

Abstract
Pancreatic carcinomas with acinar differentiation, including acinar cell carcinoma, pancreatoblastoma, and carcinomas with mixed differentiation, are distinct pancreatic neoplasms with poor prognosis. Although recent whole exome sequencing analyses have defined the somatic mutations that characterize the other major neoplasms of the pancreas, the molecular alterations underlying pancreatic carcinomas with acinar differentiation remain largely unknown. In the current study, we sequenced the exomes of 23 surgically resected pancreatic carcinomas with acinar differentiation. These analyses revealed a relatively large number of genetic alterations at both the individual base pair and chromosomal levels. There was an average of 119 somatic mutations per carcinoma. When three outliers were excluded, there was an average of 64 somatic mutations per tumor (range 12–189). The mean fractional allelic loss (FAL) was 0.27 (range 0–0.89) and heterogeneity at the chromosome level was confirmed in selected cases using fluorescent in situ hybridization (FISH). No gene was mutated in >30% of the cancers. Genes altered in other neoplasms of the pancreas were occasionally targeted in carcinomas with acinar differentiation; SMAD4 was mutated in six tumors (26%), TP53 in three (13%), GNAS in two (9%), RNF43 in one (4%) and MEN1 in one tumor (4%). Somatic mutations were identified in genes in which constitutional alterations are associated with familial pancreatic ductal adenocarcinoma, such as ATM, BRCA2, and PALB2 (one tumor each), as well as in genes altered in extra-pancreatic neoplasms, such as JAK1 in four tumors (17%) BRAF in three (13%), RB1 in three (13%), APC in two (9%), PTEN in two (9%), ARID1A in two (9%), MLL3 in two (9%), and BAP1 in one (4%). Perhaps most importantly, we found that more than a third of these carcinomas have potentially targetable genetic alterations including mutations in BRCA2, PALB2, ATM, BAP1, BRAF and JAK1.
Keywords: pancreas, carcinoma, acinar cell carcinoma, genetics, sequencing, pancreatoblastoma
Introduction
Acinar cell carcinoma is a form of pancreatic carcinoma that, like ductal adenocarcinoma, confers a generally poor prognosis. The mean survival is 18–24 months and the 3-year survival rate is only 25% [1, 2]. In contrast to ductal adenocarcinoma (which accounts for more than 80% of pancreatic malignancies), acinar cell carcinoma is rare, accounting for less than 2% of all pancreatic carcinomas. Acinar cell carcinomas are distinct from other pancreatic neoplasms, with unique clinical, morphological and immunohistochemical features [1, 2]. Clinically, 15% of patients with an acinar cell carcinoma develop subcutaneous fat necrosis [3]. Morphologically, acinar cell carcinomas are characterized by cells with significant acinar differentiation, as demonstrated histologically by acinar architecture, cytoplasmic granules and single prominent nuclei, and immunohistochemically by labeling for markers of exocrine enzymes [1]. Pancreatic carcinomas with mixed differentiation (including mixed acinar-ductal carcinoma and mixed acinar-neuroendocrine carcinoma) occur less frequently. Pancreatoblastoma, a rare pancreatic neoplasm that occurs mostly in children, also displays significant acinar differentiation [4]. In addition to cells with acinar differentiation (as determined by morphologic and immunohistochemical features), pancreatoblastomas also contain nests of squamoid cells and may have neuroendocrine, ductal, or primitive components [4].
In contrast to the other major neoplasms of the pancreas, little is known of the underlying genetic alterations that drive the development of acinar cell carcinoma and other carcinomas with acinar differentiation. In recent years, the exomes of all of the other major tumor types that arise in the pancreas have been systematically analyzed by high-throughput sequencing [5–8]. These analyses have demonstrated that each of these tumor types has its own pattern of genetic alterations. This understanding of the genetic alterations in tumors of the pancreas has not only provided insight into tumorigenesis in the pancreas, but novel targets for diagnosis and therapy have also been identified.
There have only been a handful of studies of the genetics of acinar cell carcinoma of the pancreas. Large gains and losses have been previously reported at the chromosome level, although these alterations are too large to suggest specific genetic targets [9–11]. In addition, microsatellite instability has been identified in a small subset of acinar cell carcinomas [12]. At the individual gene level, alterations in genes coding for members of the APC/β (beta)-catenin pathway have been identified in 20–25% of acinar cell carcinomas. These included inactivating mutations in APC as well as activating mutations in CTNNB1 [12]. Intriguingly, acinar cell carcinomas lack frequent alterations in genes commonly mutated in pancreatic ductal adenocarcinoma. Only rare mutations in KRAS and TP53, and only rare loss of Smad4 protein expression have been reported in acinar cell carcinomas [12–14]. Even less is known about the molecular alterations underlying acinar neoplasms with mixed differentiation and pancreatoblastoma. Like acinar cell carcinomas, pancreatoblastomas lack frequent alterations in genes commonly mutated in ductal adenocarcinoma, including KRAS, TP53, P16/CDKN2A, and SMAD4, though rare loss of Smad4 expression has been reported [15]. The majority of pancreatoblastomas have somatic alterations in the APC/β (beta)-catenin pathway, including inactivating mutations in APC and activating mutations in CTNNB1 [15]. Loss of chromosome 11p also occurs frequently in pancreatoblastomas [15, 16].
