

Abstract
The two paradigms to study aging in Saccharomyces cerevisiae are the chronological life span (CLS) and the replicative life span (RLS). The chronological life span is a measure of the mean and maximum survival time of non-dividing yeast populations while the replicative life span is based on the mean and maximum number of daughter cells generated by an individual mother cell before cell division stops irreversibly. Here we review the principal discoveries associated with yeast chronological aging and how they are contributing to the understanding of the aging process and of the molecular mechanisms that may lead to healthy aging in mammals. We will focus on the mechanisms of life span regulation by the Tor/Sch9 and the Ras/adenylate cyclase/PKA pathways with particular emphasis on those implicating age-dependent oxidative stress and DNA damage/repair.
Keywords: Chronological aging, TOR (target of rapamycin), RAS, Adaptive regrowth, Caloric restriction
Yeast as a Model for Aging Research: Two Aging Paradigms
Together with the roundworm Caenorhabditis elegans, yeast represents one of the simplest and most widely adopted model organisms to study aging. Although S. cerevisiae is by far the yeast most commonly used by gerontologists, the fission yeast Schizosaccharomyces pombe has recently joined the group of the simple model systems for aging research (Roux et al. 2006, 2009). While the life span of S. cerevisiae is measured either by monitoring the replicative potential of individual mother cells (replicative life span, RLS) or by determining the mean and maximum survival time of populations of non-dividing cells (chronological life span, CLS), the life span of S. pombe is mostly measured chronologically (Roux et al. 2006, 2009). This is due to the fact that, although subtle visual differences can be observed between two cells generated by fission, it is challenging to base a survival assay on them (Barker and Walmsley 1999). Conversely, the obvious size difference between mother and daughter cells in the budding yeast S. cerevisiae allows the counting of the total number of daughter cells generated by individual mother cells before cell division halts (RLS) (Steinkraus et al. 2008).
Replicative Life Span
A method to measure RLS was originally set up by R Mortimer and JR Johnston 50 years ago (Mortimer 1959) but it was not until 30 years later that the RLS became widely studied for aging research (Egilmez and Jazwinski 1989; Kennedy et al. 1994). Among the principal outcomes associated with the RLS is the identification of the extrachromosomal ribosomal DNA circles (ERCs) as toxic species, whose accumulation causes yeast replicative senescence (Sinclair and Guarente 1997) and of several genes implicated in life span regulation such as RTG2, LAG1, and SIR2 (Steinkraus et al. 2008; D’Mello et al. 1994; Borghouts et al. 2004). The latter, SIR2, which encodes for a NAD-dependent histone deacetylase highly conserved in higher eukaryotes, has become one of the most studied gerontogenes (Kaeberlein et al. 1999; Longo and Kennedy 2006; Longo 2009). In yeast, the activity of Sir2 is known to prevent replicative senescence by reducing recombination between rDNA repeats and consequently the formation of ERCs (Kaeberlein et al. 1999). However, recent evidence has demonstrated that Sir2 functions to extend the RLS also independently of ERCs formation by maintaining low levels of histone 4 lysine 16 acetylation and consequently inducing silencing at the telomeres and subtelomeric regions (Dang et al. 2009). The activity of the closest Sir2 homologues has been shown to promote life span extension in both C. elegans and Drosophila melanogaster (Tissenbaum and Guarente 2001; Rogina and Helfand 2004). Since ERCs accumulation occurs only in S. cerevisiae, it will be of interest to establish whether the link between Sir2, telomeric and subtelomeric DNA silencing, and life span is also found in higher eukaryotes.
Chronological Life Span
As an alternative to the replicative life span measurement, a method to monitor the chronological life span was developed in the nineties and is currently used by several laboratories worldwide (Fabrizio and Longo 2003; Longo et al. 1996). One of the advantages of the chronological life span is that it measures survival of non-dividing cells, and it can serve as both a system to model aging in post-mitotic mammalian cells but also as a very simple model for organismal aging. From a technical point of view, monitoring the CLS is extremely simple, does not require micromanipulation, and is generally based on the use of cultures of millions of yeast, which facilitates the screen of longevity mutants and allows the performance of a wide range of genetics, genomics, and biochemistry assays (Fabrizio and Longo 2007; Fabrizio et al. 2005b). In a standard CLS experiment yeast are grown in synthetic complete medium (SDC) until nutrient depletion promotes cell cycle arrest. The majority of cells stops dividing within 2–3 days from the starting of the culture and viability is usually assayed by colony forming units (CFUs) measurement beginning on day 3 until survival reaches 1–5% of the day 3 CFUs (Fabrizio and Longo 2007). In analogy with post-diauxic phase cultures (see section “The TOR/Sch9 Pathway”), chronologically aging populations are characterized by the constant presence of a small fraction of budded cells (~3–8% depending on the genetic backgrounds). Several lines of evidence suggest that these budded cells may be improperly arrested in S/G2 (starvation normally induces G1-arrest) (Weinberger et al. 2007; Allen et al. 2006). However, it is possible that a very small percentage of them may be dividing although, under the standard conditions described above, the low pH (3.5) would cause a very slow growth (Fabrizio et al. 2004a). Therefore, under these conditions, cell division is very unlikely to affect the CLS measurements. By contrast, cell division can occur and it is easily detected (population size raises up to 100 times) in chronologically aging cultures after the majority of the cells has died (Fabrizio et al. 2004a). This growth of the population, defined as “adaptive regrowth”, appears to be due to mutations or possibly epigenetic changes that allow yeast to reenter the cell cycle by consuming the nutrients released by dead cells and it shares some similarities with the bacterial Growth Advantage in Stationary Phase (GASP) phenotype (Zinser and Kolter 2004; Zambrano et al. 1993; Zambrano and Kolter 1996). The latter was originally described in stationary phase cultures of Escherichia coli grown in rich medium (LB), whose viability, after a rapid decline, remains stable for extended periods of time, reflecting cycles of death and regrowth in the populations rather than extended survival (Zambrano et al. 1993). In analogy with adaptive regrowth, the GASP phenotype also arises after the acquisition of mutations that trigger cell division by promoting the catabolism of nutrients released by dead microorganisms (Zinser and Kolter 2004).
For aging studies the period in which no cell division occurs represents the life span. In yeast this phase is characterized by a gradual increase of mortality rates and it can last up to a few weeks depending on the yeast strain (Fabrizio and Longo 2003). On the contrary, in E. coli ~99% of the culture loses viability within 2–3 days, thus providing a more limited time window to observe age-related changes.
Links Between Replicative and Chronological Life Span
The degree of overlap between the mechanisms that control CLS and RLS is only partially understood. We have known for several years that certain genes that increase CLS can actually reduce RLS, possibly by affecting growth and not aging (Fabrizio et al. 2004b) but we have also known that chronological aging can reduce the RLS of mother cells (Ashrafi et al. 1999), indicating that distinct but overlapping mechanisms are regulating the two aging paradigms. In fact, the two major yeast pro-aging pathways, TOR/Sch9 and Ras/adenylate cyclase/PKA (see next section), promote aging and early cell death in both CLS and RLS paradigms. CLS extension induced by lowering the activity of either of the two pro-aging pathways requires the activity of protein kinase Rim15 and stress resistance transcription factors Msn2/4 and Gis1 (Fabrizio et al. 2001, 2003; Wei et al. 2008). Msn2/4 and Rim15, however, limit the RLS extension of a mutant with reduced Ras/PKA activity and overexpression of Msn2 shortens the RLS of wild type yeast (Fabrizio et al. 2004b). Analogously, while mitochondrial superoxide dismutase (Sod2) is required for CLS extension, its overexpression shortens RLS (Fabrizio et al. 2003, 2004b). Intriguingly, Msn2/4 were shown to mediate the RLS extension associated with decreased TOR signaling (Medvedik et al. 2007). The key players in prolonging the RLS in TOR-deficient yeast were reported to be members of the Sir2 family (sirtuins) (Medvedik et al. 2007). Consistently, an additional copy of SIR2 prolongs RLS in wild type yeast (Kaeberlein et al. 1999; Medvedik et al. 2007). The activity of Sir2, nevertheless, reduces significantly the CLS of yeast lacking the serine/threonine kinase Sch9, which live 3-fold longer than wild type but 5-fold longer in a sir2Δ context (Fabrizio et al. 2005a). Taken together, our current knowledge suggests that the relationship between CLS and RLS is complex and that several life span determinants such as Sir2 and Msn2/4 may play opposite roles in controlling the two life span paradigms. The most likely possibility is that the pro-aging TOR/Sch9 and Ras/adenylate cyclase/PKA pathways and the downstream stress resistance transcription factors affect aging by similar mechanisms in both the RLS and CLS. However, because RLS is based on cell division and since protective enzymes and stress-resistance transcription factors can negatively affect cell division, replicatively aging cells in which protective systems are activated can stop dividing before they are severely damaged or dead (Fabrizio et al. 2004b). A comprehensive analysis of the genes regulating either CLS, RLS, or both based on a comparison between partial genome-wide datasets relative to screens for CLS- and RLS-regulatory genes has been published by Laun et al. According to this study, only a handful of genes prolongs both RLS and CLS (Laun et al. 2006). These results may depend in part on the negative role of anti-chronological aging genes on cells division discussed above but also on the threshold selected to determine whether a mutant is long-lived or not. Notably, genome-wide life span studies often yield false positive/negative results (Hansen et al. 2005; Powers et al. 2006) and the long/short-lived phenotype of mutants identified by genome-wide screen must be confirmed by measuring the life span of each mutant individually. Thus, the results of the analyses based on genome-wide data might change substantially once all the individual validation experiments are performed. Nevertheless, these analyses can be informative. For example, the consistent reduction of both life spans caused by the deletion of numerous mitochondrial genes reported by Laun et al underscores the importance of functional mitochondria for normal life span (Laun et al. 2006).
