

Abstract
Ciliary disorders share typical features, such as polydactyly, renal and biliary cystic dysplasia, and retinitis pigmentosa, which often overlap across diagnostic entities. We report on two siblings of consanguineous parents and two unrelated children, both of unrelated parents, with co-occurrence of Joubert syndrome and Jeune asphyxiating thoracic dystrophy, an association that adds to the observation of common final patterns of malformations in ciliary disorders. Using homozygosity mapping in the siblings, we were able to exclude all known genes/loci for both syndromes except for INVS, AHI1, and three genes from the previously described Jeune locus at 15q13. No pathogenic variants were found in these genes by direct sequencing. In the third child reported, sequencing of RPGRIP1L, ARL13B, AHI1, TMEM67, OFD1, CC2D2A, and deletion analysis of NPHP1 showed no mutations. Although this study failed to identify a mutation in the patients tested, the co-occurrence of Joubert and Jeune syndromes is likely to represent a distinct entity caused by mutations in a yet to be discovered gene. The mechanisms by which certain organ systems are affected more than others in the spectrum of ciliary diseases remain largely unknown.
Keywords: Joubert syndrome, Jeune syndrome, asphyxiating thoracic dystrophy, skeletal dysplasia, molar tooth sign, renal cystic dysplasia, cystic kidneys, encephalocele, brain malformation
INTRODUCTION
Joubert syndrome (JS) denotes a spectrum of ciliary disorders, all of which feature episodic hyperpnea and apnea, ataxia, cognitive impairment, abnormal eye movements, elongated midbrain tegmentum, dysplastic caudal medulla, and vermis hypo/dysplasia [Joubert et al., 1969]. Microcystic renal disease, hepatic dysplasia, and retinal disease are also recognized as part of the spectrum [Boltshauser and Isler, 1977; Pellegrino et al., 1997; Steinlin et al., 1997]. The “molar tooth sign”—a result of the triad of a deep posterior interpeduncular fossa, prominent superior cerebellar peduncles, and vermian hypoplasia/aplasia—is observed in 82% of patients [Maria et al., 1997, 1999]. Skeletal dysplasia is not recognized as a feature of JS. To date, nine causative recessive genes have been identified, all of which relate to ciliary function: AHI1 [Lagier-Tourenne et al., 2004],NPHP1 [Parisi et al., 2004],CEP290/NPHP6 [Sayer et al., 2006], TMEM67/MKS3 [Baala et al., 2007], RPGRIP1L [Arts et al., 2007; Delous et al., 2007], ARL13B [Cantagrel et al., 2008], CC2D2A [Gorden et al., 2008], INPP5E [Bielas et al., 2009; Jacoby et al., 2009], and OFD1 [Coene et al., 2009]. Each accounts for no more than 5–10% of cases [Parisi et al., 2007]. An additional locus has also been mapped to 11p12–q13 [Keeler et al., 2003; Valente et al., 2005].
Jeune asphyxiating thoracic dystrophy (JATD) is a rare autosomal recessive disorder characterized by a long, narrow thorax, short-limbed short stature, polydactyly, and renal cystic dyplasia. The skeletal dysplasia manifests as cone-shaped epiphyses in the hands and feet, irregular metaphyses, decreased iliac cephalocaudal height, and a trident-shaped acetabulum [Oberklaid et al., 1977]. Renal cystic changes, either diffuse or focused in the cortex, may be seen early in life [Barnes and Opitz, 1992]. Those who survive early childhood frequently develop chronic kidney disease in their later life because of diffuse tubulointerstitial fibrosis, tubular atrophy, periglomerular fibrosis, or glomerular sclerosis. These histopathological changes are often indistinguishable from juvenile nephronophthisis [Bernstein et al., 1974; Kozlowski and Masel, 1976; Donaldson et al., 1985]. A subset of JATD cases have mapped to locus 15q13 [Morgan et al., 2003], in which the causative gene remains unknown. Recessive mutations of IFT80 (3q25), which encodes a component of the ciliary intraflagellar transport (IFT) complex, have been found in another subset of patients with JATD [Beales et al., 2007]. Mapping of a third locus, 11q14–11q23, led to the identification of recessive mutations in DYNC2H1, which encodes a dynein complex subunit [Dagoneau et al., 2009; Merrill et al., 2009]. Mutations in DYNC2H1 were also found in families affected by short-rib polydactyly type III (SRPIII). This disorder has been proposed to lie on the severe end of a spectrum shared with JATD [Ho et al., 2000]. The short rib polydactyly group of skeletal dysplasias is characterized by typically lethal thoracic hypoplasia, short limbs, polydactyly, and visceral abnormalities. SRPIII can be quite similar to type I SRP, but in general, can be radiologically distinguished by the presence of a trident-shaped acetabular roof, a “ball-in-cone” appearance at the end of the long bones due to lateral and medial spiky metaphyseal spurs and overall decreased severity of long bone hypoplasia [Lachman, 2007]. SRPIII can be radiographically distinguished from JATD by the presence of medial and lateral metaphyseal spurs, more severe rib shortening, abnormal vertebra, and less frequent observation of phalangeal coned epiphyses [Yang et al., 1987; Lachman, 2007].
The observation of both JATD and JS occurring together in four patients described herein strongly suggests that a single ciliary gene important in both skeletal and cerebellar development could be responsible for the overlapping phenotype. The aim of our study was to search for the causative locus by screening for homozygosity of known loci and genes implicated in both disorders, as well as the Ellis Van Creveld gene (EVC) and inversin (INVS), using data from single nucleotide polymorphism (SNP) arrays (Affymetrix Genome-Wide Human 6.0 SNP array) in two siblings born of a consanguineous union. The coding exons of genes located in homozygous intervals shared between the siblings were then sequenced for mutations but none were found. Sequencing of several ciliary genes in a third child with JATD and JS did not reveal mutations.
