

Abstract
Polymorphisms in the intracellular pattern recognition receptor gene NLRP3 have been associated with susceptibility to Crohn’s disease, a type of inflammatory bowel disease (IBD). Following tissue damage or infection, NLRP3 triggers the formation of inflammasomes, containing NLRP3, ASC and caspase-1, which mediate secretion of IL-1β and IL-18. However, the precise role of NLRP3 inflammasomes in mucosal inflammation and barrier protection remains unclear. Here we show that upon infection with the attaching/effacing (A/E) intestinal pathogen Citrobacter rodentium, Nlrp3−/− and Asc−/− mice displayed increased bacterial colonization and dispersion, more severe weight loss and exacerbated intestinal inflammation. Analyses of irradiation bone marrow chimeras revealed that protection from disease was mediated through Nlrp3 activation in non-hematopoietic cells and was initiated very early after infection. Thus, early activation of Nlrp3 in intestinal epithelial cells limits pathogen colonization and prevents subsequent pathology, potentially providing a functional link between NLRP3 polymorphisms and susceptibility to IBD.
Introduction
NOD-like receptors (NLRs) are cytosolic pattern recognition receptors (PRRs) that detect a variety of pathogen associated molecular patterns (PAMPs) as well as danger associated molecular patterns (DAMPs) and signal to coordinate multiple downstream pathways.1 When a subset of NLRs are triggered they oligomerize with an adaptor protein known as ASC (Apoptosis-associated Speck-like protein containing a CARD domain), and a pro-enzyme, caspase-1 to form a multimeric protein complex known as the inflammasome.1 Inflammasomes activate caspase-1, which leads to the processing and secretion of the inflammatory cytokines IL-1β and IL-18 and to a form of inflammatory cell death termed pyroptosis.2, 3 Recently, single nucleotide polymorphisms (SNPs) in the regulatory region of NLRP3, resulting in lowered expression of the inflammasome-forming NLRP3, were linked with susceptibility to Crohn’s disease and this was attributed to decreased expression of IL-1β from monocytes.4 Moreover, several recent studies in mice reported that Nlrp3 inflammasome activation contributed to protection from chemically-induced intestinal inflammation through secretion of IL-18,5-7 although other investigators reported conflicting results.8-11
To investigate the role of the Nlrp3 inflammasome in infection-associated intestinal inflammation, we employed Citrobacter rodentium, an attaching and effacing (A/E) intestinal pathogen that is a model of enteropathogenic Escherichia coli (EPEC) infections in humans.12, 13 In order to colonize the intestinal mucosa, C. rodentium uses a type III secretion system (T3SS) to deliver effector proteins that allows attachment to epithelial cells and subversion of host signalling pathways, triggering cytoskeletal rearrangements. This results in the formation of hallmark A/E lesions on the apical epithelium, characterized by intimate bacterial attachment and effacement of the brush border microvilli 12, 13. How the innate immune system detects these virulent assaults to the epithelium and mounts an antimicrobial response towards C. rodentium is poorly characterized. In this study, we show that very early during the course of the infection, activation of Nlrp3 and Asc within non-hematopoietic cells serves to limit tissue bacterial burdens and consequently dampens intestinal inflammation.
Results
Nlrp3−/− and Asc−/− mice display exacerbated intestinal inflammation upon C. rodentium infection
To investigate the role of Nlrp3 inflammasome in bacterially-triggered intestinal inflammation, we infected cohorts of WT, Nlrp3−/− and Asc−/− mice with Citrobacter rodentium, a mouse intestinal pathogen that causes transient diarrhoea and intestinal inflammation.12, 13 It was recently reported that mice lacking a related inflammasome-forming NLR, Nlrp6−/− mice, as well as Casp1−/− and Asc−/− mice, harboured a more ‘colitogenic’ intestinal microbiota that exacerbated acute intestinal inflammation induced by dextran sulfate sodium (DSS) administration14. This ‘colitogenic’ flora and susceptibility phenotype could be transferred to WT mice by co-housing prior to DSS challenge14. Therefore, to circumvent the microbiota as a potential contributing factor to any observed differences in Nlrp3−/− and Asc−/− mice, these mice were co-housed with WT mice for at least 2 weeks prior to infection with C. rodentium. Following infection with C. rodentium, mice were either re-segregated by genotype, or were co-housed for the entire period of infection. We observed identical results irrespective of the overall period of co-housing.
Following oral infection with C. rodentium WT mice maintained their original body weight throughout the course of infection, whereas Nlrp3−/− and Asc−/− mice lost significant amounts of weight between day 7 and day 14 post-infection (p.i.), before returning to their original weight by day 21 (Figure 1A). Interestingly Nlrp3−/− mice lost less weight than Asc−/− mice after infection, suggesting the involvement of other inflammasome-forming NLRs in protective immunity against C. rodentium or inflammasome-independent, Asc-driven functions. Overall, these results indicated that signalling through Nlrp3 and Asc protected against C. rodentium-induced wasting disease.
Figure 1. Nlrp3 and Asc activation protects against C. rodentium-induced wasting disease and intestinal inflammation.

WT, Nlrp3−/−, and Asc−/− mice were infected orally with ~109 C. rodentium. Cohorts were sacrificed 8 and 14 days post-infection (p.i.) and assessed for intestinal inflammation.
(A) Body weights of WT, Nlrp3−/−, and Asc−/− mice. Symbols denote mean weights (±SEM) as a percentage of the initial body weight (n = 4-16).
(B) Representative photomicrographs depicting H&E staining of C. rodentium infected caeca (magnification x50).
(C, D) Inflammation scores in the caecum (C) and distal colon (D) were assessed as described in materials and methods (n = 9-15).
Data represent either a representative experiment (one of three independent experiments (A)) or pooled results from two to three independent experiments (C-D). Horizontal bars represent the median and each symbol represents an individual mouse. Statistical significance was determined by either the two-way ANOVA (A) or the Mann-Whitney test. (* = P < 0.05; ** = P < 0.01; *** = P < 0.001)
Consistent with the absence of systemic disease, C. rodentium infection of WT mice led only to mild intestinal inflammation with limited colonic hyperplasia, and leukocyte infiltration (Figure 1B-D). In contrast, Nlrp3−/− and Asc−/− mice developed severe typhlitis and colitis after infection with C. rodentium, characterized by crypt loss, submucosal inflammation, prominent oedema, leukocyte infiltration and increased expression of inflammatory cytokines (Figure 1B-D and Figure S1). Thus, activation of Nlrp3 and Asc limits C. rodentium-driven intestinal inflammation.