Hereafter we will review the major findings associated with yeast chronological aging with particular emphasis on those that contributed to discover the evolutionary conserved longevity pathways. We will describe the similarities between these pathways in different species. We will also discuss how CLS is contributing not only to the understanding of the genetics of aging but also to elucidate the key modifications occurring in senescent cells that might be relevant to the onset of age-related diseases such as cancer.
Conserved Life Span-Regulatory Pathways
Simple model organisms and S. cerevisiae in particular have been instrumental to the study of fundamental cellular processes, e.g. cell cycle regulation and DNA transcription, which have not diverged dramatically throughout evolution. Aging appears to be the new process that this unicellular eukaryote helps us understand. In fact, several conserved proteins/pathways were found to mediate longevity in phylogenetically distant organisms and work done in the last 15 years supports the theory that life span regulation emerged early in evolution, most likely in ancestral microorganisms to overcome periods of starvation (see section “The TOR/Sch9 Pathway”) (Longo et al. 2005; Kenyon 2005). The CLS of S. cerevisiae seems to be particularly well suited to identify conserved life span mediators because life span measurements are performed in conditions similar to those under which the longevity regulatory pathways have evolved (Longo et al. 2005). In fact, in the wild, microorganisms alternate rare periods in which they grow rapidly on glucose and other nutrients (e.g. on grapes) to others, usually lengthy ones, in which they survive under starvation conditions (Fabrizio and Longo 2003). To mimic this natural environment, CLS is monitored in populations of yeast grown in SDC medium containing a limited amount of nutrients (section “Chronological Life Span”). Once glucose and other nutrients are exhausted, cell division stops and yeast can survive for extended periods of time in different metabolic states (Fabrizio and Longo 2003).
The TOR/Sch9 Pathway
Conclusive evidence for the conservation of life span regulation throughout evolution was obtained in 2001. Prior to then, the insulin/insulin-like growth factor-like signaling (IIS) pathway had been demonstrated to control life span in C. elegans. In fact, lowering the activity of DAF-2, homolog of both human insulin and IGF-I receptors, was known to double worm life span and numerous downstream mediators activated or inhibited by DAF-2 activity had been identified as part of the IIS cascade (Kimura et al. 1997). Among these, the phosphoinositide-3-kinase AGE-1, the serine/threonine kinases AKT-1/2, PDK-1, SGK-1, and the fork-head transcription factor DAF-16 (Friedman and Johnson 1988; Morris et al. 1996; Paradis and Ruvkun 1998; Paradis et al. 1999; Hertweck et al. 2004). The latter had been shown to play a key role in antagonizing DAF-2 activity and mediating the longevity extension observed in the daf-2 mutants (Ogg et al. 1997; Lin et al. 1997) (Fig. 5.1).
Fig. 5.1.
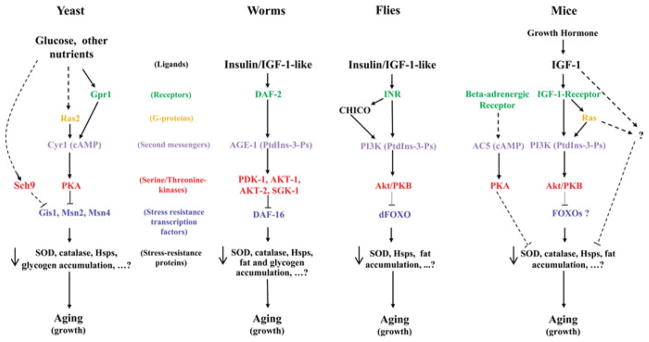
Conserved regulation of the life span-regulatory pathways. In yeast, worms, and flies the activation of the pro-growth nutrient-sensing/IIS pathways inhibits the activity of stress resistance transcription factors (Gis1, Msn2/4, DAF-16, dFOXO), reduces cell protection and accumulation of fat and/or glycogen, and promotes aging. Mutations that reduce the activity of these pathways prolong life span. In yeast and worms this longevity extension requires the activation of a stress response dependent on Gis1/Msn2/4 and Daf-16, which lead to the activation of anti-oxidant enzymes and heat-shock proteins. In flies overexpression of dFOXO extends lifespan most likely via the activation of stress response analogous to that of yeast and worms. In mice, reduction on the IIS by mutation of the IGF-1 receptor gene or by lowering growth hormone (GH) synthesis promotes longevity and stress resistance possibly by activating the transcription factors FoxOs. GH might also function to promote aging in part independently of IGF-1 signaling. Dampening the beta-adrenergic signaling by deleting the adenylate cyclase 5 gene (AC5) or reducing the activity of PKA also extends mice life span
In 2001 three articles demonstrated that the role of IIS in life span regulation was evolutionary conserved. In fact, on the one hand, it was found that lowering the activity of Sch9, a protein kinase involved in cell growth and cell cycle regulation in response to nutrients, extended CLS up to 3-fold (Fabrizio et al. 2001) and, on the other hand, it was shown how reduced IIS promoted longevity in Drosophila (Clancy et al. 2001; Tatar et al. 2001). Sch9 shares a high degree of homology with Akt and S6 kinase (S6K) in higher eukaryotes and numerous proteins activated in Sch9-deficient yeast are also activated in the daf-2 mutants and in long-lived flies, e.g. superoxide dismutase and heat-shock proteins (Longo and Finch 2003) (Fig. 5.1). Taken together, the novel results obtained in yeast combined with those from worms and flies, indicated that life span extension can be obtained through the inactivation of nutrient signaling and pro-growth pathways and the consequent activation of a “survival program” characterized by increased cellular protection (Longo and Finch 2003).
In most long-lived mutants, longevity is also associated with the accumulation of either reserve carbohydrate (glycogen), fat, or both (Longo and Fabrizio 2002) (Fig. 5.1). Some of the phenotypes observed in the long-lived mutants, e.g. fat accumulation, are shared by both worms in a state of developmental arrest (dauer larva) and flies in adult reproductive diapauses (McElwee et al. 2006; Tatar and Yin 2001). Both the dauer larva stage and the Drosophila diapause are characterized by prolonged life span and triggered by nutrient shortage or temperature shifts. Entry into dauer larva or diapause depends on IIS suggesting a significant overlap between the molecular mechanisms that promote longevity in adult worms and flies and those regulating life span in developmentally arrested worms and diapause flies (Larsen et al. 1995; Williams et al. 2006). S. cerevisiae has also evolved to enter “resting states” to prolong its life span when nutrients are limited. Depending on its ploidy yeast can enter stationary phase (haploid or diploid) or undergo spore-formation (diploid) both characterized by extended life span. Several of the phenotypes of the chronologically long-lived mutants are shared by stationary phase yeast, e.g. high resistance to cellular stress, and the same pathways can control both CLS and entry into stationary phase (Fabrizio and Longo 2003; Werner-Washburne et al. 1993). Taken together, the similarities between the life span regulatory pathways in model organisms and their link to alternative developmental programs/resting states that allow long-term survival in hostile environments has led to the hypothesis that the molecular mechanisms underlying longevity extension have evolved in ancestral microorganisms in order to overcome periods of starvation by arresting growth and allocating energy into cell maintenance and stress resistance (Longo et al. 2005).