CLINICAL REPORTS AND METHODS
Clinical Report, Patient I
Two siblings with features of both JATD and JS were born to healthy third cousin parents of Filipino ethnicity. A daughter was noted to have hypotonia, developmental delay, and oculomotor apraxia at 6 months of age, and subsequent neuroimaging revealed a “molar tooth sign,” with hypoplasia of the inferior vermis of the cerebellum and elongated superior cerebellar peduncles (Fig. 1). She had mild frontal bossing, a small thorax, and disproportionately short limbs and digits. Radiographs demonstrated short ribs with expanded anterior ends, brachydactyly with cone-shaped epiphyses, a trident-shaped acetabular roof, and abnormal proximal femoral metaphyses (Fig. 1). Echogenic kidneys with cortical cysts and dilated hepatobiliary ducts were observed on ultrasound. She sat unsupported at age 2 years, stood at age 4 years, and walked with aid of a walker at age 5 years; she could speak single words by age 3 years and sentences by age 5 years. At 31 months she developed symptomatic renal failure requiring dialysis, and she later received a renal transplant from a living related donor at 4 years of age. A gastrostomy tube was placed at age 4 years because of inadequate oral intake. She has been admitted to hospital several times for respiratory tract infections compounded by reactive airways disease; she requires bi-level positive airway pressure at night. At age 5 years she suffered sudden loss of vision bilaterally and was diagnosed with idiopathic elevated intracranial pressure on the basis of elevated opening pressure with lumbar puncture. She regained some vision following treatment with acetazolamide, further suggesting the vision loss was likely due to increased intracranial pressure. Enlarged lateral ventricles and extra-axial CSF spaces were noted on MRI. Also noted was a narrow upper cervical spinal canal with hypoplasia of the first vertebra. Ophthalmological assessment demonstrated optic disc swelling, mild myopia, and patchy choroidal atrophy of the posterior poles. Small creamy white subretinal deposits, in keeping with retinal dystrophy, were seen along the superior temporal arcade.
FIG. 1.
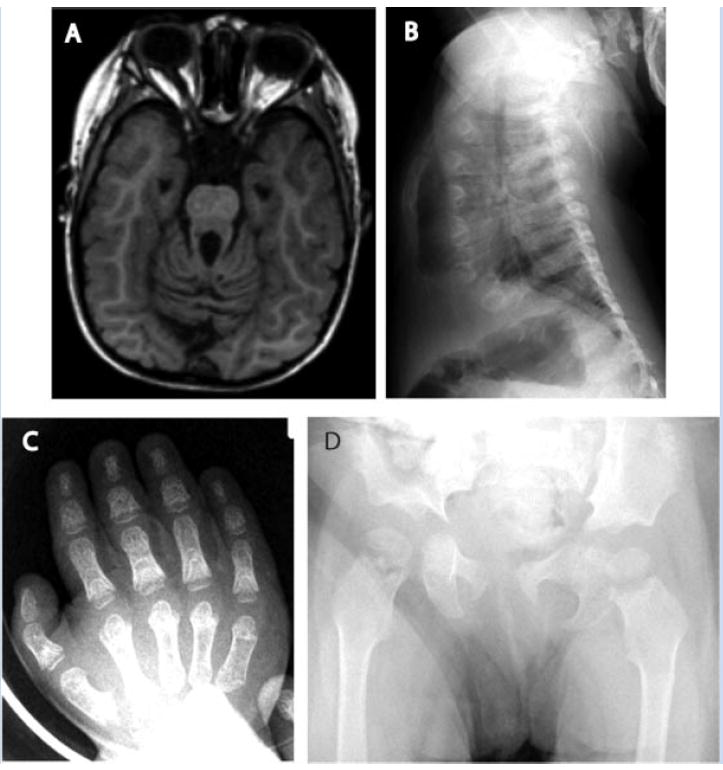
Patient I. A: Elongated superior cerebellar peduncles contribute to the molar tooth sign demonstrated in T1 MRI. B: Marked hypoplasia of the ribs, with decreased anterior–posterior diameter and thoracic volume. The anterior ends of the ribs are expanded. C: Hand radiographs demonstrate generalized brachydactyly and wide, cone-shaped epiphyses in the proximal phalanges. D: Pelvic radiograph demonstrates trident shape of horizontal acetabular roofs, bilateral coxa valga, and bilateral irregular widening of the proximal femoral epiphyses.
Clinical Report, Patient II
The parents of Patient I conceived again. Serial antenatal scans, beginning at 19 weeks gestation, showed hyperechoic kidneys and hypoplasia of the cerebellar vermis. A male infant was delivered at term. A postnatal MRI confirmed vermian hypoplasia and a molar tooth sign (Fig. 2). MRI also demonstrated hypoplasia of the first cervical vertebra that appeared to be contributing to CSF outflow obstruction and ventriculomegaly, more severe than the malformation present in his sister, Patient I. At age 3 years, he has global developmental delay (including absence of speech), hypotonia (unable to sit independently), oculomotor apraxia, short-limbed short stature (height 3.4 SD below the mean and weight at the 1st centile), hypoplastic thorax, biliary duct dilatation, and enlarged cystic dysplastic kidneys (>95th centile) with normal renal function and blood pressure thus far. He has had elevated serum trans-aminases of uncertain cause. Nutrition is largely supplied via gastrostomy tube for feeding difficulties and bi-level positive airway pressure is provided at night. A barium swallow has demonstrated an aberrant right subclavian artery partly indenting, but not obstructing, his esophagus.
FIG. 2.
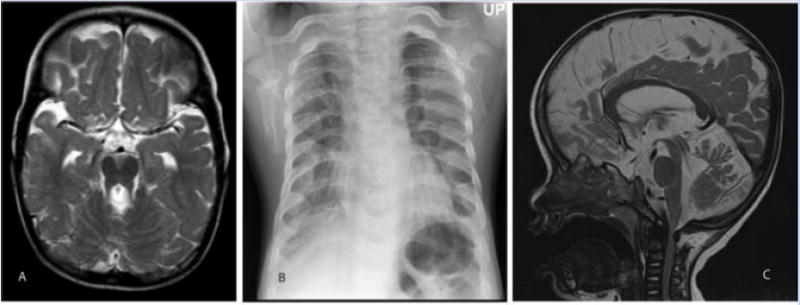
Patient II. A: T2-weighted MRI demonstrating the molar tooth sign. B: Thoracic hypoplasia with broad costochondral junctions and abnormal clavicles. C: Saggital T2 MRI view demonstrates narrowing of the subarachnoid space at the C1 level, contributing to enlargement of the subarachnoid space caudally.