Nlrp3 and Asc activation limits C. rodentium colonization and systemic translocation
To determine if the exacerbated intestinal inflammation in Nlrp3−/− and Asc−/− mice was due to an inability to control bacterial burdens we measured C. rodentium levels at the site of infection. Fitting with exacerbated intestinal inflammation, we observed significantly higher bacterial burdens in the caecum and distal colon of Nlrp3−/− and Asc−/− mice compared to WT mice, on both day 8 and day 14 p.i. (Figure 2A and B). To examine whether Nlrp3 activation limited C. rodentium attachment to the epithelium, we visualized the infection by immunofluorescence. We observed that while C. rodentium was mainly localized to the luminal surface of the epithelium in WT mice, the bacterium was able to penetrate deep into the base of the crypts in Asc−/− and Nlrp3−/− mice (Figure 2C). This indicated that activation of Nlrp3 and Asc limited bacterial burdens and restricted their localization within the intestine.
Figure 2. Activation of the Nlrp3 and Asc limits C. rodentium burdens.

WT, Nlrp3−/−, and Asc−/− mice were infected orally with ~109 C. rodentium. Cohorts were sacrificed 8 and 14 days p.i.
(A,B) C. rodentium burdens were measured in the caecum (A) and distal colon (B) as described in materials and methods (ND, no detectable bacteria).
(C) Caeca from infected (8 days p.i.) WT, Nlrp3−/−, and Asc−/− mice were stained for C. rodentium (red), E-cadherin (green) and DAPI (blue). Bar = left panel 30μm, mid panel 15μm, right panel 70μm.
Data represent pooled results from at least two independent experiments (A, B). Horizontal bars represent the medians and each symbol represents an individual mouse (n = 10). Statistical significance was determined by the Mann-Whitney test. (* = P < 0.05; ** = P < 0.01; *** = P < 0.001)
C. rodentium is thought to cause diarrhoea partly by weakening the tight junctions between epithelial cells.15 One potential consequence of this breach is translocation of bacteria past the intestinal barrier into systemic sites. To determine the degree of this translocation we measured the C. rodentium levels in the spleen. Strikingly, while we observed no translocation of C. rodentium in WT spleens, 80% of Asc−/− mice and 50% of Nlrp3−/− mice harboured detectable numbers of C. rodentium in their spleens at 8 days p.i. (Figure 3A). In accordance with the presence of C. rodentium in the spleen, we also observed significant splenomegaly in Nlrp3−/− and Asc−/− mice, but not in WT mice (Figure 3B). Splenomegaly was associated with greater accumulation of granulocytes (CD11b+GR1high) in Nlrp3−/− and Asc−/− spleens compared to WT spleens (Figure 3C, D). These results indicate that signalling via Nlrp3 and Asc is important for limiting systemic translocation of C. rodentium, and for preventing excessive systemic inflammatory responses.
Figure 3. C. rodentium infected Nlrp3−/− and Asc−/− mice cannot restrict bacterial translocation and display systemic inflammation.

WT, Nlrp3−/−, and Asc−/− mice were orally infected with ~ 109 C. rodentium and sacrificed 8 or 14 days p.i.
(A) Splenic bacterial loads 8 days p.i.
(B) Spleen weights
(C) Representative FACS plots of splenic granulocytes (CD11b+ Gr1hi)
(D) Frequency of granulocytes in the spleen (CD11b+ Gr1hi)
Each symbol represents a single animal and data represent pooled results from at least two independent experiments (n = 9-20). Horizontal bars represent group medians. Statistical significance was determined by the Mann-Whitney test (* = P < 0.05; ** = P < 0.01; *** = P < 0.001)
Caspase-1 activation is intact in the intestines of C. rodentium-infected Nlrp3−/− mice
Triggering of the Nlrp3 inflammasome in leukocytes results in the activation of caspase-1 and subsequent maturation and secretion of the cytokines IL-1β, IL-1α and IL-18 16, 17. However, we observed no induction of IL-1β during the course of infection, nor any differences in the relative IL-1β levels between WT and Asc−/− or Nlrp3−/− mice before or during infection (Figure 4A). Similarly, levels of IL-1α were also very low and we found no difference between WT and Asc−/− or Nlrp3−/− mice (Figure 4A). By contrast, we observed almost no IL-18 production by intestinal explants from Asc−/− mice throughout the course of infection, however Nlrp3−/− mice produced slightly higher levels of IL-18 than WT mice (Figure 4A). Fitting with this pattern of IL-18 production, we found equal or greater levels of active caspase-1 (p10) and mature IL-18 in the intestines of Nlrp3−/− mice compared to WT mice at 8 days p.i., while caspase-1 p10 and mature IL-18 were barely detectable in Asc−/− mice (Figure 4B). Importantly, this result suggests that the susceptibility phenotype observed in Nlrp3−/− mice may be caspase-1-independent. IL-18 has been implicated in protecting the intestinal epithelium from DSS-induced colitis,5-7 thus defective IL-18 production in Asc−/− mice may explain the slightly exacerbated disease phenotype observed in Asc−/− mice relative to Nlrp3−/− mice. However, as IL-18 production was intact in Nlrp3−/− mice, these results indicate that the major susceptibility phenotype of Nlrp3−/− mice to C. rodentium cannot be attributed to a lack of IL-18 production. Consistent with a minor role for IL-18, when we compared Il18r1−/− and WT mice after 8 days p.i. we found only a small trend towards moderate caecal inflammation in Il18r1−/− mice that was not statistically significant (Figure 4C). However, IL-18 production did contribute to limiting bacterial burdens as Il18r1−/− mice harboured significantly higher C. rodentium levels in their caeca, compared to WT mice (Figure 4D). Furthermore, similar to Asc−/− mice, we observed translocation of C. rodentium into the spleens in around 80% of Il18r1−/− mice (Figure 4D). Collectively, these data suggest that IL-18 might explain the difference in disease severity between Nlrp3−/− and Asc−/− mice.
Figure 4. Caspase-1 activation is intact in the intestines of Nlrp3−/− mice.