Is life span regulation conserved in mammals? At the time when yeast, worms, and flies were shown to “grow old together” (Strauss 2001), there was no conclusive evidence for a role of IIS in mammalian life span regulation. However, it was known that dwarf mice carrying mutations affecting the development of their pituitary gland (Prop-1 and Pit-1) had low levels of plasma growth hormone (GH), insulin, and IGF-I as well as thyroid hormones and prolactin (Longo and Finch 2003). Notably, these mice live 25–65% longer than wild type (Brown-Borg et al. 1996; Flurkey et al. 2002) and, as the long-lived worms and flies, accumulate fat. Successive work showed that lowering IGF-I signaling is important for extending the life span of mice (Fig. 5.1). In fact, mice carrying only one IGF-I receptor (IGF-IR) functional allele were shown to be long-lived (26% mean life span extension) and resistant to oxidative stress as the IIS mutants of other species (Holzenberger et al. 2003). Interestingly, this effect was not confirmed in a different genetic background (Ladiges et al. 2009), suggesting that part of the phenotypes caused by GH deficiency are independent of IGF-I signaling or that it is difficult to reach a level of IGF-I signaling that is sufficient for normal function but also for life span extension. Further evidence that IIS signaling modulate mice life span came from the discovery that overexpressing Klotho, a hormone that represses the intracellular response triggered by both insulin and IGF-I, leads to longevity extension (Kurosu et al. 2005). Notably, Klotho transgenic mice were reported to have higher levels of mitochondrial superoxide dismutase activity and to be more resistant to oxidative stress at both the cellular and organismal levels (Yamamoto et al. 2005). These effects were shown to depend on the activation and nuclear translocation of the FoxO fork-head transcription factors, which is triggered by lowering IIS but may also depend on the activity of other signaling pathways (Yamamoto et al. 2005) (Fig. 5.1).
The yeast model system has been remarkably successful and perhaps the most successful at identifying genes later shown to regulate life span in mammals. In fact, mice lacking the Sch9 homologue S6 kinase 1 (S6K1), are long-lived and are protected against several age-dependent defects (Selman et al. 2009). Furthermore, mice lacking adenylate cyclase 5 (AC5) or with a reduced activity of PKA, AC5 and PKA being orthologues of the yeast pro-aging adenylate cyclase and PKA, are long-lived and display protection against age-dependent diseases or damage/loss of function (Yan et al. 2007; Enns et al. 2009) (see section “The Ras/Adenylate Cyclase/PKA Pathway”). The additional major gene implicated in yeast life span regulation is TOR, which is believed to function upstream of Sch9 to control growth in response to nutrients (Polak and Hall 2009). TOR is a serine-threonine protein kinase conserved in organisms ranging from yeast to humans originally implicated in the regulation of life span in worms and flies. In fact, knocking down CeTOR from the first day of adulthood approximately doubles the life span of C. elegans and a similar effect is obtained by reducing the activity of Daf-15, the worm ortholog of the mammalian TOR-interacting protein raptor (Vellai et al. 2003; Jia et al. 2004). Analogously, in Drosophila the overexpression of either dominant-negative dTOR or of TOR-inhibiting dTsc1/2 prolongs life span (Kapahi et al. 2004). Recently the TOR inhibitor rapamycin has been shown to extend longevity in mice (Harrison et al. 2009). In all organisms so far investigated except yeast there is one TOR iso-form that functions in two different multiprotein complexes, TORC1 and TORC2. In yeast two TOR kinases are present, Tor1 and Tor2. Either one of them can be found in the yeast TORC1 complex but only Tor2 is found in association with the TORC2 complex (Rohde et al. 2008). Down-regulation of TORC1 activity extends both yeast life spans (Wei et al. 2008; Powers et al. 2006; Kaeberlein et al. 2005). With respect to the CLS, genetic data suggest that TORC1 and Sch9 function in the same molecular pathway to control longevity (Wei et al. 2009; Pan and Shadel 2009 and P. Fabrizio, unpublished results) in agreement with the recently demonstrated role of TORC1 in the direct activation of Sch9 via the phosphorylation of several of its C-terminal amino acid residues (Urban et al. 2007). Notably, in higher eukaryotes the IIS cascade can activate TORC1, which in turn activates S6K, known to negatively affect longevity not only in mice (see above) but also in worms and flies (Kapahi et al. 2004; Hansen et al. 2007). Interestingly, both S6K and Sch9 play important roles in the activation of translation in response to nutrients and longevity regulation and are both dependent on the TORC1 complex for their activation. Mammalian S6K shares the same degree of homology with Sch9 as Akt. Since no closer homologue to S6K or Akt has been identified on the yeast genome, it is plausible that Sch9 originated from an ancestral protein that underwent duplication throughout evolution leading to the presence of S6K and Akt in metazoans. Importantly, both of them have maintained roles in controlling life span.
The Ras/Adenylate Cyclase/PKA Pathway
The first chronological long-lived mutant identified was the ras2Δ mutant (Longo 1997). RAS2 codes for one of two highly conserved G-proteins known to activate PKA via adenylate cyclase (Cyr1). Its deletion double CLS and promotes a marked increased of heat and oxidative stress resistance (Fabrizio et al. 2003). Ras proteins directly activate Cyr1 and a mutation causing a partial loss of Cyr1 function was shown to extend CLS as well (Fabrizio et al. 2001). In presence of glucose the Ras/PKA pathway mediates the activation of a pro-growth transcriptional program which depends in part on the inhibition of the stress resistance transcription factors Msn2/4 (Zaman et al. 2009). The function of these factors is key for the CLS extension of both ras2 and cyr1 mutants (Fabrizio et al. 2001, 2003) although another transcription factor, Gis1, has been recently shown to contribute to the effect of lowering the activity of the Ras/PKA pathway on CLS and resistance to stress (Wei et al. 2008).
For a long time the role of the Ras/Cyr1/PKA pathway in the control of life span appeared to be limited to yeast, although mice lacking p66Shc, a signal transducer that might trigger mitosis via Ras activation, were shown to be resistant to oxidants and live 30% longer than wild type littermates (Migliaccio et al. 1999). However, more recently mammalian aging has been shown to depend on the adenylate cyclase/PKA signaling. In fact, Yan et al. reported the implication of one of the mammalian adenylate cyclase isoforms, AC5, in the regulation of mice longevity (Yan et al. 2007). According to these authors, AC5 knock-out mice’s median life span is approximately 30% longer than that of control littermates. AC5 is expressed mostly in brain and heart and is activated by the β-adrenergic receptor signaling (Fig. 5.1). AC5 KO mice are protected against age-dependent cardiomyopathy. Notably, in agreement with results in yeast (Fabrizio et al. 2001, 2003), both myocytes and fibroblasts isolated from AC5 KO mice are more resistant to oxidative stress than control cells. Accordingly, MnSOD levels in heart, brain, and kidney were higher in AC5 KO mice than in controls confirming the association between protection against superoxide and life span extension (Yan et al. 2007). Further evidence to support a role for the adenylate cyclase/PKA cascade in mice aging has been provided by Enns et al. who have reported that the disruption of RIIβ, which codes for one of the mammalian PKA regulatory subunits, promotes median and maximum life span extension in male mice (Enns et al. 2009).
Molecular Mechanisms Responsible for Life Span Extension
While the field of the genetics of aging is progressing rapidly, we are still lacking a comprehensive view of how aging occurs at the cellular and organismal level and how it can be delayed. The yeast CLS is providing an important tool to address this issue.
