunB-binding sequence located within the 5′-flanking region of the IRE1a gene (5′-GTGAGCTCAG-3′)38 were synthesized. The probes were labeled with digoxigenin-11-ddUTP and EMSA assays) were performed using a digoxigenin gel shift kit (Roche Applied Science). Competition experiments were performed by pre-incubating nuclear extract with excess unlabeled probes before adding labeled oligonucleotides. In supershift assays, 5 μg of anti-JunB antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) were incubated with the reaction mixture for 15 min before the addition of the digoxigenin-labeled probe. Reaction mixtures were incubated for 20 min at room temperature. Samples were subjected to electrophoresis on a native 5% polyacrylamide gel run in 0.5 × TBE (89 mmol/l Tris-HCl, 89 mmol/l boric acid, and 2 mmol/l EDTA) for 2.5 h at100 V. The signal was detected using a chemiluminescent detection system (Roche Applied Science). The method was as described previously.57, 58
Chromatin immunoprecipitation
C2C12 cells transfected with pcDNA3.1(−)-JunB plasmid or treated with BMP2 were fixed by 1% formaldehyde for 10 min before cell lysis. Cell lysates were subsequently sonicated, followed by centrifugation. The input (1% of the supernatant) was used in PCR as a positive control. The supernatant was then precleared using protein A-agarose/salmon sperm DNA for 30 min at 4 °C. After centrifugation, the supernatant was then used for immunoprecipitation using anti-JunB antibody or control IgG and incubated overnight at 4 °C. The protein–DNA complex was subsequently incubated with protein A-agarose/salmon sperm DNA for 1h at 4 °C. The immune complex was collected by centrifugation and then washed five times with the following for 5 min each: once with low salt immune complex wash buffer, once with high salt immune complex wash buffer, once with LiCl salt immune complex wash buffer, and twice with TE buffer. Histone–DNA complex was eluted from the antibody using elution buffer (1% SDS, 0.1 M NaHCO3), and 5 M NaCl was added to reverse the histone–DNA cross-link by heating for 4 h at 65 °C. The DNA was then extracted with phenol/chloroform and precipitated with ethanol in the presence of glycogen (20 mg) as a carrier. The precipitate was used as a template for PCR amplification. The primers specifically amplified two 120-bp segments spanning the JunB-binding site of the IRE1a gene promoter. The sequences of primers were as follows: sense, 5′-GCAACTCCGCCCCGACCGGAAG-3′ antisense, 5′-GGGCCGGCATGGCGAGGACTCG-3′. PCR was performed under the following conditions: 94 °C for 5 min, 35 cycles at 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 45 s.
Reporter gene assays
C2C12 cells were plated at a density of 3 × 105 cells/well in six-well tissue culture plates and transfected with IRE1a-specific reporter plasmids (pGL3-IRE1a-luc,pGL3-IRE1a(mut1)-luc, pGL3-IRE1a(mut2)-luc,pGL3-IRE1a(mut3)-luc or pGL3-IRE1a-(mut4)-luc), pcDNA3.1(−)-JunB, and pCMV-gal (an internal control for transfection efficiency). Forty-eight hours after transfection, cells were collected, and luciferase and β-galactosidase activity was measured using the Bioscan Mini-Lum luminometer. Relative transcriptional activity was expressed as a ratio of luciferase reporter gene activity from the experimental vector to that from the internal control vector. The cultures were processed and analyzed as described above.
Immunohistochemistry
Sections of postcoital day 18.5 embryos mice (GEP−/− and GEP+/+) were deparaffinized, rehydrated, and placed in Tris buffer (10 mM Tris-HCl (pH 8.0), 150 mM NaCl). Serum block was applied for 30 min at room temperature before incubation of the primary antibody. Antimouse XBP1S (BioLegend, San Diego, CA, USA) was diluted 1 : 50 and sections were incubated at room temperature for 2 h. For detection, biotinylated secondary antibody and horseradish peroxidase (HRP)–streptavidin complex (Santa Cruz Biotechnology, Inc.) were used. HRP substrate was used for visualization and sections were then counterstained with Mayer's hematoxylin.