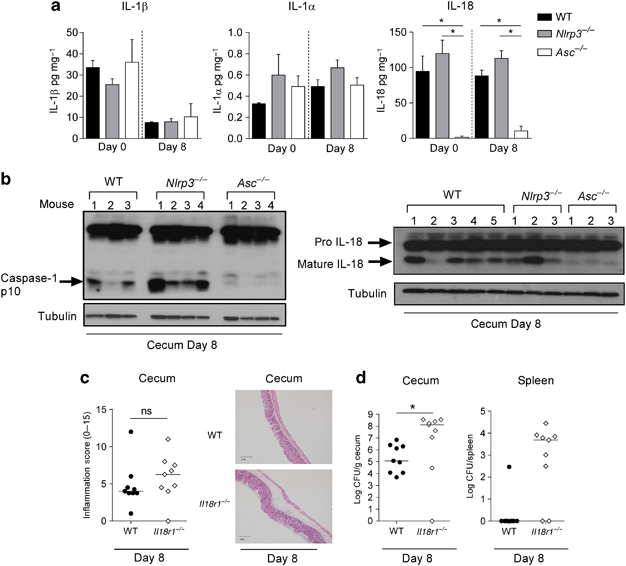
Cohorts of WT, Nlrp3−/−, Il18r1−/− and Asc−/− mice were infected with ~109 C. rodentium and sacrificed 8 days p.i.
(A) Protein levels of IL-1β, IL-1α and IL-18 in the caecal organ explants (n = 4)
(B) Immunoblot analysis of caecal protein extracts from WT, Nlrp3−/− and Asc−/− mice, probed with antibody to caspase-1, IL-18 and tubulin (n = 3-5)
(C) Inflammation scores in the caecum in WT and Il18r1−/− mice (n = 9) and representative photomicrographs depicting hematoxylin and eosin (H&E) staining of C. rodentium-infected ceca.
(D) C. rodentium burdens were measured in the caecum and spleen in WT and Il18r1−/− mice (n = 9)
Each symbol represents a single animal and is either representative of two experiments (A, B, C) or pooled from two independent experiments (D). Bar graphs represent means (±SEM). Horizontal bars represent the median. Statistical significance was determined by the Mann-Whitney test (* = P < 0.05; ** = P < 0.01; *** = P < 0.001)
Signalling via Nlrp3 and Asc in non-hematopoietic cells limits bacterial colonization and intestinal inflammation
The adaptive immune system, specifically B-cell derived IgG and Th17 cells, is crucial for protection against and clearance of C. rodentium.18-20, 21 The eventual recovery of the Nlrp3−/− and Asc−/− mice from C. rodentium suggested that essential adaptive clearance mechanisms were not significantly compromised and this was confirmed by analyses of effector Th17 cell responses at late stages of infection, which were equivalent in WT, Nlrp3−/− and Asc−/− mice (data not shown). To confirm that susceptibility of Nlrp3−/− and Asc−/− mice to C. rodentium stemmed from an inability to mount an effective innate immune response we generated Rag1−/−Nlrp3−/− and Rag1−/−Asc−/− mice, lacking mature B and T cells, and infected them with C. rodentium. After 8 days p.i., we observed that Rag1−/−Nlrp3−/− and Rag1−/−Asc−/− mice still displayed significantly higher levels of intestinal inflammation compared to Rag1−/− mice, indicating that Nlrp3 and Asc confer protection against C. rodentium independently of the adaptive immune response (Figure S2).
To determine the cell types responsible for Nlrp3/Asc-mediated protection we generated irradiation bone marrow (BM) chimeras selectively lacking Nlrp3 or Asc in distinct cellular compartments (i.e. hematopoietic or intestinal tissue cells) and infected them with C. rodentium. In agreement with our previous observations, control Asc−/−→Asc−/− and Nlrp3−/−→Nlrp3−/− BM chimeric mice displayed enhanced intestinal inflammation and weight loss compared to WT→WT mice (Figure 5A and B). Selective restoration of Nlrp3 expression in the hematopoietic compartment (WT→Nlrp3−/−) conferred no significant protection from C. rodentium-induced wasting disease, whereas mice with selective expression of Nlrp3 in the non-hematopoietic compartment (Nlrp3−/−→WT) exhibited robust protection (Figure 5A). Similarly, while mice in which Asc was selectively restored in the haematopoietic compartment (WT→Asc−/−) exhibited an intermediate phenotype, selective expression of Asc in the non-hematopoietic compartment (Asc−/−→WT) provided complete protection from weight loss (Figure 5B). An identical disease pattern was observed for intestinal pathology; mice with selective expression of Nlrp3 (Nlrp3−/−→WT) or Asc (Asc−/−→WT) in the non-hematopoietic compartment were protected from intestinal inflammation, whereas the reciprocal chimeras lacking Nlrp3 (WT→Nlrp3−/−) or Asc (WT→Asc−/−) in the non-hematopoietic compartment exhibited severe intestinal inflammation (Figure 5C and D). Consistent with a minor role for hematopoietic Asc expression, Asc−/−→WT mice had slightly more severe typhlitis than WT→WT mice, but significantly lower levels than Asc−/−→Asc−/− mice at 14 days p.i. (Figure 5D).
Figure 5. Nlrp3 and Asc signaling in non-hematopoietic cells provides protection from C. rodentium induced intestinal inflammation.

Irradiation bone marrow chimeras were generated as described in materials and methods and infected with ~109 C. rodentium and sacrificed 8 or 14 days p.i.
(A, B) Body weights of Nlrp3−/− (A) and Asc−/− (B) bone marrow chimeras. Symbols denote mean weights (±SEM) as a percentage of the initial body weight. Statistical significance was determined between chimera groups and controls i.e. Asc−/− → Asc−/− and Nlrp3−/− → Nlrp3−/− respectively.
(C, D) Inflammation scores in the caecum of Nlrp3−/− (C) and Asc−/− (D) bone marrow chimeras.
(E,F) C. rodentium loads in the caecum and distal colon of Nlrp3−/− (E) and Asc−/− (F) bone marrow chimeras 8 days p.i. (ND, no detectable bacteria).
(G) Immunoblot analysis of total caecal protein extracts from WT, Nlrp3−/− and Asc−/− chimeras 8 days p.i., probed with antibody to caspase-1 and tubulin (n = 2-4)
(H) Caeca from WT and Asc−/− mice were stained for Asc (red), E-cadherin (green) and DAPI (blue). Bar = 15μm.