Oxidative Stress Response
With few exceptions, life span extension is associated with an increased ability to withstand different types of stress (Longo and Fabrizio 2002). Oxidative stress resistance in particular has been the object of intensive study in the context of aging since the “Free radical theory of aging” was first proposed (Harman 1956). According to this theory aging is caused by the cellular damage produced by highly reactive oxygen species with unpaired electrons, which are generated mostly in the mitochondrion at complexes I and III of the electron transport chain (Longo 1997). As mentioned in the previous sections, mutations that prolong life span in different model organisms are associated with high levels of antioxidant enzymes. All the yeast mutants with prolonged CLS are highly resistant to the superoxide-generating agents menadione and paraquat and to hydrogen peroxide and mitochondrial superoxide dismutase (MnSOD) activity is required for long term survival and for the longevity extension promoted by lack of Sch9 or Ras2 (Longo et al. 1996; Fabrizio et al. 2003; Longo and Fabrizio 2002). However, the overexpression of individual antioxidant enzymes or combination of them leads to a maximum of 30% mean CLS extension, which is relatively modest when compared to the 3-fold life span extension obtained by deleting SCH9 (Fabrizio et al. 2001, 2003). Similar results were obtained in Drosophila and mice (Sun et al. 2002; Orr and Sohal 1994; Hu et al. 2007; Schriner et al. 2005) suggesting that, while MnSOD activity provides a fundamental anti-oxidant defense to mitochondria, its role in longevity extension is significant but limited. Nevertheless, work done using the yeast CLS paradigm strongly supports a causative role for oxidative-damage in aging. In fact, firstly, exploiting the ability of yeast to grow on glucose as a carbon source in absence of functional mitochondria (fermentative growth), it was possible to show that loss of mitochondrial function precedes death in both mutants lacking MnSOD activity and wild type (Longo et al. 1999). Secondly, it was reported that the mitochondrial enzymes aconitase and succinate dehydrogenase, which contain iron-sulfur clusters particularly sensitive to superoxide-dependent oxidation, are primary targets of age-dependent mitochondrial damage in yeast (Fabrizio et al. 2001, 2003; Longo et al. 1999). Thirdly, an age-dependent accumulation of oxidation-induced DNA damage/mutations has been shown in chronologically aging yeast (Madia et al. 2009) (see section “Age-Dependent Genomic Instability in Yeast”).
Metabolic Switches
In order to further understand the mechanisms that lead to life span extension, we have recently obtained the gene expression profiles of chronologically aging wild type and long-lived mutants (Wei et al. 2009). In our analysis we compared the transcriptomes of the sch9Δ, ras2Δ, and tor1Δ mutants to those of wild type yeast at day 2.5. This age was chosen to avoid both the noise that could originate from residual cell growth at younger ages and the transcriptional changes associated with reduction of metabolic rates that normally occurs at day 4–5 (Fabrizio and Longo 2003; Fabrizio et al. 2003). Our results have shown a significant degree of overlap between the genes either up- or down-regulated in the different mutants in comparison to the wild type underlying how the different life span regulatory pathways may impinge on a set of common downstream effectors controlled by the same transcriptional activators to modulate longevity (Wei et al. 2008, 2009). Importantly, yeast long-lived mutants exhibit transcriptional changes consistent with a wide range of metabolic changes. In fact, glycolytic/fermentative genes are up-regulated in all the mutants, while a vast set of mitochondrial genes, which include those coding for electron transport proteins, TCA cycle enzymes, and mitochondrial ribosomal proteins among others, are down-regulated (Wei et al. 2009). Our data also suggest that a part of dihydroxy-acetone-phosphate (DHAP), a glycolysis intermediate, is metabolized to produce glycerol. In fact, all the long-lived mutants showed a general activation of glycerol biosynthetic genes, above all GPD1 and GPD2, which code for the key enzymes required for glycerol biosynthesis from DHAP. Consistently, our in depth analysis of the chronologically aging sch9Δ mutant revealed a significant accumulation of glycerol both intracellularly and extracellularly, which was not observed in the wild type (Wei et al. 2009). The role of GPD1/2 and other glycerol biosynthetic genes in CLS extension in a sch9Δ context was confirmed by epistatis analysis, which underscored the importance of glycerol biosynthesis in promoting both life span and resistance to stress (Wei et al. 2009). Notably, in aging sch9Δ mutants the metabolic switch to glycerol biosynthesis is associated with ethanol catabolism (Wei et al. 2009). We have previously demonstrated that ethanol is accumulated in aging wild type yeast and negatively affects their CLS in spite of being a carbon source that yeast can utilize by mitochondrial respiration (Fabrizio et al. 2005a). Acetic acid is also accumulated in chronologically aging cultures and it has been proposed to play a pro-aging role (Burtner et al. 2009). However, it should be underlined that at the level generated during chronological aging (approximately 6 mM) acetic acid is a carbon source that promotes acidification, cell cycle arrest, and high respiratory rates (Fabrizio et al. 2003, 2004a, 2005a) and not a toxin, as it can be at high concentrations (Ludovico et al. 2001; Madeo et al. 2004). Thus, acetic acid, may generate the appropriate conditions (no division, high metabolism, etc.) to obtain a short and high metabolism life span that models cells from higher eukaryotes. The remarkable number of genes later shown to promote aging in higher eukaryotes identified using this CLS paradigm supports this notion. Under certain incubation conditions, the ethanol generated during log phase appears to be converted to acetic acid. Mutants lacking Sch9 but not wild type cells, appear to redirect acetic acid to glycerol production (V Longo unpublished results). We now know that ethanol as glucose represents a pro-aging carbon source. In fact, when yeast are exposed to a constant concentration of either of them under experimental conditions that do not allow cell growth (lack of an essential amino acid), their CLS is dramatically reduced when compared to that of yeast exposed to glycerol (Wei et al. 2009). In this respect, exposure to glycerol has the same CLS extending effect as carbon source removal, which is a method to promote caloric restriction (CR) in yeast (Wei et al. 2009). This is remarkable because it suggests that the molecular anti-aging strategies activated by CR, the only non genetic intervention known to prolong life span in all species so far tested (Mair and Dillin 2008), are not repressed by glycerol. Thus, the CLS extension of the sch9Δ mutant is due, in part, to key metabolic changes that mimic CR by triggering ethanol consumption and glycerol accumulation (Wei et al. 2009). Very recent evidence has implemented our data by showing that phosphoenolpyruvate carboxykinase (Pck1), a key enzyme for gluconeogenesis, is required for ethanol consumption and for both normal life span and CR-dependent CLS extension (Lin et al. 2009). Pck1 was shown to be dependent on the histone acetylase complex NuA4 for activation and on Sir2 for inactivation (Lin et al. 2009), which is consistent with our findings demonstrating that Sir2 activity limits the longevity extension promoted by CR (Fabrizio et al. 2005a). Intriguingly, our gene expression profile data did not show transcriptional activation of genes involved in gluconeogenesis in the early phase of chronological aging. By contrast, as mentioned above, they pointed to an activation of the glycolytic function in long-lived yeast (Wei et al. 2009). Further studies are needed to clarify this point and establish whether glycolysis or gluconeogenesis is predominant in yeast under CR and which enzymatic activities are key for glycerol accumulation and ethanol catabolism.
How do the metabolic switches observed in long-lived yeast translate to other long-lived organisms? Do they represent a conserved anti-aging strategy? We do not have an answer yet. However, gene expression data suggest that both glyceroneogenesis and gluconeogenesis are up-regulated in calorie restricted C. elegans (Castelein et al. 2008). Furthermore, an anti-aging role for a metabolic switch to gluconeogenesis was originally hypothesized based on gene expression profile data obtained from calorie restricted mice (Lee et al. 1999) and is supported by preliminary results showing that transgenic mice overexpressing the cytosolic form of phosphoenolpyruvate carboxykinase in the skeletal muscle are long-lived (Hakimi et al. 2007).
