

Abstract
Glioblastoma is the most common and most aggressive primary brain tumor in adults. Optimized standard treatment only confers a modest improvement in progression and overall survival, underscoring the pressing need for the development of novel therapies. Our understanding of glioblastoma (a molecularly heterogeneous disorder) has been accelerated in the setting of large scale genomic analyses, lending insight into potential actionable targets. Antiangiogenic therapies have been used in the treatment of glioblastoma, and our understanding of the means to optimize the role of these agents is continuing to evolve. Recently, immunotherapy has garnered increasing attention as a therapeutic approach in the treatment of gliomas. Promising novel approaches are under active development in the treatment of glioblastoma.
Introduction
Glioblastoma is the most common malignant primary brain tumor in adults and invariably carries a poor prognosis. Despite optimal multimodality treatment that typically includes surgery, radiation, and cytotoxic chemotherapy, recent clinical trials have reported a median survival of only 14–16 months with a 26–33% 2 year survival rate [1,2]. The challenges in developing effective treatments for patients with glioblastoma are attributed to the relative rarity of the tumor and its molecular heterogeneity. Varying susceptibility to treatment toxicities further complicates treatment planning. New therapeutic approaches are needed to improve the outcomes of patients with glioblastoma.
Current standard of care for glioblastoma
Glioblastomas are inherently aggressive tumors. Their infiltrative behavior renders them difficult to completely resect. Nevertheless, maximal safe surgical resection does improve prognosis and is therefore recommended as the initial step in the management of glioblastoma [3,4]. The addition of chemotherapy to radiation emerged as the standard of care for glioblastoma based on the seminal study performed by Stupp and colleagues [1]. Temozolomide, an alkylating cytotoxic agent, administered concurrently at a dose of 75 mg/m2 daily during the course of regional radiotherapy, followed by maintenance temozolomide given at a dose of 150–200 mg/m2 days 1–5 every 28 days for maintenance, resulted in an improvement in median overall survival from 12.1 to 14.6 months when compared to patients treated with radiation alone. The 2 year survival rate was 26.5% in patients who received chemotherapy in addition to radiotherapy compared to 10.4% in those patients who received radiotherapy alone. The addition of temozolomide to radiotherapy had clearly demonstrated a statistically significant survival benefit.
O-6-methylguanine-DNA methyltransferase (MGMT) is a DNA repair protein that reverses the damage induced by alkylating agents (such as temozolomide) and has been implicated as a major mechanism of resistance to alkylating agents [5]. Methylation of the gene promoter results in decreased expression of the enzyme, rendering tumor cells more susceptible to alkylating agents, which has been observed to translate into a striking survival benefit for those patients treated with radiotherapy and temozolomide [6]. Dose dense scheduling of temozolomide results in prolonged depletion of MGMT, suggesting that prolonged exposure to temozolomide may result in improved survival in patients with newly diagnosed glioblastoma. This hypothesis was studied in a randomized phase III clinical trial comparing standard adjuvant temozolomide (days 1–5 every 28 days) with a dose dense schedule (days 1–21 every 28 days). No statistically significant difference in either median overall survival or median progression free survival was observed between the dose dense and standard treatment arms of the study. Although dose dense temozolomide was not found to confer a survival benefit for newly diagnosed glioblastoma, the study did reaffirm the prognostic significance of MGMT methylation as evidenced by the improved overall survival, progression free survival, and response in the methylated versus the unmethylated patients [2].
Molecular heterogeneity
There is a growing body of evidence that our lack of effective therapies is related to our increasing recognition that glioblastoma is a molecularly heterogeneous disorder. This heterogeneity is both intertumoral and intratumoral, further complicated by continued molecular changes over time; this is clearly a major challenge in developing effective treatments. Large scale profiling efforts have accelerated our understanding of this complex disease. The Cancer Genome Atlas (TCGA) has attempted to catalog the spectrum of molecular abnormalities seen in glioblastoma, spurring the development of molecular subclasses. The analysis specifically designated four subclasses termed proneural, neural, classical, and mesenchymal, distinguished from each other based on shared genomic, epigenomic, and transcriptional features [7]. More recently, a similar classification schema was developed using tumor methylation arrays along with other molecular testing, and now has defined six sub-classifications if pediatric glioblastoma is included [8]. Figure 1 highlights these key genetic and epigenetic findings in six glioblastoma subgroups. Although the prognostic and predictive significance of these tumor subclasses remains unclear and currently has a limited role in treatment decisions, the recognition of molecular subclasses has piqued interest that tumor profiling may translate into the development of targeted therapy.
Figure 1. Graphical summary of key molecular and biologic characteristics of glioblastoma subgroups.

A simplified schematic representation of key genetic and epigenetic findings in six glioblastoma subgroups as identified by methylation profiling and correlations with clinical patient data. Permission obtained from Elsevier publishing group. Cancer Cell, 2012. 22(4): p. 425-37.