Data represent pooled results from two to three independent experiments, each symbol represents a single mouse with at least 6 mice per group. Horizontal bars represent the median. Statistical significance was determined by either the two-way ANOVA (A, B) or the Mann-Whitney test (C-F). (* = P < 0.05; ** = P < 0.01; *** = P < 0.001)
Earlier, we observed that the exacerbated phenotype in Asc−/− and Nlrp3−/− mice was accompanied by higher bacterial burdens at the site of infection (Figure 2A and B). Correspondingly, BM chimeras that selectively lacked Nlrp3 (WT→Nlrp3−/−) or Asc (WT→Asc−/−) in the non-hematopoietic compartment also harboured significantly higher C. rodentium levels in the caecum and distal colon (Figure 5E and F), although there again appeared to be a minor role for Asc in hematopoietic cells (Figure 5F). To determine whether Nlrp3 or Asc-mediated protection correlated with caspase-1 activation we assessed caspase-1 cleavage. Intriguingly, caspase-1 activation (p10) was absent in the caeca of mice lacking Asc in non-haematopoietic cells (WT→Asc−/−) (Figure 5G), indicating that the majority of caspase-1 activation occurs in non-haematopoietic cells in the gut. However, caspase-1 activation was unimpaired in all the Nlrp3 chimera groups (Figure 5G), confirming our earlier conclusion that caspase-1 activation is still intact in Nlrp3−/− mice. Our earlier results suggested that Asc-dependent IL-18 production partially limited C. rodentium burdens in the caecum (Figure 4D). To assess the cellular compartment responsible for intestinal IL-18 production we performed Western blotting in tissue samples from the BM chimeras. We observed that mature IL-18 expression in the intestine correlated strongly with Asc expression in non-haematopoietic cells as mature IL-18 expression was markedly reduced in WT→Asc−/− BM chimeras, but remained intact in Asc−/−→WT BM chimeras (Figure S3). Consistent with a dominant protective role for non-hematopoietic Nlrp3 and Asc activation, immunofluorescence analysis indicated that Asc was strongly and ubiquitously expressed by E-cadherin+ IECs, whereas very few cells in the lamina propria had detectable Asc expression (Figure 5H). Taken together, our findings show that activation of Nlrp3-Asc signalling in the intestinal epithelium limits C. rodentium colonization and confers protection against intestinal inflammation.
Nlrp3 and Asc activation mediates early control against C. rodentium
We next assessed the mechanism through which Nlrp3 and Asc activation conferred protection against C. rodentium. Asc−/− and Nlrp3−/− mice harboured higher C. rodentium loads compared to WT mice thus we hypothesized that impaired control of bacterial colonization might underlie the enhanced severity of intestinal inflammation in these mice. However, some enteric pathogens such as Salmonella gain a competitive advantage under inflammatory environments that allow them to outgrow the intestinal microbiota.22, 23 Thus, to determine if the outgrowth of C. rodentium in Nlrp3−/− and Asc−/− mice was the cause or the consequence of the severe intestinal inflammation, we focused on an early time point after infection. Strikingly, we found that after just 72h p.i., Nlrp3−/− and Asc−/− mice already harboured around 1000-fold more tissue-adherent C. rodentium in their caeca than WT mice (Figure 6A), with clear evidence of penetration into the crypts (Figure 6B), even though no intestinal pathology was detectable at this early time point (Figure 6C, D). Thus, Nlrp3 and Asc activation is triggered very early in response to C. rodentium and functions to limit bacterial colonization and subsequent disease.
Figure 6. Nlrp3 and Asc activation mediates early control against C. rodentium.

Cohorts of WT, Nlrp3−/−, and Asc−/− mice were infected with ~109 C. rodentium and sacrificed 72hours p.i.
(A) C. rodentium loads in the caecum at 72h p.i. (n = 17-20)
(B) Caecal tissues from infected mice (72h p.i.) were stained for C. rodentium (red), E-cadherin (green) and DAPI (blue). Bar = left panel 20μm, right panel 10μm.
(C) Representative photomicrographs depicting H&E staining of C. rodentium infected caeca (magnification x50)
(D) Inflammation scores in the caecum at 72h p.i. (n = 6-11)
(E) Frequency of CD45+ Lin− Thy1+ Sca1+ innate lymphoid cells (ILC) of total CD45+ cells in the caecum at 72h p.i. (n = 4)
(F) mRNA expression levels of Il22, Reg3g, and Il17a at uninfected or 72h p.i. (n = 3)
Data obtained from two independent experiments. Bar graphs (E and F) shown are means (±SEM). Horizontal bars (A, D) represent the medians. Statistical significance was determined by the Mann-Whitney test (* = P < 0.05: ** = P < 0.01, *** = P < 0.001, **** = P < 0.0001)
Recent studies have identified a crucial role for IL-22-producing RORγt+ innate lymphoid cells (ILC) in early innate immunity against C. rodentium 24, 25. IL-22 mediates protection by inducing the secretion of anti-microbial peptides (AMPs) such as RegIIIγ, by IECs 26. However, we observed similar frequencies of ILCs (Figure 6E) and efficient induction of Il22, Reg3g, and Il17a in the caeca of WT, Nlrp3−/− and Asc−/− mice at 72h p.i. (Figure 6F), as well as comparable expression of several other AMPs (Figure S4). Taken together with the bone marrow chimera results showing that the protection is mediated by non-hematopoietic cells, our data indicate that the early protection mediated by Nlrp3 and Asc activation is independent of the ILC-IL-22 axis. Furthermore, consistent with our previous findings at day 8 p.i. (Figure 4A), we observed no difference in the levels of IL-1α, IL-1β, IL-18 or active caspase-1 (p10) in the intestine of WT and Nlrp3−/− mice at 72h p.i. (Figure S5), indicating that early Nlrp3 signalling in IEC protects from pathogenic infection independently of caspase-1 activation.