Clinical Report, Patient III
A child unrelated to the aforementioned siblings presented prenatally with a prominent cisterna magna at 19 weeks gestation. The parents were an unrelated couple of Caribbean descent. The perinatal findings of JS in this patient have been previously described [Fluss et al., 2006], prior to an additional postnatal diagnosis of JATD. An amniocyte karyotype was normal 46,XY. Additional ultrasound findings at 22 weeks included absence of the cerebellar vermis, a smooth cortex, and moderate ventriculomegaly (14 mm); at 27 weeks, MRI demonstrated a dysgenetic vermis and molar tooth sign, undersulcation, delayed opercularization, postaxial polydactyly, and hypertelorism. Growth parameters at birth were normal (length 51 cm: 50th centile; weight 3,500 g: 25th centile; head circumference: 35.3 cm: 25th centile). The infant had a narrow, bell-shaped chest with widely spaced nipples, rhizomelic shortening of his upper and lower limbs, bilateral postaxial polydactyly of his hands and feet, and a left simian crease (Fig. 3). In addition, hypertelorism, a high forehead with bossing, depressed nasal bridge, epicanthic folds, posteriorly rotated and low-set ears with overfolded helices, a supernumerary nipple and grade I hypospadias with incomplete prepuce were observed. Episodic tachypnea was observed, which gradually resolved during infancy. A skeletal survey demonstrated only 11 ribs on the right side, rib hypoplasia with broadening at the costochondral junctions, and spurs from the medial acetabular roof (Fig. 3). Radiographs of the hands at age 10 months confirmed the presence of two phalanges in the supernumerary digits with a bifid, Y-shaped fifth metacarpal on the left and a broad distal metaphasis of the fifth metacarpal on the right (Fig. 3). Ultrasonography of the kidneys liver, biliary tracts, and heart was normal. An MRI in the neonatal period confirmed the molar tooth sign and bilateral ventriculomegaly as well as absence of the left olfactory sulcus and bulb and subependymal cysts. Hearing screening and subsequent auditory brainstem testing confirmed severe bilateral sensorineural hearing loss. Ophthalmologic assessment showed the presence of the Duane anomaly of extraocular movement; dilated retinal examination and electroretinogram showed features of retinal dystrophy. An EEG was within the normal range. He developed significant feeding difficulties and gastro-esophageal reflux with recurrent aspiration pneumonia, which necessitated gastrostomy tube insertion at 7 months of age. Despite tube feeding, he developed chronic lung disease secondary to aspiration, and failure to thrive with weight, height, and head circumference all below the 3rd centile. Global developmental delay has been most significant for gross and fine motor domains. At 3 years of age he could sit but could not crawl or stand, could scribble, could pile 4–5 blocks, and could utter single words but not phrases.
FIG. 3.
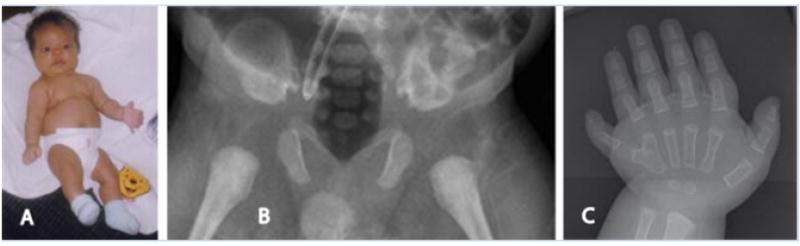
Patient III. A: Hypoplastic thorax with decreased chest circumference, protuberant abdomen, and rhizomelic limb shortening are shown. A depressed nasal root, prominent forehead, and postaxial polydactyly are also present. B: Medial spurs of the acetabular roof as well as a narrow sacroiliac notch are seen. The ilia are generally hypoplastic. C: Postaxial polydactyly of the right hand with two phalangeal bones in the extra digit, a bifid metacarpal, and brachydactyly. [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Clinical Report, Patient IV
Nonconsanguineous parents with two healthy children, and no family history of developmental disorders or congenital anomalies, presented in their third pregnancy with a female fetus with intracranial abnormalities at 20 weeks of gestation. Mild lateral ventriculomegaly (10–11 mm), a mildly dilated third ventricle, an absent septum pellucidum, cerebellar hypoplasia, and increased posterior fossa fluid spaces were observed. An MRI at 31 weeks gestation confirmed vermis hypoplasia and also detected a small occipital encephalocele/cervical meningocele, colpocephaly, and thickened and horizontally oriented superior cerebellar peduncles consistent with a molar tooth sign. Following an uncomplicated delivery via caesarean section at 39 weeks gestation, mild frontal bossing, low-set ears, mild micrognathia, redundant nuchal skin, abnormal eye movements, mild hypotonia, and episodic tachypnea and apnea were noted. Moderate central and severe obstructive apnea were documented on polysomnography. She was fed through a nasoduodenal tube because of an aspiration risk related to gastroesophageal reflux. An MRI confirmed the prenatal findings and also demonstrated dysplastic cerebellar hemispheres, absent splenium of the corpus callosum, globular-appearing basal ganglia, prominent fornices, and a bulging dorsal medulla (Fig. 4). Renal and hepatic ultrasonography was normal. A skeletal survey showed dysplasia. Thoracic abnormalities included a narrow bell shape, shortening of horizontally oriented ribs with flaring of the anterior ends, and vestigial-like hypoplasia of the 12th ribs. The clavicles were relatively long compared to the size of the thorax. Premature ossification of the epiphyses, particularly of the proximal femora, was observed. Limb lengths were mildly short and phalangeal lengths were within the normal range. The pelvis demonstrated a mild trident appearance of the acetabular margin with an inferolateral spur of the sciatic notch (Fig. 4). The pelvis also featured flared iliac bones with decreased cephalocaudal height. The vertebrae were normal with neither platyspondyly nor sagittal or coronal clefting.