Immunoblotting analysis
To examine the expression of XBP1S protein in the course of chondrogenesis, total cell extracts prepared from micromass cultures of ATDC5 cells in the presence of 300 ng/ml recombinant BMP2 protein were mixed with 5 × sample buffer (312.5 mM Tris-HCl (pH 6.8), 5% β-mercaptoethanol, 10% SDS, 0.5% bromphenol blue, 50% glycerol). Proteins were resolved on a 10% SDS polyacrylamide gel and electroblotted onto a nitrocellulose membrane. After blocking in 10% non-fat dry milk in Tris buffer, saline Tween 20 (10 mM Tris-HCl (pH8.0), 150 mM NaCl, 0.5% Tween 20), blots were incubated with mouse monoclonal anti-XBP1S antibody (diluted 1:500; BioLegend) for 1 h. After washing, the respective secondary antibody (HRP-conjugated anti-mouse immunoglobulin (Sigma-Aldrich), both 1:1000 dilution) was added and bound antibody was visualized using an enhanced chemiluminescence system (Amersham Biosciences, Amersham, UK).
RNA preparation and quantitative real-time PCR
Micromass cultures ATDC5 cells were plated at a density of 3 × 105 cells/well in six-well tissue culture plates. Total RNA was extracted from micromass cultures of ATDC5 cells infected with either control, Ad-IRE1a, its point mutant Ad-IRE1a-PM, or its C-terminal deletion mutants in the presence of 300 ng/ml recombinant BMP2 for various time points using RNAeasy kit (Qiagen, Hilden, Germany). One microgram of total RNA per sample was reverse-transcribed using the ImProm-II Reverse Transcription system (Promega, Fitchburg, WI, USA). The following sequence-specific primers were synthesized: IRE1α sense: 5′-CGGAATTCATGCCGGCCCGGCGGCTGCTGCTG-3′ IRE1α antisense: 5′-AAGCTTTTTG AGCACATGCTTCGCTG-3′ BMP2 sense: 5′-ATGGTGGCCGGGACCCGCTGTCTTG-3′ and BMP2 antisense: 5′-ACGACACCCGCAGCCCTCCACAAC-3′. The following pair of oligonucleotides was used as internal controls: 5′-AGGTCGGTGTGAACGGATTTG-3′ and 5′-TGTAGACCATGTAG TTGAGGTCA-3′ for mouse GAPDH. Reactions were performed in a 50-ml SYBRGREEN PCR volume in a 96-well optical reaction plate formatted in the 7300 Sequence Detection System (ABI PRISM, Applied Biosystems, Foster City, CA, USA) using the following PCR conditions: 40 cycles, 95 °C for 15 s, 60 °C for 1 min. The transcript of GAPDH mRNA was employed as an internal control for RNA quality.
For each gene, three independent PCRs from the same reverse transcription sample were performed. The presence of a single specific PCR product was verified by melting curve analysis and confirmed on an agarose gel and further sequenced by the Applied Biosystems sequencing.
Statistical analysis
Two-sample Student's t-test was used to determine significant differences (P<0.05) in the normal distribution of the data of the two groups. Data for multiple variable comparisons were analyzed by one-way analysis of variance or Kruskal–Wallis test. The statistical analysis was performed with SPSS10.0.1 software for Windows. Data were expressed as mean±S.D. from at least three independent experiments.
Acknowledgments
This work was aided by the National Science Foundation of China (No. 81371928 and No. 81171697); and New Century Excellent Talent Support Project of Education Ministry of China (NCET-12-1090); Returned Overseas Preferred Funded Project of Human Resources and Social Security Ministry of China (No. 2011-235).
Glossary
- IRE1α
inositol requiring enzyme 1α
- XBP1S
X-box binding protein1 spliced
- GEP
granulin-epithelin precursor
- BMP2
bone morphogenetic protein 2
- UPR
unfolded protein response
- PERK
PKR-like ER-resistant kinase
- ATF6
activating transcription factor 6
- BiP
IgH chain-binding protein
- CHOP
C/EBP homologous protein
- EDEM
ER degradation- enhancing α-mannosidase-like protein
- OSE2
osteoblast-specific cis-acting element 2
The authors declare no conflicts of interest.