To further investigate the mechanism through which Nlrp3 and Asc activation promotes early protection against C. rodentium infection, we performed genome-wide transcriptional profiling of caecal tissue at steady state and at 72h post-infection. We found that the expression of 26 genes were increased in WT ceca at 72h p.i., however, the vast majority of these were also increased in Nlrp3−/− and Asc−/− ceca (Figure S6). In fact, upon infection only 5 genes were exclusively induced in WT mice but not in Nlrp3−/− or Asc−/− mice (Figure S6). Of these 5 genes the strongest candidate that could affect intestinal immune homeostasis was Ccl5, which encodes the chemokine CCL5 that has been implicated in recruitment of T cells, eosinophils and basophils into inflammatory sites.27 However, subsequent qPCR analyses were unable to validate the differential induction of Ccl5 between WT and Nlrp3−/− and Asc−/− mice (data not shown). Somewhat surprisingly, we identified 249 genes which were induced at significantly higher levels in Nlrp3−/− and/or Asc−/− caeca at 72h p.i., but not in WT caeca (Figure S6). Approximately 50% (124) of these genes were increased in both Nlrp3−/− and/or Asc−/− mice, suggesting that this expression profile may reflect responses to the higher bacterial levels present in Nlrp3 and Asc-deficient mice at 72h p.i.. Further analyses of the caecal gene expression profiles revealed that 39 probes, encompassing 33 different genes, were expressed at significantly higher levels in WT caeca compared with Nlrp3−/− and/or Asc−/− caeca (Figure S7). Strikingly, the vast majority of these 33 genes were expressed at higher levels in WT mice both at steady state and at 72h p.i. (Figure S7), suggesting that baseline differences in gene expression in the intestine of WT and Nlrp3 and Asc-deficient mice might contribute to protection from C. rodentium.
Collectively, these data show that the major protective effect of Nlrp3 and Asc activation in the intestinal epithelium is evident very early after infection with C. rodentium, is not associated with caspase-1 activation and does not seem to be mediated through the production of AMPs. Transcriptional profiling revealed differentially expressed genes between WT and Nlrp3- and Asc-deficient mice that were conserved between steady state and 72h p.i.. These observations raise the possibility that factors induced by constitutive Nlrp3 and Asc activation in the intestinal epithelium during steady state conditions may limit early infection by mucosal pathogens.
Discussion
Accumulating evidence suggests that innate immune recognition at mucosal surfaces particularly within the intestine is a crucial mediator of intestinal homeostasis.28 Apart from basal roles at steady state, PRR signalling confers protection against a multitude of enteric pathogens.29-31 Much of the work however has centred on the consequences of hematopoietic cell microbial detection while non-hematopoietic cells such as IECs have been relatively understudied. As IECs are the first cells that enteric bacteria encounter they are well poised to act as sentinels to limit invasion and alert the immune system to infection.
We first showed that mice lacking Nlrp3 exhibit exacerbated intestinal inflammation in response to infection with C. rodentium as compared to WT mice. This finding extends Nlrp3’s previously described protective role in intestinal injury induced by chemical challenges.5, 6 We also observed that Asc−/− mice exhibited an even greater disease phenotype relative to the Nlrp3−/− mice suggesting that other NLRs utilising Asc may be involved in the protective response against C. rodentium.
To investigate which cellular compartment Nlrp3 and Asc activation provided protection against C. rodentium, we created irradiation bone marrow chimeras, which revealed a major protective role for Nlrp3 and Asc activation in non-hematopoietic cells. Furthermore, using immunofluorescence microscopy, we observed that IECs ubiquitously expressed high levels of Asc, whereas very few cells in the lamina propria had detectable Asc, suggesting that IECs are the major cell type in which this protective Nlrp3 and Asc activation is occurring. C. rodentium colonizes the intestine by intimate attachment to IECs via a T3SS which delivers numerous virulence factors to the host.13 Collectively these virulence factors enable C. rodentium to disrupt host processes, such as actin polymerization, water reabsorption and epithelial tight junction maintenance, ultimately leading to intestinal inflammation and diarrhoea.15 As IECs are the first cells to come into contact with C. rodentium they are ideally positioned to detect C. rodentium and the outcomes of these virulent assaults. Consistent with this hypothesis others have reported that MyD88 and NOD2 signalling within the non-hematopoietic compartment is important for optimal protection against C. rodentium infection.31-33 Our findings reinforce the concept that innate immune recognition at the epithelial barrier has a crucial function in the initiation of protective immunity34 and extend this paradigm to include activation of NLRs in IECs.
We next ascertained the mechanism through which Nlrp3-Asc activation conferred protection. As Asc−/− and Nlrp3−/− mice consistently harboured higher C. rodentium burdens than WT mice, we asked whether this could explain the exacerbated phenotype. Since some enteric pathogens such as S. typhimurium gain a selective advantage under inflammatory conditions,22, 23 we could not be certain that higher C. rodentium levels were not merely a consequence of greater inflammation levels in Asc−/− and Nlrp3−/− mice. We thus measured the levels of C. rodentium in the tissue after 72h of infection, before the onset of intestinal inflammation, and found that Nlrp3−/− and Asc−/− mice already harboured ~1000-fold more tissue-adherent bacteria. Together, our findings show that signalling via Nlrp3 and Asc in non-hematopoietic cells early during the course of infection limits C. rodentium levels in the caecum and consequently dampens the ensuing intestinal inflammation.
Sensing of an infection at the epithelial barrier can have two foreseeable outcomes. First, activation of an effector antimicrobial response could directly limit the invading pathogen, such as the production of antimicrobial peptides. Second, recruitment and/or conditioning of other cells that subsequently clear the pathogen, for example Nod2 sensing in stromal cells triggers the production of Ccl2, attracting inflammatory monocytes which promote C. rodentium clearance.31 The first outcome would be predicted to be engaged with rapid kinetics early on in the response while the second outcome might take relatively longer. Our result showing higher pathogen burdens after 72h in Nlrp3−/− and Asc−/− mice coupled with the non-hematopoietic phenotype, suggests that an epithelial-intrinsic mechanism of action limits pathogen loads early on during infection, fitting with the predicted kinetics. Indeed, others have reported that Nlrp3−/− caecal epithelial cells show a relatively impaired ability to kill E. coli compared to WT epithelial cells.35 While none of the AMPs that we measured were deficient in Asc−/− or Nlrp3−/− mice, it is conceivable that an AMP might be regulated through a post-translational modification by an effector that is downstream of Nlrp3 and Asc activation. S100 proteins for instance are leaderless peptides and have been postulated to require caspase-1 for their release36, however, we did not find any defect in caspase-1 activation in the intestines of Nlrp3−/− mice.