Abbreviations: CHOP, CpG hypomethylator phenotype; CIMP, CpG island methylator phenotype; EGFR, epidermal growth factor receptor; IDH, isocitrate dehydrogenase; PDGFRA, platelet derived growth factor receptor; RTK, receptor tyrosine kinase.
Targeted therapy
The efficacy of molecularly based targeted therapy has been validated in the treatment of other cancers. Chronic myeloid leukemia (CML), a myeloproliferative disorder, has a characteristic abnormality, the Philadelphia (Ph) chromosome, which results from a reciprocal translocation between the long arms of chromosomes 9 and 22. This translocation results in the generation of the fusion protein BCR-ABL, which is a constitutively activated tyrosine kinase in CML. Mutational analysis established that the tyrosine kinase activity of the protein was necessary for its oncogenic activity. This recognition of the essential role of BCR-ABL tyrosine kinase activity in CML was then exploited in the development of Imatinib which acts as an inhibitor of the BCR-ABL tyrosine kinase [9]. Imatinib revolutionized the treatment of Ph(+) CML and has emerged as a paradigm for the development of targeted therapy in cancer therapeutics.
Glioblastoma is now one of the most molecularly characterized of all human cancers. Molecular profiling efforts have resulted in the identification of molecular prognostic factors as well as identified molecular vulnerabilities that could be potentially targeted in the development of novel treatments in glioblastoma. Table 1 highlights some of the potential actionable targets in glioblastoma. One of the first abnormalities recognized in glioblastoma was alterations in the epidermal growth factor receptor (EGFR), which is seen in approximately 40–50% of patients. Amplification or overexpression of EGFR is most common, but a relatively high percent of tumors also demonstrate a unique mutation designated EGFRvIII. EGFRvIII mutation represents a partial deletion of the extracellular domain that constitutively activates the receptor of the ligand [10]. EGFR mutations are a compelling drug target in glioblastoma. The relatively high frequency of EGFR amplification, EGFRvIII mutation, and other EGFR extracellular domain mutations suggest that EGFR tyrosine kinase inhibitors would be an appropriate actionable target in glioblastoma. Unfortunately, anti-EGFR kinase therapy trials with erlotinib and gefitinib in glioblastoma have been largely unsuccessful, even in patients with tumors where the gene is overexpressed or the EGFRvIII mutation is present [11–14]. EGFR inhibitor resistance has led to an improved understanding of the mechanism mediating resistance. PTEN is an important factor in determining EGFR tyrosine kinase inhibitor response. EGFRvIII expression and loss of PTEN results in resistance to EGFR tyrosine kinase inhibitors in glioblastoma patients and preclinical models, as signal flux through the phosphatidylinositol 3-kinase (PI3K) signaling pathway is maintained [15]. Recent studies examining potential EGFR pathway inhibition suggest that agents that target the intracellular component, such as lapatinib, may have greater efficacy [16].
Table 1. Actionable molecular targets in glioblastoma.
| Molecular Targets in Glioblastoma |
|---|
Cell Surface Growth Factor Receptors
|
Receptor Tyrosine Kinase Signaling and Downstream Effectors
|
Angiogenesis Inhibition
|
Other Targets
|
PI3K pathway-activating genetic lesions occur in almost 90% of glioblastomas arising due to amplification and/or mutation in EGFR or other receptor tyrosine kinases (RTKs), PIK3CA, or PTEN loss. Protein kinase B (AKT) phosphorylation is commonly observed [17]. Recognition that PI3K/AKT signaling is hyperactivated has made PI3K, and its key downstream effector AKT, appealing targets for therapy in glioblastoma. PI3K and AKT inhibitors are currently in early phase clinical trials. Although the PI3K pathway remains an intriguing therapeutic target, early studies were complicated by unacceptable toxicities, likely related to the importance of these pathways in normal cellular homeostasis. More selective inhibitors are currently in early phase clinical trials for glioblastoma.
Identification of an actionable gene mutation or alteration in glioblastoma is exemplified in the work done by Singh and colleagues [18]. A small subset of glioblastomas harbor an oncogenic chromosomal translocation that fuse in-frame the tyrosine kinase coding domains of fibroblast growth factor receptor (FGFR) genes (FGFR1 or FGFR3) to the transforming acidic coiled-coil (TACC) coding domains of TACC1 or TACC3, respectively. This FGFR-TACC fusion protein was found to display oncogenic activity in the mouse brain. Oral administration of an FGFR inhibitor was found to prolong the survival of mice harboring the intracranial FGFR3-TACC3 initiated glioma, suggesting that targeted FGFR kinase inhibition may benefit the small subset of glioblastoma patients with FGFR3-TACC2 fusions [18]. Clinical trials specifically targeting this unique fusion protein are in development for glioblastoma. However, the very low incidence of the mutation (approximately 3%) will render accrual of patients for the clinical trial challenging.