FIG. 4.
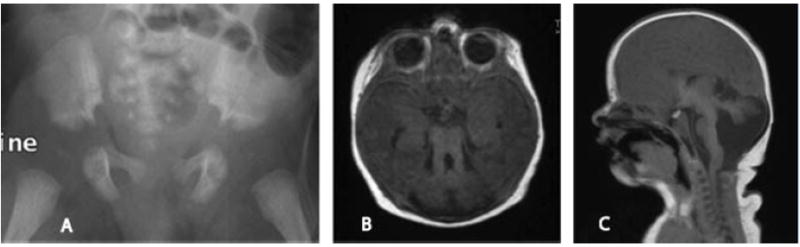
Patient IV. A: The mildly hypoplastic pelvis demonstrates narrow sacroiliac notches with inferolateral notches and flared iliac bones. B: Thickened and elongated superior cerebellar peduncles and a deep interpeduncular fossa are seen as part of the molar tooth sign displayed in an axial T1 view. C: Saggital T1 MRI demonstrates dysplastic cerebellar hemispheres and a hypoplastic corpus callosum.
Homozygosity Analysis
Affymetrix 6.0 SNP array was performed by the Affymetrix clinical laboratory (Sacramento, CA) on lymphocyte DNA from Patients I and II. Informed consent was obtained and the study was approved by an institutional review board. Regions of homozygosity that entirely encompassed the genes in question (Table I) were screened for homozygosity using the Affymetrix Genotyping Console™ software. It was also noted if the children demonstrated shared heterozygosity indicating a possibility of pathological compound heterozygosity or homozygosity not identical by descent. Genes for which the siblings did not share alleles were excluded from further analysis (CEP 290/NPHP6, TMEM67/MKS3, RPGRIP1L, ARL13B, CC2D2A, IFT80, DYNC2H1, and EVC1).
TABLE I.
Results From Homozygosity Mapping and Molecular Analyses to Rule in or Rule Out Candidate Loci Based on Previously Described Genes/Loci for JATD and JS in Patients I and II
| Region | Gene | Associated disorder(s) | Segregation status | Molecular |
|---|---|---|---|---|
| Chr9:135,345,956–139,856,799 | INPP5E | JS, MORM | No shared allelesb | |
| JBTS2 (CORS2) 11p12–q13: 46,023,436–59,656,135 | Unknown | JS | No shared alleles | |
| Chr12:86,966,921–87,060,124 | CEP290 (NPHP6) | JS, MKS, BBS, LCA, SL | No shared alleles | |
| Chr8:94,836,248–94,900,634 | TMEM67 (MKS3) | JS, MKS, BBS; COACH | No shared alleles | |
| Chr2:110,238,203–110,319,928 | NPHP1 | JS, NPH, SL, COMA | Heterozygously shared alleles | No deletion |
| Chr6:135,646,817–135,860,576 | AHI1 | JS | Homozygously shared alleles | No mutations |
| Chr3:95,181,672–95,256,813 | ARL13B | JS | No shared alleles | |
| Chr16:52,191,319–52,244,400 | RPGRIP1L | JS; MKS; COACH | No shared alleles | |
| Chr4:15,080,760–15,212,278 | CC2D2A | JS; MKS; COACH | No shared alleles | |
| Chr9:101,901,332–102,103,247 | INVS | NPH | Homozygously shared alleles | No mutations |
| Chr4: 5,750,000–5,900,000 | EVC | EVC | No shared alleles | |
| Chr3:161,427,936–161,650,320 | IFT80 | JATD | No shared alleles | |
| Chr11:102,485,370–102,855,801 | DYNC2H1 | JATD, SRP | No shared alleles | |
| Chr15: 28,947,842–29,258,267 | MTMR10a | 15q13 linked to JATD | Homozygously shared alleles | No mutations |
| MTMR15a | 15q13 linked to JATD | Homozygously shared alleles | No mutations | |
| TRPM1a | 15q13 linked to JATD | Homozygously shared alleles | No mutations |
JS, Joubert syndrome; MKS, Meckel syndrome; BBS, Bardet–Biedl syndrome; LCA, Leber congenital amaurosis; SL, Senior–Loken syndrome; COMA, Cogan-type congenital oculomotor apraxia; NPH, nephronophthisis; EVC, Ellis Van Creveld syndrome; JATD, Jeune asphyxiating thoracic dystrophy; SRP, short rib polydactyly; COACH, cerebellar vermis hypoplasia, oligophrenia, ataxia, colobomas, hepatic fibrosis syndrome; MORM, mental retardation, truncal obesity, retinal dystrophy, and micropenis.
Linkage information is also provided for EVC, the causative gene for Ellis Van Creveld syndrome, a disorder with some similar features to JATD, and INVS, a ciliary gene involved in nephronophthisis.
These candidate genes are within a region of 15q13, a JATD locus, that overlapped our linkage data and that published by Morgan et al. (2003).
Within the gene itself, there are only two SNPs represented, but analysis extending beyond the gene indicates heterozygous haplotypes not linked.
Candidate Gene Sequencing
Candidate genes in homozygous regions (INVS, AHI1, and genes within a portion of 15q13: TRPM1, MTMR10, and MTMR15) were further analyzed by Sanger sequencing of coding exons, and at least 20 bp of bordering intronic sequence, after standard PCR amplification. Primers and conditions are available upon request. Sequencher® software (Gene Codes, Ann Arbor, MI) was used to identify variants in the sequence compared to human reference sequence NCBI Build 36.1. Sequence variants were manually inspected to rule out sequencing artifacts, and then referenced to dbSNP (http://www.ncbi.nlm.nih.gov/projects/SNP) to identify known polymorphisms. Unidirectional sequencing was performed initially, followed as needed by sequencing from the opposite direction to clarify ambiguous traces. The NPHP1 region was examined for heterozygosity to exclude deletion of the region that accounts for the majority of NPHP1 mutations [Heninger et al., 2001]. In Patient III, sequencing of the coding exons was undertaken for RPGRIP1L, ARL13B, AHI1, TMEM67/MKS3, OFD1, and CC2D2A; deletion analysis was performed for NPHP1.
