

Abstract
Ideas about personalized medicine are underpinned in part by evolutionary biology's Modern Synthesis. In this essay we link personalized medicine to the efforts of the early statistical investigators who quantified the heritability of human phenotype and then attempted to reconcile their observations with Mendelian genetics. As information about the heritability of common diseases was obtained, similar efforts were directed at understanding the genetic basis of disease phenotypes. These ideas were part of the rationale driving the Human Genome Project and subsequently the personalized medicine movement. In this context, we discuss: (1) the current state of the genotype–phenotype relationship in humans, (2) the common-disease–common-variant hypothesis, (3) the current ability of ‘omic’ information to inform clinical decision making, (4) emerging ideas about the therapeutic insight available from rare genetic variants, and (5) the social and behavioural barriers to the wider potential success of personalized medicine. There are significant gaps in knowledge as well as conceptual, intellectual, and philosophical limitations in each of these five areas. We then provide specific recommendations to mitigate these limitations and close by asking if it is time for the biomedical research community to ‘stop chasing Mendel?’
 |
Michael J. Joyner, MD, is the Caywood Professor of Anaesthesiology at Mayo Clinic where he was named Distinguished Investigator in 2010. His interests include: exercise physiology, cardiovascular regulation, the physiology of world records, autonomic regulation of metabolism, and clinical transfusion practices. His undergraduate (1981) and medical (1987) degrees are from the University of Arizona with residency and research training at Mayo. He has held leadership positions at Mayo, in the extramural research community, and with leading journals. His lab has been funded by the NIH since 1993, and former fellows have established independent research programs at leading institutions throughout the world. Franklyn G. Prendergast, MD., PhD is Edmond and Marion Guggenheim Professor of Biochemistry & Molecular Biology and Professor in the Department of Molecular Pharmacology & Experimental Therapeutics at the Mayo Clinic. He received his MBBS from the University of the West Indies and was a Rhodes Scholar at Oxford where he received an M.A. in physiology. This was followed by Internal Medicine training at Mayo from 1971 to 1973 and a Ph.D. in Biochemistry at University of Minnesota/Mayo Graduate School (1977). In addition to his experimental focus on protein biophysics. Dr. Prendergast directed the Mayo Clinic Comprehensive Cancer Center for many years and was the founding director of the Mayo Center for Individualized Medicine. His leadership activities include membership on the Mayo Board of Governors and Trustees, (1992–2009) and numerous NIH Board advisory boards. Dr. Prendergast was named a Mayo Distinguished Investigator in 1987; and has received honorary degrees from Purdue University and the University West Indies along with the Musgrave Gold Medal from the Institute of Jamaica.
Introduction
In this essay we want to discuss some ideas and raise questions about how the Modern Synthesis in evolutionary biology has influenced what might loosely be described as biomedical thinking including the current enthusiasm for ‘personalized medicine’ and related ideas (Wilkins, 2011; Hood & Auffray, 2014). By ‘Modern Synthesis’ we mean ideas that emerged at the confluence of population inheritance studies, Mendelian genetics and natural selection prior to World War II that have subsequently been applied in an effort to better understand and explain human variation, including the risk of common chronic diseases. By ‘personalized medicine’ we mean the general idea that by ‘reading’ the genome or other molecular ‘omes’ of an individual human it will be possible to predict what diseases or medical conditions he or she might ultimately suffer from. Furthermore with such knowledge, it should be possible to either prevent or preempt the occurrence of the disease or to provide more targeted therapy and improve therapeutic outcomes. While the above definition is perhaps oversimplified, it has notable advocates (Hood & Auffray, 2014). Additionally, elements of personalized (or individualized) medicine are currently being pursued via large and costly initiatives by essentially every major academic medical centre, pharma, and biomedical research funding agencies worldwide.
There is no ‘aha’ moment marking when the biomedical world suddenly adopted the Modern Synthesis version of evolutionary biology and started to look for the genetic causes of diseases, including the common non-communicable or chronic diseases responsible for so much morbidity and mortality throughout the world (Murray et al. 2006; World Health Organization: http://www.who.int/mediacentre/factsheets/fs310/en/). In addition to well-known diseases like cystic fibrosis, sickle cell anaemia, and hereditary bleeding disorders with clear-cut patterns of generation-to-generation transmission, one can also see the influence of heritability estimates for things like height and intelligence made by the early biometricians and statisticians, led by Darwin's cousin Francis Galton, on what has emerged (Fisher, 1919; Forrest, 1974). Additionally, as similar heritability estimates were generated for conditions like blood pressure, heart disease, obesity and diabetes it became clear that most common diseases and medical conditions had a statistically heritable element. Of course the people in the various cohorts used to make these estimates frequently shared similar cultural and socioeconomic backgrounds, so strategies like identical vs. fraternal twin studies have also been used in an effort to control for these factors. Along these lines, ‘family history’ is a major component of many risk calculators for common diseases (National Cancer Institute Breast Cancer Risk Assessment Tool: http://www.cancer.gov/bcrisktool/; Cardiovascular Risk Calculator: http://www.patient.co.uk/doctor/cardiovascular-risk-calculator). These calculators are used by clinicians to make therapeutic decisions and advise patients.