Isocitrate dehydrogenase (IDH) mutations in gliomas are now known to be a positive prognostic factor, with an increase in overall survival noted in patients harboring an IDH mutation over those with wild-type IDH, but to date specific treatments based on this finding have not been established [19]. Targeting this mutation or its molecular or metabolic effects may be feasible. With few exceptions, the molecular findings have not impacted treatment decisions to date. However, all of these findings serve as clues that a molecular diagnosis is trending towards significantly influencing the clinical management of patients.
Antiangiogenic therapy
Angiogenesis is a pathologic hallmark of glioblastoma, with the expression of vascular endothelial growth factor (VEGF) among other pro-angiogenic cytokines as one of the most important regulators of angiogenesis [20]. VEGF is an essential regulator of angiogenesis, which has rendered it an appealing target in cancer therapeutics [21]. The expression of VEGF and other proangiogenic cytokines in glioblastoma results in the development of abnormal tumor vasculature characterized by tortuous, hyperpermeable vessels, increased vessel diameter, and abnormally thickened basement membranes. This aberrant tumor vasculature is believed to enhance tumor hypoxia and impair the delivery of cytotoxic chemotherapy.
Several studies have demonstrated that VEGF and its receptors can be antagonized by monoclonal antibodies to VEGF and small-molecule inhibitors of VEGF receptor 2 (VEGFR-2) [22–24]. Bevacizumab (Avastin, Genentech/Roche) is a humanized monoclonal antibody that binds to VEGF preventing its interaction with VEGFRs resulting in suppression of VEGF signaling. Bevacizumab was Food and Drug Administration (FDA) approved in May 2009 for use as a single agent in patients with glioblastoma with progressive disease, following frontline therapy consisting of surgical resection, radiotherapy, and temozolomide. The incorporation of antiangiogenic therapy in the treatment of glioblastoma initially generated excitement that tumor growth could be substantially inhibited, resulting in improved patient outcomes.
In the early, uncontrolled clinical studies, impressive radiographic responses and prolongation of progression free survival were noted. Promising results were seen in single arm phase II clinical trials using the oral pan-VEGFR-2 tyrosine kinase inhibitor cediranib (AZD2171, AstraZeneca) and the anti-VEGF-A antibody bevacizumab (Avastin, Genentech/Roche). In patients with recurrent glioblastoma, a randomized, phase III, placebo-controlled, partially blinded clinical trial (REGAL [Recentin in Glioblastoma Alone and With Lomustine]) was performed to determine the efficacy of cediranib either as monotherapy or in combination with lomustine versus lomustine alone. The primary endpoint of prolonging progression free survival was not reached and there was no demonstration that cediranib conferred a benefit, either as monotherapy or in combination with lomustine, compared to lomustine alone [25].
Bevacizumab was subsequently evaluated in phase III clinical trials for newly diagnosed glioblastoma, with unfortunately no effect on overall patient survival. Two recent large randomized phase III trials, AVAglio and RTOG 0825, demonstrated that the addition of bevacizumab to upfront treatment with radiation and temozolomide conferred no benefit in terms of overall survival in newly diagnosed glioblastoma patients when compared to the standard treatment arm [26,27]. Progression free survival was found to be prolonged in both studies by approximately 3–4 months, reaching statistical significance in the AVAglio study but not in the RTOG 0825 study based on pre-defined criteria [26,27]. Both trials additionally evaluated patient-reported outcomes including quality of life and neurocognitive testing as secondary endpoints, with divergent results as commented on by a recent editorial [28]. The AVAglio trial found improvement in or prolonged maintenance of quality of life and performance status while RTOG 0825 found a worsened quality of life and a decline in cognitive function over time. These findings have spurred conversation regarding the quality of life endpoints for each study and the role a measure such as quality of life could play in the approval of a new therapy for cancer, regardless of its effect on survival [28].
The sobering realization that VEGF pathway inhibitors result in only transitory clinical and radiographic benefit for a few months, prior to the inevitable resumption of disease progression, has prompted the effort to better understand the mechanistic basis underlying resistance to antiangiogenic therapy. The rapid and robust radiographic response of angiogenesis inhibitors suggests that they have little intrinsic antitumor activity, and that the main benefit is derived from the indirect effects secondary to reduction in cerebral edema and the potential to enhance the efficacy of other therapies. Figure 2 highlights the postcontrast radiographic response and the interval fluid attenuated inversion recovery (FLAIR) signal changes seen in the setting of the use of single agent bevacizumab in a patient with recurrent glioblastoma.
Figure 2. Axial magnetic resonance (MR) images in a patient with recurrent glioblastoma.