Previous studies using the DSS colitis model reported a protective effect of Nlrp3 activation in non-hematopoeitic cells and described a role for IL-18 in this protection.5, 6 In addition, a recent study reported that Nlrp3 and caspase-1 played a protective role during infection with C. rodentium and ascribed this protective effect to the production of the caspase-1-processed cytokines IL-1β and IL-18.37 However, this study focussed only on the latter stages of C. rodentium infection, from day 14 p.i. onwards, and did not identify the cell types responsible for Nlrp3-mediated protection.37 Our work has revealed that activation of Nlrp3 and Asc in intestinal epithelial cells acts very early in the course of infection to restrict bacterial replication and spread, which in turn limits the severity of subsequent intestinal infection. During this early phase of infection, we did not find any induction of IL-1β and we observed equivalent IL-18 production between Nlrp3−/− and WT mice, while IL-18 production was completely abrogated in Asc−/− mice. Consistent with this expression pattern, we found that Nlrp3−/− mice expressed equivalent levels of activated caspase-1 (p10) relative to WT mice after C. rodentium infection, whereas caspase-1 cleavage was greatly diminished in Asc−/− mice. The simplest explanation for these observations is that Nlrp3 activation protects against C. rodentium-driven disease independently of caspase-1 activation. However, it is possible that the intact caspase-1 activation observed in the absence of Nlrp3 was due to compensatory activation of alternative inflammasome-forming NLRs, although this was unable to rescue the exacerbated disease phenotype. Thus, although we cannot definitively exclude a potential role for caspase-1 in Nlrp3-mediated protection, such a hypothesis would imply that discrete, non-redundant effector pathways are triggered following caspase-1 activation by distinct inflammasomes. Furthermore, although Asc and caspase-1-dependent IL-18 production might explain the difference in disease severity between Nlrp3−/− and Asc−/− mice, it cannot account for the major susceptibility phenotype observed in Asc−/− mice compared to WT mice. This also implies that other inflammasome-forming NLRs may make a minor contribution to protection from C. rodentium through the production of IL-18. Consistent with a relatively minor role for IL-18 in immunity against C. rodentium, others have also reported that Il18−/− mice display a mild phenotype in this model.38 Additionally, we observed that Il18r1−/− mice developed an intermediate phenotype after infection with C. rodentium; they harboured greater C. rodentium levels in their caeca and spleens but did not develop significantly greater intestinal inflammation compared to WT mice. Thus, the major protective effect of epithelial Nlrp3 activation appears to be independent of caspase-1. This finding has important implications for our understanding of NLR activation in distinct cell types, as it suggests that the Nlrp3-Asc signalling circuits that have been elucidated through studies in hematopoietic cells may not be similarly hard-wired in other cell types. When taken together with the findings of Liu et al37, our results suggest that there may be two phases of Nlrp3/Asc activation that contribute to protection from C. rodentium; the very early IEC-intrinsic innate pathway described in our study; and a later circuit, most likely in myeloid cells, leading to classical caspase-1 inflammasome activation and the secretion of IL-1β and IL-18 37. Evidence of the important contribution of the latter pathway is provided by the reported susceptibility phenotypes of Casp1−/− and Il1r−/− after infection with C. rodentium.37, 38
The effector pathways triggered following Nlrp3 activation in non haematopoietic cells have not been well studied, but additional proteins linked to NLR activation, such as HMGB1 released by means of unconventional protein secretion, might also contribute to protection against C. rodentium and will be investigated in the future.17, 39, 40 Furthermore, it was recently shown the activation of the related NLR, Nlrc4, triggered caspase-1-mediated release of eicosanoids, such as leukotrienes and prostaglandins, independently of IL-1β or IL-18 secretion, highlighting that selective release of distinct classes of inflammatory mediators can occur in myeloid cells after NLR activation.41 Another consequence of inflammasome activation in myeloid cells is an inflammatory form of cell death known as pyroptosis.2, 3 An attractive hypothesis is that early Nlrp3 activation in IEC might induce pyroptosis in IEC, and this ‘altruistic’ cell death could limit the replicative niche for C. rodentium. However, we found very few numbers of TUNEL+ IECs in the intestines of Nlrp3−/−, Asc−/− and WT mice after infection with C. rodentium (data not shown). Furthermore, mice lacking caspase-11, a key upstream mediator of pyroptosis, displayed similar susceptibility to C. rodentium infection compared to WT mice.42 Thus, the potential role of pyroptosis in protection against C. rodentium infection remains to be resolved.
An open question remains as to how precisely C. rodentium triggers Nlrp3 and Asc activation. Several studies have shown that C. rodentium, upon injection of effector proteins into epithelial cells, destabilize host cell mitochondria and cause apoptosis.15 Recent studies of the mechanism of Nlrp3 activation have uncovered a role for mitochondrial dysfunction in the triggering of inflammasome activation,43, 44 suggesting that T3SS delivered effector proteins could potentiate Nlrp3 activation within IECs. In contrast, a recent study showed that the C. rodentium T3SS was dispensable for activating the inflammasome in bone marrow-derived macrophages in vitro.37 Future work will address this question using a panel of virulence factor-deficient C. rodentium mutants in vivo.
Overall our study has identified an early protective circuit in the intestinal epithelium driven by signalling though Nlrp3 and Asc that provides protection against bacterial-driven intestinal inflammation. This may constitute a key function through which Nlrp3 and Asc help maintain intestinal homeostasis and could represent the dysregulated pathway in Crohn’s disease patients with SNPs in NLRP3.
Methods
Ethics statement
Animal experiments were conducted in accordance with the UK Scientific Procedures Act of 1986 under a Project License authorized by the UK Home Office Animal Procedures Committee and approved by the Sir William Dunn School of Pathology Local Ethical Review Committee.
Mice
WT C57BL/6 (B6), 129SvEv, B6.Asc−/−, B6.Il18r1−/− (Jackson, USA) and B6.Nlrp3−/− mice were bred and maintained under specific pathogen-free conditions in accredited animal facilities at the University of Oxford, UK. Mice were older than 6 weeks of ages when used and aged and sex-matched. Experimental cohorts were co-housed for at least 2 weeks prior to infection with C. rodentium. During C. rodentium infection experiments, WT, Nlrp3−/− and Asc−/− cohorts were either co-housed throughout the entire infection period, or were re-segregated into their respective genotypes and housed separately after infection. We obtained similar results irrespective of whether co-housing was maintained or discontinued after infection with C. rodentium.
Quantitative PCR
Total RNA was isolated using Qiagen RNeasy Mini Kit. RNA was reverse transcribed using Superscript III reverse transcriptase (Invitrogen) and oligo-dT primers. Gene expression was assessed using primer and probes from Applied Biosystems TaqMan® Gene Expression Assay on a Chromo4 detection system (MJ Research). Expression levels were normalized to Hprt1 and calculations were made using the 2−ΔCt method.45
Bacterial infection
A single C. rodentium colony was transferred to nalidixic acid-supplemented LB broth and grown overnight to saturation. The next day, the culture was diluted to an optical density of 0.05 and grown to log phase before harvest by centrifugation and resuspension in PBS. Mice were orally gavaged with 200μl of PBS containing ~109 C. rodentium (nalidixic acid resistant) (ICC169). Following infection, mice were weighed every day and culled if weight loss exceeded 20% of starting weight.