It is also important to emphasize before we go on, that the concept of a gene predates DNA and was originally defined or assumed to be something having a high-level phenotypic effect per se (Johannsen, 1911; Gerstein et al. 2007; Edwards, 2011). Thus, it seems reasonable to argue that since the early Galton–Fisher statistical heritability estimates were high, then ‘genes’ in the pre DNA sense were driving the heritability. We should also point out that the shifting definition over the years of ‘what is a gene’ to the current DNA-centric version may also have contributed to an oversimiplifed view of the genotype–phenotype relationship as it applies to DNA. The problems associated with this oversimplified view were then likely to be amplified by the so-called Central Dogma of Molecular Biology positing that information transfer from DNA to proteins and by extension phenotype is essentially a one-way street (Crick, 1970). Thus, the statistical heritability estimates and fundamental approaches to making them have remained similar over many years while the concept of what is a gene has changed dramatically.
A classic example that illustrates this interpretation is found in a paper on height by Fisher (1919) nearly a century ago as he tried to reconcile the heritability of height with Mendelian genetics. Fisher, as shown in Fig. 1, focused on the proportion of variance attributable to parents and grandparents and speculated on the genetic basis of his observations. This approach also led to efforts to develop statistical approaches to dominance modifiers by Fisher and his competitor Wright, but it also led the genetics community to believe that there were clear-cut genetic explanations for these heritability findings (Crow, 2010; Edwards et al. 2011). From our perspective it is not much of an intellectual leap from efforts like this to what became known as the common-disease–common (gene)-variant hypothesis (Shields, 2011). In other words a limited number of common gene variants would explain much of the apparently heritable risk for common non-communicable diseases. Thus, the efforts of Fisher and his contemporaries along with what flowed from them give rise to our idea that biomedical research has in fact been chasing Mendel. However, it is interesting to think about what conclusions Fisher might have drawn if his reference population was from 20th century Japan, where there has been a dramatic increase in height over several generations (Japanese Height Trends: http://www.dh.aist.go.jp/en/research/centered/anthropometry/). For obesity, the heritability estimates vary by country depending on both GDP per person and also the rate of change of GDP (Minn et al. 2013).
Figure 1. Estimated contribution of ancestoral inheritance to human height (stature) calculated by Fisher (1919) as he attempted to reconcile the statistical analysis of the heritability of human variation to Mendelian genetics.
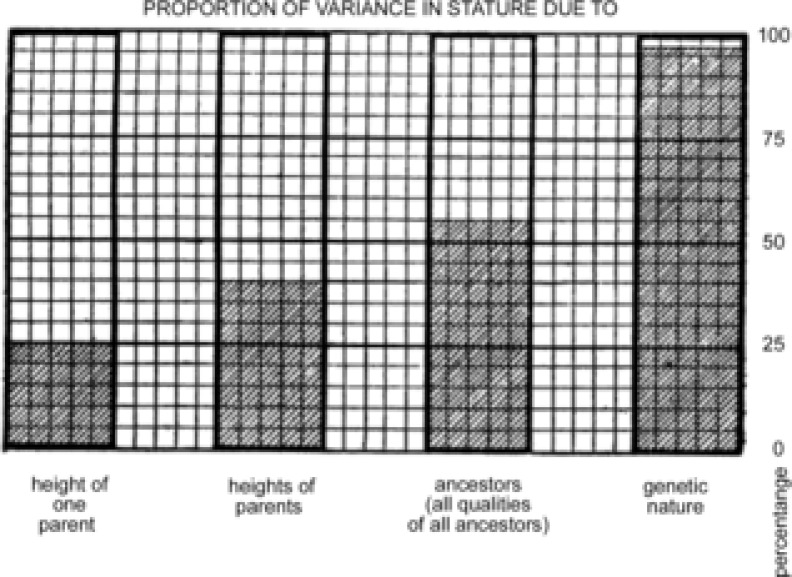
Observations and estimates such as these stimulated geneticists to believe that a potentially large number of intra- and inter-locus alleles are responsible for the phenotypic variation of complex traits, and this search has continued as the definition of gene became DNA-centric (for historical perspective see Fisher, 1919 and Edwards, 2011).