Conserved Pro-aging Genes, Genomic Instability, and Cancer
Age-Dependent Genomic Instability in Yeast
Yeast CLS extension is associated with a reduction in genomic instability (Fabrizio et al. 2004a; Madia et al. 2008, 2009). By performing simple mutation assays on chronologically aging yeast, it is possible to monitor the age-dependent accumulation of different types of DNA mutations including base substitutions, small DNA insertions/deletions, and gross chromosomal rearrangements (GCRs) (Madia et al. 2007). The frequency of all these mutations increases in an age-dependent manner (Fabrizio et al. 2004a; Madia et al. 2009). A reduction of this effect is observed in mutants lacking Sch9 (Fabrizio et al. 2004a; Madia et al. 2009) and it was shown to depend on their reduced sensitivity to the superoxide-dependent DNA damage and the inactivation of the error-prone Rev1-Polζ DNA polymerase complex, which is involved in DNA-repair by translesion synthesis (TLS) (Madia et al. 2009). As discussed in the section “Oxidative Stress Response”, oxidative damage is believed to play a key role in aging and high levels of protection against oxidants are detected in yeast long-lived mutants (Longo and Fabrizio 2002). In a sch9− context both SOD2 transcription and Sod2 activity are up-regulated during chronological aging (Fabrizio et al. 2003, 2004a) and the levels of 8-hydroxy-2′-deoxiguanidine (8-OHdG), one of the most frequent oxidative DNA lesions, are reduced in comparison with wild type (Madia et al. 2009). Importantly, the overexpression of either cytosolic or mitochondrial Sod reduces the age-dependent accumulation of mutations suggesting that superoxide is a principal mediator of DNA-damage and mutagenesis observed in aging yeast or that superoxide or Sods are regulating other poorly understood processes that lead to hypermutability states (Madia et al. 2009). By studying the mutational spectra obtained by sequencing CAN1, a gene coding for an arginine permease, in wild type and sch9− clones originated from 7-day old cells previously screened for the presence of can1 mutations, the deoxycytidyl-transferase Rev1 was identified as key for age-dependent mutagenesis (Madia et al. 2009). Rev1 is found in a complex with polymerase zeta (Polζ) and it is implicated in the repair of abasic sites in damaged DNA by TLS. The activity of Rev1-Polζ is error-prone and compatible with the mutational spectra observed in old wild type cells. Furthermore, Sch9 activity and oxidative stress were shown to promote Rev1 function, which, although crucial to prevent DNA double strand breaks, causes the generation of point mutations when aging non-dividing cells undergo the first round of replication (Madia et al. 2009). Interestingly, in sch9− mutants TLS is completely abolished suggesting that lack of Sch9 not only represses REV1 expression, which is not absolutely required for TLS in yeast (Pages et al. 2008), but it may also reduce the activity of further components of this error-prone repair system (Madia et al. 2009).
In addition to its role in regulating TLS, Sch9 was shown to contribute to the regulation of mitotic recombination, which is also important for age-dependent genomic instability (Madia et al. 2008). In fact, in yeast lacking Sgs1, the homolog of the human BLM and WRN RecQ helicases, which are mutated in the human progeroid Werner and Bloom syndromes (see next section), a dramatic increase of the age-dependent point mutations and GCRs is dampened by the deletion of SCH9. This effect is due mostly to the down-regulation of the error-prone recombination between sister chromatids (Madia et al. 2008).
In summary, our current knowledge suggests that Sch9 controls genomic instability in aging yeast by: (1) down-regulating the expression of anti-oxidant genes such as Sod2, which limits DNA oxidation, (2) activating error-prone repair systems such as Rev1-Polζ, which generates mutations to repair oxidative DNA lesions when old cells resume growth, and error-prone mitotic recombination and, (3) regulating the production and catabolism of ethanol and acetic acid and the generation of carbon sources that reduce the aging rate. The discovery of the mechanisms behind the enhanced genomic stability of the long-lived sch9Δ mutant is important not only because of its possible conservation and implication in the regulation of aging in mammals but also because, if conserved, these mechanisms might be central for tumorigenesis (see next sections).
Oxidative DNA-Damage and Cancer
Besides yeast, an age-dependent accumulation of DNA mutations has been observed in other model organisms and in human cells and tissues (Vijg 2007; Longo et al. 2008). A causative role for DNA damage/mutations in aging is supported by a number of human segmental progeroid syndromes characterized each by the premature appearance of a few aging features. Notably, many of these syndromes are caused by mutations affecting the activity of either DNA-repair or DNA-damage sensing genes and promote cancer (Hasty et al. 2003). Although to what extent genomic instability contributes to aging is still uncertain, its role in tumorigenesis is well established and several lines of evidence suggest that oxidative DNA damage plays a pivotal role in causing an age-dependent accumulation of DNA mutations and cancer. In fact, (1) a vast group of mutations detected in the anti-oncogene p53, which is mutated in ~50% of the human cancers, is generated in an attempt to repair the damage caused by reactive oxygen species (ROS) (Pfeifer 2000), (2) high levels of DNA damage/mutations and cancer have been found in mice with reduced activity of either cytosolic or mitochondrial SOD (Busuttil et al. 2005; Van Remmen et al. 2003), (3) in S. cerevisiae the lack of cytosolic Sod causes an increase of mutations and a high frequency of adaptive regrowth, a phenotype described as “cancer-like” because, as discussed in the section “Chronological Life Span”, it is characterized by the ability of aging cells to resume cell division under conditions that normally do not promote growth (Fabrizio et al. 2004a; Longo et al. 1999; Madia et al. 2007, 2009). Importantly, the adaptive regrowth phenotype is extremely rare in cultures of the long-lived ras2Δ or sch9Δ mutants consistently with their high degree of protection against superoxide (Fabrizio et al. 2004a) and the low mutation frequency detected in aging sch9Δ discussed in the previous section.
Pro-aging Genes and Cancer in Higher Eukaryotes
As discussed broadly in the section “Conserved Life Span-Regulatory Pathways”, the nutrient-sensing/insulin-IGF-like pathways promote aging in several model organisms. The expression of conserved pro-aging genes such as Sch9 causes aging, genomic instability, and a “cancer-like” phenotype in yeast. Is there a similar association between pro-aging genes/pathways and cancer in other organisms? It certainly seems to be the case. In fact, life-extending mutations that decrease IIS in worms block germ cells proliferation and induce apoptosis in a C. elegans mutant that forms germ-line tumors (Pinkston et al. 2006). Analogously, in flies reducing IIS not only extends life span but also delays the growth of germline cysts (LaFever and Drummond-Barbosa 2005).
Most interestingly, long-lived GH/IGF-I-deficient mice are long lived and show lower levels of lifetime tumor incidence (Vergara et al. 2004; Ikeno et al. 2003). A similar reduction of tumors is observed in Akt-deficient mice (Skeen et al. 2006). Human studies indicate that dampening IGF-I signaling may reduce cancer incidence likely because of the pro-growth activity of IGF-I (Longo et al. 2008). However, we hypothesize that the IIS-dependent regulation of genomic instability is also important for tumorigenesis and cancer progression (see next section).
The Role of Pro-aging Genes in Cancer
Yeast pro-aging Sch9 and Ras2 are homologues of mammalian proto-oncogenes Akt and Ras, which are activated in many human cancers (Rodriguez-Viciana et al. 1994; Yoeli-Lerner and Toker 2006). In both yeast and mammals Ras and Sch9/Akt signal through pathways that regulate cell growth and promote aging (see section “Conserved Life Span-Regulatory Pathways”). In humans oncogenic mutations that lead to the activation of Ras, Akt, or in the up-stream IGF-I-receptor are commonly believed to promote cancer by allowing the survival and growth of damaged cells, which are normally removed by apoptosis. They have also been proposed to increase genomic instability because the high proliferation rates they promote may increase the occurrence of errors during replication across sites of unrepaired damage (Pollak et al. 2004). The accumulation of further mutations in cells already hit by an initial oncogenic mutation is thought to be responsible for tumor growth and metastasis.
Our opinion, based on studies on the conserved role of oncogene homologues in life span regulation, is that the tumorigenesis scenario might be more complex. In fact, in analogy with yeast, the activity of Ras and Akt might reduce cellular protection and in particular oxidative stress resistance in human cells leading to increased mutational rates, which in turn may cause oncogenic mutations activating Ras and Akt themselves and/or the additional mutations required for cancer development. This hypothesis is supported by data showing that cells isolated from long-lived mice are resistant to several different DNA-damaging agents (Longo et al. 2008).