Colony counts of C. rodentium
To measure the C. rodentium load in infected mice, tissues were weighed and then homogenized in 600 μl of PBS. Serial dilutions of tissue lysates were plated on nalidixic acid (Sigma; final concentration 50 μg/ml) agar plates and then incubated at 37 °C overnight before counting colonies. The number of colonies were normalised to the weight of the tissue (CFU/g).
Total protein extracts and immunoblot analysis
Total protein extracts were prepared by homogenizing snap frozen caecal tissue in RIPA buffer containing a protease inhibitor cocktail (Roche). Protein levels were equalized by the Lowry assay (Biorad), resolved by SDS-PAGE and then analyzed with anti-caspase-1 p10 (Santa Cruz sc-514), anti-IL-18 (Abcam ab71495) anti-Tubulin (Santa Cruz sc5286). Visualization and imaging was then carried out using ECL solution (Pierce).
Irradiation bone marrow chimeras
Bone marrow was isolated from tibias and fibulas of either WT C57BL/6, Nlrp3−/−, or Asc−/− mice, and 10 × 106 cells were injected intravenously into γ-irradiated 11Gy, (2 × 550rad, given 4h apart) recipient mice and left for at least 6 weeks for reconstitution.
Organ explant culture
To measure cytokine production, tissues were weighed, washed in antibiotic supplemented PBS and then incubated overnight in complete RPMI at 37°C. Supernatants were harvested and the levels of IL-1α, (eBioscience, San Diego, CA; 88-5019-22), IL-1β (coating antibody: eBioscience; 14-7012-85; detection antibody: eBioscience; 13-7112-85; standard: PeproTech, Rocky Hill, NJ; 211-11b), and IL-18 (eBioscience; BMS618/2TEN) measured by enzyme-linked immunosorbent assay and IFN-γ, IL-17A, IL-22, and TNFα were quantified using Flow Cytomix Cytokine Bead Assay (Bender MedSystems).46
Assessment of intestinal inflammation
Mice were euthanized at the indicated time points during the course of infection whereupon tissue sections were cut and fixed in buffered 10% formalin. Sections were then cut and stained with haematoxylin and eosin (H&E). Sections of caecum and distal colon were then blinded and scored by two researchers. In summary, five categories were considered (each scored 0-3): epithelial hyperplasia/damage and goblet cell depletion; leukocyte infiltration in lamina propria; submucosal inflammation and edema; area of tissue affected; and markers of severe inflammation such as bleeding, crypt abscesses, and necrosis/ulceration. The sums of these five categories are presented in the figures (scored 0-15).
Isolation of leukocyte subpopulations and FACS
Cell suspensions from spleen, and the lamina propria were prepared as described previously.47 Cells were washed, incubated with anti-Fc receptor (anti CD16/32 from eBioscience) at 4°C. Cells were washed and stained for CD11b and Gr1, before being fixed overnight in at 4°C. Cells were washed twice and acquired with a Cyan (Dako) and analysis performed using FlowJo (Tree Star) software.
Gene expression analysis
Caecal RNA was extracted using the Ambion RiboPure kit (Ambion) and whole genome expression was profiled using Illumina Single Colour-Mouse WG-6_V2_0_R0_11278593 BeadChip with direct hybridization assay. Biotinylated cRNA was prepared using the Illumina TotalPrep-96 RNA Amplification Kit (#4393543 Ambion). Fluorescence emissions by Cy3 were imaged using iScan system (Illumina), and data was generated using the GenomaStudio 2011 software (Illumina). Data was imported to GeneSpring GX 12 (Agilent Technologies) for analysis.
Fluorescence microscopy
Paraformaldehyde-fixed tissue sections were deparaffinized and antigen retrieval was performed in 0.01 M sodium citrate buffer. Slides were blocked with normal goat serum and stained with rabbit anti-C. rodentium antiserum48, and mouse anti-E-cadherin (BD Bioscience), and rabbit anti-Asc (sc 22514-R) followed by an AF555-conjugated goat-anti-rabbit and an AF488-conjugated goat-anti-mouse secondary antibody (Invitrogen). Slides were mounted in DAPI-containing Vectashield (Vector Laboratories) and then visualized using an Olympus FV1000 confocal microscope.
Statistics
Statistical significance was determined by two-way ANOVA with Bonferroni post-tests for weight curves. All other statistical significance was determined either by nonparametric Mann-Whitney test or by paired t-tests. Differences were considered statistically significant when p < 0.05.
Supplementary Material
Acknowledgements
We would like to thank V. M. Dixit (Genentech Inc, USA) for providing Nlrp3−/− and Asc−/− mice; R. Stillion for histology; PSB staff for animal maintenance; High Throughput Genomics, The Wellcome Trust Centre for Human Genetics for running the microarray; and C. Schiering for advice on bioinformatic analysis. This work was supported by grants from the Wellcome Trust (to K.J.M., and G.F.), a MSD-Norman Heatley Studentship (G.X.S.), the Clarendon Fund (to G.X.S. and N.S.), and an EMBO fellowship (to J.P).
Footnotes
Disclosure The authors declare no competing financial or other conflicts of interest.