The common-disease–common-variant hypothesis was also one driver of the human genome project (HGP; Collins, 2001). The HGP and the ever-falling costs of genotyping and related technology then provided the needed ingredients and stimulus to move forward with the dream of personalized medicine (Hood, 1988). When the draft HGP was finished in 2001 optimism was high and to quote Francis Collins (Collins, 2001):
If research support continues at vigorous levels, it is hard to imagine that genomic science will not soon reveal the mysteries of hereditary factors in heart disease, cancer, diabetes, mental illness, and a host of other conditions.
However, for the linear view of personalized medicine outlined at the outset of this essay to work for common diseases, it seems that three criteria need to be met. First, a clearly identifiable genetic variant or related pathway clearly linked to a disease must be identified. Second, this variant must be modifiable or actionable by some preventive or therapeutic intervention. Third, when such information is available patients, individual clinicians, and health care systems will use it in a rational way to reduce disease risk and/or improve therapeutic decision making. With these introductory comments as a background, we now raise five questions for the general field of personalized medicine.
What is the current status of the genotype–phenotype relationship?
For most of the common non-communicable diseases that kill the vast majority of people in the developed world, the search for gene variants that account for most or much of the heritable risk of a given disease has not turned up much (World Health Organization: http://www.who.int/mediacentre/factsheets/fs310/en/). In most instances a large number of common variants have been discovered that cause clinically insignificant changes in risk for things like cardiovascular disease, hypertension and diabetes (Baker, 2010; Edwards et al. 2013; Ganesh et al. 2013). There have also been issues with population-to-population-based replication of results (Kidambi et al. 2012; Major Depressive Disorder Writing Group, 2013). These issues are clouded further by environment, culture and behaviour in the people and populations sampled. For example, common variants in the FTO obesity gene are associated with increased BMI in populations of sedentary Caucasians. However, the association is either blunted or absent in physically active groups (Rampersaud et al. 2008; Ahmad et al. 2013). There is hope that whole genome sequencing of large numbers of humans will provide more insight into genotype–phenotype relationships for common diseases, but skepticism about what additional insights are possible is understandable given that so little has come from genome-wide association studies.
Parenthetically, multiple rare genotypes have emerged as the clear-cut causes of catastrophic medical conditions. A good example is potentially lethal hypertrophic cardiomyopathy and other arrythmogenic conditions that typically kill apparently healthy young athletes with little or no warning. In contrast to the early hope that only a few rogue variants would be responsible for these conditions, huge numbers of private mutations have emerged (Landstrom & Ackerman, 2010). More importantly not all carriers of the potentially lethal genotype manifest the phenotype of concern, highlighting the general problem of variable penetrance for predictive medicine based on genotypic information. In the non-affected individuals perhaps other protective or redundant pathways are engaged that either keep expression of the potentially lethal variant in check or counteract it in other ways to mitigate lethality. Molecular pathways involved in this mitigation might include non-coding transcription, how pathways interact as networks, and the almost inevitable role of epigenetic mechanisms.
On a more positive note, rare and generally private mutations might explain some extreme cases of extreme longevity, but as is the case for phenotypes that are likely to have complex causes, clear-cut genetic explanations for extreme longevity are elusive (Christensen et al. 2009; Sebastiani & Perls, 2012). Thus, for most common non-communicable diseases, understanding the role of genotypic variation as a major driver of phenotype remains an important challenge (Ganesh et al. 2013). One possibility is that the role of genotypic variation in common non-communicable diseases is essentially overwhelmed by environmental, cultural and behavioural factors (Marmot & Syme, 1976).
A complementary expanation is that these diseases typically include age as a major risk factor and ideas about ‘evolutionary medicine’ suggest that as we evolved we lived shorter lives in environements that are radically different than the current world for most people. In this context, there has a been a search for ‘thrifty genes’ based on the idea that frequent nutritional stress favoured selection for genotypes that could gain weight when calories were plentiful and thus avoid death from starvation during famine (McDermott, 1998). Similar arguments have been made for selection of genotypes that favoured salt retention and to preserve blood volume in hot environments (Young, 2007). However, when these genotypes are exposed to the modern world, a combination of longevity plus abundant food and salt leads to an explosion of non-communicable diseases including obesity/diabetes and hypertension.
What is the current status of common-disease–common-variant hypothesis?