RESULTS
Of a total of 14 loci examined for homozygosity shared between the siblings (Patients I and II), 3 were identified (Table I). The 15q13 candidate region (ATD1) previously identified through linkage study in several JATD families spanned ~1.5 Mb (between D15S165 and D15S1010; 28,947,842–30,777,610) [Morgan et al., 2003]. The siblings in our study demonstrated homozygosity across a ~530 kb region (28,726,417–29,258,267) that overlapped ~310 kb with this previously reportedly region and contained only 3 genes: MTMR10, MTMR15, and TRPM1. Sequencing of the coding regions of INVS, AHI1, MTMR10, MTMR15, and TRPM1 in one sibling revealed no mutations. Several SNPs catalogued by dbSNP were identified, all of which were homozygous as expected. NPHP1 deletion analysis confirmed two copies. Sequencing of RPGRIP1L, ARL13B, AHI1, TMEM67, OFD1, CC2D2A in Patient III demonstrated no mutations. A heterozygous nonsynonymous SNP (rs16892095) was found in CC2D2A. Deletion analysis of NPHP1 was normal.
DISCUSSION
This clinical observation of JATD and JS co-occurrence expands the recognized phenotypic spectrum in both disorders, and suggests the involvement of a single causative ciliary gene required for both skeletal and neurological development (see Table II for comparisons). Cilia are specialized, membrane-bound, hair-like structures that project from the cell surface. Each cilium is composed of a microtubule cytoskeleton (the axoneme) surrounded by a specialized membrane and anchored in the cell by the basal body. IFT proteins including dynein and kinesin motor proteins are required for the assembly and function of cilia [reviewed in Beisson and Wright, 2003]. Primary cilia are present in most cell types, including renal tubule epithelia, retinal photoreceptors, chondrocytes, fibroblasts, and neurons. Cilia perform numerous roles in cellular functioning, such as detection of mechanical, chemical, and light stimuli, as well as mediation of multiple signaling pathways involved in development and differentiation. The ciliopathic phenotype in human disease consists of multiple typical features such as: cystic renal and liver disease, retinal dystrophy, situs inversus, polydactyly, brain anomalies of the posterior fossa, skeletal anomalies, and more. Microcystic renal disease, present in the siblings reported here, is an overlapping feature of JATD, JS, and SRP. Although retinal dystrophy, Dandy–Walker malformation, venticulomegaly, and arachnoid cysts have been described in JATD previously [Wilson et al., 1987; Singh et al., 1988; Ardura Fernández et al., 1990; Trabelsi et al., 1990; Silengo et al., 2000], classically this syndrome has been considered a skeletal-visceral disorder. The phenotypes of our four patients also overlap with the core features of Meckel syndrome: brain malformation (usually occipital encephalocele), renal cystic disease, congenital hepatic fibrosis/ductal plate malformation, and postaxial polydactyly [Seller, 1981; Salonen, 1984]. Most of the genes implicated in Meckel syndrome have also been confirmed to cause JS. In addition, Franceschini et al. [2004] described sibling fetuses with a Meckel-like phenotype and skeletal dysplasia: trident-shaped acetabular roofs, decreased vertical height of the ilia, and short, curved long bones, in addition to typical Meckel features of occipital encephalocele, bilateral polycystic kidneys, and hepatic cystic dysplasia. Similarly, Tsai et al. [1992] reported a fetus with short rib polydactyly, encephalocele, and situs inversus.
TABLE II.
Comparison of Features of Patients I–IV Versus the Features of Joubert Syndrome, Jeune Asphyxiating Thoracic Dystrophy, and Short Rib Polydactyly III
| Pt I | Pt II | Pt III | Pt IV | JATD | JS | |
|---|---|---|---|---|---|---|
| CNS | ||||||
| Dandy–Walker malformation | − | − | − | − | +a | + |
| Agenesis of the corpus callosum | − | − | − | Partial | +a | +a |
| Occipital encephalocele | − | − | − | + | − | +a |
| Molar tooth sign | + | + | + | + | − | + |
| Ventriculomegaly | + | + | + | + | +a | +a |
| Oculomotor apraxia | + | + | + | + | − | + |
| Episodic hyperpnea/apnea | + | + | + | + | − | + |
| Gross motor delay | + | + | + | + | − | + |
| Intellectual disability/developmental delay | + | + | + | NA | − | + |
| Retinal dystrophy | + | − | + | − | +a | + |
| Renal cystic dysplasia | + | + | − | − | + | + |
| Biliary dysgenesis/hepatic fibrosis | + | + | − | − | + | + |
| Situs inversus | − | − | − | − | +a | +a |
| Cardiac abnormalities | − | − | − | − | − | +a |
| Genital abnormalities | − | − | + | − | − | +a |
| Skeletal | ||||||
| Depressed nasal root | − | − | + | ? | − | − |
| Hypoplastic thorax (short, horizontal ribs) | + | + | + | + | + | − |
| Disproportionate short limbs | + | + | + | + | + | − |
| Decreased cephalocaudal height of pelvis | + | ? | + | + | + | − |
| Irregular/wide metaphyses | + | ? | − | − | + | − |
| Horizontal, trident-shaped acetabulum | + | ? | + | + | + | − |
| Lateral and medial metaphyseal spurs | + | ? | − | − | +a | − |
| Brachyphalangy | + | + | + | − | + | − |
| Postaxial polydactyly | − | − | + | + | + | + |
| Cone-shaped epiphyses in the hands and feet | + | ? | − | − | + | − |
Occasional/atypical features.
Loss of function for either of the genes responsible for JATD (IFT80 or DYNC2H1) is associated with abnormal cilia [Beales et al., 2007; Dagoneau et al., 2009; Merrill et al., 2009]. The restricted and dysplastic growth of the long bones, pelvis, and thoracic cage observed in these ciliary disorders could result from perturbed hedgehog signaling during development. This pathway is at least partially mediated through cilia [Eggenschwiler and Anderson, 2007]. In particular, Indian hedgehog requires functional IFT in order to influence proliferation of the chondrocyte lineage in long bone development [Haycraft et al., 2007]; Indian hedgehog mutant mice demonstrate limb shortening as well as hypoplasia of the axial skeleton [St. Jacques et al., 1999].