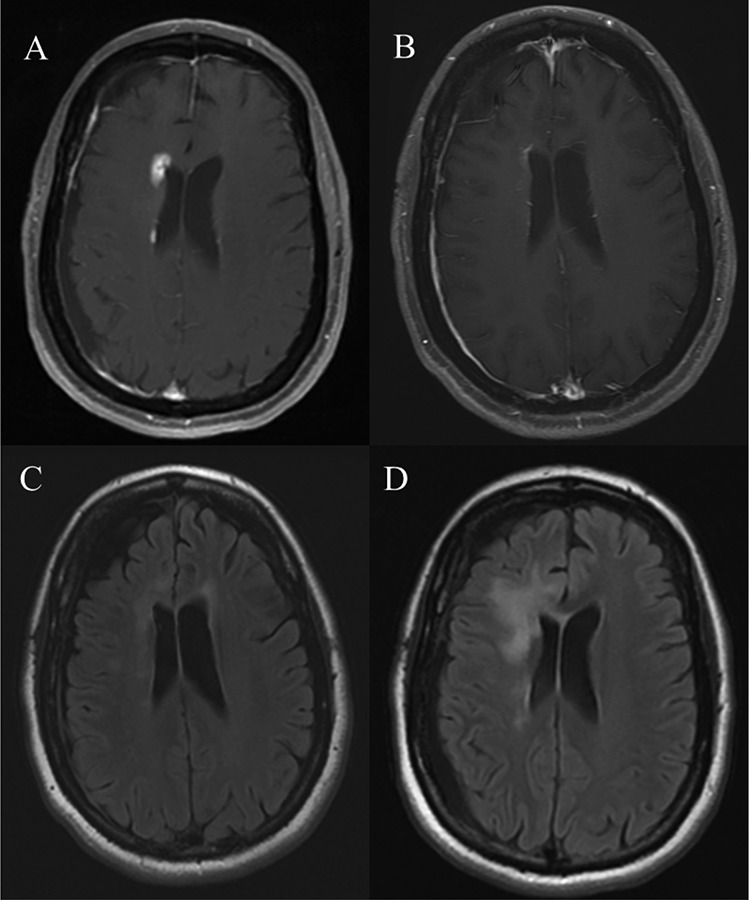
(A) Postcontrast MRI demonstrating an enhancing focus of disease adjacent to the right frontal horn. (B) Postcontrast MRI obtained 6 weeks following initiation of single agent bevacizumab demonstrating an interval reduction in contrast enhancing disease. (C) Fluid attenuated inversion recovery (FLAIR) sequence of the same patient at the same point in time as image (A) revealing subtle FLAIR hyperintensity adjacent to the anterior horns of the lateral ventricles. (D) FLAIR sequence obtained 6 weeks following initiation of single agent bevacizumab revealing interval evolution of confluent FLAIR signal changes adjacent to the right frontal horn consistent with non-enhancing disease progression.
Abbreviations: FLAIR, fluid attenuated inversion recovery; MRI, magnetic resonance imaging.
The lack of a durable response seen with the use of antiangiogenic agents has been disappointing. There is a growing body of research investigating the mechanisms underlying the resistance to anti-VEGF therapy, in an attempt to improve our current therapeutic approaches. Adaptive (evasive) resistance and intrinsic (pre-existing) non-responsiveness have emerged as the modes of resistance to antiangiogenic therapy, with multiple mechanisms believed to underlie each type [29]. Adaptive mechanisms described have included activation and/or upregulation of alternative pro-angiogenic signaling pathways within the tumor, recruitment of bone marrow derived pro-angiogenic cells, increased pericyte coverage of the tumor vasculature, and activation and enhancement of invasion and metastasis [29]. In contrast to mechanisms of adaptive resistance, it has been speculated that some glioblastoma-associated blood vessels are intrinsically resistant to antiangiogenic therapy, based on the observation of patients on clinical trials with VEGF pathway inhibitors who had no discernible reduction in contrast enhancement on magnetic resonance imaging (MRI) after treatment with antiangiogenic therapy. A subset of patients in clinical trials for bevacizumab, sorafenib, and sunitinib was identified who did not demonstrate any transitory radiographic response or clinical benefit [30]. This lack of response was characterized by no evidence of reduction in vascular permeability, no cessation of tumor growth or retardation of growth rate, no observed quality of life benefit, and no evidence of increased survival [29]. It is speculated that these tumors likely express high levels of multiple pro-angiogenic growth factors, such as placental growth factor, in addition to VEGF or that the tumor angiogenesis is completely VEGF-independent.
Immunotherapy
Immune system modulation has evolved as a promising treatment modality in many malignancies, including gliomas. Immunotherapy has long been an appealing therapeutic strategy and is based on the premise of harnessing the patient's own immune system to stimulate an antitumor response. Immunosuppression is inherently associated with glioblastoma and is mediated by a variety of mechanisms. There are a number of immunotherapy approaches being studied including the use of autologous stimulated lymphocytes, immunotherapy with cytokines and dendritic cells, and tumor or peptide based vaccines. Recent clinical trials of single peptide-based vaccine, rindopepimut (PEP-3-KLH/CDX-110), targets the 13 amino acid sequence EGFRvIII receptor antigen in EGFRvIII expressing tumors. Rindopepimut has been evaluated in two phase II clinical trials in patients with newly diagnosed glioblastoma. Following completion of concurrent chemoradiation, patients were enrolled to receive maintenance temozolomide with a rindopepimut vaccination. The results of these two trials have been encouraging with a median progression free survival of 14.2 months and 15.2 months and a median overall survival of 23.6 and 26 months suggesting that tumor-mediated immune suppression can be overcome [31,32].