Implicit in the answer to our first question is the idea that the common-disease–common-variant hypothesis is no longer viable (Shields, 2011). It is also interesting to note that the traditional statistical heritability estimates from the late 19th century are much more predictive for things like adult height than techniques based on gene variants (Aulchenko et al. 2009). This is leading to divergent views on the cause of the missing heritability. One idea is that either unknown or poorly understood genetic factors will emerge to explain this missing heritability. The other idea is that the causes are non-genetic and not due to structural differences in DNA that will be easy to understand via DNA sequencing of any sort. On a conceptual basis what comes next in this area is linked to very basic questions about whether or not DNA per se has a ‘privileged place’ in driving all elements of phenotypic expression (Omholt, 2013; Noble et al. 2014). Additionally, the computational challenges associated with understanding how genes interact, the nuances of transcription, and subsequent protein expression and function are vast (Noble, 2011; Vidal et al. 2011). This information must then be considered on a temporal basis and in the context of both genetic and cellular networks along with potentially overriding ‘whole body’ physiological control mechanisms (Joyner, 2011). Thus, at least for now it appears safe to say that the common-disease–common-variant hypothesis is no longer viable.
Can genotype inform clinical decision making for commonly used drugs?
For most common diseases addition of current generation ‘omic’ data to risk prediction has been of limited value. A good example is that the inclusion data on gene variants thought to increase risk for type 2 diabetes does little to improve the predictive value of risk scores based on traditional phenotypic risk factors (Talmud et al. 2010). As mentioned above, the data for disease prediction are consistent with the observation that addition of gene variant data does little to improve the prediction of adult height beyond the classical Galtonian techniques arising from the Victorian era (Aulchenko et al. 2009).
One area where the impact of ‘omics’ on clinical decision making has shown mixed (including positive) results is the use of genotyping to predict drug response. Examples include the determining the optimal dosing regimens for 6-mercaptopurine in acute lymphoblastic leukaemia and also the anti-platelet drug clopidogrel (Lennard et al. 1990; Scott et al. 2013). Additionally, individualized gene variant-driven treatment for tamoxifen therapy in breast cancer has shown promise, but larger trials are needed to confirm the results from smaller studies (Schroth et al. 2009; Province et al. 2014). However, the news in this area is not all positive and several recent trials of gene variant-informed Coumadin anticoagulant therapy dosing failed to show better results compared to traditional dosing schemes based on clinical phenotype (Furie, 2013).
One interesting topic related to clinical decision making, personalized medicine and drug dose is the recent controversy surrounding the ‘new’ guidelines for statin therapy released by the American Heart Association and American College of Cardiology (Stone et al. 2013). It might have been anticipated based on the expectations and promise of the HGP that new guidelines would be driven in part by individual genetic information or other advanced biomarkers. Instead the new guidelines rely on a relatively generic phenotypic risk calculator, less frequent or aggressive monitoring of blood cholesterol levels, and simpler dosing schemes. Based on these features, the new guidelines appear headed in a direction opposite to that of personalized medicine. The response to the new guidelines also highlights the individual and collective social challenges of guidelines in general.
Along these lines, the average clinician and the general public are reportedly confused by the new guidelines (New York Times, 2013). This confusion highlights the challenge of translating estimates of disease risk into both changes in behaviour and improved health care outcomes. First, medical guidelines change and are reversed at regular intervals, with about 30–40% of guidelines being superseded every 10 years (Prasad et al. 2013). So, what is ‘right’ today, might be ‘wrong’ tomorrow (Joyner, 2011). Second, there is no evidence that people will behave rationally based on what might be called gene scores (Markowitz et al. 2011; Vassy et al. 2012; Bloss et al. 2014). Will those who perceive their risk to be reduced act in a cavalier way even if the effects of a reduced risk gene score are modest in the overall scheme of things? Will those with higher risk scores become fatalistic and ignore other potentially more important health guidelines? Will the average clinician telling the average overweight and inactive person with impaired fasting glucose to exercise and diet be more effective when armed with a gene score? There is also at least some evidence that people with ‘increased’ gene-based risk sores become more medicalized and seek more drugs or preemptive procedures of questionable value. Such behaviour might then drive health care spending up vs. down with little to show for it. Thus, all of the observations outlined in response to our third question run counter to and challenge one or more of the basic tenants of personalized medicine.
Will rare variants have therapeutic implications?