We report here the results of the first whole exome sequencing analysis of pancreatic carcinomas with acinar differentiation. These analyses revealed a relatively large number of genetic alterations both at the chromosome and gene levels. Moreover, they defined the unique genomic landscape for acinar cell carcinoma, confirming that this neoplasm is genetically distinct from other pancreatic neoplasms and suggesting therapeutic targets.
Materials and Methods
Sample acquisition/preparation
This study was approved by the Institution Review Boards at Johns Hopkins University, Memorial Sloan Kettering Cancer Center, and University Medical Center Utrecht. We analyzed 23 well-characterized fresh-frozen surgically resected pancreatic neoplasms with significant acinar differentiation (Supplementary Table 1). These included 21 acinar cell carcinomas (17 pure acinar cell carcinomas, three mixed acinar-ductal carcinomas, and one mixed acinar-neuroendocrine carcinoma) and two pancreatoblastomas. In addition to acinar morphology on hematoxylin and eosin (H&E) sections, each carcinoma had immunohistochemical evidence of acinar differentiation, as demonstrated by immunolabeling for trypsin, chymotrypsin, lipase, and/or alpha-1-antitrypsin. Mixed acinar-ductal carcinomas additionally contained a component of glandular differentiation (adenocarcinoma). In addition to its morphologic evidence of acinar differentiation, the mixed acinar-neuroendocrine carcinoma also exhibited evidence of neuroendocrine differentiation with immunolabeling for chromogranin and synaptophysin. The pancreatoblastomas both arose in adults and had squamoid nests. In addition to the components with acinar differentiation, both tumors also had neuroendocrine components with immunolabeling for chromogranin and synaptophysin. Each fresh-frozen sample was macrodissected to achieve a neoplastic cellularity of >70%.
Exome capture, sequencing, and somatic mutation identification
We sequenced the exons of approximately 21,000 protein coding genes (greater than 37,000,000bp of coding sequence) in matched tumor and normal DNA. The coding sequences from individual libraries for each sample were captured using the Agilent SureSelect paired end version 4.0 human exome kit, and the captured libraries were then sequenced using the Illumina HiSeq genome analyzer. Sequencing reads were analyzed and aligned to human genome hg18 with the Eland algorithm in CASAVA 1.6 software (Illumina). A mismatched base was identified as a mutation only when the following occurred: (1) it was identified by 5 or more distinct pairs; (2) the number of distinct tags containing a particular mismatched base was at least 15% of the total distinct tags; and (3) it was not present in greater than 0.2% of the tags in the matched normal sample. See Supplementary Methods for more details on library preparation and exome capture.
Microsatellite Instability (MSI) Testing
Microsatellite instability was detected using the MSI Analysis System (Promega, Madison, WI), which is composed of 5-mononucleotide repeats (BAT-25, BAT-26, NR-21, NR-24 and MONO-27) to detect MSI and 2-pentanucleotide repeat loci to confirm identity between normal and tumor samples, following the manufacturer’s instructions. One sample (ACINAR28) was tested with additional mononucleotide repeat loci, including BAT-40, NR-22, NR-27 and CAT-25 as previously reported [17–19]. After amplification, the fluorescent PCR products were sized on an Applied Biosystems 3130 capillary electrophoresis instrument (Invitrogen, Calsbad, CA). Tumor samples were designated as MSI-high if two or more mononucleotide loci varied in length compared to the germline DNA, MSI-low if only one locus varied, and microsatellite stable (MSS) if there was no variation compared to the germline. Pentanucleotide loci confirmed identity in all cases.
Fluorescence in situ Hybridization
Fluorescence in situ hybridization (FISH) was performed on formalin-fixed, paraffin-embedded (FFPE) sections using a combination of newly designed and commercially available probes for chromosomes 11, 15, and 22. The newly designed probes consisted of two closely mapped BAC or PAC clones. The resulting probes were labeled via nick translation with either Orange-dUTP or Green-dUTP (Abbott Molecular, Abbott Park, IL). Two differentially labeled probes covered each chromosome arm. The proximal probe was always green, and the distal probe was always orange. For chromosome 11 the proximal probe (band 11q14.1) consisted of BACs RP11-7H7 and CTD-3159I7, and the distal probe (band 11q22.3) was the commercial ATM probe (Abbott Molecular, Abbott Park, IL). The proximal probes for chromosome 15 were BACs RP11-294O11 and RP11-562A8 (band 15q21.2), and the distal probes were CTD-2071N1 and RP11-285A1 (band 15q24.3-25.1). The commercial BCR probe (proximal, band 22q11.2) and the RP3-508I15 and RP3-327J16 PACs (distal, band 22q13.1) covered chromosome 22. The clones for libraries RP11 and RP3 were obtained from BACPAC Resources Center, Oakland, CA, while clones for the CTD library were purchased from Life Technologies.