In metastatic melanoma, ipilimumab, a human immunoglobulin (Ig)G1 monoclonal antibody to cytotoxic T-lymphocyte antigen 4 (CTLA-4), has been demonstrated to result in a durable response and improved overall survival when compared to non-ipilimumab containing treatment arms in randomized trials [33,34]. There have also been recent data demonstrating the effectiveness of ipilimumab in the treatment of melanoma patients with brain metastases [35,36], as evidenced by the measurable tumor reduction seen with ipilimumab used as monotherapy. Nivolumab, a fully humanized IgG4 monoclonal antibody to the immune checkpoint molecule programmed death-1 (PD-1), has also been found to have activity in metastatic melanoma as a single agent. PD-1 is a complementary checkpoint protein to CTLA-4, and Nivolumab has been shown to block PD-1, reversing the immunosuppression mediated by this molecule.
At the present time, there are no data available for ipilimumab or nivolumab in patients with malignant gliomas, but the responses seen in melanoma have generated increasing interest that the incorporation of these agents into the treatment paradigms for glioma may translate to improving patient outcomes. This hypothesis is supported by preclinical data in work done in orthotopic murine models of glioblastoma [37,38]. In these studies, ipilimumab was found to be efficacious in the central nervous system (CNS) murine models of glioma resulting in >80% of the treated animals being cured. This efficacy was associated with enhancement of CD8+ (cytotoxic) T cells without decreasing the CD4 population, suggesting successful inhibition of the immunosuppressive activity of regulatory T cells. Similar results have been demonstrated using a murine anti-PD-1 in combination with radiosurgery [39]. These results suggest that there is a therapeutic synergy in generating a robust immune response at the time of radiation-induced tumor cytotoxicity.
Recurrent disease
Unfortunately, despite initial multimodality treatment, all patients will experience recurrent disease warranting a change in therapy. The increasing understanding of the molecular changes of glioblastoma over time presents a major challenge in the development of effective treatments in the recurrent setting. The molecular profile of the tumor tissue obtained at time of diagnosis becomes less representative over time, as the tumor continues to accumulate mutations and develops resistance to treatment. There is a present lack of effective salvage therapies for recurrent glioblastoma. Nitrosoureas continue to be used commonly as salvage therapy and have been found not to be inferior to targeted therapies, underscoring their enduring relevance. In a phase III study comparing the targeted therapeutic agent enzastaurin, an oral serine threonine kinase inhibitor, to the nitrosourea, lomustine, enzastaurin was found not to be superior to lomustine suggesting that nitrosoureas still play an important role in the treatment of glioblastoma in the recurrent setting [40].
The determination of response to therapy is based on clinical manifestations and the interpretation of the MRI findings. Interpretation of the MRI becomes even more paramount, particularly in the absence of any clinical deterioration in the patient. A phenomenon termed pseudoprogression has evolved to describe radiographic changes that can mimic tumor progression but actually represents radiation induced changes. Pseudoprogression is a clinically significant entity which can affect patient management and the conduct of clinical trials. This phenomenon can most commonly be appreciated in the first few months following completion of concurrent chemoradiation, and is often manifested as a transient increase in contrast enhancement following completion of radiotherapy [41]. Radiographic changes suggestive of progression particularly within the first few months after completion of concurrent chemoradiation should be interpreted with caution and followed closely before a change in therapy is made.
Conclusion
Glioblastoma is one of the most lethal human cancers. Our current standard multimodality treatment of surgery, radiation, and cytotoxic chemotherapy results in only a modest survival benefit at best for a subset of patients with glioblastoma. Radiation therapy in glioblastoma has historically been administered as fractionated, conformal external beam photon radiation, but randomized clinical trials are currently underway evaluating proton beam radiation compared to photon radiation in glioblastoma. Genomic analyses are now furthering our understanding of this molecularly heterogeneous disorder and providing insight into potential actionable targets in the development of targeted therapeutics. Antiangiogenic therapies once held great promise in the treatment of glioblastoma and, although the results have been largely disappointing, there likely is a role for angiogenesis inhibitors in the treatment of glioblastoma. The identification of this role will be contingent upon broadening our understanding of the mechanisms of action of VEGF inhibitors, and the mechanisms of mediating resistance, to help determine how to optimally incorporate the use of antiangiogenic agents in our treatment paradigm. Immunotherapy has recently been generating increasing excitement in the treatment of glioma, with clinical trials underway and others under active development. The hope of truly transforming the treatment and outcomes of patients with glioblastoma will require a furthering of our understanding of this heterogeneous disorder, devising novel therapies to exploit identified molecular vulnerabilities, designing innovative clinical trials which incorporate clinical and molecular factors with mandatory tumor tissue collection, and collaborating on a multi-institutional level.