In view of the problems with the common-variant–common-disease hypothesis, one idea is that rare variants will be identified with clear-cut relationships (increased or decreased risk) to disease and lead to new ‘druggable’ targets. This approach is currently being used to target the PSK9 pathway in people with familial or other high cholesterol syndromes that are resistant to or intolerant of statin therapy (Raal et al. 2012). The potential for this pathway as a therapeutic target was identified in patients with familial hypercholesterolaemia. There are a number of injectable monoclonal antibodies that target this pathway undergoing clinical trials (Mullard, 2012). However, the fact that these compounds are injectable highlights a practical problem for pharma related to developing easy-to-use small molecules that target non-traditional pathways (Cohen & Hobbs, 2013). Along these lines, a high fraction of commonly used drugs act on membrane receptors, channels, reuptake mechanisms, or second messengers and are perhaps easier targets for small molecules than ‘omic’ targets. While it is too soon to tell how many novel therapeutic targets or pathways will flow from the discovery of rare variants, it is perhaps fair to say that the search for them was not a primary goal of the HGP or personalized medicine as promulgated in the 1990s. However, like many fruitful avenues of biomedical investigation and discovery, frequently things emerge that might be described as ‘right for the wrong reasons’ (Comroe & Dripps, 1974).
Will targeting dominant pathways work?
In the US (and other countries) there has been a ‘war on cancer’ that started in 1971. Some have argued that the success of this ‘war’, especially for drug therapy directed at solid tumours has been disappointing (Hanahan, 2014). In this context, the development of very fast gene sequencing and other ‘omic’ technology has led to the discovery that many solid tumours with similar clinical and histological phenotypes may be driven by or manifest different genetic mutations. At some level this is very interesting because it is another challenge to the idea that genotype is the driver of phenotype. At another level, with enough ‘omic’ information about a given tumour it might be possible to better target anti-neoplastic therapy using compounds that hit the genetic mutation, defect or gene product that is causing the problem. The success of imatinib in treating chronic myeloid leukaemia shows both the potential for success and longer term limitations with this approach (Mahon, 2012). This general approach also offers some promise for targeting rare non-neoplastic diseases by finding already approved drugs that might offer therapeutic benefit via so-called drug repurposing. However, the example of chronic myeloid leukaemia is unusual.
While individualized therapy for solid tumours is appealing, there are at least two main issues with it. The first is practical and relates to how best to design clinical trials to test whether individualized chemotherapy is more effective in comparison to standard chemotherapy regimens. The second issue is the fact that most tumours are multi-clonal and that by targeting the dominant clone, the resistant clones that survive will then emerge with a vengeance (Gatenby, 2009; Hanahan, 2014; Watson, 2013). For both these scenarios it is simply too soon to tell and we would only suggest that a range of responses to ‘omically’ targeted therapy is likely. However, because tumours are multi-clonal, the idea that drug sensitivity testing similar to that used for antibiotic treatment of microbial infections will be widely applicable to solid tumours may be a stretch.
Summary
We started this essay with our ideas about ‘where’ the intellectual underpinning for personalized medicine came from. We then pointed out that for personalized medicine to become a reality clear-cut information about the ‘omics’ of risk was needed, that this information had to be linked to actionable preventive or therapeutic interventions, and that individuals and health care systems had to choose to make the interventions happen. There are serious concerns about each of these three conditions. While things are more hopeful in relation to the identification of novel therapeutic targets and perhaps the selection of the ‘best’ drug for a given disease, how widely effective and applicable these strategies will be is unknown. Based on the issues and observations above we make the following suggestions and raise additional questions:
The potential of personalized medicine should not be over-hyped. There will be successes and there will be experimental and therapeutic dead ends. The promised broad-based revolution may not come and over-promising might lead to cynicism by the general public and ultimately loss of enthusiasm by funding agencies.
Biomedical research is more than a series of linear ‘moon shots’ to conquer specific diseases or gain certain knowledge. The successes and failures of the War on Cancer, the Human Genome Project, and other major initiatives might be judged one way based on their original goals and another way when the collateral and unanticipated benefits are considered over a longer time scale (Comroe & Dripps, 1974).
Has a medicalized version of the Modern Synthesis led to overly narrow thinking and models that have an excessive focus on the DNA version of genotype-equals-phenotype? The vast proliferation of either inbred or engineered rodent models that undergo minimal physical activity is one example of biomedical research models dominated by a sort self-fulfilling genotype-equals-phenotype world view (Safdar et al. 2011.
How will potentially useful personalized medicine information be disseminated and acted on? A major problem across many areas of medicine is not a lack of information about how to prevent, modulate or even cure disease. Frequently the real problem is at the interface of individual, organizational and societal change (Pagoto & Appelhans, 2013).
We close this essay by postulating that there has been an pervasive influence of the gene centrism inherent in the Modern Synthesis in conjunction with the Central Dogma of Molecular Biology on biomedical thinking. We believe this influence has now become counterproductive. Thus, it is critical for new ideas stemming from evolutionary biology highlighted in this special issue of The Journal of Physiology and elsewhere to more fully inform biomedical thinking about the complex relationship between DNA and phenotype (Müller, 2007; Jablonka, 2012; Noble, 2013; Omholt, 2013). The time has come to stop chasing Mendel.