Even though the parents of the siblings had a low degree of consanguinity (3rd cousins), more shared homozygosity than anticipated was encountered, making a search for new candidate genes impractical. Indeed, 3 of 14 examined loci (21%) demonstrated homozygous inheritance common to both siblings and required further molecular analysis. For comparison, runs of homozygosity around 100 kb have been estimated to constitute 13% of individual European genomes [Frazer et al., 2007]. There were 35 runs of homozygosity longer than 1 Mb shared between the siblings (none longer than 3 Mb), a length that begins to suggest recent ancestral sharing [McQuillan et al., 2008; Hildebrandt et al., 2009]. Our studies do not completely rule out causation from the linked candidate genes, as only the coding portions of the genes were sequenced; mutations in the promotor, untranslated, or intronic regions would have been missed.
Children with JATD should be evaluated for signs of cerebellar dysfunction and visual impairment, with consideration for cranial MRI; this co-occurrence may be more common than previously recognized. Although the MRI findings in all four patients are in keeping with JS, we believe in each case (and the siblings especially) that the vermis has a lace-like appearance more suggestive of atrophy rather than hypoplasia. Furthermore, the superior cerebellar peduncles are thick, but less so than usual. Another unusual MRI finding in these patients was atlas hypoplasia (Patients I and II). Symptomatic atlantoaxial instability has been reported previously in a patient with JATD [Tüysüz et al., 2009], which indicates the importance of investigation for neurological symptoms or signs in JATD patients. Our report also highlights the possible severity of the renal dysplasia in JATD and JS, and demonstrates that renal transplantation may be successfully performed after rigorous evaluation of pulmonary function.
References
- Ardura Fernández J, Alvarez González C, Rodríguez Fernández M, Andrés de Llano J. Asphyxiating thoracic dysplasia associated with proximal myopathy and arachnoid cyst. An Esp Pediatr. 1990;33:592–596. [PubMed] [Google Scholar]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler MC, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attie-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes EG, Opitz JM. Renal abnormalities in malformation syndromes. In: Edelmann CM Jr, Bernstein J, Meadow SR, Spitzer A, Travis LB, editors. Pediatric kidney disease. 2. Boston: Little Brown; 1992. pp. 1067–1119. [Google Scholar]
- Beales PL, Bland E, Tobin JL, Bacchelli C, Tüysüz B, Hill J, Rix S, Pearson CG, Kai M, Hartley J, Johnson C, Irving M, Elcioglu N, Winey M, Tada M, Scambler PJ. IFT80, which encodes a conserved intraflagellar transport protein, is mutated in Jeune asphyxiating thoracic dystrophy. Nat Genet. 2007;39:727–729. doi: 10.1038/ng2038. [DOI] [PubMed] [Google Scholar]
- Beisson J, Wright M. Basal body/centriole assembly and continuity. Curr Opin Cell Biol. 2003;15:96–104. doi: 10.1016/s0955-0674(02)00017-0. [DOI] [PubMed] [Google Scholar]
- Bernstein J, Brough AJ, McAdams AJ. The renal lesion in syndromes of congenital malformations. Birth Defects. 1974;10:35–43. [PubMed] [Google Scholar]
- Bielas SL, Silhavy JL, Brancati F, Kisseleva MV, Al-Gazali L, Sztriha L, Bayoumi RA, Zaki MS, Abdel-Aleem A, Rosti RO, Kayserili H, Swistun D, Scott LC, Bertini E, Boltshauser E, Fazzi E, Travaglini L, Field SJ, Gayral S, Jacoby M, Schurmans S, Dallapiccola B, Majerus PW, Valente EM, Gleeson JG. Mutations in INPP5E, encoding inositol polyphosphate-5-phosphatase E, link phosphatidyl inositol signaling to the ciliopathies. Nat Genet. 2009;41:1032–1036. doi: 10.1038/ng.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boltshauser E, Isler W. Joubert syndrome: Episodic hyperpnea, abnormal eye movements, retardation and ataxia, associated with dysplasia of the cerebellar vermis. Neuropadiatrie. 1977;8:57–66. doi: 10.1055/s-0028-1091505. [DOI] [PubMed] [Google Scholar]
- Cantagrel V, Silhavy JL, Bielas SL, Swistun D, Marsh SE, Bertrand JY, Audollent S, Attié-Bitach T, Holden KR, Dobyns WB, Traver D, Al-Gazali L, Ali BR, Lindner TH, Caspary T, Otto EA, Hildebrandt F, Glass IA, Logan CV, Johnson CA, Bennett C, Brancati F, Valente EM, Woods CG, Gleeson JG International Joubert Syndrome Related Disorders Study Group. Mutations in the cilia gene ARL13B lead to the classical form of Joubert syndrome. Am J Hum Genet. 2008;83:170–179. doi: 10.1016/j.ajhg.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coene KL, Roepman R, Doherty D, Afroze B, Kroes HY, Letteboer SJ, Ngu LH, Budny B, van Wijk E, Gorden NT, Azhimi M, Thauvin-Robinet C, Veltman JA, Boink M, Kleefstra T, Cremers FP, van Bokhoven H, de Brouwer AP. OFD1 is mutated in X-linked Joubert syndrome and interacts with LCA5-encoded lebercilin. Am J Hum Genet. 2009;85:465–481. doi: 10.1016/j.ajhg.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagoneau N, Goulet M, Geneviève D, Sznajer Y, Martinovic J, Smithson S, Huber C, Baujat G, Flori E, Tecco L, Cavalcanti D, Delezoide AL, Serre V, Le Merrer M, Munnich A, Cormier-Daire V. DYNC2H1 mutations cause asphyxiating thoracic dystrophy and short rib-polydactyly syndrome, type III. Am J Hum Genet. 