Conclusions and Perspectives
The study of the pathways and mechanisms regulating longevity has benefited greatly from the use of S. cerevisiae. This simple unicellular organism has been instrumental in the discovery of the conservation of the principal life span-regulatory pathways and, thanks to the amenability of the CLS to genetic, genomic, and biochemistry assays, it is providing an excellent system to gain further insights into the metabolic changes occurring in senescent cells. Recently, yeast CLS has been used successfully to study the mechanisms behind age-dependent genomic instability leading to the identification of oxidative DNA damage and error-prone DNA repair systems as key for the mutation accumulation observed during chronological aging. This is an important finding because of the possibility that these molecular mechanisms may be conserved through evolution and involved in enhancing genomic instability and promoting cancer in mammals.
We expect that in the near future additional yeast genetic determinants of aging and novel mechanisms to explain how life span can be prolonged will be discovered. These are expected to contribute to further understand aging and diseases in a wide range of species including humans.
Appendix
While this book was in production we published two articles relevant to the topic of this chapter. For the sake of completeness, we believe it is appropriate to discuss them briefly here. The first article has reported the results of a screen of the yeast deletion collection aimed at identifying novel life span determinants (Fabrizio et al. 2010). Besides confirming the importance of the mitochondrial function and the autophagic process in long-term survival, our screen has uncovered numerous novel genes involved in the process of determining yeast longevity. Among others ACB1, CKA2, and TRM9. The deletion of each of these three genes prolongs life span and increases heat resistance. ACB1 codes an acyl-coA binding protein involved in lipid biosynthesis and vesicle formation. Cka2 is the catalytic subunit of a serine-threonine kinase, CK2, which controls several cellular functions including cell growth and proliferation. Trm9 is a tRNA methylase that targets the uridine residues at the wobble position in tRNA(Glu) and tRNA(Arg3). Currently, the mechanisms by which these proteins regulate longevity have not been described. It will be important to elucidate them given the high degree of conservation of these novel life span determinants and the possibility that their role in aging extends to other organisms.
The second article concerns the role of the conserved pro-aging pathways in the regulation of genomic instability and cancer. In the section “Conserved Pro-aging Genes, Genomic Instability, and Cancer” we have discussed how the activity of the Sch9 and GH/IGF-I pathways promotes DNA damage in yeast and mice, respectively. We have also mentioned that GH/IGF-I-deficient mice show decreased rates of cancer incidence. Recently this observation has been extended to humans with growth hormone receptor deficiency who display a major reduction in cancer and diabetes, which is associated with reduced levels of several orthologs of the key yeast pro-aging genes (Guevara-Aguirre et al. 2011). Importantly, serum from GH/IGF-I signaling-deficient individuals protects cells in culture from H2O2-dependent DNA damage and down-regulates the expression of N-Ras, PKA, and TOR while activating SOD2 transcription. This suggests that a reduction of GH/IGF-I signaling may lead to cellular protection and reduced DNA damage in vivo via the inactivation of the pro-aging Ras, PKA, and TOR pathways, which in turn may contribute to lower incidence of cancer and other diseases. Thus, this new evidence from a human study further supports a causative link between the activity of the conserved pro-aging pathways, genomic instability, and diseases.
Contributor Information
Valter D. Longo, Email: vlongo@usc.edu, Department of Biological Sciences, Andrus Gerontology Center, University of Southern California, Los Angeles, CA 90089-0191, USA
Paola Fabrizio, Email: paola.fabrizio@ens-lyon.fr, Laboratory of Molecular and Cellular Biology, UMR5239 CNRS, Ecole Normale Supérieure de Lyon, Lyon, France.
References
- Allen C, Buttner S, Aragon AD, Thomas JA, Meirelles O, et al. Isolation of quiescent and nonquiescent cells from yeast stationary-phase cultures. J Cell Biol. 2006;174:89–100. doi: 10.1083/jcb.200604072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi K, Sinclair D, Gordon JI, Guarente L. Passage through stationary phase advances replicative aging in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1999;96:9100–9105. doi: 10.1073/pnas.96.16.9100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker MG, Walmsley RM. Replicative ageing in the fission yeast Schizosaccharomyces pombe. Yeast. 1999;15:1511–1518. doi: 10.1002/(sici)1097-0061(199910)15:14<1511::aid-yea482>3.3.co;2-p. [DOI] [PubMed] [Google Scholar]
- Borghouts C, Benguria A, Wawryn J, Jazwinski SM. Rtg2 protein links metabolism and genome stability in yeast longevity. Genetics. 2004;166:765–777. doi: 10.1534/genetics.166.2.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown-Borg HM, Borg KE, Meliska CJ, Bartke A. Dwarf mice and the ageing process. Nature. 1996;384:33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- Burtner CR, Murakami CJ, Kennedy BK, Kaeberlein M. A molecular mechanism of chronological aging in yeast. Cell Cycle. 2009;8:1256–1270. doi: 10.4161/cc.8.8.8287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busuttil RA, Garcia AM, Cabrera C, Rodriguez A, Suh Y, et al. Organ-specific increase in mutation accumulation and apoptosis rate in CuZn-superoxide dismutase-deficient mice. Cancer Res. 2005;65:11271–11275. doi: 10.1158/0008-5472.CAN-05-2980. [DOI] [PubMed] [Google Scholar]
- Castelein N, Hoogewijs D, De Vreese A, Braeckman BP, Vanfleteren JR. Dietary restriction by growth in axenic medium induces discrete changes in the transcriptional output of genes involved in energy metabolism in Caenorhabditis elegans. Biotechnol J. 2008;3:803–812. doi: 10.1002/biot.200800003. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, et al. Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature. 2009;459:802–807. doi: 10.1038/nature08085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Mello NP, Childress AM, Franklin DS, Kale SP, Pinswasdi C, et al. Cloning and characterization of LAG1, a longevity-assurance gene in yeast. J Biol Chem. 1994;269:15451–15459. [PubMed] [Google Scholar]
- Egilmez NK, Jazwinski SM. Evidence for the involvement of a cytoplasmic factor in the aging of the yeast Saccharomyces cerevisiae. J Bacteriol. 1989;171:37–42. doi: 10.1128/jb.171.1.37-42.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enns LC, Morton JF, Treuting PR, Emond MJ, Wolf NS, et al. Disruption of protein kinase A in mice enhances healthy aging. PLoS One. 2009;4:e5963. doi: 10.1371/journal.pone.0005963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, et al. Genome-wide screen in Saccharomyces cerevisiae identifies vacuolar protein sorting, autophagy, biosynthetic, and tRNA methylation genes involved in life span regulation. PLoS Genet. 2010;6(8):1227–1228. doi: 10.1371/journal.pgen.1001024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Battistella L, Vardavas R, Gattazzo C, Liou LL, et al. Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae. J Cell Biol. 2004a;166:1055–1067. doi: 10.1083/jcb.200404002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Gattazzo C, Battistella L, Wei M, Cheng C, et al. Sir2 blocks extreme life-span extension. Cell. 2005a;123:655–667. doi: 10.1016/j.cell.2005.08.042. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Li L, Longo VD. Analysis of gene expression profile in yeast aging chronologically. Mech Ageing Dev. 2005b;126:11–16. doi: 10.1016/j.mad.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Liou LL, Moy VN, Diaspro A, Selverstone-Valentine J, et al. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. 2003;163:35–46. doi: 10.1093/genetics/163.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Aging Cell. 2003;2:73–81. doi: 10.1046/j.1474-9728.2003.00033.x. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Longo VD. The chronological life span of Saccharomyces cerevisiae. Meth Mol Biol. 2007;371:89–95. doi: 10.1007/978-1-59745-361-5_8. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Pletcher SD, Minois N, Vaupel JW, Longo VD. Chronological aging-independent replicative life span regulation by Msn2/Msn4 and Sod2 in Saccharomyces cerevisiae. FEBS Lett. 2004b;557:136–142. doi: 10.1016/s0014-5793(03)01462-5. [DOI] [PubMed] [Google Scholar]