Additional information
Competing interests
None declared.
References
- Ahmad S, Rukh G, Varga TV, Ali A, Kurbasic A, Shungin D, Ericson U, Koivula RW, Chu AY, Rose LM, Ganna A, Qi Q, et al. Gene×physical activity interactions in obesity: combined analysis of 111,421 individuals of European ancestry. PLoS Genet. 2013;9:e1003607. doi: 10.1371/journal.pgen.1003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko YS, Struchalin MV, Belonogova NM, Axenovich TI, Weedon MN, Hofman A, Uitterlinden AG, Kayser M, Oostra BA, van Duijn CM, Janssens AC, Borodin PM. Predicting human height by Victorian and genomic methods. Aur J Hum Genet. 2009;17:1070–1075. doi: 10.1038/ejhg.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M. Genomics: The search for association. Nature. 2010;467:1135–1138. doi: 10.1038/4671135a. [DOI] [PubMed] [Google Scholar]
- Bloss CS, Schork NJ, Topol EJ. Direct-to-consumer pharmacogenomics testing is associated with increased physician utilization. J Med Genet. 2014;51:83–89. doi: 10.1136/jmedgenet-2013-101909. [DOI] [PubMed] [Google Scholar]
- Christensen K, Johnson TE, Vaupel JW. The quest for genetic determinants of human longevity: challenges and insights. Nat Rev Genet. 2009;7:436–448. doi: 10.1038/nrg1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JC, Hobbs HH. Genetics. Simple genetics for a complex disease. Science. 2013;340:689–690. doi: 10.1126/science.1239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins FS. Contemplating the end of the beginning. Genome Res. 2001;11:641–643. doi: 10.1101/gr.189801. [DOI] [PubMed] [Google Scholar]
- Comroe JH, Jr, Dripps RD. Ben Franklin and open heart surgery. Circ Res. 1974;35:661–669. doi: 10.1161/01.res.35.5.661. [DOI] [PubMed] [Google Scholar]
- Crick F. Central dogma of molecular biology. Nature. 1970;227:561–563. doi: 10.1038/227561a0. [DOI] [PubMed] [Google Scholar]
- Crow JF. Wright and Fisher on inbreeding and random drift. Genetics. 2010;184:609–611. doi: 10.1534/genetics.109.110023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards AW. Mathematizing Darwin. Behav Ecol Sociobiol. 2011;65:421–430. doi: 10.1007/s00265-010-1122-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: Illuminating the dark road from association to function. Am J Hum Genet. 2013;93:779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RA. The causes of human variability. Eugene Rev. 1919;10:213–220. [PMC free article] [PubMed] [Google Scholar]
- Forrest DW. Francis Galton: The Life and Work of a Victorian Genius. New York: Taplinger Publishing Co., Inc; 1974. p. 340. [Google Scholar]
- Furie B. Do pharmacogenetics have a role in the dosing of vitamin K antagonists? N Engl J Med. 2013;369:2345–2346. doi: 10.1056/NEJMe1313682. [DOI] [PubMed] [Google Scholar]
- Ganesh SK, Arnett DK, Assimes TL, Basson CT, Chakravarti A, Ellinor PT, Engler MB, Goldmuntz E, Herrington DM, Hershberger RE, Hong Y, Johnson JA, et al. Genetics and genomics for the prevention and treatment of cardiovascular disease: update: a scientific statement from the American Heart Association. Circulation. 2013;128:2813–2851. doi: 10.1161/01.cir.0000437913.98912.1d. [DOI] [PubMed] [Google Scholar]
- Gatenby RA. A change of strategy in the war on cancer. Nature. 2009;459:508–509. doi: 10.1038/459508a. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Bruce C, Rozowsky JS, Zheng D, Du J, Korbel JO, Emanuelsson O, Zhang ZD, Weissman S, Snyder M. What is a gene, post-ENCODE? History and updated definition. Genome Res. 2007;17:669–681. doi: 10.1101/gr.6339607. [DOI] [PubMed] [Google Scholar]
- Hanahan D. Rethinking the war on cancer. Lancet. 2014;383:558–563. doi: 10.1016/S0140-6736(13)62226-6. [DOI] [PubMed] [Google Scholar]