Abbreviations
- AKT
protein kinase B
- CML
chronic myeloid leukemia
- CTLA-4
cytotoxic T-lymphocyte antigen 4
- EGFR
epidermal growth factor receptor
- FGFR
fibroblast growth factor receptor
- IDH
isocitrate dehydrogenase
- Ig
immunoglobulin
- MGMT
O-6-methylguanine-DNA methyltransferase
- MRI
magnetic resonance imaging
- PI3K
phosphatidylinositol 3-kinase
- PD-1
programmed death-1
- Ph chromosome
Philadelphia chromosome
- RTK
receptor tyrosine kinase
- TACC
transforming acidic coiled-coil
- VEGF
vascular endothelial growth factor
- VEGFR
vascular endothelial growth factor receptor
Disclosures
Mark R. Gilbert serves on the Advisory Boards for Genentech/Roche, Abbvie, Merck and Bristol-Myers Squibb; provides research support for Genentech/Roche, Merck and GlaxoSmithKline; and receives honoraria from Genentech/Roche, Abbvie and Merck.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/6/46
References
- 1.Stupp R, Mason WP, van den Bent, Martin J, Weller M, Fisher B, Taphoorn Martin JB, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384243
- 2.Gilbert MR, Wang M, Aldape KD, Stupp R, Hegi ME, Jaeckle KA, Armstrong TS, Wefel JS, Won M, Blumenthal DT, Mahajan A, Schultz CJ, Erridge S, Baumert B, Hopkins KI, Tzuk-Shina T, Brown PD, Chakravarti A, Curran WJ, Mehta MP. Dose-dense temozolomide for newly diagnosed glioblastoma: a randomized phase III clinical trial. J Clin Oncol. 2013;31:4085–91. doi: 10.1200/JCO.2013.49.6968. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718135659
- 3.Lacroix M, Abi-Said D, Fourney DR, Gokaslan ZL, Shi W, DeMonte F, Lang FF, McCutcheon IE, Hassenbusch SJ, Holland E, Hess K, Michael C, Miller D, Sawaya R. A multivariate analysis of 416 patients with glioblastoma multiforme: prognosis, extent of resection, and survival. J Neurosurg. 2001;95:190–8. doi: 10.3171/jns.2001.95.2.0190. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384244
- 4.Stummer W, Reulen H, Meinel T, Pichlmeier U, Schumacher W, Tonn J, Rohde V, Oppel F, Turowski B, Woiciechowsky C, Franz K, Pietsch T. Extent of resection and survival in glioblastoma multiforme: identification of and adjustment for bias. Neurosurgery. 2008;62:564–76. doi: 10.1227/01.neu.0000317304.31579.17. discussion 564-76. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384245
- 5.Esteller M, Garcia-Foncillas J, Andion E, Goodman SN, Hidalgo OF, Vanaclocha V, Baylin SB, Herman JG. Inactivation of the DNA-repair gene MGMT and the clinical response of gliomas to alkylating agents. N Engl J Med. 2000;343:1350–4. doi: 10.1056/NEJM200011093431901. [DOI] [PubMed] [Google Scholar]
- 6.Hegi ME, Diserens A, Gorlia T, Hamou M, de Tribolet N, Weller M, Kros JM, Hainfellner JA, Mason W, Mariani L, Bromberg Jacoline EC, Hau P, Mirimanoff RO, Cairncross JG, Janzer RC, Stupp R. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997–1003. doi: 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384246
- 7.Verhaak Roel GW, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, Alexe G, Lawrence M, O'Kelly M, Tamayo P, Weir BA, Gabriel S, Winckler W, Gupta S, Jakkula L, Feiler HS, Hodgson JG, James CD, Sarkaria JN, Brennan C, Kahn A, Spellman PT, Wilson RK, Speed TP, Gray JW, Meyerson M, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718384247
- 8.Sturm D, Witt H, Hovestadt V, Khuong-Quang D, Jones David TW, Konermann C, Pfaff E, Tönjes M, Sill M, Bender S, Kool M, Zapatka M, Becker N, Zucknick M, Hielscher T, Liu X, Fontebasso AM, Ryzhova M, Albrecht S, Jacob K, Wolter M, Ebinger M, Schuhmann MU, van Meter T, Frühwald MC, Hauch H, Pekrun A, Radlwimmer B, Niehues T, von Komorowski G, et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell. 2012;22:425–37. doi: 10.1016/j.ccr.2012.08.024. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/717961084
- 9.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 10.Pelloski CE, Ballman KV, Furth AF, Zhang L, Lin E, Sulman EP, Bhat K, McDonald JM, Yung, WK Alfred, Colman H, Woo SY, Heimberger AB, Suki D, Prados MD, Chang SM, Barker FG, Buckner JC, James CD, Aldape K. Epidermal growth factor receptor variant III status defines clinically distinct subtypes of glioblastoma. J Clin Oncol. 2007;25:2288–94. doi: 10.1200/JCO.2006.08.0705. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384248
- 11.Kesavabhotla K, Schlaff CD, Shin B, Mubita L, Kaplan R, Tsiouris AJ, Pannullo SC, Christos P, Lavi E, Scheff R, Boockvar JA. Phase I/II study of oral erlotinib for treatment of relapsed/refractory glioblastoma multiforme and anaplastic astrocytoma. J Exp Ther Oncol. 2012;10:71–81. [PubMed] [Google Scholar]