Footnotes
Edited by A Stephanou
References
- Lai CF, Cheng SL. Signal transductions induced by bone morphogenetic protein-2 and transforming growth factor-β in normal human osteoblastic cells. J Biol Chem. 2002;277:15514–15522. doi: 10.1074/jbc.M200794200. [DOI] [PubMed] [Google Scholar]
- Chen D, Zhao M, Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004;22:233–241. doi: 10.1080/08977190412331279890. [DOI] [PubMed] [Google Scholar]
- Ogata T, Wozney JM, Benezra R, Noda M. Bone morphogenetic protein 2 transiently enhances expression of a gene, Id (inhibitor of differentiation), encoding a helix-loop-helix molecule in osteoblast-like cells. Proc Natl Acad Sci USA. 1993;90:9219–9222. doi: 10.1073/pnas.90.19.9219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Saito A, Hino S, Kondo S, Kanemoto S, Chihara K, et al. Signaling mediated by the endoplasmic reticulum stress transducer OASIS is involved in bone formation. Nat Cell Biol. 2009;11:1205–1211. doi: 10.1038/ncb1963. [DOI] [PubMed] [Google Scholar]
- Jang WG, Kim EJ, Kim DK, Ryoo HM, Lee KB, Kim SH, et al. BMP2 protein regulates osteocalcin expression via Runx2-mediated ATF6 gene transcription. J Biol Chem. 2012;287:905–915. doi: 10.1074/jbc.M111.253187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, et al. The unfolded protein response signals through high-order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Signaling the unfolded protein response from the endoplasmic reticulum. J Biol Chem. 2004;279:25935–25938. doi: 10.1074/jbc.R400008200. [DOI] [PubMed] [Google Scholar]
- Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, et al. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science. 2000;287:664–666. doi: 10.1126/science.287.5453.664. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Oku M, Suzuki M, Mori K. pXBP1(U) encoded in XBP1 pre-mRNA negatively regulates unfolded protein response activator pXBP1(S) in mammalian ER stress response. J Cell Biol. 2006;172:565–575. doi: 10.1083/jcb.200508145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Calfon M, Zeng H, Urano F, Till JH, Hubbard SR, Harding HP, et al. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP1 mRNA. Nature. 2002;415:92–96. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- Zhou J, Gao G, Crabb JW, Serrero G. Purification of an autocrine growth factor homologous with mouse epithelin precursor from a highly tumorigenic cell line. J Biol Chem. 1993;1:10863–10869. [PubMed] [Google Scholar]
- Ong CH, Bateman A. Progranulin (granulinepithelin precursor, PC-cell derived growth factor, acrogranin) in proliferation and tumorigenesis. Histol Histopathol. 2003;18:1275–1288. doi: 10.14670/HH-18.1275. [DOI] [PubMed] [Google Scholar]
- Baba T, Hoff HB, 3rd, Nemoto H, Lee H, Orth J, Arai Y, et al. Acrogranin, an acrosomal cysteinerich glycoprotein, is the precursor of the growth-modulating peptides, granulins, and epithelins, and is expressed in somatic as well as male germ cells. Mol Reprod Dev. 1993;34:233–243. doi: 10.1002/mrd.1080340302. [DOI] [PubMed] [Google Scholar]
- Sun X, Gulyas M, Hjerpe A. Mesothelial differentiation as reflected by differential gene expression. Am J Respir Mol Cell Biol. 2004;30:510–518. doi: 10.1165/rcmb.2003-0266OC. [DOI] [PubMed] [Google Scholar]
- Barreda DR, Hanington PC, Walsh CK, Wong P, Belosevic M. Differentially expressed genes that encode potential markers of goldfish macrophage development in vitro. Dev Comp Immunol. 2004;28:727–746. doi: 10.1016/j.dci.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Nishiahara M. Granulin precursor gene: a sex steroid-inducible gene involved in sexual differentiation of the rat brain. Mol Genet Metab. 2002;75:31–37. doi: 10.1006/mgme.2001.3274. [DOI] [PubMed] [Google Scholar]
- Jüsten HP, Grunewald E, Totzke G, Gouni-Berthold I, Sachinidis A, Wessinghage D, et al. Differential gene expression in synovium of rheumatoid arthritis and osteoarthritis. Mol Cell Biol Res Commun. 2000;3:165–172. doi: 10.1006/mcbr.2000.0211. [DOI] [PubMed] [Google Scholar]