Following deparaffinization, the slides were denatured at 80°C for eight minutes and hybridized for 22 hours at 37°C in a humidified atmosphere. The slides were washed in 2X SSC/0.3% NP-40 for two min at 62°C and for two min at room temperature, with agitation. Traces of detergent were removed with wash in 2X SSC at room temperature. The slides were counterstained with DAPI. The probe pattern was evaluated in 100 nuclei. The presence of two pairs of green and orange signals was recorded as the normal pattern. The loss of an orange signal or the loss of an orange and a green signal were recorded as the deletion of a chromosome arm of smaller or larger size, respectively. The presence of more than two pairs of green and orange signals was recorded as a gain of copy number of that chromosomal region.
Bisulfite Conversion and Quantitative Methylation-Specific PCR (qMSP)
Genomic DNA (gDNA) was bisulfite treated with the EZ DNA methylation kit (Zymo Research) for 16 cycles of 95°C for 10 minutes, 50°C for 60 minutes. qMSP was performed on the iCycler iQ real-time PCR detection system (Bio-Rad) using bisulfite-converted template with QuantiTect SYBR Green mix (Qiagen) reaction mix containing either the unmethylated (U) or methylated (M) primer pairs. Primer sequences and annealing temperatures are for BRCA1 unmethylated sense ATTGGGTGGTTAATTTAGAGTTTTGAGAGATG, unmethylated antisense AAATCTCAACAAACTCACACCACACAATCA, methylated sense GGGTGGTTAATTTAGAGTTTCGAGAGACG, methylated antisense AACGAACTCACGCCGCGCAATCG with an annealing temperature of 64°C and for MLH1 unmethylated sense TTTTGATGTAGATGTTTTATTAGGGTT, unmethylated antisense ACCACCTCATCATAACTACCCACA, methylated sense ACGTAGACGTTTTATTAGGGTCGC, methylated antisense CCTCATCGTAACTACCCGCG with an annealing temperature of 60°C. Standard curves for the unmethylated (U) and methylated (M) qMSP reactions were generated from serial dilutions using bisulfite treated template from normal peripheral lymphocyte gDNA (NL) and in vitro CpG methylated (IVD) Jurkat gDNA (N4002S, New England BioLabs). The percent methylation of each sample was calculated from the percentage of the amount of the M product over the sum quantity of the U and M products. Each sample was performed in duplicate. Primers were designed using MSPPrimer [20].
Results
We performed whole exome sequencing on 23 pancreatic carcinomas with significant acinar differentiation, including 17 pure acinar cell carcinomas, three mixed acinar-ductal carcinomas, one mixed acinar-neuroendocrine carcinoma and two pancreatoblastomas (Supplementary Table 1). The sample set included tumors from 16 men (70%) and 7 women (30%) and the average age at surgical resection was 61 years. The majority of patients were stage 1 or 2 at the time of resection – only one patient was classified as stage 3.
Base-pair instability
A total of 37 MB of captured DNA was sequenced with an average depth of coverage of 131-fold in the targeted region, and 91% of targeted bases were represented by at least 10 reads (Supplementary Table 2). 2745 somatic mutations were identified in 2340 genes in the 23 carcinomas (Supplementary Table 3). These carcinomas had a mean of 119 non-synonymous somatic mutations. Two of the carcinomas (ACINAR01 and ACINAR03) were microsatellite unstable as assessed by short tandem repeat length assay using five mononucleotide markers (MSI Analysis System from Promega) – these two carcinomas contained 701 and 404 non-synonymous somatic mutations, respectively. One additional carcinoma had a strikingly large number of mutations: the carcinoma ACINAR28 had 362 total non-synonymous somatic mutations (compared to 189 mutations in the next most mutated sample) but was not microsatellite unstable using the MSI Analysis System. Because ACINAR28 tested microsatellite stable yet had a large number of mutations, additional testing with mononucleotide repeat microsatellites was performed. This sample tested stable with NR-22, NR-27 and CAT-25, but showed only a shift in BAT-40, and was accordingly reclassified as MSI-Low. When these three outlier samples were eliminated, the remaining carcinomas contained an average of 64 nonsynonymous somatic mutations per tumor. This number is higher than the average number of mutations in pancreatic ductal adenocarcinoma and other primary pancreatic neoplasms but not higher than the average number of mutations in other solid tumors such as colorectal and breast cancers [5, 6, 8, 21, 22].
All of the carcinomas were enriched for C:G-to-T:A transitions (35% of mutations in remaining 20 carcinomas after the 3 outliers were excluded), and this enrichment was even more striking in the carcinomas with microsatellite instability, with C:G-to-T:A transitions accounting for 63% and 54% mutations in these two samples (ACINAR01 and ACINAR03). As expected, the carcinomas with microsatellite instability were also enriched for single base deletions, accounting for 14% and 18% of mutations in these two samples (ACINAR01 and ACINAR03), compared to 4% of the remaining 20 samples.