- 12.Yung WK Alfred, Vredenburgh JJ, Cloughesy TF, Nghiemphu P, Klencke B, Gilbert MR, Reardon DA, Prados MD. Safety and efficacy of erlotinib in first-relapse glioblastoma: a phase II open-label study. Neuro-oncology. 2010;12:1061–70. doi: 10.1093/neuonc/noq072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Raizer JJ, Abrey LE, Lassman AB, Chang SM, Lamborn KR, Kuhn JG, Yung, WK Alfred, Gilbert MR, Aldape KA, Wen PY, Fine HA, Mehta M, Deangelis LM, Lieberman F, Cloughesy TF, Robins HI, Dancey J, Prados MD. A phase II trial of erlotinib in patients with recurrent malignant gliomas and nonprogressive glioblastoma multiforme postradiation therapy. Neuro-oncology. 2010;12:95–103. doi: 10.1093/neuonc/nop015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Franceschi E, Cavallo G, Lonardi S, Magrini E, Tosoni A, Grosso D, Scopece L, Blatt V, Urbini B, Pession A, Tallini G, Crinò L, Brandes AA. Gefitinib in patients with progressive high-grade gliomas: a multicentre phase II study by Gruppo Italiano Cooperativo di Neuro-Oncologia (GICNO) Br J Cancer. 2007;96:1047–51. doi: 10.1038/sj.bjc.6603669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang Julie HY, Chute DJ, Riggs BL, Horvath S, Liau LM, Cavenee WK, Rao PN, Beroukhim R, Peck TC, Lee JC, Sellers WR, Stokoe D, Prados M, Cloughesy TF, Sawyers CL, Mischel PS. Molecular determinants of the response of glioblastomas to EGFR kinase inhibitors. N Engl J Med. 2005;353:2012–24. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384249
- 16.Vivanco I, Robins HI, Rohle D, Campos C, Grommes C, Nghiemphu PL, Kubek S, Oldrini B, Chheda MG, Yannuzzi N, Tao H, Zhu S, Iwanami A, Kuga D, Dang J, Pedraza A, Brennan CW, Heguy A, Liau LM, Lieberman F, Yung, WK Alfred, Gilbert MR, Reardon DA, Drappatz J, Wen PY, Lamborn KR, Chang SM, Prados MD, Fine HA, Horvath S, et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012;2:458–71. doi: 10.1158/2159-8290.CD-11-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/716947814
- 17.Riemenschneider MJ, Betensky RA, Pasedag SM, Louis DN. AKT activation in human glioblastomas enhances proliferation via TSC2 and S6 kinase signaling. Cancer Res. 2006;66:5618–23. doi: 10.1158/0008-5472.CAN-06-0364. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384254
- 18.Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, Liu EM, Reichel J, Porrati P, Pellegatta S, Qiu K, Gao Z, Ceccarelli M, Riccardi R, Brat DJ, Guha A, Aldape K, Golfinos JG, Zagzag D, Mikkelsen T, Finocchiaro G, Lasorella A, Rabadan R, Iavarone A. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. 2012;337:1231–5. doi: 10.1126/science.1220834. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718384255
- 19.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–73. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 21.Ebos John ML, Kerbel RS. Antiangiogenic therapy: impact on invasion, disease progression, and metastasis. Nat Rev Clin Oncol. 2011;8:210–21. doi: 10.1038/nrclinonc.2011.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grünwald V, Hidalgo M. Development of the epidermal growth factor receptor inhibitor OSI-774. Semin Oncol. 2003;30:23–31. doi: 10.1016/S0093-7754(03)70022-0. [DOI] [PubMed] [Google Scholar]
- 23.Prewett M, Huber J, Li Y, Santiago A, O'Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin DJ. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999;59:5209–18. [PubMed] [Google Scholar]
- 24.Viloria-Petit A, Crombet T, Jothy S, Hicklin D, Bohlen P, Schlaeppi JM, Rak J, Kerbel RS. Acquired resistance to the antitumor effect of epidermal growth factor receptor-blocking antibodies in vivo: a role for altered tumor angiogenesis. Cancer Res. 2001;61:5090–101. [PubMed] [Google Scholar]
- 25.Batchelor TT, Mulholland P, Neyns B, Nabors LB, Campone M, Wick A, Mason W, Mikkelsen T, Phuphanich S, Ashby LS, Degroot J, Gattamaneni R, Cher L, Rosenthal M, Payer F, Jürgensmeier JM, Jain RK, Sorensen AG, Xu J, Liu Q, van den Bent, Martin Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. J Clin Oncol. 2013;31:3212–8. doi: 10.1200/JCO.2012.47.2464. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718074022