2009;84:706–711. doi: 10.1016/j.ajhg.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, Encha-Razavi F, Boddaert N, Gonzales M, Macher MA, Nivet H, Champion G, Bertheleme JP, Niaudet P, McDonald F, Hildebrandt F, Johnson CA, Vekemans M, Antignac C, Ruther U, Schneider-Maunoury S, Attie-Bitach T, Saunier S. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Donaldson MD, Warner AA, Trompeter RS, Haycock GB, Chantler C. Familial juvenile nephronophthisis, Jeune’s syndrome, and associated disorders. Arch Dis Child. 1985;60:426–434. doi: 10.1136/adc.60.5.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggenschwiler JT, Anderson KV. Cilia and developmental signaling. Annu Rev Cell Dev Biol. 2007;23:345–373. doi: 10.1146/annurev.cellbio.23.090506.123249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluss J, Blaser S, Chitayat D, Akoury H, Glanc P, Skidmore M, Raybaud C. Molar tooth sign in fetal brain magnetic resonance imaging leading to the prenatal diagnosis of Joubert syndrome and related disorders. J Child Neurol. 2006;21:320–324. doi: 10.1177/08830738060210041001. [DOI] [PubMed] [Google Scholar]
- Franceschini P, Licata D, Guala A, Gaglioti P, Botta G, Gianotti G, Franceschini D. Cerebro-reno-digital (Meckel-like) syndrome with limb malformations and acetabular spurs in two sibs: A new MCA syndrome? Am J Med Genet Part A. 2004;131A:213–215. doi: 10.1002/ajmg.a.30313. [DOI] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM The International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. 54. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorden NT, Arts HH, Parisi MA, Coene KL, Letteboer SJ, van Beersum SE, Mans DA, Hikida A, Eckert M, Knutzen D, Alswaid AF, Ozyurek H, Dibooglu S, Otto EA, Liu Y, Davis EE, Hutter CM, Bammler TK, Farin FM, Dorschner M, Topcu M, Zackai EH, Rosenthal P, Owens KN, Katsanis N, Vincent JB, Hildebrandt F, Rubel EW, Raible DW, Knoers NV, Chance PF, Roepman R, Moens CB, Glass IA, Doherty D. CC2D2A is mutated in Joubert syndrome and interacts with the ciliopathy-associated basal body protein CEP290. Am J Hum Genet. 2008;83:559–571. doi: 10.1016/j.ajhg.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson WS, Detloff PJ, Serra R, Yoder BK. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- Heninger E, Otto E, Imm A, Caridi G, Hildebrandt F. Improved strategy for molecular genetic diagnostics in juvenile nephronophthisis. Am J Kidney Dis. 2001;37:1131–1139. doi: 10.1053/ajkd.2001.24514. [DOI] [PubMed] [Google Scholar]
- Hildebrandt F, Heeringa SF, Rüschendorf F, Attanasio M, Nürnberg G, Becker C, Seelow D, Huebner N, Chernin G, Vlangos CN, Zhou W, O’Toole JF, Hoskins BE, Wolf MT, Hinkes BG, Chaib H, Ashraf S, Schoeb DS, Ovunc B, Allen SJ, Vega-Warner V, Wise E, Harville HM, Lyons RH, Washburn J, Macdonald J, Nürnberg P, Otto EA. A systematic approach to mapping recessive disease genes in individuals from outbred populations. PLoS Genet. 2009;5:e1000353. doi: 10.1371/journal.pgen.1000353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho NC, Francomano CA, van Allen M. Jeune asphyxiating thoracic dystrophy and short-rib polydactyly type III (Verma-Naumoff) are variants of the same disorder. Am J Med Genet. 2000;90:310–314. doi: 10.1002/(sici)1096-8628(20000214)90:4<310::aid-ajmg9>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Jacoby M, Cox JJ, Gayral S, Hampshire DJ, Ayub M, Blockmans M, Pernot E, Kisseleva MV, Compère P, Schiffmann SN, Gergely F, Riley JH, Pérez-Morga D, Woods CG, Schurmans S. INPP5E mutations cause primary cilium signaling defects, ciliary instability and ciliopathies in human and mouse. Nat Genet. 2009;41:1027–1031. doi: 10.1038/ng.427. [DOI] [PubMed] [Google Scholar]
- Joubert M, Eisenring JJ, Robb JP, Anderman F. Familial agenesis of the cerebellar vermis: A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 19:1969. 813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- Keeler LC, Marsh SE, Leeflang EP, Woods CG, Sztriha L, Al-Gazali L, Gururaj A, Gleeson JG. Linkage analysis in families with Joubert syndrome plus oculo-renal involvement identifies the CORS2 locus on chromosome 11p12-q13. 3. Am J Hum Genet. 2003;73:656–662. doi: 10.1086/378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski K, Masel J. Asphyxiating thoracic dystrophy without respiratory disease. Pediatr Radiol. 1976;15:30–33. doi: 10.1007/BF00988659. [DOI] [PubMed] [Google Scholar]
- Lachman RS. Taybi and Lachman’s radiology of syndromes, Metabolic disorders, and skeletal dysplasias. Philadelphia: Mosby Elsevier; 2007. p. 5e. [Google Scholar]
- Lagier-Tourenne C, Boltshauser E, Breivik N, Gribaa M, Betard C, Barbot C, Koenig M. Homozygosity mapping of a third Joubert syndrome locus to 6q23. J Med Genet. 2004;41:273–277. doi: 10.1136/jmg.2003.014787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maria BL, Hoang KB, Tusa RJ, Mancuso AA, Hamed LM, Quisling RG, Hove MT, Fennell EB, Booth-Jones M, Ringdahl DM, Yachnis AT, Creel G, Frerking B. “Joubert syndrome” revisited: Key ocular motor signs with magnetic resonance imaging correlation. J Child Neurol. 1997;12:423–430. doi: 10.1177/088307389701200703. [DOI] [PubMed] [Google Scholar]