Chromosome level alterations
The carcinomas with acinar differentiation had relatively high numbers of large chromosomal changes, with fractional allelic losses (FAL) ranging from 0 – 0.89 (mean 0.27) (Supplementary Figure 1). This is higher than the FAL previously reported in pancreatic ductal adenocarcinoma, which had a mean FAL of 0.15 and a range of 0.015–0.32 in 82 analyzed tumors [23]. Loss of heterozygosity of chromosome 11p, which has been previously reported to be targeted in acinar cell carcinoma and pancreatoblastoma, was present in 12 of 23 tumors (52%), including both pancreatoblastomas [12, 15]. The 23 carcinomas also had frequent loss of heterozygosity at loci of known tumor suppressor genes, including TP53 on 17p (lost in 9 of 23 or 39%) and SMAD4 on 18q (lost in 13 of 23 or 57%).
A subset of chromosomal changes was confirmed with fluorescence in situ hybridization (FISH) on formalin-fixed paraffin embedded tumor sections from seven selected tumors. For these studies, we focused on three chromosomal regions that frequently exhibited loss of heterozygosity in the acinar cell carcinoma samples – chromosomes 11, 15 and 22. Both losses and gains were identified in these regions in the seven tumor samples analyzed. As might be expected, in the one assayed sample with microsatellite instability, no gains or losses were identified in the FISH assays. The other tumor samples each contained at least one area of gain or loss in the chromosomal regions analyzed. Chromosome 15 was analyzed in six of the carcinomas – two had the normal number of chromosome 15, two had loss of one of the two signals at each locus, and two had polysomic signals (Figure 2). Chromosome 11 was investigated in three tumor samples – one showed a normal FISH signal, one showed loss of one of the two signals at each locus, and one showed multiple FISH signals consistent with polysomy in this region. Chromosome 22 was assayed in five tumors – two had the normal number of chromosome 22, one showed loss of one of the two signals at each locus, and two were polysomic in this region (Figure 1). Remarkably, three (ACINAR06, ACINAR07, and ACINAR15) of the seven carcinomas analyzed showed dramatic intratumoral heterogeneity and polysomy in five of six regions assayed (Figure 1). This finding suggests that heterogeneity at the chromosome level may represent a distinct pattern of genetic alteration that occurs in a subset of acinar cell carcinomas, while other acinar cell carcinomas that are microsatellite unstable (like ACINAR03) have few large chromosomal alterations.
Figure 1. Fluorescent in situ Hybridization of Acinar Cell Carcinomas.
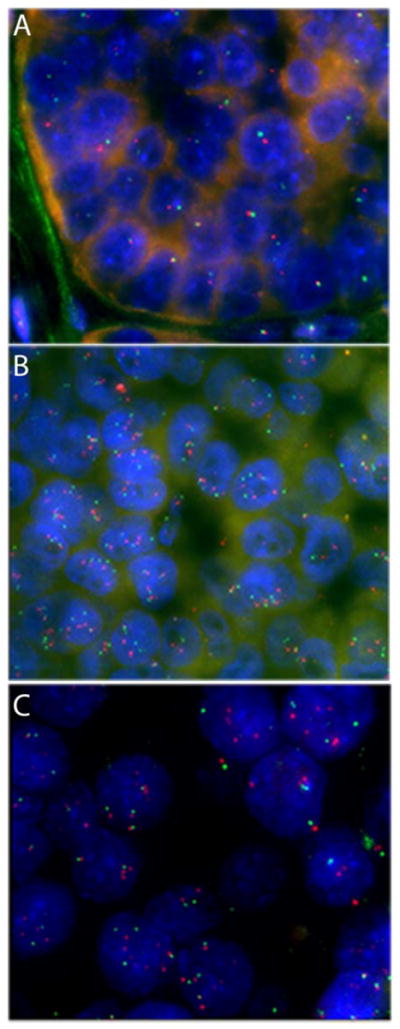
Reveals Chromosomal Losses, Gains, and Heterogeneity. A. FISH for Chr22q13.1 (red) and Chr22q11.2 (green) reveals frequent chromosomal losses. B. FISH for Chr15q24.3-25.1 (red) and Chr15q21.2 (green) reveals areas of polysomy. C. FISH for Chr11q22.3 (red) and Chr11q14.1 (green) reveals heterogeneity in the tumor.
Mechanisms underlying genetic alterations
We next sought to identify possible mechanisms that could explain the relatively large and heterogeneous number of alterations in these tumors. For this purpose, we reviewed our whole exome sequencing data to identify somatic mutations in genes known to cause base pair and chromosomal instability in other tumor types. One of the acinar cell carcinomas with microsatellite instability (ACINAR01) contained a somatic mutation in MSH2, a gene whose constitutional alteration causes Lynch syndrome (also known as hereditary non-polyposis colon cancer or HNPCC), an inherited cancer predisposition syndrome caused by mutations in DNA mismatch repair genes. As >90% of the total sequenced tags in the region contained the mutation, we concluded that MSH2 was biallelically inactivated in this tumor. Somatic mutations were also identified in MLH3, MSH3, and PMS2. The MSH3 mutation also occurred in ACINAR01, which had microsatellite instability and somatic MSH2 mutation. However, because the other tumors with these mutations did not have particularly high numbers of somatic mutations, the role of these mutations in base pair instability was unclear. No mutations were identified in POLE or POLD1, DNA polymerase genes whose constitutional alterations were recently associated with colorectal cancer predisposition [24]. We also reviewed our whole exome sequencing data for somatic mutations in genes involved in the spindle checkpoint and DNA double-strand break repair, such as MAD1, MAD2, MAD3, BUB1, BUB3, MPSI, CDC20, MRE11, RAD50, NBS1, ROD, ZW10, ZWILCH and FBXW7, as mutations in these genes have been associated with chromosomal instability in other tumor types [25, 26]. We found no somatic mutations in these genes in our samples. However, because our analyses focused on exome sequencing, these studies would not identify large deletions or other types of rearrangements.