- 26.Gilbert MR, Dignam JJ, Armstrong TS, Wefel JS, Blumenthal DT, Vogelbaum MA, Colman H, Chakravarti A, Pugh S, Won M, Jeraj R, Brown PD, Jaeckle KA, Schiff D, Stieber VW, Brachman DG, Werner-Wasik M, Tremont-Lukats IW, Sulman EP, Aldape KD, Curran WJ, Mehta MP. A randomized trial of bevacizumab for newly diagnosed glioblastoma. N Engl J Med. 2014;370:699–708. doi: 10.1056/NEJMoa1308573. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718283109
- 27.Chinot OL, Wick W, Mason W, Henriksson R, Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea D, Brandes AA, Hilton M, Abrey L, Cloughesy T. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. N Engl J Med. 2014;370:709–22. doi: 10.1056/NEJMoa1308345. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718283108
- 28.Fine HA. Bevacizumab in glioblastoma--still much to learn. N Engl J Med. 2014;370:764–5. doi: 10.1056/NEJMe1313309. [DOI] [PubMed] [Google Scholar]
- 29.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Batchelor T, Sorensen AG, Ancukiewicz M. A phase II trial of AZD2171 (cediranib), an oral pan-VEGF receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma [abstract] J Clin Oncol. 2007;2001;25(Suppl 18) [Google Scholar]
- 31.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling RJ, Shi W, Vredenburgh JJ, Bigner DD. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28:4722–9. doi: 10.1200/JCO.2010.28.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/6040956
- 32.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, Friedman HS, Gilbert MR, Herndon JE, McLendon RE, Mitchell DA, Reardon DA, Sawaya R, Schmittling R, Shi W, Vredenburgh JJ, Bigner DD, Heimberger AB. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-oncology. 2011;13:324–33. doi: 10.1093/neuonc/noq157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh Alfons JM, Lutzky J, Lorigan P, Vaubel JM, Linette GP, Hogg D, Ottensmeier CH, Lebbé C, Peschel C, Quirt I, Clark JI, Wolchok JD, Weber JS, Tian J, Yellin MJ, Nichol GM, Hoos A, Urba WJ. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/4128956
- 34.Robert C, Thomas L, Bondarenko I, O'Day S, Jeffrey Weber MD, Garbe C, Lebbe C, Baurain J, Testori A, Grob J, Davidson N, Richards J, Maio M, Hauschild A, Miller WH, Gascon P, Lotem M, Harmankaya K, Ibrahim R, Francis S, Chen T, Humphrey R, Hoos A, Wolchok JD. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364:2517–26. doi: 10.1056/NEJMoa1104621. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/11993960
- 35.Lebbé1 C, McDermott DF, Robert C, Lorigan P, Ottensmeier CH, Wolchok J, Garbe C, Messina M, Hoos A, Weber JS. Ipilimumab improves survival in previously treated, advanced melanoma patients with poor prognostic factors: subgroup analysis from a phase III trial [abstract] Annals Oncol. 2010;21:viii401. [Google Scholar]
- 36.Schartz, Noël EC, Farges C, Madelaine I, Bruzzoni H, Calvo F, Hoos A, Lebbé C. Complete regression of a previously untreated melanoma brain metastasis with ipilimumab. Melanoma Res. 2010;20:247–50. doi: 10.1097/CMR.0b013e3283364a37. [DOI] [PubMed] [Google Scholar]
- 37.Fecci PE, Ochiai H, Mitchell DA, Grossi PM, Sweeney AE, Archer GE, Cummings T, Allison JP, Bigner DD, Sampson JH. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clin Cancer Res. 2007;13:2158–67. doi: 10.1158/1078-0432.CCR-06-2070. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384257
- 38.Grauer OM, Nierkens S, Bennink E, Toonen Liza WJ, Boon L, Wesseling P, Sutmuller Roger PM, Adema GJ. CD4+FoxP3+ regulatory T cells gradually accumulate in gliomas during tumor growth and efficiently suppress antiglioma immune responses in vivo. Int J Cancer. 2007;121:95–105. doi: 10.1002/ijc.22607. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384260
- 39.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, Durham N, Meyer C, Harris TJ, Albesiano E, Pradilla G, Ford E, Wong J, Hammers H, Mathios D, Tyler B, Brem H, Tran PT, Pardoll D, Drake CG, Lim M. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86:343–9. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718014910
- 40.Wick W, Puduvalli VK, Chamberlain MC, van den Bent, Martin J, Carpentier AF, Cher LM, Mason W, Weller M, Hong S, Musib L, Liepa AM, Thornton DE, Fine HA. Phase III study of enzastaurin compared with lomustine in the treatment of recurrent intracranial glioblastoma. J Clin Oncol. 2010;28:1168–74. doi: 10.1200/JCO.2009.23.2595. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/1927958
- 41.Kruser TJ, Mehta MP, Robins HI. Pseudoprogression after glioma therapy: a comprehensive review. Expert Rev Neurother. 2013;13:389–403. doi: 10.1586/ern.13.7. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718384261