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD. Regulation of longevity and stress resistance by Sch9 in yeast. Science. 2001;292:288–290. doi: 10.1126/science.1059497. [DOI] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Harrison DE. The Snell dwarf mutation Pit1(dw) can increase life span in mice. Mech Ageing Dev. 2002;123:121–130. doi: 10.1016/s0047-6374(01)00339-6. [DOI] [PubMed] [Google Scholar]
- Friedman DB, Johnson TE. A mutation in the age-1 gene in Caenorhabditis elegans lengthens life and reduces hermaphrodite fertility. Genetics. 1988;118:75–86. doi: 10.1093/genetics/118.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guevara-Aguirre J, et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci Transl Med. 2011;3(70):70ra13. doi: 10.1126/scitranslmed.3001845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakimi P, Yang J, Casadesus G, Massillon D, Tolentino-Silva F, et al. Overexpression of the cytosolic form of phosphoenolpyruvate carboxykinase (GTP) in skeletal muscle repatterns energy metabolism in the mouse. J Biol Chem. 2007;282:32844–32855. doi: 10.1074/jbc.M706127200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005;1:119–128. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, et al. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans. Aging Cell. 2007;6:95–110. doi: 10.1111/j.1474-9726.2006.00267.x. [DOI] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol. 1956;11:298–300. doi: 10.1093/geronj/11.3.298. [DOI] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–1359. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- Hertweck M, Gobel C, Baumeister R. C. elegans SGK-1 is the critical component in the Akt/PKB kinase complex to control stress response and life span. Dev Cell. 2004;6:577–588. doi: 10.1016/s1534-5807(04)00095-4. [DOI] [PubMed] [Google Scholar]
- Holzenberger M, Dupont J, Ducos B, Leneuve P, Geloen A, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–187. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Hu D, Cao P, Thiels E, Chu CT, Wu GY, et al. Hippocampal long-term potentiation, memory, and longevity in mice that overexpress mitochondrial superoxide dismutase. Neurobiol Learn Mem. 2007;87:372–384. doi: 10.1016/j.nlm.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeno Y, Bronson RT, Hubbard GB, Lee S, Bartke A. Delayed occurrence of fatal neoplastic diseases in ames dwarf mice: correlation to extended longevity. J Gerontol A Biol Sci Med Sci. 2003;58:291–296. doi: 10.1093/gerona/58.4.b291. [DOI] [PubMed] [Google Scholar]
- Jia K, Chen D, Riddle DL. The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development. 2004;131:3897–3906. doi: 10.1242/dev.01255. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, McVey M, Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW, 3rd, Steffen KK, Westman EA, Hu D, et al. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science. 2005;310:1193–1196. doi: 10.1126/science.1115535. [DOI] [PubMed] [Google Scholar]
- Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, et al. Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol. 2004;14:885–890. doi: 10.1016/j.cub.2004.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy BK, Austriaco NRJ, Guarente L. Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span. J Cell Biol. 1994;127:1985–1993. doi: 10.1083/jcb.127.6.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Kimura KD, Tissenbaum HA, Liu Y, Ruvkun G. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277:942–946. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kurosu H, Yamamoto M, Clark JD, Pastor JV, Nandi A, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829–1833. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladiges W, Van Remmen H, Strong R, Ikeno Y, Treuting P, et al. Lifespan extension in genetically modified mice. Aging Cell. 2009;8:346–352. doi: 10.1111/j.1474-9726.2009.00491.x. [DOI] [PubMed] [Google Scholar]
- LaFever L, Drummond-Barbosa D. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science. 2005;309:1071–1073. doi: 10.1126/science.1111410. [DOI] [PubMed] [Google Scholar]
- Larsen PL, Albert PS, Riddle DL. Genes that regulate both development and longevity in Caenorhabditis elegans. Genetics. 1995;139:1567–1583. doi: 10.1093/genetics/139.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laun P, Rinnerthaler M, Bogengruber E, Heeren G, Breitenbach M. Yeast as a model for chronological and reproductive aging – a comparison. Exp Gerontol. 2006;41:1208–1212. doi: 10.1016/j.exger.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Lee CK, Klopp RG, Weindruch R, Prolla TA. Gene expression profile of aging and its retardation by caloric restriction. Science. 1999;285:1390–1393. doi: 10.1126/science.285.5432.1390. [DOI] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–1322. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lin YY, Lu JY, Zhang J, Walter W, Dang W, et al. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo V. Thesis. University of California; Los Angeles, CA: 1997. The chronological life span of Saccharomyces cerevisiae. Studies of superoxide dismutase, Ras and Bcl-2. [Google Scholar]
- Longo VD. Linking sirtuins, IGF-I signaling, and starvation. Exp Gerontol. 2009;44:70–74. doi: 10.1016/j.exger.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Longo VD, Fabrizio P. Regulation of longevity and stress resistance: a molecular strategy conserved from yeast to humans? Cell Mol Life Sci. 2002;59:903–908. doi: 10.1007/s00018-002-8477-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longo VD, Finch CE. Evolutionary medicine: from dwarf model systems to healthy centenarians. Science. 2003;299:1342–1346. doi: 10.1126/science.1077991. [DOI] [PubMed] [Google Scholar]
- Longo VD, Gralla EB, Valentine JS. Superoxide dismutase activity is essential for stationary phase survival in Saccharomyces cerevisiae. Mitochondrial production of toxic oxygen species in vivo. J Biol Chem. 1996;271:12275–12280. doi: 10.1074/jbc.271.21.12275. [DOI] [PubMed] [Google Scholar]
- Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Longo VD, Lieber MR, Vijg J. Turning anti-ageing genes against cancer. Nat Rev Mol Cell Biol. 2008;9:903–910. doi: 10.1038/nrm2526. [DOI] [PubMed] [Google Scholar]
- Longo VD, Liou LL, Valentine JS, Gralla EB. Mitochondrial superoxide decreases yeast survival in stationary phase. Arch Biochem Biophys. 1999;365:131–142. doi: 10.1006/abbi.1999.1158. [DOI] [PubMed] [Google Scholar]
- Longo VD, Mitteldorf J, Skulachev VP. Programmed and altruistic ageing. Nat Rev Genet. 2005;6:866–872. doi: 10.1038/nrg1706. [DOI] [PubMed] [Google Scholar]
- Ludovico P, Sousa MJ, Silva MT, Leao C, Corte-Real M. Saccharomyces cerevisiae commits to a programmed cell death process in response to acetic acid. Microbiology. 2001;147:2409–2415. doi: 10.1099/00221287-147-9-2409. [DOI] [PubMed] [Google Scholar]
- Madeo F, Herker E, Wissing S, Jungwirth H, Eisenberg T, et al. Apoptosis in yeast. Curr Opin Microbiol. 2004;7:655–660. doi: 10.1016/j.mib.2004.10.012. [DOI] [PubMed] [Google Scholar]
- Madia F, Gattazzo C, Fabrizio P, Longo VD. A simple model system for age-dependent DNA damage and cancer. Mech Ageing Dev. 2007;128(1):45–49. doi: 10.1016/j.mad.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madia F, Gattazzo C, Wei M, Fabrizio P, Burhans WC, et al. Longevity mutation in SCH9 prevents recombination errors and premature genomic instability in a Werner/Bloom model system. J Cell Biol. 2008;180:67–81. doi: 10.1083/jcb.200707154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madia F, Wei M, Yuan V, Hu J, Gattazzo C, et al. Oncogene homologue Sch9 promotes age-dependent mutations by a superoxide and Rev1/Polzeta-dependent mechanism. J Cell Biol. 2009;186:509–523. doi: 10.1083/jcb.200906011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mair W, Dillin A. Aging and survival: the genetics of life span extension by dietary restriction. Annu Rev Biochem. 2008;77:727–754. doi: 10.1146/annurev.biochem.77.061206.171059. [DOI] [PubMed] [Google Scholar]
- McElwee JJ, Schuster E, Blanc E, Thornton J, Gems D. Diapause-associated metabolic traits reiterated in long-lived daf-2 mutants in the nematode Caenorhabditis elegans. Mech Ageing Dev. 2006;127:458–472. doi: 10.1016/j.mad.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Medvedik O, Lamming DW, Kim KD, Sinclair DA. MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biol. 2007;5:e261. doi: 10.1371/journal.pbio.0050261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G. A phospatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhbditis elegans. Nature. 1996;382:536–539. doi: 10.1038/382536a0. [DOI] [PubMed] [Google Scholar]