- Maria BL, Boltshauser E, Palmer SC, Tran TX. Clinical features and revised diagnostic criteria in Joubert syndrome. J Child Neurol. 1999;14:583–590. doi: 10.1177/088307389901400906. [DOI] [PubMed] [Google Scholar]
- McQuillan R, Leutenegger AL, Abdel-Rahman R, Franklin CS, Pericic M, Barac-Lauc L, Smolej-Narancic N, Janicijevic B, Polasek O, Tenesa A, Macleod AK, Farrington SM, Rudan P, Hayward C, Vitart V, Rudan I, Wild SH, Dunlop MG, Wright AF, Campbell H, Wilson JF. Runs of homozygosity in European populations. Am J Hum Genet. 2008;83:359–372. doi: 10.1016/j.ajhg.2008.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill AE, Merriman B, Farrington-Rock C, Camacho N, Sebald ET, Funari VA, Schibler MJ, Firestein MH, Cohn ZA, Priore MA, Thompson AK, Rimoin DL, Nelson SF, Cohn DH, Krakow D. Ciliary abnormalities due to defects in the retrograde transport protein DYNC2H1 in short-rib polydactyly syndrome. Am J Hum Genet. 2009;84:542–549. doi: 10.1016/j.ajhg.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan NV, Bacchelli C, Gissen P, Morton J, Ferrero GB, Silengo M, Labrune P, Casteels I, Hall C, Cox P, Kelly DA, Trembath RC, Scambler PJ, Maher ER, Goodman FR, Johnson CA. A locus for asphyxiating thoracic dystrophy, ATD, maps to chromosome 15q13. J Med Genet. 2003;40:431–435. doi: 10.1136/jmg.40.6.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberklaid F, Danks DM, Mayne V, Campbell P. Asphyxiating thoracic dysplasia. Clinical, radiological, and pathological information on 10 patients. Arch Dis Child. 1977;52:758–765. doi: 10.1136/adc.52.10.758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Doherty D, Chance PF, Glass IA. Joubert syndrome (and related disorders) (OMIM 213300) Eur J Hum Genet. 2007;15:511–521. doi: 10.1038/sj.ejhg.5201648. [DOI] [PubMed] [Google Scholar]
- Pellegrino JE, Lensch MW, Muenke M, Chance PF. Clinical and molecular analysis in Joubert syndrome. Am J Med Genet. 1997;72:59–62. doi: 10.1002/(sici)1096-8628(19971003)72:1<59::aid-ajmg12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Salonen R. The Meckel syndrome: Clinicopathological findings in 67 patients. Am J Med Genet. 1984;18:671–689. doi: 10.1002/ajmg.1320180414. [DOI] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Seller MJ. Phenotypic variation in Meckel syndrome. Clin Genet. 1981;20:74–77. doi: 10.1111/j.1399-0004.1981.tb01811.x. [DOI] [PubMed] [Google Scholar]
- Silengo M, Gianino P, Longo P, Battistoni G, Defilippi C. Dandy-Walker complex in a child with Jeune’s asphyxiating thoracic dystrophy. Pediatr Radiol. 2000;30:430. doi: 10.1007/s002470050779. [DOI] [PubMed] [Google Scholar]
- Singh M, Ray D, Paul VK, Kumar A. Hydrocephalus in asphyxiating thoracic dystrophy. Am J Med Genet. 1988;29:391–395. doi: 10.1002/ajmg.1320290221. [DOI] [PubMed] [Google Scholar]
- St Jacques B, Hammerschmidt M, McMahon AP. Indian hedgehog signaling regulates proliferation and differentiation of chondrocytes and is essential for bone formation. Genes Dev. 1999;13:2072–2086. doi: 10.1101/gad.13.16.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinlin M, Schmid M, Landau K, Boltshauser E. Follow-up in children with Joubert syndrome. Neuropediatrics. 1997;28:204–211. doi: 10.1055/s-2007-973701. [DOI] [PubMed] [Google Scholar]
- Trabelsi M, Hammou-Jeddi A, Kammoun A, Bennaceur B, Gharbi HA. Asphyxiating thoracic dysplasia associated with hepatic ductal hypoplasia, agenesis of the corpus callosum and Dandy-Walker syndrome. Pediatrie. 1990;45:35–38. [PubMed] [Google Scholar]
- Tsai YC, Chang JM, Changchien CC, Eng HL, Chen WJ, Lui CC, Huang CB. Unusual short rib-polydactyly syndrome. Am J Med Gen. 1992;44:31–36. doi: 10.1002/ajmg.1320440108. [DOI] [PubMed] [Google Scholar]
- Tüysüz B, Baris S, Aksoy F, Madazli R, Ungür S, Sever L. Clinical variability of asphyxiating thoracic dystrophy (Jeune) syndrome: Evaluation and classification of 13 patients. Am J Med Genet Part A. 2009;149A:1727–1733. doi: 10.1002/ajmg.a.32962. [DOI] [PubMed] [Google Scholar]
- Valente EM, Marsh SE, Castori M, Dixon-Salazar T, Bertini E, Al-Gazali L, Messer J, Barbot C, Woods CG, Boltshauser E, Al-Tawari AA, Salpietro CD, Kayserili H, Sztriha L, Gribaa M, Koenig M, Dallapiccola B, Gleeson JG. Distinguishing the four genetic causes of Joubert syndrome-related disorders. Ann Neurol. 2005;57:513–519. doi: 10.1002/ana.20422. [DOI] [PubMed] [Google Scholar]
- Wilson DJ, Weleber RG, Beals RK. Retinal dystrophy in Jeune’s syndrome. Arch Ophthalmol. 1987;105:651–657. doi: 10.1001/archopht.1987.01060050069040. [DOI] [PubMed] [Google Scholar]
- Yang SS, Langer LO, Jr, Cacciarelli A, Dahms BB, Unger ER, Roskamp J, Dinno ND, Chen H. Three conditions in neonatal asphyxiating thoracic dysplasia (Jeune) and short rib-polydactyly syndrome spectrum: A clinicopathologic study. Am J Med Genet. 1987;(Suppl 3):191–207. doi: 10.1002/ajmg.1320280523. [DOI] [PubMed] [Google Scholar]