Because genes governing DNA repair can also be epigenetically inactivated in tumors with genomic instability, we analyzed the tumors for methylation of the MLH1 and BRCA1 genes, which have been reported to be methylated in tumors with microsatellite and chromosomal instability, respectively [27]. Two different methylation-specific PCR assays were performed for each gene. None of the 23 carcinomas were methylated at the BRCA1 locus. One carcinoma (ACINAR06) exhibited methylation of MLH1 in both PCR assays, while another carcinoma (ACINAR18) had an equivocal result with methylation in only one of the assays. Methylation at MLH1 did not correlate with microsatellite instability or with increased number of mutations in the whole exome sequencing data. Therefore, mechanisms other than methylation at these loci are responsible for the acquisition of the microsatellite instability phenotype in these carcinomas.
Mutated genes
The 23 pancreatic carcinomas with acinar differentiation harbored somatic mutations in a variety of genes (Supplementary Table 4). No gene was mutated in >30% of the cancers. Mutations were identified in genes altered in other pancreatic neoplasms, including SMAD4 in six tumors (26%), TP53 in three tumors (13%), GNAS in two tumors (9%, both at the previously described oncogenic hotspot in codon 201), RNF43 in one tumor (4%) and MEN1 in one tumor (4%) (Table 1). The MEN1 mutation occurred in a mixed acinar-ductal carcinoma, but there were no features of neuroendocrine differentiation in this carcinoma. Except for MEN1, mutations in genes altered in other pancreatic neoplasms did not occur in the mixed acinar-ductal carcinomas, and no carcinoma in our study was associated with a cystic lesion such as an intraductal papillary mucinous neoplasm or mucinous cystic neoplasm, which have previously been reported to harbor GNAS and RNF43 mutations [7, 8]. However, two of the six SMAD4 mutations occurred in pancreatoblastomas. Acinar cell carcinomas also contained somatic mutations in genes whose constitutional alterations are associated with familial pancreatic ductal adenocarcinoma, including ATM, BRCA2 and PALB2 in one tumor each (4%) (Table 1). None of the patients with somatic alterations in these genes had known constitutional disease-causing mutations.
Table 1.
Genes with somatic mutations and known roles in tumorigenesis
| Gene Symbol | Mutation Frequency |
|---|---|
| SMAD4 | 6/23 (26%) |
| JAK1 | 4/23 (17%) |
| BRAF | 3/23 (13%) |
| RB1 | 3/23 (13%) |
| TP53 | 3/23 (13%) |
| APC | 2/23 (9%) |
| ARID1A | 2/23 (9%) |
| GNAS | 2/23 (9%) |
| MLL3 | 2/23 (9%) |
| PTEN | 2/23 (9%) |
| ATM | 1/23 (4%) |
| BAP1 | 1/23 (4%) |
| BRCA2 | 1/23 (4%) |
| PALB2 | 1/23 (4%) |
| MEN1 | 1/23 (4%) |
| RNF43 | 1/23 (4%) |
The 23 carcinomas also harbored somatic mutations in genes altered in other extra-pancreatic tumor types, including JAK1 in four tumors (17%), BRAF or RB1in three (13%), APC, PTEN, MLL3 or ARID1A in two each (9%), and BAP1 in one tumor (4%) (Table 1). Importantly, the types of mutations in some of these genes concurred with their previously described roles in tumorigenesis, such as mutations in the oncogenic hotspot (codon 600) in BRAF and inactivating mutations in the tumor suppressor gene RB1. In addition, many other genes had somatic mutations at a frequency of 10–20% (Supplementary Table 4). However, it is difficult to determine whether these genes are drivers or passengers based on mutational data alone, and further studies will be needed to determine the role of these genes, if any, in tumorigenesis in the pancreas. Importantly, more than a third of carcinomas with acinar differentiation had potentially targetable genetic alterations, including BRCA2 (4%), PALB2 (4%), ATM (4%), BAP1 (4%), BRAF (13%) and JAK1 (17%).
In order to identify copy number alterations in known driver genes, we examined the ratio of tumor to normal coverage in coding exons of individual genes from the exome sequencing data. No amplifications were identified in previously described driver genes. However, CDKN2A, which encodes the p16 cell cycle regulator and is frequently deleted in pancreatic ductal adenocarcinoma, was homozygously deleted in four of the 23 carcinomas, including two of the three that had a mixed acinar-ductal morphology.