- Hood L. Biotechnology and medicine of the future. JAMA. 1988;25:1837–1844. [PubMed] [Google Scholar]
- Hood L, Auffray C. Participatory medicine: a driving force for revolutionizing healthcare. Genome Med. 2014;5:110. doi: 10.1186/gm514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka E. Epigenetic variations in heredity and evolution. Clin Pharmacol Ther. 2012;92:683–688. doi: 10.1038/clpt.2012.158. [DOI] [PubMed] [Google Scholar]
- Johannsen W. The genotype conception of heredity. Am Naturalist. 1911;XLV:129–159. doi: 10.1093/ije/dyu063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner MJ. Why physiology matters in medicine. Physiology. 2011;26:72–75. doi: 10.1152/physiol.00003.2011. [DOI] [PubMed] [Google Scholar]
- Kidambi S, Ghosh S, Kotchen JM, Grim CE, Krishnaswami S, Kaldunski ML, Cowley AW, Jr, Patel SB, Kotchen TA. Non-replication study of a genome-wide association study for hypertension and blood pressure in African Americans. BMC Med Genet. 2012;13:27. doi: 10.1186/1471-2350-13-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landstrom AP, Ackerman MJ. Mutation type is not clinically useful in predicting prognosis in hypertrophic cardiomyopathy. Circulation. 2010;122:2441–2449. doi: 10.1161/CIRCULATIONAHA.110.954446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lennard L, Lilleyman JS, Van Loon J, Weinshilboum RM. Genetic variation in response to 6-mercaptopurine for childhood acute lymphoblastic leukaemia. Lancet. 1990;336:225–229. doi: 10.1016/0140-6736(90)91745-v. [DOI] [PubMed] [Google Scholar]
- McDermott R. Ethics, epidemiology and the thrifty gene: biological determinism as a health hazard. Soc Sci Med. 1998;47:1189–1195. doi: 10.1016/s0277-9536(98)00191-9. [DOI] [PubMed] [Google Scholar]
- Mahon F-X. Is going for cure in chronic myeloid leukemia possible and justifiable? Hematology Am Soc Hematol Educ Program. 2012;2012:122–128. doi: 10.1182/asheducation-2012.1.122. [DOI] [PubMed] [Google Scholar]
- Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. Ripke S, Wray NR, Lewis CM, Hamilton SP, Weissman MM, Breen G, Byrne EM, Blackwood DH, Boomsma DI, Cichon S, Heath AC, et al. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry. 2013;18:497–511. doi: 10.1038/mp.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz SM, Park ER, Delahanty LM, O'Brien KE, Grant RW. Perceived impact of diabetes genetic risk testing among patients at high phenotypic risk for type 2 diabetes. Diabetes Care. 2011;34:568–573. doi: 10.2337/dc10-1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marmot MG, Syme SL. Acculturation and coronary heart disease in Japanese-Americans. Am J Epidemiol. 1976;104:225–247. doi: 10.1093/oxfordjournals.aje.a112296. [DOI] [PubMed] [Google Scholar]
- Minn J, Chiu DT, Wang Y. Variation in the heritability of body mass index based on diverse twin studies: a systematic review. Obes Rev. 2013;14:871–882. doi: 10.1111/obr.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullard A. Cholesterol-lowering blockbuster candidates speed into Phase III trials. Nat Rev Drug Discov. 2012;11:817–819. doi: 10.1038/nrd3879. [DOI] [PubMed] [Google Scholar]
- Müller GB. Evo-devo: extending the evolutionary synthesis. Nat Rev Genet. 2007;8:943–949. doi: 10.1038/nrg2219. [DOI] [PubMed] [Google Scholar]
- Murray CJL, Kulkarni S, Michaud C, Tomijima N, Bulzacchelli MT, Iandiorio TJ, Ezzati M. Eight Americas: investigating mortality disparities across races, counties, and race-counties in the United States. PLoS Med. 2006;3:1513–1524. doi: 10.1371/journal.pmed.0030260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- New York Times. Bumps in the road to new cholesterol guidelines. 2013. http://www.nytimes.com/2013/11/26/health/heart-and-stroke-study-hit-by-a-wave-of-criticism.html.