References
- 1.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 2.Miao EA, et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010;11:1136–1142. doi: 10.1038/ni.1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kayagaki N, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 4.Villani AC, et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 2009;41:71–76. doi: 10.1038/ng285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaki MH, et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity. 2010;32:379–391. doi: 10.1016/j.immuni.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dupaul-Chicoine J, et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity. 2010;32:367–378. doi: 10.1016/j.immuni.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 7.Allen IC, et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010;207:1045–1056. doi: 10.1084/jem.20100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bauer C, et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut. 2010;59:1192–1199. doi: 10.1136/gut.2009.197822. [DOI] [PubMed] [Google Scholar]
- 9.Bauer C, et al. The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-alpha mRNA expression. Dig. Dis. Sci. 2007;52:1642–1652. doi: 10.1007/s10620-007-9802-8. [DOI] [PubMed] [Google Scholar]
- 10.Siegmund B, et al. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. U. S. A. 2001;98:13249–13254. doi: 10.1073/pnas.231473998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sivakumar PV, et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut. 2002;50:812–820. doi: 10.1136/gut.50.6.812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Luperchio SA, Schauer DB. Molecular pathogenesis of Citrobacter rodentium and transmissible murine colonic hyperplasia. Microbes. Infect. 2001;3:333–340. doi: 10.1016/s1286-4579(01)01387-9. [DOI] [PubMed] [Google Scholar]
- 13.Mundy R, et al. Citrobacter rodentium of mice and man. Cell. Microbiol. 2005;7:1697–1706. doi: 10.1111/j.1462-5822.2005.00625.x. [DOI] [PubMed] [Google Scholar]
- 14.Elinav E, et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garmendia J, et al. Enteropathogenic and enterohemorrhagic Escherichia coli infections: translocation, translocation, translocation. Infect. Immun. 2005;73:2573–2585. doi: 10.1128/IAI.73.5.2573-2585.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinon F, et al. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 17.Gross O, et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity. 2012;36:388–400. doi: 10.1016/j.immuni.2012.01.018. [DOI] [PubMed] [Google Scholar]
- 18.Maaser C, et al. Clearance of Citrobacter rodentium requires B cells but not secretory immunoglobulin A (IgA) or IgM antibodies. Infect. Immun. 2004;72:3315–3324. doi: 10.1128/IAI.72.6.3315-3324.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vallance BA, et al. Mice lacking T and B lymphocytes develop transient colitis and crypt hyperplasia yet suffer impaired bacterial clearance during Citrobacter rodentium infection. Infect. Immun. 2002;70:2070–2081. doi: 10.1128/IAI.70.4.2070-2081.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torchinsky MB, et al. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature. 2009;458:78–82. doi: 10.1038/nature07781. [DOI] [PubMed] [Google Scholar]
- 21.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 22.Stecher B, et al. Salmonella enterica serovar typhimurium exploits inflammation to compete with the intestinal microbiota. PLoS. Biol. 2007;5:2177–2189. doi: 10.1371/journal.pbio.0050244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Winter SE, et al. Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature. 2010;467:426–429. doi: 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sonnenberg GF, et al. CD4(+) lymphoid tissue-inducer cells promote innate immunity in the gut. Immunity. 2011;34:122–134. doi: 10.1016/j.immuni.2010.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tumanov AV, et al. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell. Host. Microbe. 2011;10:44–53. doi: 10.1016/j.chom.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zheng Y, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008;14:282–289. doi: 10.1038/nm1720. [DOI] [PubMed] [Google Scholar]
- 27.D’Ambrosio D, et al. Chemokine receptors in inflammation: an overview. J. Immunol. Methods. 2003;273:3–13. doi: 10.1016/s0022-1759(02)00414-3. [DOI] [PubMed] [Google Scholar]
- 28.Abreu MT. Toll-like receptor signalling in the intestinal epithelium: how bacterial recognition shapes intestinal function. Nat. Rev. Immunol. 2010;10:131–144. doi: 10.1038/nri2707. [DOI] [PubMed] [Google Scholar]
- 29.Muller AJ, et al. The S. Typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell. Host. Microbe. 2009;6:125–136. doi: 10.1016/j.chom.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 30.Jarchum I, et al. Critical Role for MyD88-Mediated Neutrophil Recruitment during Clostridium difficile Colitis. Infect. Immun. 2012;80:2989–2996. doi: 10.1128/IAI.00448-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim YG, et al. The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity. 2011;34:769–780. doi: 10.1016/j.immuni.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson DL, et al. MyD88 signalling plays a critical role in host defence by controlling pathogen burden and promoting epithelial cell homeostasis during Citrobacter rodentium-induced colitis. Cell. Microbiol. 2008;10:618–631. doi: 10.1111/j.1462-5822.2007.01071.x. [DOI] [PubMed] [Google Scholar]
- 33.Lebeis SL, et al. TLR signaling mediated by MyD88 is required for a protective innate immune response by neutrophils to Citrobacter rodentium. J. Immunol. 2007;179:566–577. doi: 10.4049/jimmunol.179.1.566. [DOI] [PubMed] [Google Scholar]
- 34.Fritz JH, et al. Innate immune recognition at the epithelial barrier drives adaptive immunity: APCs take the back seat. Trends. Immunol. 2008;29:41–49. doi: 10.1016/j.it.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 35.Hirota SA, et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel. Dis. 2010;17:1359–1372. doi: 10.1002/ibd.21478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamkanfi M. Emerging inflammasome effector mechanisms. Nat. Rev. Immunol. 2011;11:213–220. doi: 10.1038/nri2936. [DOI] [PubMed] [Google Scholar]
- 37.Liu Z, et al. Role of Inflammasomes in Host Defense against Citrobacter rodentium Infection. J. Biol. Chem. 2012;287:16955–16964. doi: 10.1074/jbc.M112.358705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lebeis SL, et al. Interleukin-1 receptor signaling protects mice from lethal intestinal damage caused by the attaching and effacing pathogen Citrobacter rodentium. Infect. Immun. 2009;77:604–614. doi: 10.1128/IAI.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Keller M, et al. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 40.Lamkanfi M, et al. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010;185:4385–4392. doi: 10.4049/jimmunol.1000803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.von Moltke J, et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature. 2012;490:107–111. doi: 10.1038/nature11351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurung P, et al. TRIF-mediated caspase-11 production integrates TLR4- and Nlrp3 inflammasome-mediated host defense against enteropathogens. J. Biol. Chem. 2012;287:34474–34483. doi: 10.1074/jbc.M112.401406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou R, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 44.Shimada K, et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity. 2012;36:401–414. doi: 10.1016/j.immuni.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Boulard O, et al. TLR2-independent induction and regulation of chronic intestinal inflammation. Eur. J. Immunol. 2010;40:516–524. doi: 10.1002/eji.200939669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahern PP, et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity. 2010;33:279–288. doi: 10.1016/j.immuni.2010.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Uhlig HH, et al. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 2006;177:5852–5860. doi: 10.4049/jimmunol.177.9.5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mundy R, et al. Comparison of colonization dynamics and pathology of mice infected with enteropathogenic Escherichia coli, enterohaemorrhagic E. coli and Citrobacter rodentium. FEMS. Microbiol. Lett. 2006;265:126–132. doi: 10.1111/j.1574-6968.2006.00481.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.