Comparison of different types of acinar neoplasms
Although the numbers of samples of pancreatoblastoma and mixed acinar-ductal carcinomas are too small to make definitive conclusions, some intriguing observations were made. The two pancreatoblastoma samples (ACINAR17 and ACINAR19) had fewer mutations than almost all acinar cell carcinomas – these two tumors contained 18 and 17 somatic mutations, compared to an average of 131 for the remaining acinar cell carcinomas. In addition, both pancreatoblastomas contained somatic mutations in CTNNB1, which was not mutated in any of the other acinar neoplasms. The mixed acinar-ductal carcinomas were also unique. Two of the three mixed acinar-ductal carcinomas harbored a CDKN2A homozygous deletion and two of the three contained a BRAF mutation, compared to the acinar cell carcinomas without ductal differentiation in which two of 18 harbored a CDKN2A homozygous deletion and one harbored a BRAF mutation. The mixed acinar-ductal carcinomas did not contain mutations in the other genes that are mutated in pancreatic ductal adenocarcinoma and intraductal papillary mucinous neoplasms, including SMAD4, TP53, RNF43, and GNAS.
Discussion
Pancreatic carcinomas with significant acinar differentiation are characterized by a relatively large number of alterations at the chromosome level. There was also a striking degree of intratumoral heterogeneity demonstrated by FISH. These data are consistent with the hypothesis that these tumors are chromosomally unstable, though instability is a rate rather than a state and cannot be measured through evaluation of tumors at a single point in time [28]. Although the mechanism(s) underlying this presumptive chromosomal instability in acinar cell carcinomas is unclear, it may, in part, explain the aggressive behavior and resistance to therapy exhibited by most acinar cell carcinomas [2, 29].
We were able to identify a number of genes that are mutated in acinar cell carcinomas, although at relatively low frequencies. Although there is some overlap with ductal adenocarcinomas (SMAD4 in six tumors (26%), TP53 in three tumors (13%)), the genomic landscape of acinar cell carcinomas differs significantly from the other neoplasms of the pancreas. For example, in contrast to ductal adenocarcinomas which almost universally harbor KRAS gene mutations, none of the acinar cell carcinomas in this series had a KRAS mutation [12–14].
Now that all of the major tumor types of the pancreas have been sequenced, it is clear that each tumor type has its own mutational profile. Acinar cell carcinomas harbor large numbers of chromosomal changes, ductal adenocarcinomas are characterized by SMAD4, TP53, KRAS and CDKN2A mutations, pancreatic neuroendocrine tumors by MEN-1, DAXX, ATRX and mTOR pathway gene mutations, solid-pseudopapillary neoplasms by CTNNB1 mutations, serous cystadenomas by VHL mutations, intraductal papillary mucinous neoplasms by GNAS, RNF43, TP53, SMAD4 and CDKN2A mutations, and mucinous cystic neoplasms by RNF43, TP53, SMAD4 and CDKN2A mutations [5–8]. These findings have diagnostic implications as they suggest that sequencing of difficult-to-classify pancreatic neoplasms may help guide diagnoses.
Through whole exome sequencing we also identified potentially therapeutically targetable mutations, such as those in genes coding for members of the Fanconi anemia pathway, in 43% of the carcinomas. These included mutations in BRCA2 (4%), PALB2 (4%), BAP1 (4%), ATM (4%), BRAF (13%) and JAK1 (17%). Mutations in Fanconi anemia pathway genes such as BRCA1 and BRCA2 have also been reported previously in acinar cell carcinomas [30, 31]. We and others have previously shown that ductal adenocarcinomas of the pancreas with inactivating mutations in genes coding for members of this pathway can be exquisitely sensitive to DNA cross-linking agents and to Poly (ADP-ribose) polymerase (PARP) inhibitors [32–34]. Similarly, it has been suggested that ATM mutant neoplasms may be more sensitive to PARP inhibitors and inhibitors of protein kinase DNA-activated catalytic polypeptide (DNA-PKcs) [35, 36]. In addition, targeted inhibitors for BRAF and JAK1 are currently in human clinical trials with promising results, and a BRAF inhibitor was recently approved by the United States Food and Drug Administration (FDA) for treatment of melanoma with the BRAF V600E mutation [37, 38]. While the sensitivity of acinar cell carcinomas with potentially targetable mutations has to first be established in the clinic, we can speculate that acinar cell carcinomas will be good candidates for a personalized approach to therapy based on the genetic changes in these patients’ cancers.
Supplementary Material
Supplementary Methods
Supplementary Figure 1 – Fractional Allelic Loss in Carcinomas with Acinar Differentiation
Supplementary Table 1 – Clinical Information on Pancreatic Neoplasms with Acinar Differentiation
Supplementary Table 2 – Summary of Exome Sequencing Data on Pancreatic Neoplasms with Acinar Differentiation
Supplementary Table 3 – Summary of Mutation Spectrum of Pancreatic Neoplasms with Acinar Differentiation
Supplementary Table 4 – Somatic Mutations in Pancreatic Neoplasms with Acinar Differentiation
Acknowledgments
The authors wish to acknowledge Stacy Mosier for expert technical assistance. This work was supported by The Virginia and D. K. Ludwig Fund for Cancer Research, The Lustgarten Foundation for Pancreatic Cancer Research, The Blum-Kovler Foundation, The Stringer Foundation, The Sol Goldman Pancreatic Cancer Research Center, and NIH grants CA62924 and CA43460.