- Noble D. Differential and integral views of genetics in computational systems biology. Interface Focus. 2011;1:7–15. doi: 10.1098/rsfs.2010.0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble D. Physiology is rocking the foundations of evolutionary biology. Exp Physiol. 2013;98:1235–1243. doi: 10.1113/expphysiol.2012.071134. [DOI] [PubMed] [Google Scholar]
- Noble D, Jablonka E, Joyner MJ, Müller G, Omholt S. Evolution evolves: physiology returns to centre stage. J Physiol. 2014;592:2237–2244. doi: 10.1113/jphysiol.2014.273151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omholt SW. From sequence to consequence and back. Prog Biophys Mol Biol. 2013;111:75–82. doi: 10.1016/j.pbiomolbio.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Pagoto SL, Appelhans BM. A call for an end to the diet debates. JAMA. 2013;310:687–688. doi: 10.1001/jama.2013.8601. [DOI] [PubMed] [Google Scholar]
- Prasad V, Vandross A, Toomey C, Cheung M, Rho J, Quinn S, Chacko SJ, Borkar D, Gall V, Selvarai S, Ho N, Cifu A. A decade of reversal: an analysis of 146 contradicted medical practices. Mayo Clin Proc. 2013;88:790–798. doi: 10.1016/j.mayocp.2013.05.012. [DOI] [PubMed] [Google Scholar]
- Province MA, Goetz MP, Brauch H, Flockhart DA, Hebert JM, Whaley R, Suman VJ, Schroth W, Winter S, Zembutsu H, Mushiroda T, Newman WG, et al. CYP2D6 genotype and adjuvant tamoxifen: meta-analysis of heterogeneous study populations. Clin Pharmacol Ther. 2014;95:216–227. doi: 10.1038/clpt.2013.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, Stein EA. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease inpatients with heterozygous familial hypercholesterolemia: the reduction of LDL-C with PCSK9 inhibition in heterozygous familial hypercholesterolemia disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–2417. doi: 10.1161/CIRCULATIONAHA.112.144055. [DOI] [PubMed] [Google Scholar]
- Rampersaud E, Mitchell BD, Pollin TI, Fu M, Shen H, O'Connell JR, Ducharme JL, Hines S, Sack P, Naglieri R, Shuldiner AR, Snitker S. Physical activity and the association of common FTO gene variants with body mass index and obesity. Arch Intern Med. 2008;168:1791–1797. doi: 10.1001/archinte.168.16.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A. 2011;108:4135–4140. doi: 10.1073/pnas.1019581108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroth W, Goetz MP, Hamann U, Fasching PA, Schmidt M, Winter S, Fritz P, Simon W, Suman VJ, Ames MM, Safgren SL, Kuffel MJ, et al. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA. 2009;302:429–436. doi: 10.1001/jama.2009.1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott SA, Sangkuhl K, Stein CM, Hulot J-S, Mega JL, Roden DM, Klein TE, Sabatine MS, Johnson JA, Shuldiner AR. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin Pharmacol Ther. 2013;94:317–323. doi: 10.1038/clpt.2013.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani P, Perls TT. The genetics of extreme longevity: lessons from the new England centenarian study. Front Genet. 2012;3:277. doi: 10.3389/fgene.2012.00277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields R. Common disease: are causative alleles common or rare? PLoS Biol. 2011;9:e1001009. doi: 10.1371/journal.pbio.1001009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone NJ, Robinson J, Lichtenstein AH, Bairey Merz CN, Blum CB, Eckel RH, Goldberg AC, Gordon D, Levy D, Lloyd-Jones DM, McBride P, Schwartz JS, Shero ST, Smith SC, Jr, Watson K, Wilson PWF. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2013 doi: 10.1016/j.jacc.2013.11.002. http://circ.ahajournals.org/content/early/2013/11/11/01.cir.0000437738.63853.7a.citation. [DOI] [PubMed] [Google Scholar]
- Talmud PJ, Hingorani AD, Cooper JA, Marmot MG, Brunner EJ, Kumari M, Kivimaki M, Humphries SE. Utility of genetic and non-genetic risk factors in prediction of type 2 diabetes: Whitehall II prospective cohort study. Br Med J. 2010;340:b4838. doi: 10.1136/bmj.b4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassy JL, O'Brien KE, Waxler JL, Park ER, Delahanty LM, Florez JC, Meigs JB, Grant RW. Impact of literacy and numeracy on motivation for behavior change after diabetes genetic risk testing. Med Decis Making. 2012;32:606–615. doi: 10.1177/0272989X11431608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal M, Cusick ME, Barabási AL. Interactome networks and human disease. Cell. 2011;144:986–98. doi: 10.1016/j.cell.2011.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013;3:120144. doi: 10.1098/rsob.120144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins A. Why did the modern synthesis give short shrift to ‘soft inheritance’? In: Gissis SB, Jablonka E, editors. Transformations of Lamarckism: From Subtle Flids to Molecular Biology. USA: MIT Press; 2011. pp. 127–132. [Google Scholar]
- Young JH. Evolution of blood pressure regulation in humans. Curr Hypertens Rep. 2007;9:13–18. doi: 10.1007/s11906-007-0004-8. [DOI] [PubMed] [Google Scholar]