- Guo F, Lai Y, Tian Q, Lin EA, Kong L, Liu C. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum. 2010;62:2023–2036. doi: 10.1002/art.27491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Zhang Y, Ilalov K, Carlson CA, Feng JQ, Di Cesare PE, et al. Cartilage oligomeric matrix protein associates with granulin-epithelin precursor (GEP) and potentiates GEP-stimulated chondrocyte proliferation. J Biol Chem. 2007;282:11347–11355. doi: 10.1074/jbc.M608744200. [DOI] [PubMed] [Google Scholar]
- Wang W, Hayashi J, Kim WE, Serrero G. PC cell-derived growth factor (granulin precursor) expression and action in human multiple myeloma. Clin Cancer Res. 2003;9:2221–2228. [PubMed] [Google Scholar]
- Barreda DR, Hanington PC, Walsh CK, et al. Differentially expressed genes that encode potential markers of goldfish macrophage development in vitro. Dev Comp Immunol. 2004;28:727–746. doi: 10.1016/j.dci.2003.11.005. [DOI] [PubMed] [Google Scholar]
- He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225–229. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- Justen HP, Grunewald E, Totzke G, et al. Differential gene expression in synovium of rheumatoid arthritis and osteoarthritis. Mol Cell Biol Res Commun. 2000;3:165–172. doi: 10.1006/mcbr.2000.0211. [DOI] [PubMed] [Google Scholar]
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med. 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- Feng JQ, Guo FJ, Jiang BC, Zhang Y, Frenkel S, Wang DW, et al. Granulin epithelin precursor: a bone morphogenic protein 2-inducible growth factor that activates Erk1/2 signaling and JunB transcription factor in chondrogenesis. FASEB J. 2010;24:1879–1892. doi: 10.1096/fj.09-144659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YP, Tian QY, Frenkel S, Liu CJ. The promotion of bone healing by progranulin, a downstream molecule of BMP-2, through interacting with TNF/TNFR signaling. Biomaterials. 2013;34:6412–6421. doi: 10.1016/j.biomaterials.2013.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Bai X, Tian Q, Lai Y, Edward A, Shi LY, et al. GEP constitutes a negative feedback loop with MyoD and acts as a novel mediator in controlling skeletal muscle differentiation. Cell Mol Life Sci. 2012;69:1855–1873. doi: 10.1007/s00018-011-0901-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, et al. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda S, Gorman AM, Hori O, Samali A. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074. doi: 10.1155/2010/214074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Zhou J, Zhao W, Li X, Guo FJ. XBP1S associates with RUNX2 and regulates chondrocyte hypertrophy. J Biol Chem. 2012;287:34500–34513. doi: 10.1074/jbc.M112.385922. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Kim YJ, Lee MH, Wozney JM, Cho JY, Ryoo HM. Bone morphogenetic protein-2-induced alkaline phosphatase expression is stimulated by Dlx5 and repressed by Msx2. J Biol Chem. 2004;279:50773–50780. doi: 10.1074/jbc.M404145200. [DOI] [PubMed] [Google Scholar]
- Wang YK, Yu X, Cohen DM, Wozniak MA, Yang MT, Gao L, et al. Bone morphogenetic protein-2-induced signaling and osteogenesis is regulated by cell shape, RhoA/ROCK, and cytoskeletal tension. Stem Cells Dev. 2012;21:1176–1186. doi: 10.1089/scd.2011.0293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohmonda T, Miyauchi Y, Ghosh R, Yoda M, Uchikawa S, Takito J, et al. The IRE1alpha-XBP1 pathway is essential for osteoblast differentiation through promoting transcription of Osterix. EMBO Rep. 2011;12:451–457. doi: 10.1038/embor.2011.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha KM, Xin Z. Genetic and molecular control of Osterix in skeletal formation. J Cell Biochem. 2013;114:975–984. doi: 10.1002/jcb.24439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliazer S, Spencer J, Ye D, Olson E, Ilaria RL., Jr Alteration of mesodermal cell differentiation by EWS/FLI-1, the oncogene implicated in Ewing's sarcoma. Mol Cell Biol. 2003;23:482–492. doi: 10.1128/MCB.23.2.482-492.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katagiri T, Yamaguchi A, Komaki M, Abe E, Takahashi N, Ikeda T, et al. Bone morphogenetic protein-2 converts the differentiation pathway of C2C12 myoblasts into the osteoblast lineage. J Cell Biol. 1994;127 (6 Pt 1:1755–1766. doi: 10.1083/jcb.127.6.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou JH, Han XF, Liu YN, Zhang P, Guo FJ. [The effect on BiP regulation of IRE1a promoter transcription activity and protein expression] Yi Chuan. 2013;35:343–351. doi: 10.3724/sp.j.1005.2013.00343. [DOI] [PubMed] [Google Scholar]