Footnotes
Statement of Author Contributions
Y.J. conceived and carried out experiments and analyzed data. R.Y., W.P., and L.P. carried out experiments and analyzed data. G.J.A.O., D.S.K., A.M., J.R.E., J.G.H., C.L.W., B.V., K.W.K., R.H.H., N.P., and L.D.W. conceived experiments and analyzed data. All authors were involved in writing the paper and had final approval of the submitted and published versions.
Conflict of Interest: Under agreements between the Johns Hopkins University, Genzyme, Myriad Genetics, Exact Sciences, Inostics, Qiagen, Invitrogen and Personal Genome Diagnostics, N.P., B.V., K.W.K., and R.H.H. are entitled to a share of the royalties received by the University on sales of products related to genes and technologies described in this manuscript. N.P., B.V. and K.W.K. are co-founders of Inostics and Personal Genome Diagnostics, are members of their Scientific Advisory Boards, and own Inostics and Personal Genome Diagnostics stock, which is subject to certain restrictions under Johns Hopkins University policy.
References
- 1.Klimstra DS, Heffess CS, Oertel JE, et al. Acinar cell carcinoma of the pancreas. A clinicopathologic study of 28 cases. Am J Surg Pathol. 1992;16:815–837. doi: 10.1097/00000478-199209000-00001. [DOI] [PubMed] [Google Scholar]
- 2.La Rosa S, Adsay V, Albarello L, et al. Clinicopathologic study of 62 acinar cell carcinomas of the pancreas: insights into the morphology and immunophenotype and search for prognostic markers. Am J Surg Pathol. 2012;36:1782–1795. doi: 10.1097/PAS.0b013e318263209d. [DOI] [PubMed] [Google Scholar]
- 3.Burns WA, Matthews MJ, Hamosh M, et al. Lipase-secreting acinar cell carcinoma of the pancreas with polyarthropathy. A light and electron microscopic, histochemical, and biochemical study. Cancer. 1974;33:1002–1009. doi: 10.1002/1097-0142(197404)33:4<1002::aid-cncr2820330415>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 4.Klimstra DS, Wenig BM, Adair CF, et al. Pancreatoblastoma. A clinicopathologic study and review of the literature. Am J Surg Pathol. 1995;19:1371–1389. doi: 10.1097/00000478-199512000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–1806. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science. 2011;331:1199–1203. doi: 10.1126/science.1200609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A. 2011;108:21188–21193. doi: 10.1073/pnas.1118046108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohori NP, Khalid A, Etemad B, et al. Multiple loss of heterozygosity without K-ras mutation identified by molecular analysis on fine-needle aspiration cytology specimen of acinar cell carcinoma of pancreas. Diagn Cytopathol. 2002;27:42–46. doi: 10.1002/dc.10147. [DOI] [PubMed] [Google Scholar]
- 10.Taruscio D, Paradisi S, Zamboni G, et al. Pancreatic acinar carcinoma shows a distinct pattern of chromosomal imbalances by comparative genomic hybridization. Genes Chromosomes Cancer. 2000;28:294–299. doi: 10.1002/1098-2264(200007)28:3<294::aid-gcc7>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 11.Rigaud G, Moore PS, Zamboni G, et al. Allelotype of pancreatic acinar cell carcinoma. Int J Cancer. 2000;88:772–777. doi: 10.1002/1097-0215(20001201)88:5<772::aid-ijc14>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 12.Abraham SC, Wu TT, Hruban RH, et al. Genetic and immunohistochemical analysis of pancreatic acinar cell carcinoma: frequent allelic loss on chromosome 11p and alterations in the APC/beta-catenin pathway. Am J Pathol. 2002;160:953–962. doi: 10.1016/s0002-9440(10)64917-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoorens A, Lemoine NR, McLellan E, et al. Pancreatic acinar cell carcinoma. An analysis of cell lineage markers, p53 expression, and Ki-ras mutation. Am J Pathol. 1993;143:685–698. [PMC free article] [PubMed] [Google Scholar]
- 14.Terhune PG, Heffess CS, Longnecker DS. Only wild-type c-Ki-ras codons 12, 13, and 61 in human pancreatic acinar cell carcinomas. Mol Carcinog. 1994;10:110–114. doi: 10.1002/mc.2940100209. [DOI] [PubMed] [Google Scholar]
- 15.Abraham SC, Wu TT, Klimstra DS, et al. Distinctive molecular genetic alterations in sporadic and familial adenomatous polyposis-associated pancreatoblastomas : frequent alterations in the APC/beta-catenin pathway and chromosome 11p. Am J Pathol. 2001;159:1619–1627. doi: 10.1016/s0002-9440(10)63008-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kerr NJ, Fukuzawa R, Reeve AE, et al. Beckwith-Wiedemann syndrome, pancreatoblastoma, and the wnt signaling pathway. Am J Pathol. 2002;160:1541–1542. doi: 10.1016/s0002-9440(10)62580-1. author reply 1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bianchi F, Galizia E, Catalani R, et al. CAT25 is a mononucleotide marker to identify HNPCC patients. J Mol Diagn. 2009;11:248–252. doi: 10.2353/jmoldx.2009.080155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buhard O, Cattaneo F, Wong YF, et al. Multipopulation analysis of polymorphisms in five mononucleotide repeats used to determine the microsatellite instability status of human tumors. J Clin Oncol. 2006;24:241–251. doi: 10.1200/JCO.2005.02.7227. [DOI] [PubMed] [Google Scholar]