- Mortimer RK. Life span of individual yeast cells. Nature. 1959;183:1751–1752. doi: 10.1038/1831751a0. [DOI] [PubMed] [Google Scholar]
- Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, et al. The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature. 1997;389:994–999. doi: 10.1038/40194. [DOI] [PubMed] [Google Scholar]
- Orr WC, Sohal RS. Extension of life-span by overexpression of superoxide dismutase and catalase in Drosophila melanogaster. Science. 1994;263:1128–1130. doi: 10.1126/science.8108730. [DOI] [PubMed] [Google Scholar]
- Pages V, Bresson A, Acharya N, Prakash S, Fuchs RP, et al. Requirement of Rad5 for DNA polymerase zeta-dependent translesion synthesis in Saccharomyces cerevisiae. Genetics. 2008;180:73–82. doi: 10.1534/genetics.108.091066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Shadel GS. Extension of chronological life span by reduced TOR signaling requires down-regulation of Sch9p and involves increased mitochondrial OXPHOS complex density. Aging. 2009;1:131–145. doi: 10.18632/aging.100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Ailion M, Toker A, Thomas JH, Ruvkun G. A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev. 1999;13:1438–1452. doi: 10.1101/gad.13.11.1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12:2488–2498. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeifer GP. p53 mutational spectra and the role of methylated CpG sequences. Mutat Res. 2000;450:155–166. doi: 10.1016/s0027-5107(00)00022-1. [DOI] [PubMed] [Google Scholar]
- Pinkston JM, Garigan D, Hansen M, Kenyon C. Mutations that increase the life span of C. elegans inhibit tumor growth. Science. 2006;313:971–975. doi: 10.1126/science.1121908. [DOI] [PubMed] [Google Scholar]
- Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21:209–218. doi: 10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–518. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- Powers RW, 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S. Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev. 2006;20:174–184. doi: 10.1101/gad.1381406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Viciana P, Warne PH, Dhand R, Vanhaesebroeck B, Gout I, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras [see comments] Nature. 1994;370:527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohde JR, Bastidas R, Puria R, Cardenas ME. Nutritional control via Tor signaling in Saccharomyces cerevisiae. Curr Opin Microbiol. 2008;11:153–160. doi: 10.1016/j.mib.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux AE, Leroux A, Alaamery MA, Hoffman CS, Chartrand P, et al. Pro-aging effects of glucose signaling through a G protein-coupled glucose receptor in fission yeast. PLoS Genet. 2009;5:e1000408. doi: 10.1371/journal.pgen.1000408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux AE, Quissac A, Chartrand P, Ferbeyre G, Rokeach LA. Regulation of chronological aging in Schizosaccharomyces pombe by the protein kinases Pka1 and Sck2. Aging Cell. 2006;5:345–357. doi: 10.1111/j.1474-9726.2006.00225.x. [DOI] [PubMed] [Google Scholar]
- Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, et al. Extension of murine lifespan by overexpression of catalase targeted to mitochondria. Science. 2005;308(5730):1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, et al. Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science. 2009;326:140–144. doi: 10.1126/science.1177221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair DA, Guarente L. Extrachromosomal rDNA circles – a cause of aging in yeast. Cell. 1997;91:1033–1042. doi: 10.1016/s0092-8674(00)80493-6. [DOI] [PubMed] [Google Scholar]
- Skeen JE, Bhaskar PT, Chen CC, Chen WS, Peng XD, et al. Akt deficiency impairs normal cell proliferation and suppresses oncogenesis in a p53-independent and mTORC1-dependent manner. Cancer Cell. 2006;10:269–280. doi: 10.1016/j.ccr.2006.08.022. [DOI] [PubMed] [Google Scholar]
- Steinkraus KA, Kaeberlein M, Kennedy BK. Replicative aging in yeast: the means to the end. Annu Rev Cell Dev Biol. 2008;24:29–54. doi: 10.1146/annurev.cellbio.23.090506.123509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss E. Longevity. Growing old together. Science. 2001;292:41–43. doi: 10.1126/science.292.5514.41. [DOI] [PubMed] [Google Scholar]
- Sun J, Folk D, Bradley TJ, Tower J. Induced overexpression of mitochondrial Mn-superoxide dismutase extends the life span of adult Drosophila melanogaster. Genetics. 2002;161:661–672. doi: 10.1093/genetics/161.2.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, et al. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Tatar M, Yin C. Slow aging during insect reproductive diapause: why butterflies, grasshoppers and flies are like worms. Exp Gerontol. 2001;36:723–738. doi: 10.1016/s0531-5565(00)00238-2. [DOI] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Urban J, Soulard A, Huber A, Lippman S, Mukhopadhyay D, et al. Sch9 is a major target of TORC1 in Saccharomyces cerevisiae. Mol Cell. 2007;26:663–674. doi: 10.1016/j.molcel.2007.04.020. [DOI] [PubMed] [Google Scholar]
- Van Remmen H, Ikeno Y, Hamilton M, Pahlavani M, Wolf N, et al. Life-long reduction in MnSOD activity results in increased DNA damage and higher incidence of cancer but does not accelerate aging. Physiol Genomics. 2003;16:29–37. doi: 10.1152/physiolgenomics.00122.2003. [DOI] [PubMed] [Google Scholar]
- Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, et al. Genetics: influence of TOR kinase on lifespan in C. elegans. Nature. 2003;426:620. doi: 10.1038/426620a. [DOI] [PubMed] [Google Scholar]
- Vergara M, Smith-Wheelock M, Harper JM, Sigler R, Miller RA. Hormone-treated snell dwarf mice regain fertility but remain long lived and disease resistant. J Gerontol A Biol Sci Med Sci. 2004;59:1244–1250. doi: 10.1093/gerona/59.12.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijg J. Aging of the genome. Oxford University Press; Oxford: 2007. [Google Scholar]
- Wei M, Fabrizio P, Hu J, Ge H, Cheng C, et al. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet. 2008;4:e13. doi: 10.1371/journal.pgen.0040013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Fabrizio P, Madia F, Hu J, Ge H, et al. Tor1/Sch9-regulated carbon source substitution is as effective as calorie restriction in life span extension. PLoS Genet. 2009;5:e1000467. doi: 10.1371/journal.pgen.1000467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger M, Feng L, Paul A, Smith DL, Jr, Hontz RD, et al. DNA replication stress is a determinant of chronological lifespan in budding yeast. PLoS One. 2007;2:e748. doi: 10.1371/journal.pone.0000748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner-Washburne M, Braun E, Johnston GC, Singer RA. Stationary phase in the yeast Saccharomyces cerevisiae. Microbiol Rev. 1993;57:383–401. doi: 10.1128/mr.57.2.383-401.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KD, Busto M, Suster ML, So AK, Ben-Shahar Y, et al. Natural variation in Drosophila melanogaster diapause due to the insulin-regulated PI3-kinase. Proc Natl Acad Sci USA. 2006;103:15911–15915. doi: 10.1073/pnas.0604592103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M, Clark JD, Pastor JV, Gurnani P, Nandi A, et al. Regulation of oxidative stress by the anti-aging hormone klotho. J Biol Chem. 2005;280:38029–38034. doi: 10.1074/jbc.M509039200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, et al. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–258. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- Yoeli-Lerner M, Toker A. Akt/PKB signaling in cancer: a function in cell motility and invasion. Cell Cycle. 2006;5:603–605. doi: 10.4161/cc.5.6.2561. [DOI] [PubMed] [Google Scholar]
- Zaman S, Lippman SI, Schneper L, Slonim N, Broach JR. Glucose regulates transcription in yeast through a network of signaling pathways. Mol Syst Biol. 2009;5:245. doi: 10.1038/msb.2009.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambrano MM, Kolter R. GASPing for life in stationary phase. Cell. 1996;86:181–184. doi: 10.1016/s0092-8674(00)80089-6. [DOI] [PubMed] [Google Scholar]
- Zambrano MM, Siegele DA, Almiron M, Tormo A, Kolter R. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science. 1993;259:1757–1760. doi: 10.1126/science.7681219. [DOI] [PubMed] [Google Scholar]
- Zinser ER, Kolter R. Escherichia coli evolution during stationary phase. Res Microbiol. 2004;155:328–336. doi: 10.1016/j.resmic.2004.01.014. [DOI] [PubMed] [Google Scholar]