- Peng Y, Kang Q, Luo Q, Jiang W, Si W, Liu BA, et al. He TC.Inhibitor of DNA binding/differentiation helix-loophelix proteins mediate bone morphogenetic protein-induced osteoblast differentiation of mesenchymal stem cells. J Biol Chem. 2004;279:32941–32949. doi: 10.1074/jbc.M403344200. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–1647. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, et al. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:1–16. doi: 10.1007/s11154-006-9001-5. [DOI] [PubMed] [Google Scholar]
- Miyazono K, Kusanagi K, Inoue H. Divergence and convergence of TGF-beta/BMP signaling. J Cell Physiol. 2001;187:265–276. doi: 10.1002/jcp.1080. [DOI] [PubMed] [Google Scholar]
- Saito A, Imaizumi K. Endoplasmic reticulum stress response in osteogenesis. Clin Calcium. 2013;23:1569–1575. [PubMed] [Google Scholar]
- Saito A, Ochiai K, Kondo S, Tsumagari K, Murakami T, Cavener DR, et al. Endoplasmic reticulum stress response mediated by the PERK-eIF2(alpha)-ATF4 pathway is involved in osteoblast differentiation induced by BMP2. J Biol Chem. 2011;286:4809–4818. doi: 10.1074/jbc.M110.152900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, et al. The unfolded protein response signals through high order assembly of Ire1. Nature. 2009;457:687–693. doi: 10.1038/nature07661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhu H, Huang H, Jiang R, Zhao W, Liu Y, et al. Study on the effect of IRE1α on cell growth and apoptosis via modulation PLK1 in ER stress response. Mol Cell Biochem. 2012;365:99–108. doi: 10.1007/s11010-012-1248-4. [DOI] [PubMed] [Google Scholar]
- Justen HP, Grunewald E, Totzke G, Gouni-Berthold I, Sachinidis A, Wessinghage D, et al. Differential gene expression in synovium of rheumatoid arthritis and osteoarthritis. Mol Cell Biol Res Commun. 2000;3:165–172. doi: 10.1006/mcbr.2000.0211. [DOI] [PubMed] [Google Scholar]
- Olsen BR, Reginato AM, Wang W. Bone development. Annu Rev Cell Dev Biol. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- Chalaux E, Lopez-Rovira T, Rosa JL, Bartrons R, Ventura F. JunB is involved in the inhibition of myogenic differentiation by bone morphogenetic protein-2.J. Biol Chem. 1998;273:537–543. doi: 10.1074/jbc.273.1.537. [DOI] [PubMed] [Google Scholar]
- Locklin RM, Riggs BL, Hicok KC, Horton HF, Byrne MC, Khosla S. Assessment of gene regulation by bone morphogenetic protein 2 in human marrow stromal cells using gene array technology. J Bone Miner Res. 2001;16:2192–2204. doi: 10.1359/jbmr.2001.16.12.2192. [DOI] [PubMed] [Google Scholar]
- Liu CJ. Progranulin: a promising therapeutic target for rheumatoid arthritis. FEBS Lett. 2011;585:3675e80. doi: 10.1016/j.febslet.2011.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Bennett HP. The granulin gene family: from cancer to dementia. Bioessays. 2009;31:1245e54. doi: 10.1002/bies.200900086. [DOI] [PubMed] [Google Scholar]
- He Z, Ong CH, Halper J, Bateman A. Progranulin is a mediator of the wound response. Nat Med. 2003;9:225e9. doi: 10.1038/nm816. [DOI] [PubMed] [Google Scholar]
- Chen H, Yang C, Yu L, Xie L, Hu J, Zeng L, et al. Adenovirus-mediated RNA interference targeting FOXM1 transcription factor suppresses cell proliferation and tumor growth of nasopharyngeal carcinoma. J Gene Med. 2012;14:231–240. doi: 10.1002/jgm.2614. [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan Y, Yu XP, Yang N, Frenkel S, Chen L, Liu CJ. p204 protein overcomes the inhibition of core binding factor alpha-1-mediated osteogenic differentiation by Id helix-loop-helix proteins. Mol Biol Cell. 2008;19:2113–2126. doi: 10.1091/mbc.E07-10-1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Lin EA, Liu P, Lin J, Liu C. XBP1U inhibits the XBP1S-mediated upregulation of the iNOS gene expression in mammalian ER stressresponse. Cell Signal. 2010;22:1818–1828. doi: 10.1016/j.cellsig.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Kong L, Carlson CS, Liu CJ. Cbfa1-dependent expression of an interferon- inducible p204 protein is required forchondrocyte differentiation. Cell Death Differ. 2008;15:1760–1771. doi: 10.1038/cdd.2008.112. [DOI] [PubMed] [Google Scholar]