- 19.Pagin A, Zerimech F, Leclerc J, et al. Evaluation of a new panel of six mononucleotide repeat markers for the detection of DNA mismatch repair-deficient tumours. Br J Cancer. 2013;108:2079–2087. doi: 10.1038/bjc.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brandes JC, Carraway H, Herman JG. Optimal primer design using the novel primer design program: MSPprimer provides accurate methylation analysis of the ATM promoter. Oncogene. 2007;26:6229–6237. doi: 10.1038/sj.onc.1210433. [DOI] [PubMed] [Google Scholar]
- 21.Wood LD, Parsons DW, Jones S, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–1113. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 22.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 23.Iacobuzio-Donahue CA, van der Heijden MS, Baumgartner MR, et al. Large-scale allelotype of pancreaticobiliary carcinoma provides quantitative estimates of genome-wide allelic loss. Cancer Res. 2004;64:871–875. doi: 10.1158/0008-5472.can-03-2756. [DOI] [PubMed] [Google Scholar]
- 24.Palles C, Cazier JB, Howarth KM, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013;45:136–144. doi: 10.1038/ng.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang Z, Cummins JM, Shen D, et al. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004;64:2998–3001. doi: 10.1158/0008-5472.can-04-0587. [DOI] [PubMed] [Google Scholar]
- 26.Rajagopalan H, Jallepalli PV, Rago C, et al. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004;428:77–81. doi: 10.1038/nature02313. [DOI] [PubMed] [Google Scholar]
- 27.Esteller M, Herman JG. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J Pathol. 2002;196:1–7. doi: 10.1002/path.1024. [DOI] [PubMed] [Google Scholar]
- 28.Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature. 1998;396:643–649. doi: 10.1038/25292. [DOI] [PubMed] [Google Scholar]
- 29.Butturini G, Pisano M, Scarpa A, et al. Aggressive approach to acinar cell carcinoma of the pancreas: a single-institution experience and a literature review. Langenbecks Arch Surg. 2011;396:363–369. doi: 10.1007/s00423-010-0706-2. [DOI] [PubMed] [Google Scholar]
- 30.Skoulidis F, Cassidy LD, Pisupati V, et al. Germline Brca2 heterozygosity promotes Kras(G12D) -driven carcinogenesis in a murine model of familial pancreatic cancer. Cancer Cell. 2010;18:499–509. doi: 10.1016/j.ccr.2010.10.015. [DOI] [PubMed] [Google Scholar]
- 31.Lowery MA, Klimstra DS, Shia J, et al. Acinar cell carcinoma of the pancreas: new genetic and treatment insights into a rare malignancy. Oncologist. 2011;16:1714–1720. doi: 10.1634/theoncologist.2011-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10:3–8. doi: 10.1158/1535-7163.MCT-10-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van der Heijden MS, Brody JR, Dezentje DA, et al. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin Cancer Res. 2005;11:7508–7515. doi: 10.1158/1078-0432.CCR-05-1048. [DOI] [PubMed] [Google Scholar]
- 34.Fogelman DR, Wolff RA, Kopetz S, et al. Evidence for the efficacy of Iniparib, a PARP-1 inhibitor, in BRCA2-associated pancreatic cancer. Anticancer Res. 2011;31:1417–1420. [PubMed] [Google Scholar]
- 35.Williamson CT, Kubota E, Hamill JD, et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol Med. 2012;4:515–527. doi: 10.1002/emmm.201200229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riabinska A, Daheim M, Herter-Sprie GS, et al. Therapeutic targeting of a robust non-oncogene addiction to PRKDC in ATM-defective tumors. Sci Transl Med. 2013;5:189ra178. doi: 10.1126/scitranslmed.3005814. [DOI] [PubMed] [Google Scholar]
- 37.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366:787–798. doi: 10.1056/NEJMoa1110556. [DOI] [PubMed] [Google Scholar]
- 38.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods
Supplementary Figure 1 – Fractional Allelic Loss in Carcinomas with Acinar Differentiation
Supplementary Table 1 – Clinical Information on Pancreatic Neoplasms with Acinar Differentiation
Supplementary Table 2 – Summary of Exome Sequencing Data on Pancreatic Neoplasms with Acinar Differentiation
Supplementary Table 3 – Summary of Mutation Spectrum of Pancreatic Neoplasms with Acinar Differentiation
Supplementary Table 4 – Somatic Mutations in Pancreatic Neoplasms with Acinar Differentiation